
Abstract
Purpose
The aim of this study was to assess the performance of a noninvasive prenatal screening (NIPS) assay for accurate fetal genotyping of pregnancies at genetic risk for autosomal recessive nonsyndromic hearing loss (ARNSHL).
Methods
A total of 80 pregnant couples carrying known mutations in either the GJB2 or SLC26A4 genes associated with a risk for ARNSHL were recruited to the study. Fetal amniocyte samples were genotyped by invasive prenatal screening (IPS), whereas the cell-free fetal DNA present in maternal plasma samples was genotyped using a novel NIPS method based on circulating single-molecule amplification and resequencing technology (cSMART).
Results
IPS of the 80 at-risk pregnancies identified 20 normal homozygote, 42 heterozygote, 5 affected homozygote, and 13 affected compound heterozygote fetuses. Benchmarking against IPS, 73 of 80 fetuses (91.3%) were correctly genotyped by the cSMART NIPS assay. A low fetal DNA fraction (<6%) was identified as the main contributing factor in five of seven discordant NIPS results. At fetal DNA fractions >6%, the sensitivity and specificity of the cSMART assay for correctly diagnosing ARNSHL were 100 and 96.5%, respectively.
Conclusion
Based on key performance indicators, the cSMART NIPS assay has clinical potential as an alternative to traditional IPS of ARNSHL.
Similar content being viewed by others
Introduction
Prelingual hearing loss is a common sensory disorder, affecting approximately 1 in every 1,000 infants.1 In around 50% of individuals diagnosed with severe to profound hearing loss, a genetic cause is identifiable.2 Of the different inheritance modes, autosomal recessive nonsyndromic hearing loss (ARNSHL) accounts for the vast majority of hereditary hearing loss.3 Extensive carrier population testing and family pedigree DNA studies across different ethnicities have identified over 700 mutations in 42 genes associated with ARNSHL.2, 4 In the Chinese population, the most frequent pathogenic ARNSHL mutations reside in the gap junction beta 2 (GJB2) and the solute carrier family member 4 (SLC26A4) genes.5, 6 Invasive prenatal screening (IPS) by amniocentesis and molecular genotyping is commonly undertaken by Chinese couples who are known carriers of pathogenic ARNSHL mutations to determine the genetic status of their fetus.7
As an alternative to IPS for single-gene diseases, which poses a small but significant risk to the mother and fetus, a major goal of research is to develop noninvasive prenatal screening (NIPS) methods.8 Recently, for autosomal recessive Wilson disease, we reported a fetal genotyping strategy using circulating single-molecule amplification and resequencing technology (cSMART) which barcodes and quantitates the percentage of fetal mutation alleles in plasma relative to the background levels of the maternal alleles.9 In a subsequent study, we developed a prototype cSMART assay for ARNSHL and in validation studies of 25 pregnancies showed that it was reliable and accurate for identifying the correct paternally and maternally inherited GJB2 or SLC26A4 fetal alleles.10
In order to develop a comprehensive and accurate NIPS assay for determining fetal ARNSHL genotypes in at-risk pregnancies, we modified the prototype assay10 by incorporating several new diagnostic features such as expansion and redesign of the targeting primers to cover all hotspot GJB2 and SLC26A4 mutations and the addition of primers for genome-wide single-nucleotide polymorphisms (SNPs) to measure fetal DNA fraction.11 In this study, we applied this second-generation NIPS cSMART assay to maternal plasma samples from 80 pregnancies at genetic risk for ARNSHL and blindly evaluated its performance against gold-standard IPS of the fetal amniocytes collected from the same pregnancies.
Materials and methods
Study design
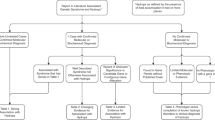
The clinical research study (Figure 1) was approved by the ethics committee of the Chinese People’s Liberation Army (PLA) General Hospital (approval number S2016-120-02), and couples provided written informed consent for participation. The final study cohort comprised 80 couples at risk of having a child with ARNSHL. In the second trimester (average gestational age of 18 weeks, range 16–23 weeks), the established fetuses from these 80 pregnancies were genotyped by IPS and NIPS for pathogenic parental GJB2 and SLC26A4 mutations to determine the overall diagnostic performance of the cSMART ARNSHL assay.

Study design. ARNSHL, autosomal recessive nonsyndromic hearing loss; IPS, invasive prenatal screening; cSMART, circulating single-molecule amplification and resequencing technology; NIPS, noninvasive prenatal screening.
Genotyping family samples for GJB2 and SLC26A4 mutations
Genomic DNA was prepared from parental blood and fetal amniocyte samples using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). For ARNSHL genotyping of the trios (maternal, paternal, and proband DNA), exon/intronic regions containing the pathogenic GJB2 and SLC26A4 mutations (Figure 2) were targeted with specific primers and amplified by PCR. Purified amplicons were subjected to Sanger sequencing using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and analysis performed on the ABI 3,130 Genetic analyzer using Chromas 2.4.1 software. Genotypes were assigned based on consensus mutations identified by sequencing both the sense and the antisense strands.

Common pathogenic mutations in the GJB2 and SLC26A4 genes. Primer pairs designed for the inverse PCR step of the cSMART assay are indicated by red and blue arrows.
cSMART assay for detecting GJB2 and SLC26A4 mutations in maternal cell-free DNA
A previous cSMART assay10 was redesigned to analyze a wider range of pathogenic GJB2 and SLC26A4 mutations in the Chinese population (Figure 2). For the GJB2 gene, 7 pairs of inverse primers were designed to target 12 exon 2 hotspot mutations.12 Similarly, for the SLC26A4 gene, 27 sets of inverse primers were required to target 22 exonic and 5 intronic mutations. In addition, as reported in previous validation studies,11 we incorporated a further 76 primer sets into the multiplex to amplify genome-wide SNPs for the purpose of simultaneously measuring the fetal DNA fraction in the same assay. Thus, the design of the cSMART diagnostic assay for ARNSHL genotyping and measurement of fetal DNA fraction was based on a multiplex of 110 targeting primer pairs.
Blood samples were collected in Streck tubes (Streck, Omaha, NE, USA) and cell-free DNA (cfDNA) isolated using the QIAamp Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany). The cSMART assay was then performed as previously described10 with minor modifications designed to increase the efficiency of mutation detection and thus the number of allelic sequencing reads for statistical analysis. First, the volume of starting plasma amount was increased twofold (from 1 to 2 ml), and the total cfDNA extracted from each plasma sample used as the input template for the assay.11 For the 80 pregnancy plasma samples, the median input cfDNA into the assay was 28.9 ng with a range of 10.3–62.9 ng. Second, where possible, the targeting inverse primer pairs were strategically located 1–5 nucleotides from the mutation sites to maximize the number of plasma DNA templates targeted and amplified (Figure 2). Third, we used the high-efficiency Phusion High-Fidelity DNA Polymerase (New England Biolabs, Ipswich, UK) to increase the yield of targeted alleles in the inverse PCR step. From receipt of sample to reporting the fetal genotype, the turnaround time of the ARNSHL cSMART assay was 2.5 days involving cfDNA extraction (1.5 h) and library preparation (9.0 h) on day 1, paired end sequencing on the Illumina MiniSeq platform (17 h) on day 2, and data analysis (0.5 h) and reporting (0.5 h) on the morning of day 3.
Analysis of cSMART data and assignment of fetal GJB2 and SLC26A4 genotypes
Following targeted sequencing, paired end reads (2 × 150 bp) were reconstructed using the FAST-Q program13 and allelic reads with sequences encompassing the 12 GJB2 and 27 SLC26A4 mutation sites retrieved for analysis. For quality control, sequences were aligned to the hg19 reference genome and filtered to remove low quality reads and any reads with sequencing errors. Duplicate and higher order reads with identical end start and stop positions were identified by their unique barcode signature and removed from the data set to reduce bias by counting the read only once. PCR failure of the 110 multiplex primer pairs was routinely monitored to avoid false negatives by checking that sufficient reads were generated for the 39 targeted alleles of the GJB2 and SLC26A4 genes and the 76 genome-wide SNPs. The maternal plasma mutation percentages for each GJB2 and SLC26A4 mutation were finally calculated by counting the uniquely barcoded allelic molecules (average 1,289 reads; range of 330–3,182 reads per mutation site). In addition, the fetal allelic fraction (FAF), which is equivalent to half the fetal DNA fraction (FF), was also calculated from the plasma allelic ratios determined from the subset of informative genome-wide SNPs and expressed as the median value, as previously described.11
For designating fetal GJB2 and SLC26A4 genotypes, we used standard nomenclature where A represents the normal or wild-type allele and a represents the mutant allele. We further used subscripts 1 and 2 to refer to different allelic variants within the same gene. Fetal genotypes were assigned according to the change in the percentage of the background maternal plasma mutations (a1 and/or a2) due to the specific inheritance patterns of the fetal alleles, as follows:
For carrier couples with the same maternal (A1a1) and paternal genotypes (A1a1);
If the fetus inherits both A1 alleles: the a1 allelic % in maternal plasma = 50% - FAF%
If the fetus inherits an A1 and an a1allele: the a1 allelic % in maternal plasma = 50%
If the fetus inherits both a1 alleles: the a1 allelic % in maternal plasma = 50% + FAF%
For carrier couples with different maternal (A1a1) and paternal (A2a2) genotypes;
If fetus inherits the maternal A1allele: the a1 allele % in maternal plasma=50% - FAF%.
If the fetus inherits the maternal a1allele: the a1 allele % in maternal plasma = 50%.
If the fetus inherits the paternal A2 allele: the a2 allele % in maternal plasma = 0%.
If the fetus inherits the paternal a2allele: the a2allelic % in maternal plasma = 0% + FAF%.
Results
GJB2 and SLC26A4 genotyping of family trios
A total of 80 carrier couples at risk of having a child with ARNSHL (Figure 1) and who had achieved a pregnancy were selected for the study. Genotyping for pathogenic variants in the GJB2 and SLC26A4 genes (Figure 2) identified 25 couples as heterozygous carriers of the same risk mutation A (maternal A1a1/paternal A1a1) and 55 couples as heterozygous carriers of two different risk mutations A and B in the same gene (maternal A1a1/paternal A2a2) (Table 1). IPS of these pregnancies identified 20 normal homozygotes (A1A1 or A1A2), 42 heterozygous carriers (A1a1, A1a2 or a1A2), 5 affected homozygotes (a1a1), and 13 affected compound heterozygotes (a1a2). Refer to Supplementary Table S1 online for full details of the paternal, maternal, and fetal GJB2/SLC26A4 genotypes.
Performance of NIPS for fetal GJB2 and SLC26A4 genotyping



Matching archived plasma samples from 80 pregnancies were coded and NIPS performed in an independent laboratory using the cSMART 110 SNP multiplex assay. After decoding the NIPS samples, the determined mutation ratios and FAF were used to deduce the fetal GJB2 and SLC26A4 genotypes (Figure 3a, Supplementary Table S2 for all NIPS results). Overall, 73 of the 80 (91.3%) fetal genotypes assigned by NIPS were concordant with IPS (Table 1). There was a 100% concordance rate of NIPS for correct prediction of the A1A1, A1a1, a1a1, and A1a2 fetal genotypes. In contrast, there was a significantly lower concordance rate of NIPS for correct prediction of the A1A2 (94.1%), a1A2 (78.6%), and a1a2 genotypes (76.9%). Concordant cSMART results for one representative example of each of the seven different possible modes of fetal inheritance (A1A1, A1a1, a1a1, A1A2, A1a2, a1A2, and a1a2), are shown in Table 2.

NIPS results for 80 couples at genetic risk for autosomal recessive nonsyndromic hearing loss. Plots of mutation percentages in plasma vs. patient sample number. (a) A1a1/ A1a1 carrier couples (n=25). Red squares denote a GJB2 mutation, red circles (•) denote a SLC26A4 mutation. (b) A1a1/A2a2 carrier couples (n=55). Red and blue squares denote the maternally and paternally inherited GJB2 alleles, respectively. Red (•) and blue (•) circles denote the maternally and paternally inherited SLC26A4 alleles, respectively. Black horizontal bars show the FAF % (0.5 × FF) and samples with a low FF (<6%) are indicated by asterisks. The observed (NIPS) genotypes deduced for each plasma sample and the expected (IPS) fetal genotypes are shown in boxes. Gray and red shading of the boxes indicates concordance and discordance between NIPS and IPS, respectively. IPS, invasive prenatal screening; NIPS, noninvasive prenatal screening.
Analysis of NIPS discordant fetal genotypes
We further investigated the seven discordant ARNSHL genotypes assigned by NIPS to identify possible contributing factors (Table 2). All seven discordant results were exclusively associated with couples in whom the maternal and paternal risk mutations were different. In all cases, there were no specific types of mutations (point, deletion, insertion) or combinations of mutations associated with discordance. Further, the cfDNA amounts used as the input starting template for the cSMART assay were close to the median cfDNA value of 28.9 ng for the entire set of 80 samples, indicating that low cfDNA was an unlikely confounding factor. However, we identified a trend of low FF with discordant NIPS results.
Five of the seven discordant samples (K050, K052, K074, K078, and K097) all had a low FF of <6% (Table 2). For samples K052 (a1A2) and K078 (A1A2), there was a slightly biased overrepresentation of the percentage of the maternally inherited mutant allele by NIPS, leading to incorrect fetal genotype calls of a1a1 and a1A2, respectively (Figure 3b). In contrast, for samples K050, K074, and K097 with a1a2 genotypes, there was a slightly biased underrepresentation of the percentage of the maternally inherited normal allele by NIPS, leading to incorrect fetal genotype calls of A1a2 in all cases (Figure 3b). These biases were observed exclusively in the percentage measurement of the maternally inherited alleles. The remaining two plasma samples, K005 and K095, had moderate FFs of 8.21 and 9.75%, respectively (Table 2). In both cases, the measured percentages of the maternally inherited fetal alleles were significantly overrepresented, indicating an a1a1 genotype instead of the expected a1A2 genotype. To exclude the possibility of a misdiagnosis by IPS or a sample mix-up, we independently resequenced the original archived amniocyte DNA. Sanger sequencing again confirmed the original IPS result of a heterozygous carrier status of the maternally inherited allele, indicating a misdiagnosis by NIPS.
Sensitivity and specificity of cSMART NIPS assay for ARNSHL
The overall performance of NIPS was assessed for sensitivity and specificity of disease detection, benchmarking against IPS-assigned genotypes a1a1 and a1a2 as true ARNSHL positives and genotypes A1A1, A1A2, A1a1, A1a2, A1a2 as true ARNSHL negatives (Figure 3a). For the 68 plasma samples with moderate to high FF (>6%), of the expected 13 positives and 55 negatives, there were no false negatives and two false positives (K005, K095) giving a sensitivity of 100% and a specificity of 96.5%, respectively. In contrast, for the 12 plasma samples with low FAF (<6%), of the expected 5 positives and 7 negatives, there were 3 false negatives (K050, K074, K097) and 1 false positive (K052) giving a significantly lower sensitivity and specificity of 62.5 and 87.5%, respectively.
Discussion
In a blind study benchmarking against gold-standard IPS, we evaluated the diagnostic performance of a novel cSMART assay for NIPS of 80 pregnancies at genetic risk for ARNSHL. The multiplex cSMART assay correctly genotyped 73 of 80 fetuses with a diagnostic concordance rate of 91.3%. The main cause identified for the seven discordant fetal genotypes was a low FF (<6%), which significantly skewed the maternally inherited allele percentage, resulting in an incorrect call of the fetal genotype. Overall, for plasma samples with moderate to high FF (>6%), the NIPS assay was highly accurate for detection of fetuses affected with ARNSHL, with a sensitivity of 100% and a specificity of 96.5%. However, at low FF (<6%), the assay performed poorly, with a low sensitivity of 62.5% and a moderate specificity of 87.5%.
Based on the performance of NIPS demonstrated in this study, the cSMART ARNSHL assay may have potential clinical utility as an alternative to IPS for some couples. Nonetheless, the test would still need to be offered using strict clinical guidelines to ensure maximum accuracy of the diagnosis. First, the pathogenic parental mutations must be known so that the final genotype can be interpreted from the cSMART results, taking into consideration the possible allelic inheritance patterns. Second, as FF generally increases by 1% per gestational week,14 the test would best be offered in the second trimester to ensure that the vast majority of samples have an adequate FF for accurate fetal genotyping. As observed in this study, 12 of the 80 women (15%) fortuitously had low FFs (between 5 and 6%). Thus, in such cases, the couple could elect to have the fetus retested by NIPS at a more advanced gestational age, where the FF is expected to increase.15, 16 Third, for all fetuses diagnosed as affected with ARNSHL, couples should be offered confirmatory IPS, which is currently the recommended procedure for confirmation of fetal aneuploidies detected by NIPS.17
For universal clinical application of the technology, additional modifications to the design of the cSMART ARNSHL assay are still needed to further increase the accuracy of fetal genotyping. The main limitation of the cSMART assay remains the total reliance on the accurate measurement of the maternally inherited allele and any significant bias in the measured mutation percentage could potentially lead to an incorrect genotype. One possible solution is to further increase the starting volume of plasma from 2 to 4 ml, to provide a twofold increase in mutation reads for even higher statistical power and reduce the impact of allelic biases. Another approach to increase the accuracy of fetal genotyping would be to substitute the 76-genome-wide SNPs with common population SNPs located within and around the GJB212, 18, 19 and SLC26A412, 20 genes. This would still provide an accurate measurement of FF, but also if the intragenic and intergenic SNPs were linked to the mutations, then this would provide additional confirmation of the fetal genotype predicted by the mutation SNPs. On similar principles, it would also be possible to design reliable and accurate cSMART assays for other significant causes of hereditary hearing loss, including autosomal dominant21 and X-linked22 NSHL, by targeting hotspot mutations in common risk alleles in the population.
In conclusion, prenatal diagnosis is an important step for couples with an established pregnancy at risk for ARNSHL to determine at an early stage whether their fetus is affected with a sensory disability, allowing the couple to make an informed decision on pregnancy termination. If the couple decide to continue the pregnancy, their clinician will be much better informed to manage and treat the condition from birth.23, 24, 25 For this goal, the availability of a reliable and accurate NIPS genotyping method would provide a more convenient prenatal option and reduce risks posed to the mother and fetus by invasive test procedures. While this second-generation cSMART assay demonstrated a high performance for NIPS of ARNSHL in this study of 80 pregnancies, further incremental improvements to increase the accuracy of fetal genotyping are warranted before the assay can be considered for clinical implementation as a stand-alone diagnostic test.
References
Morton CC, Nance WE . Newborn hearing screening—a silent revolution. N Engl J Med 2006;354:2151–2164.
Duman D, Tekin M . Autosomal recessive nonsyndromic deafness genes: a review. Front Biosci (Landmark Ed) 2012;17:2213–2236.
Petersen MB, Willems PJ . Non-syndromic, autosomal-recessive deafness. Clin Genet 2006;69:371–392.
Yan D, Tekin D, Bademci G et al, Spectrum of DNA variants for non-syndromic deafness in a large cohort from multiple continents. Hum Genet 2016;135:953–961.
Dai P, Yu F, Han B et al, GJB2 mutation spectrum in 2063 Chinese patients with nonsyndromic hearing impairment. J Trans Med 2009;7:26.
Ouyang XM, Yan D, Yuan HJ et al, The genetic bases for non-syndromic hearing loss among Chinese. J Hum Genet 2009;54:131–140.
Han MY, Lu YP, Bian XM et al, Prenatal genetic test and clinical guidance for 213 hereditary deaf families. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 2012;47:127–131.
Liu L, Li K, Fu X, Chung C, Zhang K . A forward look at noninvasive prenatal testing. Trends Mol Med 2016;22:958–968.
Lv W, Wei X, Guo R et al, Noninvasive prenatal testing for Wilson disease by use of circulating single-molecule amplification and resequencing technology (cSMART). Clin Chem 2015;61:172–181.
Chen Y, Liu Y, Wang B et al, Development and validation of a fetal genotyping assay with potential for noninvasive prenatal diagnosis of hereditary hearing loss. Prenat Diagn 2016;36:1233–1241.
Song Y, Zhou X, Huang S et al, Quantitation of fetal DNA fraction in maternal plasma using single molecule amplification and re-sequencing technology (cSMART). Clin Chim Acta 2016;456:151–156.
Yuan Y, You Y, Huang D et al, Comprehensive molecular etiology analysis of nonsyndromic hearing impairment from typical areas in China. J Transl Med 2009;7:79.
Aronesty E . Comparison of sequencing utility programs. Open Bioinformatics J 2013;7:1–8.
Canick JA, Palomaki GE, Kloza EM, Lambert-Messerlian GM, Haddow JE . The impact of maternal plasma DNA fetal fraction on next generation tests for common fetal aneuploidies. Prenat Diag 2013;33:667–676.
Shi X, Zhang Z, Cram DS, Liu C . Feasibility of noninvasive prenatal testing for common fetal aneuploidies in an early gestational window. Clin Chim Acta 2015;439:24–28.
Song Y, Huang S, Zhou X et al, Non-invasive prenatal testing for fetal aneuploidies in the first trimester of pregnancy. Ultrasound Obstet Gynecol 2015;45:55–60.
Gregg AR, Skotko BG, Benkendorf JL et al, Noninvasive prenatal screening for fetal aneuploidy, 2016 update: a position statement of the American College of Medical Genetics and Genomics. Genet Med 2016;18:1056–1065.
Cheng HB, Chen ZB, Wei QJ, Lu YJ, Xing GQ, Cao X . Single nucleotide polymorphisms and haplotypes analysis of DFNB1 locus in Chinese sporadic hearing impairment population. Chin Med J (Engl) 2009;122:1549–1553.
Grillo AP, de Oliveira FM, de Carvalho GQ et al, Single nucleotide polymorphisms of the GJB2 and GJB6 genes are associated with autosomal recessive nonsyndromic hearing loss. Biomed Res Int 2015;2015:318727.
Wu CC, Lu YC, Chen PJ et al, Phenotypic analyses and mutation screening of the SLC26A4 and FOXI1 genes in 101 Taiwanese families with bilateral nonsyndromic enlarged vestibular aqueduct (DFNB4) or Pendred syndrome. Audiol Neurootol 2010;15:57–66.
Peterson MB . Non-syndromic autosomal–dominant deafness. Clin Genet 2002;62:1–13.
Peterson MB, Wang Q, Willems PJ . Sex-linked deafness. Clin Genet 2008;73:14–23.
Han B, Dai P, Qi QW et al, Prenatal diagnosis for hereditary deaf families assisted by genetic testing. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 2007;42:660–663.
Fu S, Dong J, Wang C, Chen G . Parental attitudes toward genetic testing for prelingual deafness in China. Int J Pediatr Otorhinolaryngol 2010;74:1122–1125.
Fu X, Cai Y, Hu Y, Liu J, Yang T . Attitudes toward carrier screening and prenatal diagnosis for recessive hereditary deafness among the educated population in urban China. Am J Med Genet A 2016;170:3180–3184.
Acknowledgements
We thank Min Dong, Xin Zhang, and Xiaofei Yan for their help with conducting the study. We also thank Yin Wang for his assistance in coordinating the project and the statistical analysis of the data. This work was supported by the grants from the Project of the National Natural Science Foundation of China (81230020, 81200751, 81371098, 81371096, and 81570929), Natural Science Foundation of Hainan Province (817345), China Postdoctoral Science Foundation (2013T60947), National Basic Research Program of China (973 Program) (2014CB541706 and 2014CB541701), and the Reproductive Health and Serious Birth Defect Prevention Research Project (National Key Research Project, 2016YFC1000700, 2016YFC1000704, and 2016YFC1000706).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
Z.L., W.W., L.L., Y.L., M.X., and D.S.C. are employees of Berry Genomics Corporation. None of the authors holds any stocks or bonds in the company. The other authors declare no conflict of interest.
Additional information
Supplementary material is linked to the online version of the paper at
Supplementary information
Rights and permissions
About this article

Cite this article
Han, M., Li, Z., Wang, W. et al. A quantitative cSMART assay for noninvasive prenatal screening of autosomal recessive nonsyndromic hearing loss caused by GJB2 and SLC26A4 mutations. Genet Med 19, 1309–1316 (2017). https://doi.org/10.1038/gim.2017.54
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2017.54
Keywords
This article is cited by
-
Noninvasive prenatal diagnosis (NIPD) of non-syndromic hearing loss (NSHL) for singleton and twin pregnancies in the first trimester
Orphanet Journal of Rare Diseases (2025)
-
Preimplantation genetic testing for hereditary hearing loss in Chinese population
Journal of Assisted Reproduction and Genetics (2023)
-
Clinical evaluation of non-invasive prenatal screening for the detection of fetal genome-wide copy number variants
Orphanet Journal of Rare Diseases (2022)
-
Insights into phenotypic differences between humans and mice with p.T721M and other C-terminal variants of the SLC26A4 gene
Scientific Reports (2021)
-
One-step noninvasive prenatal testing (NIPT) for autosomal recessive homozygous point mutations using digital PCR
Scientific Reports (2018)
