
Abstract
Chimeric Antigen Receptor (CAR)-T-cell therapy has revolutionized cancer immune therapy. However, challenges remain including increasing efficacy, reducing adverse events and increasing accessibility. Use of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) technology can effectively perform various functions such as precise integration, multi-gene editing, and genome-wide functional regulation. Additionally, CRISPR screening using large-scale guide RNA (gRNA) genetic perturbation provides an unbiased approach to understanding mechanisms underlying anti-cancer efficacy of CAR T-cells. Several emerging CRISPR tools with high specificity, controllability and efficiency are useful to modify CAR T-cells and identify new targets. In this review we summarize potential uses of the CRISPR system to improve results of CAR T-cells therapy including optimizing efficacy and safety and, developing universal CAR T-cells. We discuss challenges facing CRISPR gene editing and propose solutions highlighting future research directions in CAR T-cell therapy.
Similar content being viewed by others


Introduction
Chimeric antigen receptor (CAR)-T-cell therapy showed significant potential in certain hematological malignancies, solid tumors, as well as autoimmune and infectious diseases, particularly in B-cell malignancies [1]. To date, six CAR T-cell clinical products have been approved by the U.S. Food and Drug Administration (FDA) [2,3,4,5,6,7]. Despite the impressive clinical efficacy of CAR T-cell therapy, several obstacles remain: (i) The manufacturing process for autologous CAR T-cell is complex and individualized, leading to high production costs and limited accessibility, thereby hindering large-scale production [8]. (ii) Some patients still resistant to CAR T-cell therapy or experience relapse following treatment, resulting in limited efficacy [9]. (iii) CAR T-cell therapy is associated with side effects including cytokine release syndrome (CRS), immune effector cell-associated neurotoxicity syndrome (ICANS), and on-target off-tumor (OTOT) toxicity, which raise safety concerns [10]. Therefore, enhancing the efficacy, safety and accessibility of CAR T-cell therapy are crucial areas that require further improvement to expand its application.
The traditional protein engineering of Zinc Finger Nuclease (ZFN) and Transcription Activator-Like Effector Nucleases (TALENs) requires a laborious design, construction, and testing cycle to determine the amino acid substitutions that selectively bind to the desired genomic sequences. However, the simplicity and predictability of the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) gene editing technology transformed gene editing from a complex protein engineering problem to an RNA coding problem, making CRISPR an attractive tool for both basic research and clinical applications immediately [11, 12] (Fig. 1). Modified CRISPR tools can be applied to various directions including gene regulation, epigenetic regulation, and base editing [13] (Fig. 1). Moreover, benefit from flexibility and efficiency, high throughput CRISPR screening enables for large-scale specific gene perturbation and has grown to be an essential strategy for biological discovery. Given these capabilities and advantages of CRISPR system, incorporating this technology into CAR T-cell therapy hold the potential to increase the efficacy, safety, and accessibility of CAR T-cells in tumor treatment. In this review, we discuss the latest advances in utilizing CRISPR system for optimizing the effectiveness and safety of CAR T-cell therapy, manufacturing of universal CAR T (UCAR T)-cells, and investigating its clinical applications. We also highlight the role of CRISPR screening as a tool for target discovery in the study of CAR T-cell therapy biology. Finally, we address the challenges and corresponding advancements of CRISPR system in CAR T-cell therapy, providing potential directions for future research.

A The mechanism and advantage of CRISPR-Cas9. In CRISPR-Cas9 system, the Cas9 nuclease is guided by a sgRNA and directed to the desired target sites. When Cas9 hybridizes to the target site, it generates DSBs, then the DSB was repaired through endogenous pathways including non-homologous end-joining (NHEJ) and homology-directed repair (HDR). ZFNs and TALENs rely on protein domains to recognize target sequences, which are built by assembling amino acid modules. B CRISPR tools within CAR T-therapy. CRISPR interference (CRISPRi) or CRISPR activation (CRISPRa) are consist of dCas9 fused with transcriptional repressors or activators, respectively. The epigenome-modifying enzyme enables the epigenetic regulation. Base editors are typically a fusion protein combining a nCas nickase with a deaminase ___domain, which catalyses the substitution of a single nucleotide at the PAM strand in the R-loop. Prime editors consist of a nCas fused with a reverse transcriptase ___domain. A prime editing guide RNA (pegRNA) contains a template of the desired sequence at the 3′ end as well as a target-specific spacer sequence. Cas12a is guided to the target DNA sequence by self-processed mature crRNA. Cas12a recognizes a PAM motif matching the target sequence, 5’-TTTN-3’, and forms a more reversible binding with the DNA target sequence than Cas9. Subsequently, Cas12a cleaves the non-target strand (NTS) followed by the target strand (TS), creating a DSB. Cas13d is capable of self-processing the crRNA precursor into mature crRNA, which then guides the cleavage of target RNA, enabling the regulation of gene expression at the transcriptional level. Therefore, by constructing multi-crRNA expression arrays, Cas13d also possesses the functional characteristic of regulating the expression of multiple genes. Cas13d has no limitations from the protospacer flanking sequence (PFS), and can almost target any RNA sequence, possessing a broad editing range. Created with BioRender.com.
CRISPR gene editing technology
The classification of CRISPR tools within CAR T-cell therapy
Cas9 and Cas12a systems
The CRISPR system was originally discovered in bacteria and archaea as part of their immune system, where it employs a novel mechanism in which RNA molecules guide Cas proteins to destroy viral nucleic acids [14]. Currently, a large number of high-performance and functionally specific CRISPR gene-editing tools have been developed, with the Class 2 system—including Types II, V, and VI—garnering the most attention and application. These systems typically consist of a single Cas protein acting as a nuclease effector. The basic strategy of gene editing involves activating the cleavage ___domain of nuclease at the target site, which requires not only the pairing of the crRNA guide sequence with the DNA target but also the presence of a matching protospacer adjacent motif (PAM) on the non-target strand (NTS) downstream of the target site. The Cas nuclease then cleaves the double-stranded DNA (dsDNA) upstream of the PAM sequence, generating a DNA double-strand break (DSB), which ultimately leads to gene knockout or insertion through the DNA repair mechanisms of cells. DSB is repaired through two main pathways: non-homologous end joining (NHEJ) or homology-directed repair (HDR). NHEJ is an effective and prevalent DNA repair mechanism in human cells. It rejoins the two broken DNA ends together at the site of the break, but this cutting and repair process may lead to insertions or deletions (indels) that disrupt the recognition of the target site by the nuclease. Indels integration in protein-coding genes cause frameshift or exon skipping, thereby disrupting gene function [15]. Alternatively, in the presence of the repair template, HDR introduces exogenous gene fragments from the donor to the cutting site by homologous recombination (HR), allowing for the integration of exogenous genes [16].
CRISPR-associated protein 9 (Cas9), derived from Streptococcus pyogenes (SpCas9), belongs to the Type II CRISPR-Cas system. It was the first CRISPR system developed for genome editing and comprises two components: Cas9 nuclease and a synthetic designed single guide RNA (sgRNA), formed by the fusion of a CRISPR RNA (crRNA) and a trans-activating crRNA (tracrRNA) [17] (Fig. 1). The guide RNA (gRNA) recognizes the target sequence and directs the Cas9 protein to cleave the DNA at the third base upstream of an NGG (N = A, T, C, or G) protospacer-adjacent motif (PAM) sequence, resulting in a DSB. In addition, by delivering a single form of the Cas9 protein and various sgRNAs, CRISPR-Cas9 are able to target multiple genes simultaneously [18].
The Cas12a/Cpf1 protein, which originates from Acidaminococcus and belongs to the Type V CRISPR-Cas system [19], is also favored in CAR T-cell preparation (Fig. 1). Similar to Cas9, Cas12a generate DSB for gene editing. However, this system possesses several intrinsic differences from the Cas9 protein [17, 20]. Firstly, Stringent crRNA pairing. Cas12a can loosely bind to the genomic target sites, strictly inspecting almost every base pair before moving on to the next base. During the R-loop formation, it demonstrates good reversibility in the binding of enzyme to the target sequence. In contrast, Cas9 tightly binds to DNA after moving 7 to 8 base pairs, becoming less sensitive to subsequent mismatched sequences [21]. Additionally, while crRNA can tolerate single or double mismatches in the 4-6 nucleotides at the 3’ end, it is highly sensitive to mismatches in the remaining regions [22]. Off-target mismatches disrupt its unique pairing/unwinding balance, causing a stall in the formation of R-loops and inhibiting off-target cleavage [23]. These characteristics result in a very low off-target rate for Cpf1, with whole-genome analysis revealing over nine times fewer off-target sites caused by designed Cas12a compared to Cas9 [24]. However, despite its high specificity, Cas12a typically exhibits lower editing efficiency compared to Cas9 [25]. Strategies such as base modifications of crRNA can be attempted to enhance the affinity between the editor and the target, thereby improving editing efficiency [26]. Using Cas9 and modified Cas12a for multi-gene editing in CAR T-cells, the results demonstrated that Cas12a was more efficient than Cas9 in knocking out CD3, granulocyte-macrophage colony-stimulating factor (GM-CSF), and programmed death protein 1 (PD-1) [27]. Secondly, the molecular weight of Cas proteins and gRNA structure. Cas12a (~130 kDa) is smaller than Cas9 (~160 kDa). Additionally, Cas9 requires a combination of tracrRNA and crRNA to recognize and cleave the target sequence, as its associated sgRNA (~100 nt) necessitates this dual-component structure. In contrast, the crRNA (~42 nt) of Cas12a has a naturally occurring secondary structure, which allows it to directly bind and activate the Cas12a protein [19]. This lowers cloning costs and circumvents the challenges of multiple plasmid delivery and vector capacity limitations [28]. Additionally, Cas12a can autonomously process precursor crRNA, simplifying the process of editing multiple genes [28,29,30]. Thirdly, Cas9 recognizes the PAM sequence 5’-NGG and cleaves near the PAM site, whereas Cas12a strictly recognizes the sequence 5’-TTTV and cleaves at a position further away from the PAM site [30]. This results in a relatively narrower range of target site selection for Cas12a, which somewhat limits the flexibility in target design. Lastly, Cas9 generates blunt-ended DSBs upon cleavage, while Cas12a produces sticky ends upon cleaving the target DNA, which is conducive to the insertion of the CAR transgene [31]. Cas12a and Cas9 were used to insert CD19 CAR and CD22 CAR genes into the PDCD1 and TRAC loci, and the proportion of T cells expressing bispecific CARs on the 8th day post-editing was 35.8% and 3.2%, respectively, demonstrating the superior multi-gene knock-in capability of Cas12a [32]. Several gene knock-in systems based on Cas12a, such as the SeLection by Essential-gene Exon Knock-in (SLEEK) platform, have already been developed and will be introduced in subsequent sections. These characteristics position CRISPR-Cas12a as a powerful and promising tool for editing CAR T-cells. These characteristics make CRISPR-Cas12a a powerful and promising tool for editing CAR T-cells.
Transcriptional and Post-Transcriptional modulation
Transcription initiation strength and epigenetics can influence gene expression, resulting in phenotypes that cannot be controlled simply by indel formation. To address this issue, DNA cleavage-deficient Cas9 (dCas9) has been developed for regulating gene transcription program [33]. Based on this discovery, dCas molecules can be creatively coupled to transcriptional or epigenetic modulators, to precisely target relevant therapeutic regulatory domains to specific regions of the genome without creating DNA damage or a DNA edit. This approach also mitigates the risks associated with DNA damage, off-target editing and abnormal chromosomal rearrangements (Fig. 1).
CRISPR interference (CRISPRi) system fuses the transcriptional repression ___domain Kruppel-associated box (KRAB) to dCas9, then binds to the transcription start site (TSS) of target genes, which inhibits transcription initiation and suppresses expression the target gene [34]. CRISPR activation (CRISPRa) recruits transcriptional activators like VP64 to the TSS using dCas9, which specifically activated endogenous gene transcription for the first time in the human genome [34]. Subsequently, dCas9 fused with various transcriptional activator domains including RTA, VP64, HSF1, and p65 to upregulate specific gene in multiple cell types [34]. Furthermore, the epigenetic editing system is created when dCas9 is combined with epigenetic modifiers, such as DNA methyltransferase (DNMT) and Tet methylcytosine dioxygenase (TET) for controlling DNA methylation as well as p300 and LSD1 for managing histone acetylation and methylation [35]. This approach utilizes the targeting capabilities to enable precise epigenome editing at selected DNA regions [36].
Cas13d belongs to a new member of the Type VI CRISPR system and possesses the ability to precisely target and degrade mRNA (Fig. 1). It exhibits interference efficiency in gene expression reaching up to 96%, exceeding the 65% efficiency of shRNA [37, 38]. Similar to Cas12a, Cas13d can self-process precursor crRNA into mature crRNA, which then guides the cleavage of target RNA, thereby regulating gene expression at the transcriptional level. Therefore, by constructing arrays of multiple crRNAs, Cas13d also possesses the capability to regulate the expression of multiple genes. Furthermore, Cas13d does not have restrictions on the protospacer flanking sequence, allowing it to target almost any RNA sequence and thus having a broad editing range. Additionally, as a small RNA editing system, Cas13d consists of about 930 amino acids, providing significant convenience for vector delivery [37]. The multifunctional transcriptional regulation platform based on Cas13d, termed Multiplexed Effector Guide Arrays (MEGA), utilizes lentiviral transduction of the Cas13d expression gene and single pre-crRNA guide array in primary T cells. This enables quantitative, reversible, and multiplex transcriptional regulation and screening in CAR T-cells [39]. However, it is notable that the potential immunogenicity arising from the constitutive expression of Cas13d may be a limiting factor, particularly for autologous CAR T-cells with intact antigen-presenting functions. Potential strategies to address immunogenicity include engineering Cas13d proteins to create smaller Cas proteins [40], thereby reducing the number of antigenic epitopes and the exposure area. Additionally, introducing spacers targeting B2M in the guide array to reduce the presentation of Cas protein antigenic peptides could also be an effective approach.
Base editing
The random process of indel formation is difficult to correct precise mutations, as the amount or identity of added nucleotides cannot be controlled. This may lead to low efficiency of gene knockout and increase the possibility of carcinogenic mutations. To fill this gap, CRISPR fusion for precise genetic changes has been developed and employed in CAR T-cells (Fig. 1).
Base editors (BEs) combine Cas nickase (nCas) with a deaminase to achieve precise base conversions at target sites. Cytosine base editors (CBEs) and adenine base editors (ABEs) are the two main types of DNA base editors, which use cytosine deaminase and adenine deaminase, respectively, to efficiently mediate C:G-to-T: A (CBEs) and A:T-to-G: C (ABEs) transition mutations at target sites [41, 42]. However, the utility of single-type base mutations is limited by their specificity. The novel glycosylase base editor (CGBE) system can effectively induce multiple types of base conversions, including C-to-G, C-to-T, and C-to-A. A new type of dual deaminase-mediated base editor system, named AGBE, is capable of simultaneously introducing four types of base conversions (C-to-G, C-to-T, C-to-A, and A-to-G) [43]. Additionally, deaminase-free base editors, such as cytosine (DAF-CBE) and thymine (DAF-TBE), can achieve C-to-G and T-to-G conversions [44]. These advanced base editors will significantly expand the diversity of target mutations, thereby facilitating the optimization of CAR T-cell applications. BE completes gene knockout by precisely replacing base pairs in the target DNA sequence, installing premature stop codons and resulting in loss of protein function [45, 46]. BE can also modulate splicing and force exon skipping by targeting splice acceptor [47], or mutating splice donor leading to nonsense-mediated mRNA decay (NMD), thereby degrading mRNA and terminating protein translation [48]. However, BEs do not directly interfere with splicing processes like some newer technologies, such as CRISPR-mediated RNA trans-splicing, which may provide a more immediate approach to controlling RNA splicing [49]. CRISPR-Cas9 induces highly variable indels through DSB-induced NHEJ, some of which may do not introduce frameshift mutations, thus contributing to the unpredictable editing outcomes [48]. In contrast, the precise base mutations generated by BE produce known outcomes, which explains the higher gene knockout efficiency of BE compared to DSB-induced NHEJ [48]. Furthermore, BE only induces single-strand breaks by nCas9, which avoids genome rearrangement effected by DSBs of conventional Cas9 cleavage. However, BE can also produce low rates of indels and other detrimental transcriptional responses [50]. These findings suggest that further investigation into the safety of BE in clinical applications is necessary.
Prime editing
Prime Editing achieves precise genome editing through the combination of a Prime editing guide RNA (pegRNA) and the Prime Editor protein, enabling efficient and specific editing without relying on DSBs or HDR (Fig. 1) [42], currently advancing to version 7 [51]. Because of the pegRNA template-guided editing, PE can achieve various types of edits with high precision without directly forming DSBs. This includes all 12 types of transition mutations, as well as precise insertion and deletion of multiple bases [42]. Using PE in zebrafish embryo cells, the highest editing efficiency for deletions was approximately 30% [52]. PE was also used to edit the endogenous TRAC and B2M loci in human T cells, achieving a disruption efficiency of over 90% [53]. This suggests that the editing efficiency of PE may be related to some factors such as the selection of editing sites, cell type, and the modifications of pegRNA. Compared to base editing or nucleases HDR-mediated DNA sequence replacement, PE exhibits high specificity, and editing purity in directly re-editing the target DNA, thereby holding the potential to further enhance the competitiveness of CAR T-cell products [53]. There are also several other emerging template insertion technologies, such as DNA Polymerase Editors (DPEs) and Click Editing, which utilize high-fidelity DNA-dependent DNA polymerases along with exogenous DNA templates. Both have achieved efficient and precise genome writing [54, 55].
The advantages of CRISPR for CAR T-cells
Site-specific integration
CAR transgenes are typically introduced into T cells using gammaretrovirus or lentivirus (RV/LV). Despite enabling stable insertion of the CAR gene, semi-random integration of viral vectors exhibits preferences for transcriptionally active regions, which may increase the risk of tumor induction, loss of critical gene structure and function, and the generation of suboptimal CAR T-cell products [56, 57]. As opposed to the random integration of RV/LV vectors, CRISPR system transports the CAR gene into the site-specific genome of T cells via HDR, enabling stable and homogeneous CAR expression (Fig. 2). Employing CRISPR-Cas12a, which generates sticky-end DNA cleavage, is advantageous for this purpose [29]. The selection of integration sites for CAR molecules typically include the following considerations: (i) Integration into safe sites, such as the adeno-associated virus integration site 1 (AAVS1) and eGSh6, ensuring stable CAR expression and avoiding interference with adjacent genetic loci [58,59,60]. (ii) Integration into specific sites to reduce graft-versus-host disease (GVHD) and produce UCAR T-cells. Integrating CAR into the T cell receptor Alpha constant (TRAC) locus, for instance, subjects its expression to be controlled by a powerful endogenous promoter, while avoiding activating tonic signaling [61]. The same concept has recently been applied to the CD3ζ gene, and its antitumor efficacy is comparable to that of CAR T-cells constructed by insertion into the TRAC locus or by lentiviral transduction [62]. This induces internalization and re-expression of CAR after single or repeated exposure to antigens, delaying T cell differentiation and exhaustion, and preventing GVHD [61]. (iii) Integration into specific sites to enhance CAR T-therapy efficacy, such as inhibitory molecule sites. T cells with CAR integration at the PD-1 locus demonstrate more robust and durable cytotoxic effects compared to PD-1+ CAR T-cells, and related studies have already entered clinical trial stages [63]. (iv) Integration into specific sites within an exon of essential genes, retaining essential gene function while also ensuring the long-term stable expression of transgenes. The SLEEK knock-in technology, developed based on AsCas12a nuclease, has achieved over 90% efficiency in knocking in multiple genes at essential gene sites, such as GAPDH [64]. (v) Integration into TRAC or CD3ε to create novel CAR architectures. Direct integration of the CAR antigen-binding ___domain into the TCRα and TCRβ constant regions or CD3ε can preserve the function of the endogenous T cell receptor (TCR) and enhance T cell sensitivity in recognizing tumor cells with low antigen expression levels [65,66,67]. This may represent a promising strategy for constructing autologous CAR T-cells to address antigen escape.

The application of the CRISPR system in CAR T-cell therapy mainly includes site-specific integration and multi-gene editing. By transferring CAR HDR templates encoded in the form of electroporation or AAV into T cells, the CAR sequence can be specifically integrated into the TRAC site, reducing graft-versus-host reactions and being used to produce universal CAR T-cells. This has achieved the “one stone two birds” gene editing effect of knocking in and knocking out, avoiding the oncogenic risk of random integration of viral vectors. Since the Cas protein responsible for DNA cutting is always the same, only the corresponding gRNA needs to be designed according to the target site, enabling the Cas enzyme to be expressed simultaneously with various gRNAs in the cell, thus achieving the purpose of multi-gene targeting and editing. By using electroporation or lipid particle encapsulation, Cas mRNA and gRNA or pre-formed ribonucleoprotein complexes (RNP) can be directly delivered into cells. This will transiently express the CRISPR editing system, with many advantages such as low cost, low off-target probability, and no oncogenic mutagenesis risk of viral vectors. Created with BioRender.com.
Targeted integration not only achieves the dual benefits of simultaneously inactivating TCR and introducing CAR, but also importantly avoids the oncogenic risks associated with semi-random integration of viral vectors. Currently, six CAR T-products approved by the FDA are based on viral vector-mediated CAR delivery, all of which have reported cases of post-CAR T-cell therapy malignancies involving T cells harboring the CAR gene [68]. While the specific reasons for oncogenic mutations remain unclear, targeted integration of CAR molecules undoubtedly represents a safer and more effective preferred strategy.
Multi-gene editing
As the demands for efficacy, safety, and accessibility of CAR T-cells continue to increase, the construction of these cells often requires editing multiple target genes. Compared with ZFNs and TALENs those traditional gene editing technology which need tremendous protein engineering for multi-gene editing, CRISPR system only requires designing corresponding gRNAs for each target site [69] (Fig. 2). Through the co-expression of multiple complexes of Cas proteins and mature gRNAs, the CRISPR system has truly acquired the capability for multi-gene editing. Traditional strategies involve transducing cells with viruses carrying expression vectors containing multiple gRNA concatemers, expressing CRISPR arrays containing multiple spacers under promoter transcriptional regulation, and relying on naive CRISPR mechanisms or RNA cleavage mechanisms to complete the concatenation transcription and processing of gRNAs [70]. Another straightforward strategy commonly used for CAR T-cell is directly delivering Cas mRNA and gRNA or pre-formed ribonucleoprotein complexes (RNPs) into cells through electroporation or lipid nanoparticle encapsulation [71], which allows for transient expression of the CRISPR editing system, avoiding the virus usage and insertional mutagenesis. CRISPR multi-gene editing technology greatly enhances the scope and efficiency of gene editing and transcriptional regulation, particularly suitable for intervening in the functions of multiple genetic or epigenetic targets in CAR T-cells to enhance their efficacy, safety, and accessibility [71]. Successful examples of multi-gene editing applications include CAR gene targeted knock-ins and triple-gene knockouts of TRAC, PD-1, and cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4), achieving an editing efficiency of 39.5% for triple knockout using Cas12a [30, 72]. The combination of Cas12a Ultra, used for CAR-targeted knock-in, with a base editor based on nCas9 for gene knockouts, has also efficiently generated triple-edited CAR T-cells while avoiding the adverse consequences of chromosomal translocations [73]. The MEGA system based on Cas13d possesses stronger multi-gene expression regulation capabilities. Researchers have achieved simultaneous knockdown of 5-8 genes in their upper limit tests of multi-transcriptional regulation and successfully achieved RNA-specific knockdown of LAG-3, PD-1, and TIM-3 genes in CAR T-cells, with no significant off-target or bystander activity [39]. Notably, accomplishing stable and efficient multi-gene editing faces various challenges, including the demand of customize efficient and specific gRNAs for each target, restricted vector capacity for component delivery, size constraints of inserted sequences, cell toxicity from off-target effects and DNA damage, and the immunogenicity of Cas proteins, gRNAs, and delivery vectors, especially during long-term intracellular expression of CRISPR components [74]. Furthermore, the simultaneous DSBs associated with conventional nucleases are thought to increase the risk of chromosomal translocations. Therefore, sequential editing or multiplexed base editing, as well as other non-DSB tools, may be the optimal choice when targeting multiple genes. These advanced CRISPR tools are progressively enhancing the efficiency and safety of multi-site targeting, which will be discussed further in the subsequent text.
CRISPR screening applied for CAR T-cell therapy
High-throughput CRISPR screening, as an unbiased target discovery platform, provides a comprehensive tool for characterizing the interactions between CAR T-cells and tumor cells [75]. CRISPR screening involves the transduction of a constructed sgRNA library into a population of cells, followed by specific perturbation based on the individual sgRNAs received by each cell. Following the application of specific selection criteria, interested phenotypic cell populations are selected from the pool of cells with gene perturbations for readout and sgRNA sequencing, thereby identifying potential positive and negative drivers of the underlying process [76]. Additionally, known genes relevant to the research objectives can be combined to construct various multi-gene crRNA array libraries, allowing for the screening of cell populations with synergistic gene combinations in order to obtain improved optimization schemes for T cells [39]. Traditional sgRNA library screening based on Cas9 typically results in mutations with multiple nucleotide insertions or deletions, making it unsuitable for identifying the functions of single nucleotide variants (SNVs). Base editing technology have been utilized for large-scale, high-resolution mutational screening in human T cells, enabling the introduction of specific nucleotide mutations. This approach allows for the mapping of gene variants related to the anti-tumor properties of T cells, such as amino acid residues, protein domains, and protein-protein interaction sites that affect cell activation and efficacy. These findings provide a precisely controlled mutational strategy for target screening [77, 78]. In CAR T-cells, CRISPR screening can identify new regulatory mechanisms that affect CAR T-cell efficacy under different physiological backgrounds. In tumor cells, CRISPR screening aids in elucidating resistance mechanisms to tumor killing.
Although genetic engineering targeting existing CAR T-cell antigens has profoundly altered the treatment landscape of many hematologic malignancies, the efficacy for numerous malignancies, particularly solid tumors, remains unsatisfactory, necessitating the discovery of more effective intervention targets. Thanks to its high-throughput, high editing efficiency, and high flexibility characteristics, large-scale CRISPR screening has emerged as a promising strategy for identifying potential targets for CAR T-cell anti-tumor functions [79]. CRISPR screening is performed by perturbing libraries in the T cell pool and imposing selection conditions like activation or co-culture with tumors to reveal positive and negative regulators for T cell survival, proliferation, or adaptability [80], with the screened genes targeted for perturbation in CAR T-cells to validate their functions (Fig. 3A). CRISPR screening can be classified into loss-of-function (LOF) and gain-of-function (GOF) screening based on different methods for gene perturbation. LOF screening is typically constructed using CRISPR knockout or CRISPRi systems. Differed from LOF, GOF screening reveals functional boosters via typically using the CRISPRa system to achieve gene overexpression. Whereas, conducting CRISPRa screening still poses difficulties as viral vectors are challenging to simultaneously introduce dCas9, transactivators, and sgRNAs into primary T cells [81, 82]. The dead-guide RNA (dgRNA), which adds MS2 binding loops to the sgRNA, binds to the MS2-P65-HSF1 (MPH) transcriptional activation complex, thereby guiding the catalytically active Cas9 to activate transcription [83]. In addition, CRISPR screening combined with single-cell sequencing can distinguish characteristics of different CAR T-cell subgroups, allowing for the identification of potential targets for enhancing T cell efficacy at the single-cell resolution [84].

A CRISPR screening can be classified into loss-of-function (LOF) and gain-of-function (GOF) screening based on different strategies for gene perturbation. The commonly used method for CAR T-cell functional targets identification involves introducing Cas9 protein via electroporation and transducing a lentivirus sgRNA library into the T cell pools, while GOF screening uses lentivirus packaging dgRNA library into Cas9-expressing T cells. The newly identified targets are validated in CAR T-cells with gene knockout or activation. It is also possible to conduct CRISPR screening directly in CAR T-cell pools, but performing so after lentivirus-mediated CAR gene introduction into T cells could lead to interference. Therefore, the CAR gene and the gene-edited crRNA library can be inserted into the same TRAC locus using the CLASH system (AAV library, Cas12a), achieving effective CRISPR screening in CAR T-cells. Marker expression can be employed for sorting by comparing the sgRNA sequencing between population with higher and lower expression levels, or sorting by comparing the sequencing results with a control group through co-culturing with tumor cells under immune challenge. B In Cas9-expressing tumor cells, introducing an sgRNA lentivirus library and screening through marker expression or immune challenge of CAR T-cells can identify tumor immune escape mechanisms and drug-resistant genes targeting CAR T-cell therapy. Created with BioRender.com.
CRISPR screening can also be performed directly in CAR T-cells (Fig. 3A). However, it is worth noting that applying CRISPR screening directly to CAR T-cells is still challenging as the CAR gene needs to be integrated into genome of T cells by lentiviral or retroviral vectors, which can hinder the efficiency of subsequent viral delivery of sgRNA libraries and affect the accuracy of phenotypic readout in subsequent CRISPR screening [80]. Furthermore, random integration of the CAR gene and CRISPR component often leads to insertional mutagenesis and translational silencing and the possibility of variable position effects during high throughput screening [85]. A promising strategy is introducing the CAR gene and gene editing tools into T cells simultaneously. The Cas12a-based AAV-Cpf1 KIKO (knock-in knock-out) system, known as CRISPR-based library-scale AAV perturbation with simultaneous HDR knock-in (CLASH), delivers Cas12a mRNA into human T cells via electroporation, followed by AAV infection carrying two crRNAs and the gene encoding CAR [86]. One crRNA guides specific integration of the CAR sequence, while the other is responsible for knocking out the desired gene. When replaced with a crRNA library, the CLASH system becomes an ideal platform for CRISPR screening and identifying functional genes regulating CAR T-cell activity [86].
As for tumor screening with CAR T-cell pressure, tumor cells are typically selected after library transduction to identify specific phenotypes, and then the phenotype-driving factors are screened and co-cultured with CAR T-cells for validation. Alternatively, library-transduced tumor cell populations can be challenged with CAR T-cells to analyze the gRNA of surviving tumor cells, identifying sensitive genes that affect CAR T-cell toxicity (Fig. 3B). CRISPR screening has revealed various intrinsic resistance mechanisms of different tumor types to CAR T-cells [87, 88], such as mechanisms involving tumor downregulation of antigen expression to evade recognition by CAR T-cells [89, 90]. CRISPR screening of tumor cells can also elucidate the mechanisms of action of combination therapies with CAR T-cells [91]. These newly discovered resistance genes can help design as corresponding targeted drugs to be used in combination with CAR T-cells, potentially enhancing the efficacy of CAR T-cells.
Application of CRISPR for CAR T-cells
Generally, the process of preparing CAR T-cells includes collecting peripheral blood cells from patients, selecting, enriching, and activating T cells, transducing CAR genes, expanding cells, and finally infusing them back into the patient’s body [92]. For CAR T-cell preparation, HDR-mediated targeted integration of CAR genes generates CAR T-cells, effectively avoiding the oncogenic risk associated with traditional LV-transduced CAR genes and reaping the benefits of “hitting two birds with one stone” (as in 2.2.1). For optimizing the efficacy and safety of CAR T-cells, multiplex gene editing is applied to T cells. Knocking out immune checkpoints, epigenetic modification sites, and genes that promote exhaustion addresses efficacy issues. Silencing CRS-related genes and mediating off-target-related shared antigens, such as CD7 and CD5, can help resolve safety concerns (Fig. 4). Additionally, incorporating CRISPR screening technology can help identify new regulatory mechanisms affecting the efficacy of CAR T-cells under different physiological backgrounds, as well as unknown mechanisms in tumor cells that contribute to resistance and immune escape (Table 1).

(i) Optimize the efficacy of CAR T-cells. The inhibitory pathways, the immunosuppressive factors caused by relative cells and hypoxia in TME, as well as the epigenetics of T cells, can all contribute to T cell exhaustion and decrease the effector function of CAR T-cells. Therefore, it is possible to maximize the therapeutic efficacy of CAR T-cells in the treatment of malignancies by employing CRISPR system to knock out immune checkpoints, TME-responsive receptors, and other molecules (such as Fas for promoting apoptosis, and ATG5 for increasing autophagy). Additionally, eliminating negative regulatory factors of cytokines, inflammatory factors, and CAR molecule expression using CRISPR system can also enhance the efficacy. (ii) Control adverse events of CAR T-cell therapy. GM-CSF knockout CAR T-cells can effectively alleviate CRS and ICANS, while depletion of shared antigens on T cell surface (CD7, CD5), and CD33 on HPSC can tackle OTOT toxicity. Created with BioRender.com.
Optimizing the efficacy of CAR T-cells
Knocking out immune checkpoints
Tumor cells may evade immune response through multiple immune checkpoints that hinder the anti-tumor attack of CAR T-cells [93]. CAR T-cells combined with various immune checkpoint inhibitors (ICI) block the inhibition pathway while showing notably anti-tumor responses in patients with hematological malignancies [94]. However, this combined therapy can activate the immune system, thereby increasing the risk of autoimmune diseases [95]. The immune checkpoints knockout CAR T-cells generated by CRISPR system avoid this problem (Fig. 4). The most frequently used strategy for anti-tumor effect enhancement and reduced functional exhaustion of CAR T-cells is PD-1 knockout [96,97,98]. In leukemia mouse model with TCR, B2M, and PD-1 simultaneously edited CD19 CAR T-cells demonstrated intrinsic PD-1 destruction promotes faster and more robust anti-tumor activity as compared to PD-1+ CAR T-cells [96]. In addition, PD-1 acts as an ideal locus for integrating CAR gene transfer with simultaneously knockout of PD-1. PD-1-integrated CAR T-cells showed more potent and durable effects in vitro and in lymphoma mouse models, and completed clinical trials Phase I with encouraging outcomes that a complete remission (CR) rate of 87.5% and an overall response rate (ORR) of 100% in 8 patients with relapsed/refractory B-NHL after infusion, associated with minor CRS and no ICANS events (NCT04213469) [63]. Knocking out CTLA-4, LAG-3 and CD70 has also shown strong antigen-specific anti-tumor activity in in vitro and in vivo models [99,100,101] (Table 1). The intracellular checkpoint RASA2, newly defined through CRISPR screening, negatively regulates the T cell Ras signaling pathway, thereby promoting tumor immune evasion [102]. Besides, CD5, a glycoprotein on the surface of T cells, negatively regulates T cell activation and maintains immune tolerance [103]. BTLA, by trans-binding with HVEM, recruits tyrosine phosphatases SHP-1 and SHP-2, inhibiting T cell anti-tumor activity [104]. The ablation of these genes has been shown to effectively enhance CAR T-cell signaling and cytotoxic activity. Other genes identified with immune checkpoint properties, such as Siglec receptors, TIGIT, and VISTA, also merit exploration in optimizing CAR T-cells [105,106,107]. This evidence demonstrate that knocking out inhibitory checkpoints in CAR T-cells contributes to further unleashing the potential of CAR T-cells.
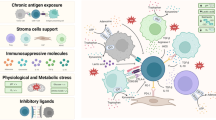
Circumventing the immunosuppressive TME
One of the largest barriers against the efficacy and persistence of CAR T-cells is the complex composition of the tumor microenvironment (TME), including a range of soluble factors, various immunosuppressive cells, and physical barriers of extracellular matrix [108,109,110]. Multiple therapeutic drug, antibodies and small molecule inhibitors, for instance, can effectively circumvent the immunosuppressive pathways in the TME, while their side effects and long development process hinder the clinical application [94, 111]. CRISPR system provides a new approach to explore these inhibitory pathways and overcome the micro-environmental obstacles (Fig. 4). As a key regulatory factor secreted by tumor cells, immune cells, and stromal cells in the TME, transforming growth factor-beta (TGF-β) binds to TGF-β receptor II (TGF-βRII) and elicits immunosuppressive responses[112]. The CRISPR-Cas9 mediated knockout of endogenous TGF-βRII has proven to prevent CAR T-cell exhaustion and reduce polarization towards Tregs [113, 114]. When tested against several solid tumor xenografts, TGF-βRII edited CAR T-cells exhibited a more pronounced ability to eliminate tumor cells [113, 114]. Additionally, diacylglycerol kinase (DKG) knockout CAR T-cells generated via CRISPR-Cas9 promoted resistance to soluble inhibitory factors of TGF-βand prostaglandin E2, resulting in significant tumor regression in a glioblastoma mouse model [115]. The purinergic signaling pathway relies on four key surface proteins, CD39, CD73, A2AR, and A2BR, to rapidly convert extracellular pro-inflammatory ATP into immunosuppressive adenosine (ADO), thereby inhibiting T cell function [116, 117]. In mouse models of breast cancer and pancreatic cancer, the CRISPR-Cas9-mediated deletion of A2AR in CAR T-cells has been shown to increase the production of cytokines such as interferon-gamma (IFN-γ) and tumor necrosis factor (TNF), and enhance the anti-tumor effects of CAR T-cells [118, 119]. Additionally, leveraging the MEGA system to simultaneously knock down the encoding genes of the above four proteins in CAR T-cells has been found to promote the secretion of IFN-γ and IL-2 factors, significantly enhancing cell proliferation and tumor killing capability [39]. Moreover, the hypoxia microenvironment affects the metabolism of immune cells, suppresses immune responses by upregulating the autophagy pathway [120]. It has been demonstrated that the knockout of autophagy related protein 5 (ATG5) in CAR T-cells enhances their metabolic activity and anti-tumor capability in clear cell ovarian cancer [120] (Table 1).
Modulating the epigenetics of T cells
Epigenetic modifications are crucial regulatory factors for CAR T-cell function and significant contributors to cancer progression. They profoundly impact T cell terminal differentiation, proliferation capacity, and effector function, thereby exerting a significant influence on the clinical efficacy of CAR T-cell therapy [121]. Using CRISPR gene editing to target genes involved in DNA and histone epigenetic modifications in CAR T-cells, T cell function can be stably reprogrammed (Fig. 4). The differentiation capacity of CAR T-cells in vivo is a critical factor affecting their persistence [122], with less differentiated CD8+ T cells demonstrating superior anti-tumor efficacy [123]. Plastic memory T cells, such as central memory T cells (Tcm) and stem cell memory T cells (Tscm), exhibit greater effector function and proliferative capacity, leading to higher tumor eradication rates in preclinical model systems and better persistence in clinical applications [124, 125]. It’s noteworthy that cytotoxic T lymphocytes (CTLs) undergo epigenetic changes, including DNA methylation and histone modifications, under chronic antigen stimulation, leading to T cell exhaustion. This impairs their proliferative and cytokine-producing abilities, often accompanied by upregulation of various immune checkpoint molecules, severely limiting immune efficacy. Epigenetic reprogramming can modulate the developmental trajectory of CAR T-cells, offering the potential to prevent or reverse terminal differentiation of cells.
To address DNA epigenetic modifications, CRISPR system can be employed to knock out epigenetic regulatory genes associated with the terminal differentiation stage of T cells (Table 1). For instance, knocking out the DNA methyltransferase 3α (DNMT3A) gene can inhibit the methylation of several key genes, such as TCF7 and LEF1, which regulate human T cell differentiation. This enables CAR T-cells to maintain memory and proliferative capacity under prolonged antigen exposure, exhibiting sustained cytotoxicity against chronic tumors in both in vitro and in vivo [126]. In terms of histone modifications, knocking out the PR ___domain zinc finger protein 1 (PRDM1), which encodes Blimp1, disrupts its ability to bind to histone H4 and regulate downstream genes. As a result, upregulation of several memory-related transcription factors and surface molecules such as TCF7, LEF1, and STAT3 was observed, along with significant downregulation of multiple effector differentiation-induced genes including KLRG1, EOMES, and ID2 and increased expression of the immune suppression-related transcriptional regulator TOX. This approach inhibits excessive T cell activation and helps maintain an early phenotype of CAR T-cells while promoting the secretion of multifunctional cytokines [127]. SUV39H1, which mediates H3K9 methylation, inhibits effector T cell function during the terminal differentiation stage [128]. Disrupting SUV39H1 can fine-tune the expression of several genes it represses simultaneously, such as TCF7, LEF1, and CCR7, thereby extending the lifespan of CAR T-cells and maintaining their cytotoxicity against tumors [129]. Additionally, isocitrate dehydrogenase 2 (IDH2), responsible for the reduction carboxylation of glutamine in the mitochondria, can be disrupted to activate compensatory metabolic pathways, alter the activity of histone-modifying enzymes, increase chromatin accessibility, and drive differentiation towards a memory T cell phenotype [130]. Other strategies to avoid exhaustion include preventing or reversing inhibitory epigenetic modifications. Disrupting Regnase, Tet2, and LSD1 to enhance immune efficacy is also worth exploring [131,132,133].
T cells derived from cancer patients may exhibit epigenetic dysregulation compared to T cells from healthy individuals [134,135,136]. Moreover, terminal patients often suffer from harsh chemotherapy, leading to impaired T cell function and a bias toward a more differentiated subset of effector memory T cells [123]. Creating an epigenomic map of isolated T cells from patients and reprogramming epigenetically dysregulated T cells into fully functional CAR T-cells could be a potential solution [121]. This approach may reduce the need for cell collection and treatment doses, expanding the applicable patient population and lowering the occurrence of adverse reactions [129].
Other CAR T-cell regulation targets
With profound mechanisms of resistance and relapse of CAR T-cell therapy, more therapeutic targets for regulating T-cellular activity have been identified (Fig. 4). Inflammatory cytokines is considered to be a significant pathway in destroying tumor cells, while suppressed by inflammatory regulatory factors, such as Roquin-1 and Regnase-1, to maintain immune homeostasis [137]. Roquin-1 and Regnase-1 dual edited CAR T-cells generated by the CRISPR-Cas9 demonstrated their capability of optimizing anti-tumor effects [137] (Table 1). Repeated antigen stimulation over-activates CAR T-cells and triggers Fas-mediated activation-induced cell death (AICD), which can be addressed by knocking out Fas using CRISPR-Cas9, thereby enhancing the anti-apoptotic capacity and prolonging the persistence of CAR T-cells [138]. CAR T-cell exhaustion caused by repeated antigen stimulation can also be counteracted by modulating transcription factors [139, 140]. For example, utilizing CRISPR-Cas9 to knockout the transcription factor Ikaros zinc finger (IKZF) 3 associated with immune cell development and cytokine secretion can enhance the secretion of IL-2 and other multiple cytokines, thereby improving the efficacy of CAR T-cells in solid tumors [141]. Ubiquitination of T cells also plays a role in exhaustion, which could be resisted via knocking out the Cbl-b ubiquitin ligase in carcinoembryonic antigen (CEA) CAR T-cell [142]. Metabolic abnormalities are also closely associated with T cell exhaustion, where continuous transition from oxidative phosphorylation (OXPHOS) to aerobic glycolysis can lead to T cell exhaustion. Targeting the PI3K/Akt signaling axis using the MEGA platform to simultaneously knock down the expression of four key genes, AKT1, AKT2, HK1, and HK2, effectively promoted CAR T-cell OXPHOS metabolism. The results showed no significant cellular or genetic toxicity but a notable enhancement in the tumor-killing ability of CAR T-cells both in vitro and in vivo [39, 143].
CRISPR screening for discovering novel targets
The above examples have confirmed that genetic knockout can enhance the efficacy of CAR T-cell therapy, albeit limited to known targets. More potential targets regulating the anti-tumor activity of CAR T-cells are being explored through CRISPR screening (Table 2). LOF screening has identified and validated positive and negative drivers that affect anti-tumor function in the whole genome of T cells, and these drivers have been further validated in CAR T-cells, including RASA2 (inhibits rapid proliferation of CAR T-cells), SOCS1 (inhibits viability and persistence of CAR T-cells), ST3GAL1 (decreases CAR T-cell migration towards tumors), cBAF (induces terminal differentiation and exhaustion of CAR T-cells), PDIA3 (suppresses functions in glioblastoma CAR T-cells), and NAD+ (a key factor in T cell activation) [102, 144,145,146,147]. Within the scope of GOF screening in T cells, a system based on the aforementioned dgRNA library and Cas9 transgenic mice for activation screening has avoided the challenging delivery of Cas9 and trans-activating agents, and identified the target proline dehydrogenase 2 (PRODH2) in primary CD8 T cells, which increases cell cytotoxicity. Overexpression of PRODH2 in CAR T-cells promotes mitochondrial proliferation and increases oxidative phosphorylation levels by reprogramming proline metabolism [81]. Using BE, high-resolution screening can identify variants that regulate functions such as T cell activation and cytokine production, including sgRNAs targeting multiple PIK3CD alleles (comprising both LOF and GOF variants) [77]. By engineering CD19 CAR T-cells with a PIK3CD GOF mutation identified through this screening process, it is possible to endow the cells with consistently enhanced signaling and effector functions [78]. Other genes obtained from T cell screening are also valuable for efficacy validation and application in CAR T-therapy. For instance, knocking out both ETS1 and RBPJ, as revealed by single-cell RNA sequencing (scRNA-seq) in CRISPR screening, can block terminal differentiation, leading to the accumulation of intermediate T cells and significantly enhancing anti-tumor activity [148].
When directly performing CRISPR screening on CAR T-cells, PD-1+ CAR T-cells identified TLE4 and IKZF2, which mediate CAR T-cell exhaustion following antigen stimulation [84]. Using the CLASH platform, a unique crRNA was discovered that can generate an exon 3 skip mutant of PRDM1 in CAR T-cells, which exhibited to enhance proliferation, persistence, and anti-tumor efficacy in various tumor models [86]. Additionally, the MEGA platform identified through transcriptomic-level dual-gene combination screening that depletion-related CBLB and FAS double-gene knockout significantly enhanced CAR T-cell cytokine secretion and anti-tumor activity [39]. Targeting the above-mentioned targets, engineered CAR T-cells can be developed, providing new avenues for unleashing the efficacy of CAR T-cells.
Immune escape, antigen loss, and other factors often lead to poor efficacy, tumor resistance, and disease relapse in CAR T-cell-based cancer therapies [149]. Therefore, in addition to screening for targets enhancing T cell anti-tumor activity, it is also essential to screen for targets in resistance pathways of the tumor cells [76, 150]. Newly discovered resistance genes can be designed into corresponding small molecule targeted drugs for combination therapy with CAR T-cells, potentially further enhancing the efficacy of CAR T-cells. Firstly, CRISPR screening can reveal the intrinsic resistance mechanisms of different tumor types against CAR T-cells. Unlike hematologic tumors, CRISPR screening has shown that the loss of IFNγ signaling reduces the adhesion of CAR T-cells to solid tumor cells, promoting resistance of glioblastoma and other solid tumors to CAR T-cell-mediated cell lysis [88]. In B-ALL tumors, CRISPR screening confirmed that the loss of CD58 inhibits the formation of the immune synapse with CAR T-cells, resulting in impaired function [87]. Furthermore, tumors evade CAR T-cell recognition by downregulating antigen expression through various mechanisms. Pancreatic cancer cells subjected to library knockout and CAR T-cell challenge revealed that loss of GPI anchoring leads to the reduction of Mesothelin (MSLN) on the surface of tumor cells, evading attack by MSLN CAR T-cells [90]. Library knockout screening of B-Acute Lymphocytic Leukemia (ALL) cells enriched for nudix hydrolase 21 (NUDT21), which evades targeting by CD19 CAR T-cells in CD19+ B-ALL by transcription control [89]. Although CRISPR screening has selected intrinsic regulatory factors in tumor cells that mediate evasion of T cell killing [151, 152], the resistance mechanisms of tumor cells to CAR T-cell killing are not exactly the same as those of T cells due to different structures and non-MHC-dependent killing forms. Therefore, it is still necessary to screen for resistance mechanisms in the context of CAR T-cells. Lastly, CRISPR screening of tumor cells can also elucidate the mechanism of action of combination drugs when used in conjunction with CAR T-cells. A systematic understanding of the regulatory functions of drugs facilitates the determination of the priority sequence for combination drugs during immunotherapy, further unleashing the anti-tumor potential. For instance, a study using over 500 small molecule drugs and CRISPR LOF screening to investigate the pharmacological mechanisms of CAR T-cell cytotoxicity in B-ALL. They found that smac mimetic drugs may activate death receptor signaling, a necessary pathway that increases cancer cell sensitivity to CAR T-cell cytotoxicity [91].
Controlling adverse events of CAR T-cell therapy
Alleviating CRS and ICANS
Despite the robust and durable anti-tumor responses achieved by CAR T-cells, the CAR T-cell-associated adverse events need to be taken seriously [10, 153, 154]. The most common adverse event is CRS, a systemic inflammatory response mediated by excessive activation of effector cells and the release of a large amount of cytokines. Another one is ICANS, which is a toxic brain disorder characterized by a wide range of neurological symptoms [10, 155]. Currently, the popular pharmacological management strategies for toxicity include high-dose corticosteroids and tocilizumab (an IL-6 receptor antagonist). The former may interfere with CAR T-cell effector function, while the latter, though FDA-approved for treating severe CRS, has limited efficacy in treating neurotoxicity [10]. GM-CSF secreted predominantly by myeloid cells and T cells is associated with CRS and ICANS [156, 157]. CRISPR-Cas9 knockout of GM-CSF can prevent GM-CSF-mediated therapeutic toxicity (Fig. 4) [158]. GM-CSF-/- CAR T-cells decreased levels of inflammatory cytokines and chemokines in ALL mouse, effectively reducing the risk of CRS and ICANS [158], while their intensive anti-tumor activity is worth mentioning [158]. A possible reason is that GM-CSF-depleted CAR T-cells inhibit the intrinsic apoptosis pathway by diminishing the expression of BH3 interacting-___domain death agonist (Bid) [10]. Monocytes stimulate the secretion of pro-inflammatory cytokines through the CD40L-CD40R axis or by uptaking GM-CSF. CRISPR-mediated knockout of CD40L and/or CSF2 in CAR T-cells can significantly reduce the secretion of IL-6 by bystander monocytes [159]. Additionally, CAR T-cells with GM-CSF knockout and autonomous co-expression of IL-6 and IL-1 blockers were used in three patients with hematologic malignancies; two of these patients experienced no CRS, and only one developed grade II CRS (ChiCTR2000032124). This approach may represent a strategy to minimize GM-CSF-related toxicity [160] (Table 1).
Tackling on-target off-tumor toxicity
The ideal target protein should be selectively and stably expressed at high levels on all diseased cells with low or no expression on the surface of normal cells [161]. Nevertheless, the antigens recognized by CAR T-cells are often expressed on both normal and malignant cells, leading to life-threatening OTOT toxicity [162]. For example, in anti-CD19 CAR T-cell therapy, B cell aplasia is often observed for the CD19 expression on normal B cell membrane. In order to tackle the off-tumor toxicity, specific gene editing of CAR T-cells or hematopoietic stem cells using CRISPR system may be required in certain cases to prevent fratricide and hematopoietic toxicity (Fig. 4).
Served as shared antigens of T cells, CD7 and CD5 expressed not only on pathogenic T cells but also on normal T cells (including designing CAR T-cells), contributing to fratricide of CAR T-cells with poor therapeutic outcomes. It have demonstrated that knocking out CD5 or CD7 on CAR T-cells by CRISPR system can achieve potent expansion and anti-tumor capabilities when combating T-ALL [163,164,165]. In addition to the challenge of self-fratricide, contamination of CAR T-products by malignant T cells poses a significant obstacle to clinical applications [166]. Therefore, CAR T-products specifically targeting T-cell malignancies and entering clinical trials are scarce. UCAR T-cell therapy demonstrates an advantage in this context [167]. CRISPR systems suitable for multi-gene editing can simultaneously knock out CD7 and TRAC, enabling the preparation of universal CD7 CAR T-cells for clinical therapy. This approach has demonstrated promising therapeutic outcomes (Table 1). Following the infusion of CD7 UCAR T-cells in 11 patients with T-ALL, T-NHL, and CD7+ acute myeloid leukemia (AML), 82% achieved overall response (NCT04538599) [168]. Utilizing base editing to knock out TCR, CD52 and CD7, CD7 UCAR T-cells were produced and infused. Following infusion, all three pediatric T-ALL patients achieved sustained leukemia remission [169]. No GVHD or other severe complications were observed in these cases.
CD33 is expressed on both myeloid leukemia cells and normal myeloid cells, so that CD33-targeting CAR T-cells attack newly transplanted hematopoietic stem cells. Therefore, when CD33 CAR T-cells are treated for AML, it is often necessary to knock out CD33 in Hematopoietic Stem and Progenitor Cell (HSPC) by CRISPR system to prevent the attack of CD33 CAR T-cells and protect normal hematopoietic function [170,171,172]. CD45-targeting CAR T-cells used for hematologic tumors faces more problems. CD45 is generally expressed in blood cells including T cells and hematopoietic stem cells, so CD45 CAR T-cell may cause fratricide and severe hematopoietic toxicity. On the other hand, CD45 influences the development of T cells and the hematopoietic function of hematopoietic stem cells, which means that it should not be knocked out completely. Epitope-base editing inserts nonsense mutations into the epitope in the extracellular ___domain of CD45, which enables T-cells and hematopoietic stem cells to avoid the attack of CD45 CAR T-cells, while still expressing CD45 and exercising intracellular phosphatase function. This modified CD45 CAR T-cell has achieved favorable antitumor activity in AML, T-ALL, and B-cell lymphoma [173].
Application of CRISPR for UCAR T-cells
Although autologous CAR T-cells have made groundbreaking progress in tumor treatment, their clinical application is severely hindered by several challenges, including poor quality of autologous T cells, complex manufacturing processes, long production cycles, and high costs [174]. UCAR T-cell products, also known as “off-the-shelf” CAR T-cells, derived from healthy donors and requiring multiple gene editing steps, are currently being widely researched and developed for mass production to be used for different patients (Fig. 5). Due to the innovation of gene editing technology in CAR T-cell production, mass-produced UCAR T-cell products are expected to overcome the aforementioned issues associated with autologous CAR T-cells, achieving accessibility in safety, efficacy, and affordability [8, 175].

A UCAR T-cells can be generated from healthy allogeneic donors to benefit multiple recipients. The manufacturing process for UCAR T-cell products starts with a source of third-party healthy T lymphocytes collected by leukapheresis. CRISPR/Cas-mediated precision editing of T cells or induced pluripotent stem cells (IPSCs) has the potential to eliminate expression of endogenous αβ-TCR, HLA and PD-1 etc, and insert a recombinant DNA coding for a CAR gene simultaneously. T cells or differentiated IPSC-T cells are then expanded using anti-CD3/anti-CD28 beads and cytokines. The remaining αβ TCR-positive cells are magnetically removed using anti-αβ TCR antibodies. The product is then packed and shipped to hospitals for using. B Combining CAR transfer with CRISPR-Cas-mediated genome editing offers the strategies to enhance CAR T-cells. Endogenous αβ TCR is removed to prevent GVHD, and HLA class I and class II molecules are also removed to prevent HVGD (through deletion of B2M/HLA-A, B and CIITA respectively). Persistence can also be achieved by deleting CD52 (allow cells to persist in the presence of alemtuzumab for lymphodepletion), or by deleting the NK cell activator Poliovirus receptor (PVR) and addition of a natural killer (NK) cell inhibitor (such as HLA-E, HLA-G, CD47 and CD300a TASR) for resistance to NK cells attack. Disrupting programmed cell death protein 1 (PD-1) enables UCAR T-cell to counteract some mechanisms of immunosuppression from tumor. Created with BioRender.com.
Overcoming the challenges of UCAR T-cells
The primary difference between UCAR T-cells and CAR T-cells lies in the donor source; UCAR T-cells are derived from allogeneic T cells of healthy donors rather than autologous T cells (Fig. 5A), which requires UCAR T-cells to address the challenges of immune rejection faced in clinical applications. One challenge is safety, as allogeneic CAR T-cells attacking host tissues can lead to life-threatening GVHD [8]. Another challenge is efficacy, as allogeneic CAR T-cells may be cleared by the host immune system, resulting in host-versus-graft rejection (HVGR) [176].
Safety issues of UCAR T-cells
The recognition of host cell antigens by the TCR protein complex on αβ T cell is central to the pathogenesis of GVHD. One promising approach to address this issue is to disrupt the expression of TCR on T cells using gene editing techniques (Fig. 5B) [8]. Early studies demonstrated that TALENs, ZFNs, and CRISPR-Cas9 can all disrupt TRAC in CAR T-cells, minimizing the risk of GVHD. However, experiments targeting TRAC disruption have indicated that CRISPR-Cas9 exhibits the highest disruption efficiency compared to other editing technologies such as TALENs and ZFNs, while maintaining low levels of toxicity and off-target cleavage [177]. In practice, the CAR gene cassette is often directly integrated into the TRAC locus. This allows for simultaneous CAR gene knock-in and TRAC knockout, which benefits UCAR T-cell stability and efficacy while avoiding the occurrence of GVHD [61]. Nevertheless, it is worth noting that while current technologies achieve an editing efficiency of over 85% for TRAC, they still cannot ensure the complete elimination of TCR on CAR T-cells [178, 179]. This may necessitate further magnetic purification of the T cell product prior to infusion.
Durability issues of UCAR T-cells
The efficacy and prognosis of UCAR T-cell therapy are closely associated with the persistence of the cells in vivo [180]. Compared to autologous CAR T-cells, UCAR T-cells may exhibit relatively weaker persistence and proliferative capacity in vivo, possibly due to HVGR [175]. One approach to prevent HVGR is modifying the lymphodepletion regimen. UCAR T-cells require more intense lymphocyte depletion compared to autologous CAR T-cells, such as the addition of alemtuzumab to the lymphodepletion regimen. However, this strategy relies on the destruction of CD52, which confers resistance to alemtuzumab in UCAR T-cells [181]. Preclinical and clinical trials have shown that simultaneous knockout of TRAC and CD52 allows UCAR T-cells to engraft and proliferate in the presence of alemtuzumab, exhibiting potent anti-tumor efficacy [179, 182]. Another approach to address HVGR is to reduce the immunogenicity of UCAR T-cells. Abrogating HLA-I molecules on UCAR T-cells can prevent CD8+ T cell-mediated immune rejection. A common target is β2-microglobulin (B2M), which is an essential component of HLA-I. However, HLA-I-negative UCAR T-cells is susceptible to NK cell-mediated killing. Knocking out the NK cell activating receptors on CAR T-cells [183], as well as inserting or overexpressing NK cell inhibitory receptors such as HLA-E, HLA-G, CD47, and CD300a TASR [184,185,186,187], are potential strategies to address this issue. However, the engineering of inhibitory molecules may complicate the production process. It has shown that deleting CD54 and CD58 in B2M-deficient CAR T-cells can universally limit the activation of all NK cell subsets [188]. Additionally, by selectively knocking out HLA-A and HLA-B in donor T cells while retaining HLA-C, partial editing and reduced expression of HLA can be achieved [189, 190]. This approach not only reduces the recognition of donor cells by host T cells but also mitigates NK cell-mediated rejection, and the UCAR T-cells exhibit significantly enhanced persistence and efficacy in preclinical and clinical research [189, 190]. Additionally, eliminating HLA-II molecules on T cells can circumvent CD4+ T cell-mediated immune rejection. This is often achieved by disrupting the transcription factor Class II transactivator (CIITA), which regulates the expression of HLA-II molecules [191] (Fig. 5B). Finally, addressing HVGR can also be achieved through a “proactive approach.” UCAR T-cells co-expressing CAR molecules and allogeneic defense receptors (ADRs) that selectively recognize 4-1BB can eliminate activated host T cells and NK cells while preserving resting lymphocytes, thereby ensuring long-term therapeutic benefits [192].
Expanding the sources of UCAR T-cells
UCAR T-cells are typically derived from T cells of healthy donors, yet the limited expansion capacity of peripheral blood T cells makes it difficult to meet the quantity demands of large-scale production. Relying on excellent expansion and tolerance to multi-gene editing, IPSCs serve as an appropriate source for constructing UCAR T-cells [193]. iPSCs are generated by reprogramming donor T cells to regain pluripotency, and then undergo gene editing to produce CAR-iPSCs. These CAR-iPSCs are subsequently induced to differentiate into CAR T-cells for clinical use (Fig. 5A). CAR-iPSCs have the ability to self-renew, and those CAR T-cells generated from the same engineered pluripotent cell line, resulting in higher homogeneity. Gene editing is an essential step in generating iPSC-derived UCAR T-cells. By using CRISPR-Cas9 to disrupt B2M, CIITA, and CD155 (the activating ligand for NK cells) in iPSCs and transducing HLA-E via lentivirus, iPSC-derived UCAR T-cell are protected from attacks by CD8+ T cells, CD4+ T cells, and NK cells [183]. The first iPSC-derived UCAR T-cell, FT-819, was created by CRISPR-Cas9 to insert the CAR into the TRAC locus, and related clinical trials are underway [194, 195]. Preliminary results from FT-819 trials in 15 patients with lymphoma, CLL, and ALL showed a complete response rate of 20% with no occurrence of GVHD, providing initial evidence of the safety of using iPSCs as a source for UCAR T-cells [196]. The application of iPSCs provides an opportunity to select the optimal cell line for industrial-scale production, effectively enhancing homogeneity across different batches and the therapeutic efficacy, and holding the promise of further reducing costs.
Ongoing clinical trials
As mentioned above, GVHD and HVGR are the primary obstacles to the clinical application of UCAR T-cells. Factors closely related to efficacy, such as limited expansion and poor persistence, also somewhat restrict their development [8, 197, 198]. Encouragingly, UCAR T-cells edited by the CRISPR system have largely overcome these challenges, and clinical trials are widely underway [175, 199]. The majority of these trials have focused on various hematological malignancies, while clinical progress in solid tumors is still in the early stages. Among them, the most widely applied UCAR T-therapy is still targeted at CD19 for B-cell malignancies, with over 10 CRISPR-edited products already entering clinical studies (Table 3). The disclosed trial data indicate that UCAR T-therapies targeting B-cell or T-cell lineage leukemias demonstrate the highest efficacy, with CR or CR with incomplete count recovery (CRi) rates ranging from 60% to 85%. Aside from isolated cases of low-grade GVHD (GvHD ≤ II: 16.7% [1/6], NCT04557436), the vast majority of trials have not observed GVHD. Adverse events such as CRS are relatively common, reaching up to 100%, but effective management of side effects has been achieved through monoclonal antibody treatment (Table 3). The CR rate for UCAR T-therapies in NHL also exceeds 60%, while efficacy is comparatively weaker for B/T-cell lymphomas, with CR rates ranging between 20% and 35%. Similarly, no GVHD has been observed, although other adverse reactions have been noted, such as ICANS ≥ III: 6.3% (NCT04035434). The significant differences in the therapeutic efficacy may be related to variations in indications, cohort sizes, and the quality of cell products across different trials. In terms of safety, it is encouraging that no trials have reported gene-editing-related adverse events, aside from a few cases of varying degrees of CRS or ICANS. GVHD was not observed in any patients except for two cases (Table 3), providing preliminary validation of the reliable safety of UCAR T-therapy. The two patients who had undergone allo-SCT experienced low-grade GVHD, which was quickly alleviated with steroid treatment. However, it remains unclear whether this was due to the cell transplantation or was mediated by UCAR T-cells [169, 182]. In terms of efficacy, HVGR significantly compromises the persistence of UCAR T-cells, largely limiting their effectiveness. As allogeneic cells, UCAR T-cells express multiple immunogenic molecules. Some trials observed that non-responding patients had significantly more CD3+ T cells compared to CR/CRi patients, and these rejection reactions are believed to be closely associated with reduced cell expansion and treatment failure [179]. Additionally, differences in the production methods of cell products and the selection of gene-editing targets may also influence efficacy, warranting further exploration in larger cohort trials in the future. In terms of solid tumors, clinical trials have been initiated for solid tumors such as renal cell carcinoma (RCC), non-small cell lung cancer (NSCLC), ovarian cancer (OV) [200, 201]. Among 15 patients infused with PD-1 and TCR knockout mesothelin-specific CAR T-cells (MPTK-CAR-T) for the treatment of pancreatic cancer, biliary tract cancer, gastric cancer, fallopian tube cancer, esophageal cancer, OV, cervical cancer, and breast cancer, 7 patients achieved stable disease within 3-4 weeks, while 8 patients experienced disease progression, with most dying within 2 months after infusion. No signs of GVHD, CRS, or ICANS were observed in any of the 15 patients, providing preliminary validation of the safety of CRISPR-edited UCAR T-cells in solid tumors [200]. In another trial targeting RCC, the disease control rate (DCR) reached 77% with no reported GVHD and severe CRS or ICANS (NCT04438083) [201]. Beyond advancements in the oncology field, the application of CAR T-therapy has also expanded to non-tumor diseases such as autoimmune diseases [202]. The CRISPR-mediated quadruple-editing product TYU19, targeting TRAC, HLA-A/B, CIITA, and PD-1, was used globally for the first time in the treatment of rheumatic autoimmune diseases. One patient with refractory immune-mediated necrotizing myopathy and two patients with diffuse cutaneous systemic sclerosis all achieved profound symptom relief following treatment, with no adverse events such as CRS or GVHD reported (NCT05859997) [203]. Overall, UCAR T-cells edited by CRISPR system have shown promising safety profiles in preventing GVHD and effective antitumor activity in hematologic malignancies. However, in terms of efficacy, UCAR T-therapy still falls short of the superior outcomes seen with autologous CAR T-therapies. For instance, in a clinical study, involving 7 pediatric and 14 adult patients, the ORR for UCAR T-therapy was 67%, whereas the initial ORR for most current autologous CAR T-cell-therapies is around 90% [199]. This disparity may be attributed to the reduced persistence of CAR T-cells lacking TCR [8, 200]. It is worth noting that another advantage of donor-derived T cells is their enhanced tolerance to multiple gene edits. This offers the opportunity to explore more genetic modifications aiming to improve cell persistence and efficacy. In the future, further improvements to UCAR T-cells are necessary to enhance the efficacy and safety of UCAR T-therapy, and CRISPR can play a crucial role in achieving these advancements [175].
Challenges and corresponding strategies of CRISPR in CAR T-cells
Off-target effects
While the CRISPR gene editing system has shown enormous potential in the field of CAR T immunotherapy, it has also introduced additional genetic toxicity risks [12, 18, 96]. Due to the similarity of the genomes [204, 205], Cas proteins may cause off-target DSBs at non-target gene loci, resulting in unintended mutations, known as off-target effects, which may lead to unpredictable alterations in non-target gene function or regulation [18]. A study targeting TRAC, T cell receptor Beta constant (TRBC), and PD-1 in engineered T-cells using CRISPR-Cas9 revealed off-target mutations at multiple sites during the in vitro manufacturing process. Using TRAC gRNA as an example, off-target editing was detected within 100bp upstream and downstream of the editing site, such as low-abundance mutations in the transcriptional region of the CLIC2 gene. Since CLIC2 is not expressed in T cells, it is expected to have no impact on cellular function. For the TRBC gRNA, off-target editing was found in genes encoding transcriptional regulators (ZNF609) and long intergenic non-protein coding RNA (LINC00377) [206]. Although no oncogenic transformation of cells was observed in these trials, off-target disruptions can occur randomly, including but not limited to tumor suppressor genes, anti-tumor or survival-related genes, potentially posing considerable adverse effects on the quality of CAR T-cells. Therefore, measures should be taken to improve targeting specificity and minimize off-target effects [207] (Fig. 6A).

A Adverse events and feasible solutions. Off-target mutations are caused by sgRNA recognizing the off-target locus in the genome and making unnecessary cleavage at the wrong locus. Cas-CLOVER, fuses the catalytically inactive dCas9 with the Clo51 (CLOVER) nuclease ___domain and uses two gRNAs to only cleave when the Clo51 nuclease dimerizes. Another approach “spacer-nick” combines the nCas9 with a pair of PAM-out sgRNAs at a long spacer distance. Hence Cas9n is guided by the spacer-nick sgRNAs to two target sequences on opposite strands and nicks both DNA strands at an optimal distance to preserve efficient HDR while minimizing NHEJ events. Chromosomal aberrations, such as large deletions and translocations, are indeed frequent phenomena. Here shows possible instances of chromosomal loss and translocations. The key factor leading to chromosomal aberrations is the generation of DSBs. BE develops precise single-base substitutions at multiple sites enabling DSB-free gene editing, thereby reducing the risk of gene rearrangements. B Traditional delivery methods include viral vectors and electroporation. Viral vectors mainly include LV, AV, and AAV. CRISPR components delivered by viral vectors in the form of DNA may increase the risk of off-target, and viral vectors also have other disadvantages including immunogenicity, cargo size limitations, and lower transduction efficiency. The delivery of electroporation in CAR T-cells may cause cell damage. Peptide delivery is an emerging delivery system for CRISPR components in CAR T-cells. LNP and VLP have the potential for in vivo delivery, but two basic issues need to be considered, that is, whether the relevant cell types can be targeted and avoid cell damage and immune rejection in vivo. Created with BioRender.com.
Since the off-target effects of the CRISPR system are primarily determined by target specificity, chromatin state, and the properties and concentration of nucleases [208]. Thus, the primary approach is to enhance the specificity and accuracy of the CRISPR system recognition. Selecting high-fidelity engineered nucleases can improve the accuracy of DNA cleavage and minimize Cas-dependent off-target effects. Cas-CLOVER, a dimeric system similar to TALENs, fuses the catalytically inactive dCas9 with the Clo51 (CLOVER) nuclease ___domain and uses two gRNAs to only cleave when the Clo51 nuclease dimerizes, making the DNA cleavage more stringent with higher specificity and fidelity [209] (Fig. 6A). Cas-CLOVER can be used to generate multi-gene edits in TRAC, B2M, and PDCD1 in CAR T-cells to produce allogenic BCMA CAR T-products with controllable off-target activity [209]. Another approach “spacer-nick” combines the nCas9 with a pair of PAM-out sgRNAs at a long spacer distance, mirroring effects seen with Cas-CLOVER [210]. Other high-fidelity Cas nucleases such as HF1, HiFi, and HypaCas9 can also reduce the occurrence of off-target events [211,212,213]. These newly discovered, more specific cleavage enzymes have not yet been applied in the CAR T-cell field, so their effects await substantiation in future studies. On the other hand, improving the sgRNA sequence is considered a crucial factor for target specificity. This can be achieved by shortening the length of the sgRNA, reducing its GC content, or chemically modifying the sgRNA to lower the affinity of the Cas protein and sgRNA complex for DNA, thus avoiding off-target risks [208, 214, 215]. Additionally, reducing the concentration of nucleases in cells and delivering nucleases transiently to limit their action time is also an effective way to decrease off-target activity [208]. With the assistance of artificial intelligence in the future, we will be able to accurately simulate complex genome editing scenarios, predict targeted and off-target editing outcomes, and design superior genome editors, further enhancing the safety of cell therapies [216].
Chromosomal aberrations
CRISPR-induced DSBs in the genome can lead to chromosomal aberrations, including large segment deletions, inversions, and translocations [18, 96], which are even prevalent phenomena (Fig. 6A). Through CRISPR screening and single-cell analysis using a gRNA library covering all chromosomes, it was discovered that editing guided by 55% of the gRNAs resulted in chromosome loss [217]. For the production of UCAR T-cells, TRAC gene knockout is a commonly used approach, but it has been reported to frequently cause chromosomal abnormalities. Editing of the TRAC locus in human primary T cells using CRISPR RNPs has shown that approximately 5–20% of T cells exhibit large segmental or whole deletions of chromosome 14 [217]. Transcriptome sequencing also revealed that these chromosomal deletions lead to abnormal expression of various genes, such as CD70 and MDM2. This Cas9-induced chromosome loss triggers a p53-related DNA damage response, activation of the apoptosis pathway, and p53-induced cell cycle arrest, thereby impairing the survival advantage of CAR T-cells [217]. Chromosomal translocations also pose a serious threat to genome stability and are a hallmark of carcinogenesis, especially in HLA-knockout cell products lacking any functional antigen processing. Primer-extension-mediated sequencing (PEM-seq) analysis has revealed that chromosomal translocations commonly occur between multiple Cas9 target sites. During gene editing, the perfect repair of DSBs exists, which is the main molecular mechanism leading to repeated cleavage of Cas9 at both on-target and off-target sites, as well as the abundant occurrence of chromosomal translocations [218]. It is worth noting that the multiple gene editing induced by CRISPR on the genome will simultaneously induce multiple DSBs. This may significantly increase the likelihood of chromosomal translocations by connecting unrelated broken ends through NHEJ. When using CRISPR-Cas9 to target TRAC, TRBC, and PD-1 in engineered T cells, chromosomal translocations were detected in approximately 4% of T cells, with the most prominent being a 9.3kB deletion mutation caused by TRBC1:TRBC2 translocation recombination. This adverse phenomenon persisted for up to 170 days or longer after infusion into lymphoma patients, but the proportion of cells with genomic aberrations gradually decreased over time, suggesting that these cells do not possess a growth advantage during multiple rounds of expansion [206]. Future clinical trials will need more experience, with CAR T products requiring higher editing efficiency and longer post-infusion observation periods to fully assess safety.
The DSBs generated during gene knockout by CRISPR system are key drivers of chromosomal aberrations [217]. The preparation of CAR T-cells involves steps such as gene editing and CAR T-cell activation. A study has shown that the sequence of T cell activation relative to CRISPR gene editing is associated with the occurrence of chromosomal deletion events. When gene editing induces DSBs, it triggers an increase in p53 expression to repair the unstable genome. However, upon T cell activation, the PI3K/AKT signaling pathway is activated, and MDM2 (a negative regulator of p53) is upregulated, which suppresses p53 activity and expression. Thus, performing gene editing before T cell activation is beneficial in reducing the frequency of chromosomal deletions [217]. Additionally, using an expanded CRISPR toolbox to avoid generating DSBs is an effective measure to prevent chromosomal abnormalities (Fig. 6A). BE enables precise single-base substitutions at multiple sites without causing DSBs, thereby reducing the risk of gene rearrangements. By employing base editing to convert amino acid codons at the TRAC, B2M, and PDCD1 gene loci into premature stop codons (pmSTOPs) or to disrupt their splice donor and splice acceptor sequences, triple gene knockout CD19 CAR T-cells are generated. These cells exhibite enhanced proliferation compared to those modified with CRISPR-Cas9 [48]. And the frequency of DSB-induced translocations is significantly reduced in CD19 CAR T-cells edited through base editing [48]. Similarly, TRAC, PD-1, CD52, and CD7 knockout CAR T-cells via CBE neither affect T cell proliferation nor lead to detectable translocations or karyotypic abnormalities [219]. The first universal cell therapy based on BE in the world has undergone clinical trials. The CD7 CAR T-cells used in this therapy, with multiple edits targeting TRAC, TRBC, CD52, and CD7, exhibit the same chromosomal translocation rate as non-edited cells (<0.1%) [169]. However, base editing is limited to single-base mutations and is challenging to achieve large-scale sequence replacement in CAR T-cell DNA. The Prime-Assisted Site-Specific Integrase Gene Editing (PASSIGE) system combines PE with Bxb1 integras, enabling precise and flexible replacement of multiple large DNA fragments at specific loci without generating DSBs. In one study, the PASSIGE system is utilized to produce CD19 CAR T-cells by co-delivering non-viral DNA donor templates with PASSIGE editing components, achieving targeted integration of the CD19-CAR transgene expression cassette into the TRAC locus with an editing efficiency exceeding 60%. The resulting CAR T-cells demonstrate exceptional anti-tumor efficacy both in vitro and in vivo, with no detectable off-target activity, large deletions, or translocations [53]. Due to the frequent repetitive cleavage by Cas9 occurs especially during the process of multiple genome editing, resulting in DSB ends that can fuse to form chromosomal translocations [218]. By fusing Cas9 with 3’-repair exonuclease 2 (TREX2) to generate an exogenous nuclease called Cas9 exonuclease (Cas9TX), TRAC, TRBC, and PDCD1 knockout CAR T-cells were constructed. The results demonstrated that Cas9TX effectively suppressed repetitive cleavage at target sites and reduced the proportion of chromosomal translocations to background levels without compromising antitumor efficacy [218]. Finally, since the safety risks associated with permanent DNA editing, such as genotoxicity, cannot be completely eliminated. CRISPRi at the epigenetic level, when coupled with HER2 CAR and PD-1 sgRNA expression, can suppress intracellular PD-1 expression in a HER2+ environment. The first clinically relevant product constructed based on CRISPRi, RB-340-1 CAR T-cells, achieved effective and sustained PD-1 gene inhibition and antitumor activity in HER2-expressing cancer xenograft models [220, 221]. Similarly, Cas13d, a reversible and flexible editor targeting transcriptomic mRNA levels, also shows potential for clinical application [39].
Delivery methods for CRISPR components
Delivery is a major bottleneck for somatic-cell genome editing [12]. Although traditional viral vector-mediated delivery of CRISPR components has demonstrated high efficiency, it is constrained by limited cargo capacity and safety risks, making it difficult to meet the increasing demands for improved quality and reduced costs of CAR T-cell production. Additionally, in vivo delivery offers the advantages of simplicity, cost-effectiveness, and direct clinical application compared to ex vivo methods. However, it still faces significant challenges in transitioning to practical applications. These challenges include cargo degradation during delivery, inability to release cargo after endocytosis, and uptake and editing of off-target cells. Continuous optimization of delivery systems is required to achieve optimal cargo capacity, delivery efficiency, target specificity, and safety (Fig. 6B).
Currently, electroporation is the most popular non-viral delivery method. Unlike the complex process of constructing viral vectors, electroporation is relatively cost-effective and operationally simple [63, 222, 223]. Moreover, it is not limited by the form and size of cargo, allowing the introduction of CRISPR components into T cells in the form of RNA or RNP. Components such as RNA or RNP can rapidly convert into Cas9 protein, inducing genome cleavage more quickly and exhibiting higher gene editing efficiency. The transient nature of delivered components results in their rapid degradation within the cells, thereby reducing the occurrence of off-target effects [224]. Furthermore, electroporation allows the simultaneous delivery of various forms of HDR templates encoding CAR, providing a tool for the development of non-viral CAR T-cell products based on CRISPR system [63, 225,226,227,228,229]. It is worth noting that electroporation delivery still carries some degree of toxicity. On the one hand, electroporation may induce cell death due to the perforation of the cell membrane. To minimize damage, it is important to rigorously control the magnitude of the electric current [230]. Inhibiting the expression of caspase-3 has also been shown to alleviate CAR T-cell apoptosis caused by electroporation [231]. On the other hand, when using plasmid or linear long double-stranded DNA (dsDNA) as the electroporation-encoded HDR template for CAR, the existing DNA toxicity can significantly reduce T cell activity [230]. Research has discovered that dsDNA can induce the death of primary T cells through activating the cGAS-STING immune pathway [223]. By adjusting the osmotic pressure of the electroporation buffer to an isotonic level, the affinity between DNA and cGAS is reduced, subsequently lowering the activation of cGAS-STING. This makes the efficient construction of CAR T-cells possible [223]. Moreover, compared to the sole electroporation transfection of long dsDNA, co-electroporation of RNP and long dsDNA can generate CAR T-cells with higher activity. Another approach to avoid DNA toxicity is using single-stranded DNA (ssDNA) templates. Considering the relatively low integration efficiency of ssDNA templates, a study encoded CAR in ssDNA templates with two double-stranded ends added for Cas9 RNP attachment. This increases CAR insertion efficiency considerably and permits the generation of plenty of CAR T-cells that target multiple myeloma [222]. Additionally, cell-penetrating peptides (CPPs), such as TAT, facilitate the internalization of gene editing tools through endocytosis into cellular vesicles. Fusogenic peptides, like HA2, release the editing enzymes into the cytoplasm by disrupting the endosome [232, 233]. In one study, Cas9 RNPs or Cas12a RNPs are combined with nuclear localization signals (NLS), TAT, and HA2, then co-incubating with CAR T-cells for a short period of time, enables safe and non-toxic delivery of Cas9 RNP to CAR T-cell nuclei for gene knockout without unwanted perturbation [232, 233]. Peptides possess favorable biocompatibility and low cytotoxicity, and peptide-assisted gene editing minimizes cell handling steps, making it a promising delivery method for the future [234].
The above non-viral delivery methods are limited to ex vivo cell processing or local administration. Novel synthetic vectors, however, offer the potential for in vivo editing and production of CAR T-cell. This approach avoids the complex and costly ex vivo manufacturing processes [202, 216]. Lipid Nanoparticles (LNPs) are a common means for in vivo delivery of CRISPR system. LNPs encapsulate CRISPR components in the form of RNA and are internalized by cells through endocytosis, causing minimal damage to edited cells [235]. Surface modification of LNPs targeting CD5 has successfully delivered FAP CAR mRNA in vivo, leading to the generation of FAP CAR T-cells in mouse T cells [236]. Using LNPs to deliver Cas9, CD19 CAR mRNA, and gRNAs enables the dual knockout of TCR and CD52 in vitro, resulting in the production of highly functional UCAR T-cells [237]. The potential for in vivo delivery warrants further validation. Additionally, enveloped delivery vehicles (EDVs), such as virus-like particles (VLPs) hidden within cell membrane fragments, are also important vectors for in vivo delivery. They combine the safety and reliability of non-viral carriers with the efficient infection capabilities of viral carriers [238, 239]. Conceptual validation has demonstrated that Cas9-VLPs, with the surface addition of the Human Immunodeficiency Virus (HIV)-1 envelope glycoprotein (Env), can achieve preferential and specific transduction of CD4+ T cells [240]. Recently, the same research team has engineered Cas9-EDV to package Cas9 RNP, successfully generating genetically edited CAR T-cells in mice with a humanized immune system. Importantly, off-target delivery to bystander cells, including liver cells, was avoided [239]. Cas9-EDVs incorporate a mutated vesicular stomatitis virus glycoprotein (VSVGmut) and an antibody-derived single-chain variable fragment on the particle surface. This enables transient delivery to homologous target cells while maintaining endosomal fusion activity. The predictable antibody-antigen interaction selectively delivers genome editing components to target cells, presenting a novel strategy for in vivo complex genetic engineering with significant therapeutic potential [239, 240].
Discussion
Currently, CAR T-cell therapy faces three major roadblocks: efficacy, safety, and accessibility. The CRISPR system, as a simple, efficient, and versatile gene editing tool, provides potential solutions. Firstly, limited efficacy of CAR T-cell therapy is still observed in most solid tumors and some hematological malignancies. Various factors including tumor antigen escape, intrinsic resistance, poor quality and exhaustion of T cells, and inhibition of tumor microenvironment contribute to suboptimal efficacy. CRISPR system-mediated knockout of inhibitory targets on CAR T-cells partially unleashes their anti-tumor potential. To date, clinical evidence on CRISPR system-edited CAR T-cells remain limited, and large-scale clinical trials are still required to validate their efficacy. Secondly, developing CAR T-cells with more controlled pro-inflammatory cytokine secretion is a solution to mitigate CAR T-cell therapy-related toxicity. Gene-engineered CAR T-cells based on CRISPR system have advantages in terms of safety and indications. For on-target, off-tumor toxicities of CAR T-cells, CRISPR system-mediated knockout of shared antigens in specific cases can partially avoid off-target killing. Thirdly, UCAR T-cells has significantly enhanced the clinical accessibility of CAR T-cell therapy. However, before entering clinical applications, UCAR T-cells must avoid TCR-mediated allogeneic reactions. The use of CRISPR for multi-gene editing, combined with HDR-mediated targeted insertion of CAR expression cassette, has markedly addressed the challenges of inefficiency and high risk in the production process of UCAR T-cells. The key issue of GVHD in UCAR T clinical applications has been largely addressed through the CRISPR system and stringent production screening. Therefore, the limited proliferation and persistence of UCAR T-cells have become significant limiting factors. Most clinical trial results also indicate a disparity in efficacy between UCAR T and CAR T-cell therapies. This may be attributed to the complex in vivo immune rejection environment, which imposes inherent immune resistance against UCAR T-cells. Currently, CRISPR-mediated gene editing can only partially address HVGR, primarily serving as a necessary adjunct to lymphodepletion regimens, or requiring more complex gene-editing strategies to better adapt to the allogeneic immune environment. Moving forward, greater attention is needed to advance solutions to this issue. Notably, CRISPR provides technical support for exploring more cost-effective and controllable cell sources, such as iPSCs. iPSCs exhibit a higher tolerance for multiple genetic edits and can be precisely selected and expanded to produce homogeneous products with knockout of immunogenic genes, thereby enhancing tissue compatibility and avoiding HVGR. However, the efficacy of iPSC-derived CAR T-cell therapies remains a critical area requiring further focus and development.
CRISPR is often used as an “execution tool” to edit the identified targets when addressing the challenges mentioned above. However, CRISPR can also serve as a “screening tool” that provides an unbiased and precise method for genome-wide functional gene and regulatory element targeting, with great potential for identifying immune regulatory targets that are directly clinically relevant in tumors and CAR T-cells. Based on RNA editing, multi-gene combinatorial screening can also uncover multiple synergistic targets, providing stronger gains for the engineering optimization of CAR T-cells. Nevertheless, current CRISPR screening analyses used in CAR T-cell therapy mainly focus on the overall sgRNA frequency in target phenotype cell samples, neglecting cell-to-cell heterogeneity. Single-cell CRISPR (scCRISPR) refers to CRISPR screening at the single-cell level. Single-cell sequencing used for CRISPR screening can effectively address the issue of genetic and phenotypic heterogeneity of cells during large-scale perturbations. For CAR T-cells, starting phenotype of individual cells as well as their dynamic changes after antigen stimulation differ considerably. Applying scCRISPR can connect the characteristics of different CAR T-cell subpopulations with large-scale perturbations, unraveling the specific molecular mechanisms of sgRNA affecting the activation and exhaustion of CAR T-cells at the single-cell level. Tumor cells exhibit high heterogeneity due to genomic instability and epigenetic modifications. scCRISPR screening of tumor cells can be used to elucidate the specific regulatory mechanisms of tumor cell survival and proliferation, as well as the intrinsic genetic changes experienced after being targeted by CAR T-cells, providing better insights into resistance to CAR T-cell therapy.
Notably, certain challenges still exist in CRISPR gene editing technology, such as unpredictable off-target mutations. To address practical issues in CAR T-cell therapy applications, improvements are required to be posed to the CRISPR system itself. Crucial factors include optimizing the CRISPR system for highly specific targeting and lessening damage to genome caused by Cas proteins. Moreover, it is necessary to detect off-target mutations sensitively and efficiently through high-throughput sequencing technologies and to predict the targeting efficiency and off-target spectrum using machine learning or deep learning models. Furthermore, the hurdles encountered by delivery systems are impeding the swift progression of CRISPR components into applications, necessitating the optimization of existing delivery strategies and continuous innovation in delivery methods. Currently, viral-free production methods are a hot topic for improving CAR T-cell production, and may further simplify the production process in the future. Since the editing of CAR T-cells primarily occurs ex vivo, practical, efficient, and capacity-unlimited delivery methods such as electroporation are evidently more favored. Additionally, with advancements in the development of synthetic vectors, there is potential for CRISPR components to achieve in vivo delivery, enabling the generation of CAR T-cells and genome editing within the body, thereby further enhancing the accessibility of CAR T-cells.
In conclusion, the utilization of the CRISPR system offers a novel approach to address the significant scientific challenges of efficacy, safety, and accessibility in CAR T-cell therapy. The application of CRISPR-based CAR T-cell therapy organically integrates preparation and optimization, closely linking basic research with clinical research. This presents robust rationale and opportunities for designing and applying CAR T-cell therapy to enhance therapeutic benefits.
References
June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361–5.
US Food and Drug Administration: KYMRIAH (tisagenlecleucel). 2024. https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/kymriah-tisagenlecleucel. Accessed 9 Oct 2024.
US Food and Drug Administration: YESCARTA (axicabtagene ciloleucel). 2024. https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/yescarta-axicabtagene-ciloleucel. Accessed 9 Oct 2024.
US Food and Drug Administration: TECARTUS (brexucabtagene autoleucel). 2024. https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/tecartus-brexucabtagene-autoleucel. Accessed 9 Oct 2024.
US Food and Drug Administration: BREYANZI (lisocabtagene maraleucel). 2024. https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/breyanzi-lisocabtagene-maraleucel. Accessed 9 Oct 2024.
US Food and Drug Administration: ABECMA (idecabtagene vicleucel). 2024. https://www.fda.gov/vaccines-blood-biologics/abecma-idecabtagene-vicleucel. Accessed 9 Oct 2024.
US Food and Drug Administration: CARVYKTI (ciltacabtagene autoleucel). 2024. https://www.fda.gov/vaccines-blood-biologics/carvykti. Accessed 9 Oct 2024.
Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. 2020;19:185–99.
Amini L, Silbert SK, Maude SL, Nastoupil LJ, Ramos CA, Brentjens RJ, et al. Preparing for CAR T cell therapy: patient selection, bridging therapies and lymphodepletion. Nat Rev Clin Oncol. 2022;19:342–55.
Morris EC, Neelapu SS, Giavridis T, Sadelain M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat Rev Immunol. 2022;22:85–96.
Zhang H-X, Zhang Y, Yin H. Genome Editing with mRNA Encoding ZFN, TALEN, and Cas9. Mol Ther. 2019;27:735–46.
Doudna JA. The promise and challenge of therapeutic genome editing. Nature. 2020;578:229–36.
Pickar-Oliver A, Gersbach CA. The next generation of CRISPR-Cas technologies and applications. Nat Rev Mol Cell Biol. 2019;20:490–507.
Wang JY, Doudna JA. CRISPR technology: A decade of genome editing is only the beginning. Science. 2023;379:eadd8643.
Wang D, Zhang F, Gao G. CRISPR-Based Therapeutic Genome Editing: Strategies and In Vivo Delivery by AAV Vectors. Cell. 2020;181:136–50.
Yeh CD, Richardson CD, Corn JE. Advances in genome editing through control of DNA repair pathways. Nat Cell Biol. 2019;21:1468–78.
Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, et al. Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol. 2020;18:67–83.
Liu X, Zhang Y, Cheng C, Cheng AW, Zhang X, Li N, et al. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2017;27:154–7.
Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–71.
Hille F, Richter H, Wong SP, Bratovič M, Ressel S, Charpentier E. The Biology of CRISPR-Cas: Backward and Forward. Cell. 2018;172:1239–59.
Strohkendl I, Saifuddin FA, Rybarski JR, Finkelstein IJ, Russell R. Kinetic Basis for DNA Target Specificity of CRISPR-Cas12a. Mol Cell. 2018;71:816–24.e3
Kleinstiver BP, Tsai SQ, Prew MS, Nguyen NT, Welch MM, Lopez JM, et al. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat Biotechnol. 2016;34:869–74.
Singh J, Liu KG, Allen A, Jiang W, Qin PZ. A DNA unwinding equilibrium serves as a checkpoint for CRISPR-Cas12a target discrimination. Nucleic Acids Res. 2023;51:8730–43.
Kim D, Kim J, Hur JK, Been KW, Yoon S, Kim J-S. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat Biotechnol. 2016;34:863–8.
Swarts DC, Jinek M. Cas9 versus Cas12a/Cpf1: Structure-function comparisons and implications for genome editing. Wiley Interdiscip Rev RNA. 2018;9:e1481.
Xun G, Zhu Z, Singh N, Lu J, Jain PK, Zhao H. Harnessing noncanonical crRNA for highly efficient genome editing. Nat Commun. 2024;15:1–13.
Siegler EL, Simone BW, Sakemura R, Tapper EE, Horvei P, Cox MJ, et al. Efficient Gene Editing of CART Cells with CRISPR-Cas12a for Enhanced Antitumor Efficacy. Blood. 2020;136:6–7.
Zetsche B, Heidenreich M, Mohanraju P, Fedorova I, Kneppers J, DeGennaro EM, et al. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat Biotechnol. 2017;35:31–4.
Dai X, Park JJ, Du Y, Kim HR, Wang G, Errami Y, et al. One-step generation of modular CAR-T cells with AAV–Cpf1. Nat Methods. 2019;16:247–54.
Li T, Zhu L, Xiao B, Gong Z, Liao Q, Guo J. CRISPR-Cpf1-mediated genome editing and gene regulation in human cells. Biotechnol Adv. 2019;37:21–27.
Paul B, Montoya G. CRISPR-Cas12a: Functional overview and applications. Biomed J. 2020;43:8–17.
Dai X, Park JJ, Du Y, Kim HR, Wang G, Errami Y, et al. One-step generation of modular CAR-T cells with AAV-Cpf1. Nat Methods. 2019;16:247–54.
Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, et al. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell. 2013;152:1173–83.
Liu G, Lin Q, Jin S, Gao C. The CRISPR-Cas toolbox and gene editing technologies. Mol Cell. 2022;82:333–47.
Nakamura M, Gao Y, Dominguez AA, Qi LS. CRISPR technologies for precise epigenome editing. Nat Cell Biol. 2021;23:11–22.
Goell JH, Hilton IB. CRISPR/Cas-Based Epigenome Editing: Advances, Applications, and Clinical Utility. Trends Biotechnol. 2021;39:678–91.
Konermann S, Lotfy P, Brideau NJ, Oki J, Shokhirev MN, Hsu PD. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell. 2018;173:665–76.e14
Kim VN. RNA-targeting CRISPR comes of age. Nat Biotechnol. 2018;36:44–5.
Tieu V, Sotillo E, Bjelajac JR, Chen C, Malipatlolla M, Guerrero JA, et al. A versatile CRISPR-Cas13d platform for multiplexed transcriptomic regulation and metabolic engineering in primary human T cells. Cell. 2024;187:1278–95. e20
Zhao F, Zhang T, Sun X, Zhang X, Chen L, Wang H, et al. A strategy for Cas13 miniaturization based on the structure and AlphaFold. Nat Commun. 2023;14:5545.
Porto EM, Komor AC, Slaymaker IM, Yeo GW. Base editing: advances and therapeutic opportunities. Nat Rev Drug Discov. 2020;19:839–59.
Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. 2020;38:824–44.
Liang Y, Xie J, Zhang Q, Wang X, Gou S, Lin L, et al. AGBE: a dual deaminase-mediated base editor by fusing CGBE with ABE for creating a saturated mutant population with multiple editing patterns. Nucleic Acids Res. 2022;50:5384–99.
Ye L, Zhao D, Li J, Wang Y, Li B, Yang Y, et al. Glycosylase-based base editors for efficient T-to-G and C-to-G editing in mammalian cells. Nat Biotechnol. 2024; https://doi.org/10.1038/s41587-023-02050-w.
Rees HA, Liu DR. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet. 2018;19:770–88.
Kuscu C, Parlak M, Tufan T, Yang J, Szlachta K, Wei X, et al. CRISPR-STOP: gene silencing through base-editing-induced nonsense mutations. Nat Methods. 2017;14:710–2.
Gapinske M, Luu A, Winter J, Woods WS, Kostan KA, Shiva N, et al. CRISPR-SKIP: programmable gene splicing with single base editors. Genome Biol. 2018;19:107.
Webber BR, Lonetree C-L, Kluesner MG, Johnson MJ, Pomeroy EJ, Diers MD, et al. Highly efficient multiplex human T cell engineering without double-strand breaks using Cas9 base editors. Nat Commun. 2019;10:5222.
Fiflis DN, Rey NA, Venugopal-Lavanya H, Sewell B, Mitchell-Dick A, Clements KN, et al. Repurposing CRISPR-Cas13 systems for robust mRNA trans-splicing. Nat Commun. 2024;15:2325.
Fiumara M, Ferrari S, Omer-Javed A, Beretta S, Albano L, Canarutto D, et al. Genotoxic effects of base and prime editing in human hematopoietic stem cells. Nat Biotechnol. 2024;42:877–91.
Yan J, Oyler-Castrillo P, Ravisankar P, Ward CC, Levesque S, Jing Y, et al. Improving prime editing with an endogenous small RNA-binding protein. Nature. 2024;628:639–47.
Petri K, Zhang W, Ma J, Schmidts A, Lee H, Horng JE, et al. CRISPR prime editing with ribonucleoprotein complexes in zebrafish and primary human cells. Nat Biotechnol. 2022;40:189–93.
Pomeroy EJ, Anzalone AV, Podracky CJ, Bloch NB, Chang R, Dwivedi AA, et al. Multiplex Prime Editing and PASSIGE TM for Non-Viral Generation of an Allogeneic CAR-T Cell Product. Blood. 2023;142:4803.
Liu B, Dong X, Zheng C, Keener D, Chen Z, Cheng H, et al. Targeted genome editing with a DNA-dependent DNA polymerase and exogenous DNA-containing templates. Nat Biotechnol. 2024;42:1039–45.
Ferreira da Silva J, Tou CJ, King EM, Eller ML, Rufino-Ramos D, Ma L, et al. Click editing enablesprogrammable genome writing using DNA polymerases and HUH endonucleases. NatBiotechnol. 2024; https://doi.org/10.1038/s41587-024-02324-x. online ahead of print.
Wagner DL, Koehl U, Chmielewski M, Scheid C, Stripecke R. Review: Sustainable Clinical Development of CAR-T Cells - Switching From Viral Transduction Towards CRISPR-Cas Gene Editing. Front Immunol. 2022;13:865424.
Milone MC, O’Doherty U. Clinical use of lentiviral vectors. Leukemia. 2018;32:1529–41.
Chen W, Tan L, Zhou Q, Li W, Li T, Zhang C, et al. AAVS1 site-specific integration of the CAR gene into human primary T cells using a linear closed-ended AAV-based DNA vector. J Gene Med. 2020;22:e3157.
Hale M, Lee B, Honaker Y, Leung W-H, Grier AE, Jacobs HM, et al. Homology-Directed Recombination for Enhanced Engineering of Chimeric Antigen Receptor T Cells. Mol Ther Methods Clin Dev. 2017;4:192–203.
Odak A, Yuan H, Feucht J, Cantu VA, Mansilla-Soto J, Kogel F, et al. Novel extragenic genomic safe harbors for precise therapeutic T-cell engineering. Blood. 2023;141:2698–712.
Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543:113–7.
Kath J, Franke C, Drosdek V, Du W, Glaser V, Fuster-Garcia C, et al. Integration of ζ-deficient CARs into the CD3ζ gene conveys potent cytotoxicity in T and NK cells. Blood. 2024;143:2599–611.
Zhang J, Hu Y, Yang J, Li W, Zhang M, Wang Q, et al. Non-viral, specifically targeted CAR-T cells achieve high safety and efficacy in B-NHL. Nature. 2022;609:369–74.
Allen AG, Khan SQ, Margulies CM, Viswanathan R, Lele S, Blaha L, et al. A highly efficient transgene knock-in technology in clinically relevant cell types. Nat Biotechnol. 2024;42:458–69.
Mansilla-Soto J, Eyquem J, Haubner S, Hamieh M, Feucht J, Paillon N, et al. HLA-independent T cell receptors for targeting tumors with low antigen density. Nat Med. 2022;28:345–52.
Manske K, Dreßler L, Fräßle SP, Effenberger M, Tschulik C, Cletiu V, et al. Miniaturized CAR knocked onto CD3ε extends TCR function with CAR specificity under control of endogenous TCR signaling cascade. J Immunol Methods. 2024;526:113617.
Du W, Noyan F, McCallion O, Drosdek V, Kath J, Glaser V, et al. Gene editing of CD3 epsilon gene to redirect regulatory T cells for adoptive T cell transfer. bioRxiv. 2024. https://www.biorxiv.org/content/10.1101/2024.03.18.584896v1. Accessed 9 Oct 2024.
Verdun N, Marks P. Secondary Cancers after Chimeric Antigen Receptor T-Cell Therapy. N. Engl J Med. 2024;390:584–6.
Razeghian E, Nasution MKM, Rahman HS, Gardanova ZR, Abdelbasset WK, Aravindhan S, et al. A deep insight into CRISPR/Cas9 application in CAR-T cell-based tumor immunotherapies. Stem Cell Res Ther. 2021;12:428.
Yuan Q, Gao X. Multiplex base- and prime-editing with drive-and-process CRISPR arrays. Nat Commun. 2022;13:2771.
McCarty NS, Graham AE, Studená L, Ledesma-Amaro R. Multiplexed CRISPR technologies for gene editing and transcriptional regulation. Nat Commun. 2020;11:1281.
Ling X, Chang L, Chen H, Gao X, Yin J, Zuo Y, et al. Improving the efficiency of CRISPR-Cas12a-based genome editing with site-specific covalent Cas12a-crRNA conjugates. Mol Cell. 2021;81:4747–56. e7
Glaser V, Flugel C, Kath J, Du W, Drosdek V, Franke C, et al. Combining different CRISPR nucleases for simultaneous knock-in and base editing prevents translocations in multiplex-edited CAR T cells. Genome Biol. 2023;24:89.
Smith CJ, Castanon O, Said K, Volf V, Khoshakhlagh P, Hornick A, et al. Enabling large-scale genome editing at repetitive elements by reducing DNA nicking. Nucleic Acids Res. 2020;48:5183–95.
Bock C, Datlinger P, Chardon F, Coelho MA, Dong MB, Lawson KA, et al. High-content CRISPR screening. Nat Rev Methods Prim. 2022;2:9.
Shi H, Doench JG, Chi H. CRISPR screens for functional interrogation of immunity. Nat Rev Immunol. 2023;23:363–80.
Schmidt R, Ward CC, Dajani R, Armour-Garb Z, Ota M, Allain V, et al. Base-editing mutagenesis maps alleles to tune human T cell functions. Nature. 2024;625:805–12.
Walsh ZH, Shah P, Kothapalli N, Shah SB, Nikolenyi G, Brodtman DZ, et al. Mapping variant effects on anti-tumor hallmarks of primary human T cells with base-editing screens. Nat Biotechnol. 2024;
Przybyla L, Gilbert LA. A new era in functional genomics screens. Nat Rev Genet. 2022;23:89–103.
Liu D, Zhao X, Tang A, Xu X, Liu S, Zha L, et al. CRISPR screen in mechanism and target discovery for cancer immunotherapy. Biochim Biophys Acta Rev Cancer. 2020;1874:188378.
Ye L, Park JJ, Peng L, Yang Q, Chow RD, Dong MB, et al. A genome-scale gain-of-function CRISPR screen in CD8 T cells identifies proline metabolism as a means to enhance CAR-T therapy. Cell Metab. 2022;34:595–614. e14
Schmidt R, Steinhart Z, Layeghi M, Freimer JW, Bueno R, Nguyen VQ, et al. CRISPR activation and interference screens decode stimulation responses in primary human T cells. Science. 2022;375:eabj4008.
Dahlman JE, Abudayyeh OO, Joung J, Gootenberg JS, Zhang F, Konermann S. Orthogonal gene knockout and activation with a catalytically active Cas9 nuclease. Nat Biotechnol. 2015;33:1159–61.
Wang D, Prager BC, Gimple RC, Aguilar B, Alizadeh D, Tang H, et al. CRISPR Screening of CAR T Cells and Cancer Stem Cells Reveals Critical Dependencies for Cell-Based Therapies. Cancer Discov. 2021;11:1192–211.
CLASH enables large-scale parallel knock-in for cell engineering. Nat Biotechnol. 2023; 41:1202-3.
Dai X, Park JJ, Du Y, Na Z, Lam SZ, Chow RD, et al. Massively parallel knock-in engineering of human T cells. Nat Biotechnol. 2023;41:1239–55.
Yan X, Chen D, Ma X, Wang Y, Guo Y, Wei J, et al. CD58 loss in tumor cells confers functional impairment of CAR T cells. Blood Adv. 2022;6:5844–56.
Larson RC, Kann MC, Bailey SR, Haradhvala NJ, Llopis PM, Bouffard AA, et al. CAR T cell killing requires the IFNγR pathway in solid but not liquid tumours. Nature. 2022;604:563–70.
Witkowski MT, Lee S, Wang E, Lee AK, Talbot A, Ma C, et al. NUDT21 limits CD19 levels through alternative mRNA polyadenylation in B cell acute lymphoblastic leukemia. Nat Immunol. 2022;23:1424–32.
Hagel KR, Arafeh R, Gang S, Arnoff TE, Larson RC, Doench JG, et al. Systematic Interrogation of Tumor Cell Resistance to Chimeric Antigen Receptor T-cell Therapy in Pancreatic Cancer. Cancer Res. 2023;83:613–25.
Dufva O, Koski J, Maliniemi P, Ianevski A, Klievink J, Leitner J, et al. Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood. 2020;135:597–609.
Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer. 2021;21:145–61.
He X, Xu C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020;30:660–9.
Xiao X, Wang Y, Zou Z, Yang Y, Wang X, Xin X, et al. Combination strategies to optimize the efficacy of chimeric antigen receptor T cell therapy in haematological malignancies. Front Immunol. 2022;13:954235.
Wang D-R, Wu X-L, Sun Y-L. Therapeutic targets and biomarkers of tumor immunotherapy: response versus non-response. Signal Transduct Target Ther. 2022;7:331.
Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin Cancer Res. 2017;23:2255–66.
Nakazawa T, Natsume A, Nishimura F, Morimoto T, Matsuda R, Nakamura M, et al. Effect of CRISPR/Cas9-Mediated PD-1-Disrupted Primary Human Third-Generation CAR-T Cells Targeting EGFRvIII on In Vitro Human Glioblastoma Cell Growth. Cells. 2020;9:998.
Choi BD, Yu X, Castano AP, Darr H, Henderson DB, Bouffard AA, et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J Immunother Cancer. 2019;7:304.
Zhang Y, Zhang X, Cheng C, Mu W, Liu X, Li N, et al. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front Med. 2017;11:554–62.
De Munter S, Buhl JL, De Cock L, Van Parys A, Daneels W, Pascal E, et al. Knocking Out CD70 Rescues CD70-Specific NanoCAR T Cells from Antigen-Induced Exhaustion. Cancer Immunol Res. 2024;12:1236–51.
Zhang W, Shi L, Zhao Z, Du P, Ye X, Li D, et al. Disruption of CTLA-4 expression on peripheral blood CD8 + T cell enhances anti-tumor efficacy in bladder cancer. Cancer Chemother Pharm. 2019;83:911–20.
Carnevale J, Shifrut E, Kale N, Nyberg WA, Blaeschke F, Chen YY, et al. RASA2 ablation in T cells boosts antigen sensitivity and long-term function. Nature. 2022;609:174–82.
Patel RP, Ghilardi G, Zhang Y, Chiang Y-H, Xie W, Guruprasad P, et al. CD5 deletion enhances the antitumor activity of adoptive T cell therapies. Sci Immunol. 2024;9:eadn6509.
Guruprasad P, Carturan A, Zhang Y, Cho JH, Kumashie KG, Patel RP, et al. The BTLA-HVEM axis restricts CAR T cell efficacy in cancer. Nat Immunol. 2024;25:1020–32.
Wieboldt R, Sandholzer M, Carlini E, Lin C-W, Börsch A, Zingg A, et al. Engagement of sialylated glycans with Siglec receptors on suppressive myeloid cells inhibits anticancer immunity via CCL2. Cell Mol Immunol. 2024;21:495–509.
Jackson Z, Hong C, Schauner R, Dropulic B, Caimi PF, de Lima M, et al. Sequential Single-Cell Transcriptional and Protein Marker Profiling Reveals TIGIT as a Marker of CD19 CAR-T Cell Dysfunction in Patients with Non-Hodgkin Lymphoma. Cancer Discov. 2022;12:1886–903.
Mortezaee K, Majidpoor J, Najafi S. VISTA immune regulatory effects in bypassing cancer immunotherapy: Updated. Life Sci. 2022;310:121083.
Hou AJ, Chen LC, Chen YY. Navigating CAR-T cells through the solid-tumour microenvironment. Nat Rev Drug Discov. 2021;20:531–50.
Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharm Ther. 2021;221:107753.
de Visser KE, Joyce JA. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell. 2023;41:374–403.
Bejarano L, Jordāo MJC, Joyce JA. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021;11:933–59.
Chen W. TGF-β Regulation of T Cells. Annu Rev Immunol. 2023;41:483–512.
Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, et al. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. 2020;5:e133977.
Alishah K, Birtel M, Masoumi E, Jafarzadeh L, Mirzaee HR, Hadjati J, et al. CRISPR/Cas9-mediated TGFβRII disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells in vitro. J Transl Med. 2021;19:482.
Jung I-Y, Kim Y-Y, Yu H-S, Lee M, Kim S, Lee J. CRISPR/Cas9-Mediated Knockout of DGK Improves Antitumor Activities of Human T Cells. Cancer Res. 2018;78:4692–703.
Mastelic-Gavillet B, Navarro Rodrigo B, Décombaz L, Wang H, Ercolano G, Ahmed R, et al. Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8+ T cells. J Immunother Cancer. 2019;7:257.
Allard B, Allard D, Buisseret L, Stagg J. The adenosine pathway in immuno-oncology. Nat Rev Clin Oncol. 2020;17:611–29.
Giuffrida L, Sek K, Henderson MA, Lai J, Chen AXY, Meyran D, et al. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nat Commun. 2021;12:3236.
Li N, Tang N, Cheng C, Hu T, Wei X, Han W, et al. Improving the anti-solid tumor efficacy of CAR-T cells by inhibiting adenosine signaling pathway. OncoImmunology. 2020;9:1824643.
Ho CJ, Samarasekera G, Rothe K, Xu J, Yang KC, Leung E, et al. Puncta intended: connecting the dots between autophagy and cell stress networks. Autophagy. 2021;17:1028–33.
Akbari B, Ghahri-Saremi N, Soltantoyeh T, Hadjati J, Ghassemi S, Mirzaei HR. Epigenetic strategies to boost CAR T cell therapy. Mol Ther. 2021;29:2640–59.
Huang Y, Hu KJ, Hu YX, Huang H. [Universal chimeric antigen receptor T cells therapy: current status and future perspectives]. Zhonghua Xue Ye Xue Za Zhi. 2021;42:782–6.
Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30:492–500.
Zebley CC, Youngblood B. Mechanisms of T cell exhaustion guiding next-generation immunotherapy. Trends Cancer. 2022;8:726–34.
Chen GM, Chen C, Das RK, Gao P, Chen C-H, Bandyopadhyay S, et al. Integrative Bulk and Single-Cell Profiling of Premanufacture T-cell Populations Reveals Factors Mediating Long-Term Persistence of CAR T-cell Therapy. Cancer Discov. 2021;11:2186–99.
Prinzing B, Zebley CC, Petersen CT, Fan Y, Anido AA, Yi Z, et al. Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Sci Transl Med. 2021;13:eabh0272.
Yoshikawa T, Wu Z, Inoue S, Kasuya H, Matsushita H, Takahashi Y, et al. Genetic ablation of PRDM1 in antitumor T cells enhances therapeutic efficacy of adoptive immunotherapy. Blood. 2022;139:2156–72.
Ito Y, Kagoya Y. Epigenetic engineering for optimal chimeric antigen receptor T cell therapy. Cancer Sci. 2022;113:3664–71.
Jain N, Zhao Z, Koche RP, Antelope C, Gozlan Y, Montalbano A, et al. Disruption of SUV39H1-Mediated H3K9 Methylation Sustains CAR T-cell Function. Cancer Discov. 2024;14:142–57.
Jaccard A, Wyss T, Maldonado-Pérez N, Rath JA, Bevilacqua A, Peng J-J, et al. Reductive carboxylation epigenetically instructs T cell differentiation. Nature. 2023;621:849–56.
Liu Y, Debo B, Li M, Shi Z, Sheng W, Shi Y. LSD1 inhibition sustains T cell invigoration with a durable response to PD-1 blockade. Nat Commun. 2021;12:1–16.
Wei J, Long L, Zheng W, Dhungana Y, Lim SA, Guy C, et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature. 2019;576:471–6.
Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. 2018;558:307–12.
Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. 2018;24:563–71.
Arcangeli S, Falcone L, Camisa B, De Girardi F, Biondi M, Giglio F, et al. Next-Generation Manufacturing Protocols Enriching TSCM CAR T Cells Can Overcome Disease-Specific T Cell Defects in Cancer Patients. Front Immunol. 2020;11:1217.
Das RK, Vernau L, Grupp SA, Barrett DM. Naïve T-cell Deficits at Diagnosis and after Chemotherapy Impair Cell Therapy Potential in Pediatric Cancers. Cancer Discov. 2019;9:492–9.
Mai D, Johnson O, Reff J, Fan T-J, Scholler J, Sheppard NC, et al. Combined disruption of T cell inflammatory regulators Regnase-1 and Roquin-1 enhances antitumor activity of engineered human T cells. Proc Natl Acad Sci USA. 2023;120:e2218632120.
Ren J, Zhang X, Liu X, Fang C, Jiang S, June CH, et al. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget. 2017;8:17002–11.
Kasakovski D, Xu L, Li Y. T cell senescence and CAR-T cell exhaustion in hematological malignancies. J Hematol Oncol. 2018;11:91.
Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. 2019;16:372–85.
Zou Y, Liu B, Li L, Yin Q, Tang J, Jing Z, et al. IKZF3 deficiency potentiates chimeric antigen receptor T cells targeting solid tumors. Cancer Lett. 2022;524:121–30.
Kumar J, Kumar R, Kumar Singh A, Tsakem EL, Kathania M, Riese MJ, et al. Deletion of Cbl-b inhibits CD8 + T-cell exhaustion and promotes CAR T-cell function. J Immunother Cancer. 2021;9:e001688.
O’Sullivan D, Sanin DE, Pearce EJ, Pearce EL. Metabolic interventions in the immune response to cancer. Nat Rev Immunol. 2019;19:324–35.
Guo A, Huang H, Zhu Z, Chen MJ, Shi H, Yuan S, et al. cBAF complex components and MYC cooperate early in CD8+ T cell fate. Nature. 2022;607:135–41.
Sutra Del Galy A, Menegatti S, Fuentealba J, Lucibello F, Perrin L, Helft J, et al. In vivo genome-wide CRISPR screens identify SOCS1 as intrinsic checkpoint of CD4+ TH1 cell response. Sci Immunol. 2021;6:eabe8219.
Hong Y, Walling BL, Kim H-R, Serratelli WS, Lozada JR, Sailer CJ, et al. ST3GAL1 and βII-spectrin pathways control CAR T cell migration to target tumors. Nat Immunol. 2023;24:1007–19.
Ye L, Park JJ, Dong MB, Yang Q, Chow RD, Peng L, et al. In vivo CRISPR screening in CD8 T cells with AAV–Sleeping Beauty hybrid vectors identifies membrane targets for improving immunotherapy for glioblastoma. Nat Biotechnol. 2019;37:1302–13.
Zhou P, Shi H, Huang H, Sun X, Yuan S, Chapman NM, et al. Single-cell CRISPR screens in vivo map T cell fate regulomes in cancer. Nature. 2023;624:154–63.
Xu X, Sun Q, Liang X, Chen Z, Zhang X, Zhou X, et al. Mechanisms of Relapse After CD19 CAR T-Cell Therapy for Acute Lymphoblastic Leukemia and Its Prevention and Treatment Strategies. Front Immunol. 2019;10:2664.
Yin H, Xue W, Anderson DG. CRISPR-Cas: a tool for cancer research and therapeutics. Nat Rev Clin Oncol. 2019;16:281–95.
Lawson KA, Sousa CM, Zhang X, Kim E, Akthar R, Caumanns JJ, et al. Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature. 2020;586:120–6.
Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science. 2018;359:770–5.
Singh AK, McGuirk JP. CAR T cells: continuation in a revolution of immunotherapy. Lancet Oncol. 2020;21:e168–78.
Ragoonanan D, Khazal SJ, Abdel-Azim H, McCall D, Cuglievan B, Tambaro FP, et al. Diagnosis, grading and management of toxicities from immunotherapies in children, adolescents and young adults with cancer. Nat Rev Clin Oncol. 2021;18:435–53.
Xiao X, Huang S, Chen S, Wang Y, Sun Q, Xu X, et al. Mechanisms of cytokine release syndrome and neurotoxicity of CAR T-cell therapy and associated prevention and management strategies. J Exp Clin Cancer Res. 2021;40:367.
Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20:31–42.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl J Med. 2017;377:2531–44.
Sterner RM, Sakemura R, Cox MJ, Yang N, Khadka RH, Forsman CL, et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood. 2019;133:697–709.
Dibas A, Rhiel M, Patel VB, Andrieux G, Boerries M, Cornu TI, et al. Cell-Based Models of ‘Cytokine Release Syndrome’ Endorse CD40L and Granulocyte–Macrophage Colony-Stimulating Factor Knockout in Chimeric Antigen Receptor T Cells as Mitigation Strategy. Cells. 2023;12:2581.
Yi Y, Chai X, Zheng L, Zhang Y, Shen J, Hu B, et al. CRISPR-edited CART with GM-CSF knockout and auto secretion of IL6 and IL1 blockers in patients with hematologic malignancy. Cell Discov. 2021;7:1–11.
Huang S, Wang X, Wang Y, Wang Y, Fang C, Wang Y, et al. Deciphering and advancing CAR T-cell therapy with single-cell sequencing technologies. Mol Cancer. 2023;22:80.
Flugel CL, Majzner RG, Krenciute G, Dotti G, Riddell SR, Wagner DL, et al. Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours. Nat Rev Clin Oncol. 2023;20:49–62.
Gomes-Silva D, Srinivasan M, Sharma S, Lee CM, Wagner DL, Davis TH, et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. 2017;130:285–96.
Raikar SS, Fleischer LC, Moot R, Fedanov A, Paik NY, Knight KA, et al. Development of chimeric antigen receptors targeting T-cell malignancies using two structurally different anti-CD5 antigen binding domains in NK and CRISPR-edited T cell lines. OncoImmunology. 2018;7:e1407898.
Dai Z, Mu W, Zhao Y, Jia X, Liu J, Wei Q, et al. The rational development of CD5-targeting biepitopic CARs with fully human heavy-chain-only antigen recognition domains. Mol Ther. 2021;29:2707–22.
Safarzadeh Kozani P, Safarzadeh Kozani P, Rahbarizadeh F. CAR-T cell therapy in T-cell malignancies: Is success a low-hanging fruit? Stem Cell Res Ther. 2021;12:527.
Cooper ML, Choi J, Staser K, Ritchey JK, Devenport JM, Eckardt K, et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia. 2018;32:1970–83.
Hu Y, Zhou Y, Zhang M, Zhao H, Wei G, Ge W, et al. Genetically modified CD7-targeting allogeneic CAR-T cell therapy with enhanced efficacy for relapsed/refractory CD7-positive hematological malignancies: a phase I clinical study. Cell Res. 2022;32:995–1007.
Chiesa R, Georgiadis C, Syed F, Zhan H, Etuk A, Gkazi SA, et al. Base-Edited CAR7 T Cells for Relapsed T-Cell Acute Lymphoblastic Leukemia. N Engl J Med. 2023;389:899–910.
Kim MY, Yu K-R, Kenderian SS, Ruella M, Chen S, Shin T-H, et al. Genetic Inactivation of CD33 in Hematopoietic Stem Cells to Enable CAR T Cell Immunotherapy for Acute Myeloid Leukemia. Cell. 2018;173:1439–53. e19
Borot F, Wang H, Ma Y, Jafarov T, Raza A, Ali AM, et al. Gene-edited stem cells enable CD33-directed immune therapy for myeloid malignancies. Proc Natl Acad Sci USA. 2019;116:11978–87.
Liu Y, Wang S, Schubert M, Lauk A, Yao H, Blank MF, et al. CD33 ‐directed immunotherapy with third‐generation chimeric antigen receptor T cells and gemtuzumab ozogamicin in intact and CD33 ‐edited acute myeloid leukemia and hematopoietic stem and progenitor cells. Int J Cancer. 2022;150:1141–55.
Wellhausen N, O’Connell RP, Lesch S, Engel NW, Rennels AK, Gonzales D, et al. Epitope base editing CD45 in hematopoietic cellsenables universal blood cancer immune therapy. Sci Transl Med. 2023;15:eadi1145.
Xu X, Huang S, Xiao X, Sun Q, Liang X, Chen S, et al. Challenges and Clinical Strategies of CAR T-Cell Therapy for Acute Lymphoblastic Leukemia: Overview and Developments. Front Immunol. 2020;11:569117.
Chen S, Zhang Y, Fang C, Zhang N, Wang Y, Chen R, et al. Donor-derived and off-the-shelf allogeneic anti-CD19 CAR T-cell therapy for R/R ALL and NHL: A systematic review and meta-analysis. Crit Rev Oncol Hematol. 2022;179:103807.
Wagner DL, Fritsche E, Pulsipher MA, Ahmed N, Hamieh M, Hegde M, et al. Immunogenicity of CAR T cells in cancer therapy. Nat Rev Clin Oncol. 2021;18:379–93.
Osborn MJ, Webber BR, Knipping F, Lonetree C, Tennis N, DeFeo AP, et al. Evaluation of TCR Gene Editing Achieved by TALENs, CRISPR/Cas9, and megaTAL Nucleases. Mol Ther. 2016;24:570–81.
Kamiya T, Wong D, Png YT, Campana D. A novel method to generate T-cell receptor–deficient chimeric antigen receptor T cells. Blood Adv. 2018;2:517–28.
Hu Y, Zhou Y, Zhang M, Ge W, Li Y, Yang L, et al. CRISPR/Cas9-Engineered Universal CD19/CD22 Dual-Targeted CAR-T Cell Therapy for Relapsed/Refractory B-cell Acute Lymphoblastic Leukemia. Clin Cancer Res. 2021;27:2764–72.
Hematopoietic Stem Cell Application Group, Chinese Society of Hematology, Chinese Medical Association, China Association for the Prevention of Hematology Diseases. Hematopoietic Stem Cell Application Group, Chinese Society of Hematology, Chinese Medical Association, China Association for the Prevention of Hematology Chinese consensus on the diagnosis and management of chronic graft1903 versus-host disease. Zhonghua Xue Ye Xue Za Zhi. 2021;42:265–75.
Benjamin R, Graham C, Yallop D, Jozwik A, Mirci-Danicar OC, Lucchini G, et al. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: results of two phase 1 studies. Lancet. 2020;396:1885–94.
Ottaviano G, Georgiadis C, Gkazi SA, Syed F, Zhan H, Etuk A, et al. Phase 1 clinical trial of CRISPR-engineered CAR19 universal T cells for treatment of children with refractory B cell leukemia. Sci Transl Med. 2022;14:eabq3010.
Wang B, Iriguchi S, Waseda M, Ueda N, Ueda T, Xu H, et al. Generation of hypoimmunogenic T cells from genetically engineered allogeneic human induced pluripotent stem cells. Nat Biomed Eng. 2021;5:429–40.
Martin KE, Hammer Q, Perica K, Sadelain M, Malmberg K-J. Engineering immune-evasive allogeneiccellular immunotherapies. Nat Rev Immunol. 2024;24:680–93.
Degagné É, Donohoue PD, Roy S, Scherer J, Fowler TW, Davis RT, et al. High-Specificity CRISPR-Mediated Genome Engineering in Anti-BCMA Allogeneic CAR T Cells Suppresses Allograft Rejection in Preclinical Models. Cancer Immunol Res. 2024;12:462–77.
Hu X, Manner K, DeJesus R, White K, Gattis C, Ngo P, et al. Hypoimmune anti-CD19 chimeric antigen receptor T cells provide lasting tumor control in fully immunocompetent allogeneic humanized mice. Nat Commun. 2023;14:2020.
Zhang S-Q, Thomas F, Fang J, Austgen K, Cowan C, Welstead GG Universal protection of allogeneic cell therapies from natural killer cells via CD300a agonism. bioRxiv. 2024. https://www.biorxiv.org/content/10.1101/2024.05.05.592600v1. Accessed 9 Oct 2024.
Hammer Q, Perica K, Mbofung RM, van Ooijen H, Martin KE, Momayyezi P, et al. Genetic ablation of adhesion ligands mitigates rejection of allogeneic cellular immunotherapies. Cell Stem Cell. 2024;S1934-5909(24)00219-4.
Winterhalter PM, Warmuth L, Hilgendorf P, Schütz JM, Dötsch S, Tonn T, et al. HLA reduction of human T cells facilitates generation of immunologically multicompatible cellular products. Blood Adv. 2024;8:3416–26.
Chen X, Tan B, Xing H, Zhao X, Ping Y, Zhang Z, et al. Allogeneic CAR-T cells with of HLA-A/B and TRAC disruption exhibit promising antitumor capacity against B cell malignancies. Cancer Immunol Immunother. 2024;73:13.
Kagoya Y, Guo T, Yeung B, Saso K, Anczurowski M, Wang C-H, et al. Genetic Ablation of HLA Class I, Class II, and the T-cell Receptor Enables Allogeneic T Cells to Be Used for Adoptive T-cell Therapy. Cancer Immunol Res. 2020;8:926–36.
Mo F, Watanabe N, McKenna MK, Hicks MJ, Srinivasan M, Gomes-Silva D, et al. Engineered off-the-shelf therapeutic T cells resist host immune rejection. Nat Biotechnol. 2021;39:56–63.
Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell. 2018;23:181–92. e5
Clarke R, Van Der Stegen S, Chang C-W, Husain M, Lai Y-S, Peralta E, et al. Pluripotent Cell-Derived Off-the-Shelf TCR-Less CAR-Targeted Cytotoxic T Cell Therapeutic for the Allogeneic Treatment of B Cell Malignancies. Blood. 2018;132:4546.
Mandal M, Clarke R, van der Stegen S, Chang C-W, Lai Y-S, Witty A, et al. Abstract 3245: FT819 path to IND: First-of-kind off-the-shelf CAR19 T-cell for B cell malignancies. Cancer Res. 2020;80:3245.
Mehta A, Farooq U, Chen A, McGuirk JP, Ly T, Wong L, et al. Interim Phase I Clinical Data of FT819-101, a Study of the First-Ever, Off-the-Shelf, iPSC-Derived TCR-Less CD19 CAR T-Cell Therapy for Patients with Relapsed/Refractory B-Cell Malignancies. Blood. 2022;140:4577–8.
Qasim W. Genome-edited allogeneic donor “universal” chimeric antigen receptor T cells. Blood. 2023;141:835–45.
Zhao J, Lin Q, Song Y, Liu D. Universal CARs, universal T cells, and universal CAR T cells. J Hematol Oncol. 2018;11:132.
DiNofia AM, Grupp SA. Will allogeneic CAR T cells for CD19+ malignancies take autologous CAR T cells “off the shelf”? Nat Rev Clin Oncol. 2021;18:195–6.
Wang Z, Li N, Feng K, Chen M, Zhang Y, Liu Y, et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell Mol Immunol. 2021;18:2188–98.
Pal S, Tran B, Haanen J, Hurwitz M, Sacher A, Agarwal N, et al. CTX130 allogeneic CRISPR-Cas9–engineered chimeric antigen receptor (CAR) T cells in patients with advanced clear cell renal cell carcinoma: Results from the phase 1 COBALT-RCC study. J Immunother Cancer. 2022; https://doi.org/10.1136/jitc-2022-SITC2022.0558.
Baker DJ, Arany Z, Baur JA, Epstein JA, June CH. CAR T therapy beyond cancer: the evolution of a living drug. Nature. 2023;619:707–15.
Wang X, Wu X, Tan B, Zhu L, Zhang Y, Lin L, et al. Allogeneic CD19-targeted CAR-T therapy in patients with severe myositis and systemic sclerosis. Cell. 2024;S0092-8674(24)00701–3.
Kang S-H, Lee W-J, An J-H, Lee J-H, Kim Y-H, Kim H, et al. Prediction-based highly sensitive CRISPR off-target validation using target-specific DNA enrichment. Nat Commun. 2020;11:3596.
Manghwar H, Li B, Ding X, Hussain A, Lindsey K, Zhang X, et al. CRISPR/Cas Systems in Genome Editing: Methodologies and Tools for sgRNA Design, Off-Target Evaluation, and Strategies to Mitigate Off-Target Effects. Adv Sci (Weinh). 2020;7:1902312.
Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367:eaba7365.
Chehelgerdi M, Chehelgerdi M, Khorramian-Ghahfarokhi M, Shafieizadeh M, Mahmoudi E, Eskandari F, et al. Comprehensive review of CRISPR-based gene editing: mechanisms, challenges, and applications in cancer therapy. Mol Cancer. 2024;23:9.
Kim D, Luk K, Wolfe SA, Kim J-S. Evaluating and Enhancing Target Specificity of Gene-Editing Nucleases and Deaminases. Annu Rev Biochem. 2019;88:191–220.
Madison BB, Patil D, Richter M, Li X, Tong M, Cranert S, et al. Cas-CLOVER is a novel high-fidelity nuclease for safe and robust generation of TSCM-enriched allogeneic CAR-T cells. Mol Ther Nucleic Acids. 2022;29:979–95.
Tran NT, Danner E, Li X, Graf R, Lebedin M, de la Rosa K, et al. Precise CRISPR-Cas-mediated gene repair with minimal off-target and unintended on-target mutations in human hematopoietic stem cells. Sci Adv. 2022;8:eabm9106.
Ikeda A, Fujii W, Sugiura K, Naito K. High-fidelity endonuclease variant HypaCas9 facilitates accurate allele-specific gene modification in mouse zygotes. Commun Biol. 2019;2:371.
Vakulskas CA, Dever DP, Rettig GR, Turk R, Jacobi AM, Collingwood MA, et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat Med. 2018;24:1216–24.
Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–5.
Wang Y, Cheng X, Shan Q, Zhang Y, Liu J, Gao C, et al. Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat Biotechnol. 2014;32:947–51.
Liang P, Xie X, Zhi S, Sun H, Zhang X, Chen Y, et al. Genome-wide profiling of adenine base editor specificity by EndoV-seq. Nat Commun. 2019;10:67.
Pacesa M, Pelea O, Jinek M. Past, present, and future of CRISPR genome editing technologies. Cell. 2024;187:1076–100.
Tsuchida CA, Brandes N, Bueno R, Trinidad M, Mazumder T, Yu B, et al. Mitigation of chromosome loss in clinical CRISPR-Cas9-engineered T cells. Cell. 2023;186:4567–82. e20
Yin J, Lu R, Xin C, Wang Y, Ling X, Li D, et al. Cas9 exo-endonuclease eliminates chromosomal translocations during genome editing. Nat Commun. 2022;13:1204.
Diorio C, Murray R, Naniong M, Barrera L, Camblin A, Chukinas J, et al. Cytosine base editing enables quadruple-edited allogeneic CART cells for T-ALL. Blood. 2022;140:619–29.
Li L, Gao Y, Srivastava R, Wang W, Xiong Q, Fang Z, et al. Lentiviral delivery of combinatorial CAR/CRISPRi circuit into human primary T cells is enhanced by TBK1/IKKɛ complex inhibitor BX795. J Transl Med. 2020;18:363.
Yang Z, Li L, Turkoz A, Chen P, Harari-Steinfeld R, Bobbin M, et al. Contextual reprogramming of CAR-T cells for treatment of HER2+ cancers. J Transl Med. 2021;19:459.
Shy BR, Vykunta VS, Ha A, Talbot A, Roth TL, Nguyen DN, et al. High-yield genome engineering in primary cells using a hybrid ssDNA repair template and small-molecule cocktails. Nat Biotechnol. 2023;41:521–31.
An J, Zhang C-P, Qiu H-Y, Zhang H-X, Chen Q-B, Zhang Y-M, et al. Enhancement of the viability of T cells electroporatedwith DNA via osmotic dampening of the DNA-sensing cGAS–STING pathway. NatBiomed Eng. 2024;8:149–64.
Seki A, Rutz S. Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J Exp Med. 2018;215:985–97.
Song X, Liu C, Wang N, Huang H, He S, Gong C, et al. Delivery of CRISPR/Cas systems for cancer gene therapy and immunotherapy. Adv Drug Deliv Rev. 2021;168:158–80.
Wang S-W, Gao C, Zheng Y-M, Yi L, Lu J-C, Huang X-Y, et al. Current applications and future perspective of CRISPR/Cas9 gene editing in cancer. Mol Cancer. 2022;21:57.
Moretti A, Ponzo M, Nicolette CA, Tcherepanova IY, Biondi A, Magnani CF. The Past, Present, and Future of Non-Viral CAR T Cells. Front Immunol. 2022;13:867013.
Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature. 2018;559:405–9.
Oh SA, Senger K, Madireddi S, Akhmetzyanova I, Ishizuka IE, Tarighat S, et al. High-efficiency nonviral CRISPR/Cas9-mediatedgene editing of human T cells using plasmid donor DNA. J Exp Med. 2022;219:e20211530.
Sinclair F, Begum AA, Dai CC, Toth I, Moyle PM. Recent advances in the delivery and applications of nonviral CRISPR/Cas9 gene editing. Drug Deliv Transl Res. 2023;13:1500–19.
Wang C, Chang C-C, Wang L, Yuan F. Inhibition of Caspases Improves Non-Viral T Cell Receptor Editing. Cancers. 2020;12:2603.
Zhang Z, Baxter AE, Ren D, Qin K, Chen Z, Collins SM, et al. Efficient engineering of human and mouse primary cells using peptide-assisted genome editing. Nat Biotechnol. 2024;42:305–15.
Foss DV, Muldoon JJ, Nguyen DN, Carr D, Sahu SU, Hunsinger JM, et al. Peptide-mediated delivery of CRISPR enzymes for the efficient editing of primary human lymphocytes. Nat Biomed Eng. 2023;7:647–60.
Ibrahim NE-S. Peptide-mediated CRISPR engineering of cells. Nat Rev Bioeng. 2023;1:388–388.
Mitchell MJ, Billingsley MM, Haley RM, Wechsler ME, Peppas NA, Langer R. Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discov. 2021;20:101–24.
Rurik JG, Tombácz I, Yadegari A, Méndez Fernández PO, Shewale SV, Li L, et al. CAR T cells produced in vivo to treat cardiac injury. Science. 2022;375:91–6.
Geczy R, Balgi A, Park S, Zhao R, Watt E, Wong M, et al. Genome Editing of Human Primary T Cells With Lipid Nanoparticles. 2024. https://www.bioprocessonline.com/doc/genome-editing-of-human-primary-t-cells-with-lipid-nanoparticles-0001. Accessed 9 Oct 2024.
Banskota S, Raguram A, Suh S, Du SW, Davis JR, Choi EH, et al. Engineered virus-like particles for efficient in vivo delivery of therapeutic proteins. Cell. 2022;185:250–65. e16
Hamilton JR, Chen E, Perez BS, Sandoval Espinoza CR, Kang MH, Trinidad M, et al. In vivo human T cell engineering with enveloped delivery vehicles. Nat Biotechnol. 2024; https://doi.org/10.1038/s41587-023-02085-z.
Hamilton JR, Tsuchida CA, Nguyen DN, Shy BR, McGarrigle ER, Sandoval Espinoza CR, et al. Targeted delivery of CRISPR-Cas9 and transgenes enables complex immune cell engineering. Cell Rep. 2021;35:109207.
Guo X, Jiang H, Shi B, Zhou M, Zhang H, Shi Z, et al. Disruption of PD-1 Enhanced the Anti-tumor Activity of Chimeric Antigen Receptor T Cells Against Hepatocellular Carcinoma. Front Pharm. 2018;9:1118.
Yi Y, Chai X, Zheng L, Zhang Y, Shen J, Hu B, et al. CRISPR-edited CART with GM-CSF knockout and auto secretion of IL6 and IL1 blockers in patients with hematologic malignancy. Cell Discov. 2021;7:27.
Dai Z, Mu W, Zhao Y, Cheng J, Lin H, Ouyang K, et al. T cells expressing CD5/CD7 bispecific chimeric antigen receptors with fully human heavy-chain-only domains mitigate tumor antigen escape. Sig Transduct Target Ther. 2022;7:85.
Georgiadis C, Rasaiyaah J, Gkazi SA, Preece R, Etuk A, Christi A, et al. Base-edited CAR T cells for combinational therapy against T cell malignancies. Leukemia. 2021;35:3466–81.
Liao C, Wang Y, Huang Y, Duan Y, Liang Y, Chen J, et al. CD38-Specific CAR Integrated into CD38 Locus Driven by Different Promoters Causes Distinct Antitumor Activities of T and NK Cells. Adv Sci (Weinh). 2023;10:e2207394.
Gurusamy D, Henning AN, Yamamoto TN, Yu Z, Zacharakis N, Krishna S, et al. Multi-phenotype CRISPR-Cas9 Screen Identifies p38 Kinase as a Target for Adoptive Immunotherapies. Cancer Cell. 2020;37:818–33. e9
Wang Y, Wang F, Wang L, Qiu S, Yao Y, Yan C, et al. NAD+ supplement potentiates tumor-killing function by rescuing defective TUB-mediated NAMPT transcription in tumor-infiltrated T cells. Cell Rep. 2021;36:109516.
Yan X, Chen D, Wang Y, Guo Y, Tong C, Wei J, et al. Identification of NOXA as a pivotal regulator of resistance to CAR T-cell therapy in B-cell malignancies. Sig Transduct Target Ther. 2022;7:98.
Ramkumar P, Abarientos AB, Tian R, Seyler M, Leong JT, Chen M, et al. CRISPR-based screens uncover determinants of immunotherapy response in multiple myeloma. Blood Adv. 2020;4:2899–911.
Li S, Wang X, Yuan Z, Liu L, Li Y, Liu J, et al. Abstract CT196: Early results of a safety and efficacy study of allogeneic TruUCARTM GC502 in patients with relapsed/refractory B-cell acute lymphoblastic leukemia (r/r B-ALL). Cancer Res. 2022;82:CT196.
Caribou Biosciences, Inc. Caribou Biosciences Reports Positive Clinical Data from Dose Escalation of CB-010 ANTLER Phase 1 Trial in r/r B-NHL. 2023. https://investor.cariboubio.com/news-releases/news-release-details/caribou-biosciences-reports-positive-clinical-data-dose/.
McGuirk JP, Tam CS, Kröger N, Riedell PA, Murthy HS, Ho PJ, et al. CTX110 Allogeneic CRISPR-Cas9-Engineered CAR T Cells in Patients (Pts) with Relapsed or Refractory (R/R) Large B-Cell Lymphoma (LBCL): Results from the Phase 1 Dose Escalation Carbon Study. Blood. 2022;140:10303–6.
Li S, Gao L, Yuan Z, Wu K, Liu L, Luo L, et al. Abstract LB147: Updates on clinical safety and efficacy result of GC027, the first-in-human, “Off-the-Shelf” CD7 CAR-T stand-alone therapy for adult patients with relapsed/refractory T-cell lymphoblastic leukemia (r/r T-ALL). Cancer Res. 2021;81:LB147.
Ghobadi A, Aldoss I, Maude SL, Bhojwani D, Wayne AS, Bajel A, et al. Phase 1/2 Dose-Escalation/Dose-Expansion Study of Anti-CD7 Allogeneic CAR-T Cells (WU-CART-007) in Relapsed or Refractory (R/R) T-Cell Acute Lymphoblastic Leukemia/ Lymphoblastic Lymphoma (T-ALL/LBL). Blood. 2023;142:770.
Iyer SP, Sica RA, Ho PJ, Hu B, Zain J, Prica A, et al. S262: THE COBALT-LYM STUDY OF CTX130: A PHASE 1 DOSE ESCALATION STUDY OF CD70-TARGETED ALLOGENEIC CRISPR-CAS9–ENGINEERED CAR T CELLS IN PATIENTS WITH RELAPSED/REFRACTORY (R/R) T-CELL MALIGNANCIES. HemaSphere. 2022;6:163.
Acknowledgements
Figures in this manuscript are created with BioRender.com. XX, YW, YZ, TL, YY, JCao, JH, JChen, HC and JZ are the Small Small bird (SSb) group members and acknowledge the strong support from SSb. We also sincerely appreciate the support of our best friends Shengkang Huang, Zichen Wu, Yushan Li, and Yukun Luo with this manuscript. This research was supported by grants from the National Natural Science Foundation of China (Nos. 823B2006, 82170181, 82370188), National Students’ Platform for Innovation and Entrepreneurship Training Program of China (No. 202212121023), Beijing Physician Scientist Training Project (No. BJPSTP-2024-01), Beijing Natural Science Foundation (No. 7222027), and the National Key R&D Program of China (No. 2022YFF1502000).
Author information
Authors and Affiliations
Contributions
XX contributed to conception and manuscript design. RPG and Liang Wang supervised this manuscript. TL, YW and YZ drafted the manuscript. TL, YW, YZ, and YY prepared the tables and figures. YZ and TL collected the related references. XX, YW, TL, YZ, JCao, JH, JChen, HC, JZ and Luzheng Wang participated in the revision of the manuscript. XX and Liang Wang were involved in funding acquisition. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
RPG is a consultant to Antengene Biotech LLC, Ascentage Pharma Group and NexImmune Inc.; Medical Director, FFF Enterprises Inc.; A speaker for Janssen Pharma and Hengrui Pharma; Board of Directors: Russian Foundation for Cancer Research Support; and Scientific Advisory Boards, Nanexa AB and StemRad Ltd. AH receives research support from Novartis, BMS, Incyte and Pfizer.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lei, T., Wang, Y., Zhang, Y. et al. Leveraging CRISPR gene editing technology to optimize the efficacy, safety and accessibility of CAR T-cell therapy. Leukemia 38, 2517–2543 (2024). https://doi.org/10.1038/s41375-024-02444-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-024-02444-y
This article is cited by
-
NKG2D/CD28 chimeric receptor boosts cytotoxicity and durability of CAR-T cells for solid and hematological tumors
Experimental Hematology & Oncology (2025)
-
Insights into next-generation immunotherapy designs and tools: molecular mechanisms and therapeutic prospects
Journal of Hematology & Oncology (2025)
-
Barriers and solutions for CAR-T therapy in solid tumors
Cancer Gene Therapy (2025)
-
Sustainability, intelligence, and more immunology: time to get back to the future!
Immunologic Research (2025)
