
Abstract
The polycomb protein EZH2 is up-regulated in Chronic Myeloid Leukaemia (CML) and associated with transcriptional reprogramming. Here we tested whether EZH2 might also act as a modulator of the mRNA splicing landscape to elicit its oncogenic function in CML. We treated CML cell lines with EZH2 inhibitors and detected differential splicing of several hundreds of events, potentially caused by the transcriptional regulation of splicing factors. Amongst those genes, CELF2 was identified as a candidate to mediate part of the EZH2 inhibitor induced phenotype. Upon over-expression, we observed (1) reduced cell growth, viability, and colony formation of CML cell lines, (2) a change in the splicing landscape, partially overlapping with EZH2 mediated changes, (3) the down-regulation of MYC signalling. Importantly, these findings were successfully validated in a cohort of CML patient samples, confirming the role of CELF2 as EZH2-regulated tumour-suppressor, contributing to the severe splicing de-regulation present in CML. Based on this we propose that EZH2 exerts part of its oncogenic function in CML through the transcriptional repression of splicing factors. Finally, analysis of publicly available datasets suggests that splicing modulation by EZH2 might not be restricted to CML.
Similar content being viewed by others


Introduction
Alternative mRNA splicing (AS) is a common mechanism that organisms use to expand the pool of available transcripts in different cell types and throughout differentiation. The process of mRNA splicing is performed by the spliceosome, a macromolecule consisting of five core subunits, and a multitude of auxiliary splicing regulators (SRs), modulating its activity. The efficacy of individual splicing events is determined by a complex interplay of factors, including nucleotide motifs on the mRNA itself, the regulation of SRs, transcriptional dynamics, and the regulation of chromatin structure [1,2,3]. Mutation or disruption in any of those pathways will lead to alterations in the normal splicing programme and are often associated with malignant transformations, most prominently cancers, where cancer-specific splicing events contribute to disease development and progression [4,5,6]. The importance of AS as oncogenic driver was first established by the discovery of frequently mutated core spliceosome genes in myelodysplastic neoplasm. Subsequent work showed that splicing mis-regulation is a common feature found in many different cancer types, often driven by SR de-regulation [7].
EZH2 is the enzymatically active subunit of the Polycomb Repressive Complex 2 (PRC2), a multi-protein complex mediating the formation and maintenance of heterochromatin. It deposits methylation marks at lysine 27 of histone H3 (H3K27), leading to transcriptional repression of target genes. EZH2 plays an essential role in many developmental processes, and is commonly mutated or de-regulated in numerous cancers, rewiring transcriptional networks in a cell-type specific manner. Currently, EZH2 inhibitors are in clinical use to treat cancers harbouring activating EZH2 mutants (follicular lymphoma, diffuse large B-cell lymphoma), but a more wide-spread use including cancers over-expressing the wild-type protein is an active area of interest in the field [8, 9].
One of the cancers that have been reported to show elevated levels of EZH2 is Chronic Myeloid Leukaemia (CML) [10, 11]. CML is an interesting cancer model, as it depends on a single oncogene as the principal driver of the disease, the constitutively active kinase BCR::ABL1, which arises due to a chromosomal translocation event (Philadelphia chromosome). It is also a model example of targeted therapy since the introduction of specific tyrosine kinase inhibitors (TKIs), which has transformed patient outlook. However, acquired drug resistance and the persistence of a pool of TKI insensitive leukaemic stem cells (LSCs) still pose major challenges to disease eradication and long-term management [12]. Therefore, additional treatment options targeting pathways independent of BCR::ABL1 are needed. Recent work suggests that EZH2 might be an interesting target in this context, due to its drugability and importance in leukaemic stem cells to regulate apoptotic and cell-cycle related genes [10]. In addition, there is growing evidence showing that splicing factors are de-regulated in CML, and might be new potential targets for treatment [13,14,15].
In this study we investigate a potential crosstalk between EZH2 and mRNA splicing in CML. We describe how EZH2 modulates cellular splicing patterns through the transcriptional regulation of splicing factors. We identify one factor as important target of EZH2 involved in the mediation of downstream effects such as mRNA splicing patterns, cell growth, viability, and cell signalling.
Material and methods
Patient samples
CML patient samples were obtained from the bone marrow or peripheral blood of CML patients, live cells were sorted for CD34 and maintained in HSC growth medium containing the following cytokines: SCF, FLT3-L, TPO, IL-6, GM-CSF, CCL3/MIP1α, LIF, G-CSF. CD34+ control bone marrow samples from healthy donors were purchased from Lonza and cultured in the same medium.
Colony formation assay, and viability assay
For Colony Formation Assays, K562 and human primary CD34 + CML cells were plated in semi-solid methylcellulose-based media (MethoCult), and scored after 10-15 days. To assess viability, we employed the CellTiter-Glo (CTG) assay.
RNA-seq, gene expression analysis
NGS sequencing data was processed using the CSI NGS pipeline, described previously [16]. Gene ontology (GO) analysis for differentially expressed genes (DEGs) was performed using the DAVID online tool [17].
Splicing analysis
Splicing levels and differences between groups were calculated using an in-house perl script [18] as well as rMATS [19]. To determine the enrichment of RNA binding protein motifs at differentially spliced regions, we used the rMAPS2 online tool [20].
Western blot and qRT-PCR
Quantification of proteins and mRNA levels were performed using standard procedures. A list of primers used can be found within the Supplementary Material section in Supplementary Table 1.
Datasets
RNA-seq and ChIP-seq datasets have been deposited at the European Genome-Phenome Archive: PRJEB51336, PRJEB51550, PRJEB72776, PRJEB72797-8. Previously published datasets used in this study are listed in Supplementary Table 2.
A comprehensive version of the material and methods section can be found online within the Supplementary Material.
Results
EZH2 inhibition induces splicing changes
To test a potential role of EZH2 in the regulation of mRNA splicing, we treated the CML cell line K562 with the EZH2 inhibitors GSK126 or EPZ-6438, leading to loss of H3K27me3 (Supplementary Fig. 1A). RNA-seq was performed at high depth, before submitting samples to splicing analysis using two bioinformatics tools: a perl-based in-house developed tool [18] as well as the popular splicing analysis tool rMATS [19]. Using the perl-based script and setting a change in the splice-index (ΔSI) of 5% as cut-off, we detected several hundreds of alternatively spliced events after GSK126 or EPZ-6438 treatment (1025, or 765 respectively, Supplementary Fig. 1B, C), with a good correlation between the two different drugs (Supplementaryl Fig. 1D). Focusing on high-confidence events generally enriched upon EZH2 inhibitor treatment (GSK126 and EPZ-6438), we detected 295 events in 256 genes (Fig. 1A). Figure 1B shows the genome-tracks of five regions associated with differential splicing events. In order to experimentally validate our findings, we performed qRT-PCR for nine genes, using primers specifically targeting either the normal, or the alternatively spliced isoform. As shown in Fig. 1C we successfully confirmed alternative splicing of six genes for both EZH2 inhibitors, of one gene for GSK126, but not EPZ-6438, while only two genes did not reproduce our in silico results. To complement our analysis, we also employed another bioinformatic tool for splicing analysis, the widely used rMATS. Comparison of EZH2 inhibitor treated samples versus DMSO-treated controls led to the discovery of 879 differentially spliced events, with the main proportion comprising SE events (Supplementary Fig. 2A–C). In line with previous reports comparing different splicing algorithms, the overlap between rMATS and perl-script target genes was limited [21] (Supplementary Fig. 2D). To assure that our findings are not a specific feature of K562 cells, we also treated two additional CML cell lines, KCL22 and AR230-s, with EPZ-6438 and performed splicing analysis, leading to the detection of comparable numbers of differential splicing events (Supplementary Fig. 3). In summary, using two independent approaches, we observe significant changes to the splicing landscape upon inhibition of EZH2 in CML cell lines.

A Plot on the left panel shows splicing events significantly de-regulated in K562 cells upon GSK126 and EPZ-6438 treatment (2 µM, 72 h), table on the right summarises up- and down-regulated events, as well as the number of associated genes. ISO: annotated isoform, SE: skipped exon, RI: retained intron, ΔSI: differential splice-index. B Genome tracks of RNA-seq signals of differentially spliced genes ZMIZ1, HERC2, IFFO2, MST1, C8orf33 in EZH2i-treated (GSK126, EPZ-6438: red) and control samples (DMSO: black). Exons are shown as blue boxes, relevant intron-exon junctions are indicated with black arrows, the strand direction with blue arrows. C Experimental validation of differential splicing via qRT-PCR. Primers specifically amplifying the spliced or non-spliced variant of nine putative target genes identified in (A) were used to examine changes in exon skipping or intron retention upon EZH2i treatment. The schematic view on the right side indicates the ___location of primers (black arrows) in relation to relevant exons (boxes) and introns (lines). Differential splicing events are highlighted in red. Results are based on 4 biological replicates, significance was calculated using t-test (paired, 2-tailed, * indicates p value < 0.05). SE ↑ , SE ↓ , and IR↓ indicates the type and direction of alternative splicing determined in (A).
Differential splicing is not associated with H3K27me3 and EZH2-binding
In order to understand the mechanistic basis for EZH2-regulated alternative splicing, we first examined to what degree EZH2 splicing targets overlap with EZH2 transcriptional targets. As expected, EZH2 inhibition led predominantly to transcriptional up-regulation, with enrichment for PRC2 targets (Fig. 2A, B, Supplementary Fig. 4A) and pathways including “pathways in cancer”, and “developmental genes” (Fig. 2C). In contrast, analysis of splicing targets did not reveal any enrichment of functional categories, and when we overlapped differentially expressed genes (DEGs) with differentially spliced genes (DSGs), we only observed a surprisingly low overlap (Fig. 2D, Supplementary Fig. 4B). We also performed EZH2 ChIP-seq in K562 and utilized a corresponding H3K27me3 dataset to assess the association of H3K27me3 and EZH2 binding with PRC2 target genes, DEGs and DSGs. As seen in Fig. 2E, metagene analysis showed that H3K27me3 as well as EZH2 are enriched at PRC2 targets and DEGs, but mostly absent from DSGs (Fig. 2E, Supplementary Fig. 5). To exclude that our comparison is skewed by a potential bias caused by the varying expression levels of genes in the three categories, we also analysed H3K27me3 and EZH2 enrichment exclusively at DSGs, comparing differentially spliced regions with corresponding control regions. As seen in Fig. 2F, H3K27me3 and EZH2 were present at comparable levels. This, together with the analysis above suggests that EZH2 transcriptional targets are distinct from EZH2 splicing targets, and differential splicing is not primarily mediated by the presence or absence of H3K27me3 and EZH2.

A Volcano plot showing preferential up-regulation of differentially expressed genes upon EZH2 inhibition in K562 cells (GSK126 and EPZ64-38 vs DMSO, 2 µM, 3 d). B GSEA analysis reveals enrichment of PRC2 target genes amongst DEGs. C Gene ontology (GO) analysis of differentially expressed genes (DEGs) or differentially spliced genes (DSGs) determined either with the in-house perl script or rMATS. D Venn diagrams show the overlap of genes found differentially expressed or spliced upon EZH2 inhibitor treatment. E Metagene analysis depicting the mean enrichment of H3K27me3 (upper panels) or EZH2 (lower panels) across PRC2 target genes (left), DEGs (middle) or DSGs (right). ChIP-seq signals were normalized to corresponding inputs before plotting. TSS: transcript start site, TES: transcript end site. F Metagene analysis of H3K27me3 or EZH2 enrichment at DSGs only, presenting either normalized mean values for all exon/intron junctions (left), or focusing exclusively on differentially spliced junctions (right).
EZH2 as transcriptional regulation of splicing factors
We explored alternative models how EZH2 might indirectly regulate mRNA splicing, and hypothesized that EZH2 acts as transcriptional regulator of splicing genes. Therefore, we tested if genes annotated in the category “RNA splicing” were enriched among DEGs, but in contrast to genes found in the category “Development”, we did not notice enrichment (Supplementary Fig. 6A). Nonetheless, 11 splicing genes were significantly up-regulated upon EZH2 inhibition in K562 (Fig. 3A, B, Supplementary Fig. 6B). Strikingly, 10/11 of genes found in K562 displayed low basal expression levels in untreated cells, in line with their potential transcriptional repression by EZH2 (Fig. 3C). Analysis of SRs commonly up-regulated across K562, KCL22, and AR230-s revealed a conserved group of splicing factors silenced by EZH2, that are shared between CML cell lines (Supplementary Fig. 6C). To investigate their mode of regulation, we generated H3K27ac and H3K4me3 ChIP-seq datasets in K562, that allowed us – together with H3K27me3 and EZH2 ChIP-seq datasets – to visualize their chromatin status at transcriptional start sites (TSSs). Indeed, we detected H3K27me3 as well as EZH2 signals at all 11 splicing genes responsive to EZH2 inhibition in K562, at levels broadly mirroring their basal expression levels (Supplementary Fig. 7). This indicates a direct role of EZH2 mediated H3K27 tri-methylation in their transcriptional repression.

A Heatmap showing differential expression of RNA splicing genes after GSK126 and EPZ-6438 treatment relative to DMSO controls. B Significantly up-regulated genes in both conditions with log2 fold changes <0.5 and p values < 0.05 are shown in detail. C To estimate basal expression levels of genes from (B) their normalized counts in control samples (DMSO) were plotted. D mRNA expression of CELF2, MBNL3, KHDRBS3, HABP4, RBM11 in K562 cell lines transduced with lentiviral constructs containing their cDNAs or an empty vector (pL), determined via qRT-PCR. Expression values are normalized to GAPDH, and represent the mean of 4 biological replicates. * p value < 0.05 (t test, paired, 2-tailed). E Western blot showing the presence of the FLAG-tagged protein of over-expressed splicing factors in K562 cells (upper panel). B-ACTIN served as loading control (lower panel). F Cell viability of K562 cell lines stably over-expressing indicated SRs. CTG assays were performed to determine ATP levels as indicator of metabolically active cells. * p value < 0.05 (t test, paired, 2-tailed). G Validation of differential splicing upon over-expression of SRs. qRT-PCR analysis for 6 genes shown to be differentially spliced upon EZH2i treatment (See Fig. 1C). Given are the relative levels of retained introns / skipped exons versus non-retained / non-skipped splice variants. Genes exhibiting similar behaviour as after EZH2i treatment are highlighted with black arrows. Biological replicates = 4, * p value < 0.05 (t test, paired, 2-tailed).
Next, we tested if some of those splicing genes were responsible to mediate the splicing changes observed upon EZH2 inhibition. We picked five candidates for further analysis: CELF2, MBML3, KHDRBS3, HABP4, and RBM11 and generated K562 lines stably over-expressing their human FLAG-tagged proteins (Fig. 3D, E). Over-expression caused varying effects on cell viability, with CELF2 exhibiting the most pronounced phenotype (Fig. 3F). Finally, we also analysed their effect on EZH2 splicing targets, using the six previously successfully validated genes. While CELF2, MBML3 and HABP4 modulated splicing patterns to some degree, only CELF2 induced splicing changes partially mirroring the effects observed upon EZH2 inhibition, i.e. reduced exon skipping or intron retention in 3/6 target genes (Fig. 3G). Finally, we also performed RNA binding motif analysis of differentially skipped exons (rMATS) using the rMAPS database which contains more than 100 binding motifs of RNA binding factors. The most significantly enriched motifs identified were GU-rich sequences associated with CELF protein binding, specifically at down-regulated skipping events (Supplementaru Fig. 8). This suggested that CELF2 could be involved in the mediation of EZH2 inhibition-induced splicing changes.
The splicing factor CELF2 acts as tumour-suppressor in leukaemia cell lines
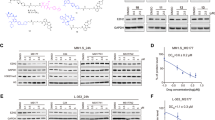
CELF2 is a broadly expressed RNA-binding protein that has been implicated in mRNA splicing [22, 23] and the regulation of mRNA polyadenylation [24]. More recently it has also been identified as tumour-suppressor in a growing number of cancers [25,26,27,28,29,30]. Our results from cell viability-, growth curve- as well as colony formation-assays suggest a similar role in CML (Figs. 3F, 4A–C). To test if EZH2 inhibition could be an effective approach to modulate CELF2 protein levels (not only mRNA abundance) we performed Western blot analysis of K562 cells lacking EZH2 activity, either mediated by EZH2 inhibitor treatment, or CRISR/Cas9 gene knock-out [31]. In both cases we detected elevated levels of the CELF2 protein (Fig. 4E).

A Stable over-expression of CELF2 in K562 cells. B Growth curve assay of K562 cell lines stably expressing CELF2 (pL- CELF2) or an empty control plasmid (pL). Cells were seeded at 200 000 cells per ml of growth medium, and cell numbers were measured on the 3 following days. Results summarize 2 independent experiments. C The ability to form colonies was determined in CELF2 over-expressing or control cells. Colony forming units (CFUs) are given relative to control cells. D To determine the effect of EZH2 inhibitor treatment on CFUs, K562 cells were treated with 2 µM for 3 days prior to seeding in Matrigel. E Western blots showing that EZH2 inhibitor treatment (2 µM, 3 days, right panel) as well as genetic inactivation of EZH2 via CRISPR/Cas9 (left panel) leads to loss of H3K27me3 and CELF2 protein up-regulation. GAPDH, LAMIN A/C, and H3 proteins served as loading controls. F CELF2 over-expressing K562 cells remain sensitive to Imatinib and CELF2 over-expression together with TKI treatment elicits increased effects on colony formation (left panel) and cell viability (right panel). Cells were grown for 3 days with or without Imatinib, then either seeded on Matrigel, or subjected to CTG assays. Experiments were performed in triplicates. G Expression of CELF2 in the TKI sensitive CML cell line AR230-s, as well as in its TKI resistant counterpart AR230-r, leads to decreased cell viability. CTG assays were conducted in triplicates. H TF1-GFP or TF1-BCR::ABL1 leukaemia cells were used as models to examine a potential dependence of CELF2 on the presence of BCR::ABL1. Cell lines over-expressing CELF2 or an empty control vector were generated and Western blot was performed to confirm CELF2 over-expression. I CTG assay was used to assess cell viability of growth factor dependent TF1-GFP or BCR::ABL1-driven TF1-BCR::ABL1 cells upon CELF2 expression. J CTG assays of TF1 cell lines treated with increasing concentrations of the TKI imatinib for 72 h. K The leukaemia cell line THP-1 served as additional model to test the role of CELF2 in a non BCR::ABL1 driven setting. Overexpression leads to reduced cell viability as seen in the right panel (CTG assay, biological triplicates). All statistical analysis presented in this figure were conducted employing the t test (paired, 2-sided). * indicates a p value < 0.05.
Another important aspect in CML biology is the dependence of tumour-suppressors on the BCR::ABL1 oncogene, the primary driver of the disease. Acquired resistance to drugs targeting the tyrosine kinase function of the ABL1-kinase (TKIs) usually leads to the activation of alternative oncogenic pathways, therefore it is crucial to identify tumour-relevant targets acting independently of BCR::ABL1, as they might represent useful targets even after the development of resistance. First, we established that treatment of K562 cells with Imatinib is still effective to reduce cell viability and colony formation ability upon CELF2 expression in K562 (Fig. 4F). Then we over-expressed CELF2 in a pair of CML cell lines: AR230-s, a BCR::ABL1 dependent cell line, that is sensitive to TKI treatment, and AR230-r, a subclone with acquired TKI resistance. As seen in Fig. 4G, CELF2 expression reduced cell viability in both scenarios. We also conducted CELF2 over-expression experiments in two subclones of the TF1 leukaemia cell line: one, that is dependent on the addition of the growth factor GM-CSF, and another, that is dependent on BCR::ABL1 activity. As seen in Fig. 4H-J, CELF2 reduced cell viability under both conditions. Finally, we generated a CELF2 over-expressing THP-1 leukemia cell line, that is not driven by BCR::ABL1, and again observed reduced cell viability (Fig. 4K). Therefore, using different cell models, we conclude, that CELF2 mediates its tumour-suppressor activity independently of BCR::ABL1.
CELF2 is a splicing regulator, modulating CML-relevant gene expression patterns
To better understand the mechanism of action underlying CELF2’s role in CML, we analysed genome wide splicing patterns upon CELF2 over-expression in K562 cells. As seen in Fig. 5A, Supplementary Fig. 9A–C, CELF2 over-expression had a severe impact on global splicing patterns with 1379 (perl) and 3971 (rMATS) significant differential splicing events. Interestingly, we noticed an enrichment of genes with kinase and transferase function as well as cell cycle/division amongst CELF2 splicing targets (Supplementary Fig. 9D). RNA binding motif analysis, using differentially skipped exons (rMATS), revealed the enrichment of the same GU-rich motifs typically associated with CELF proteins as found after EZH2 inhibitor treatment (Supplementary Fig. 9E). Enrichment was specific to down-regulated skipping events, indicating a role of CELF2 as splicing enhancer. To assess the contribution of CELF2 to EZH2 mediated splicing changes, we compared ΔSI values of EZH2 splicing targets with genes unaffected by EZH2 inhibition, and found EZH2 splicing targets more prone to differential splicing upon CELF2 over-expression than control targets (Supplementary Fig. 9F). Heatmaps revealed that >20% of EZH2 splicing targets exhibit the same direction of de-regulation (up/down) after CELF2 over-expression, in line with a model where CELF2 mediates part of the splicing changes. Interestingly, we also saw a proportion of splice events regulated in an inverse fashion, potentially caused by the differential expression of multiple splicing factors after EZH2 inhibitor treatment, leading to complex splicing pattern changes (Fig. 5B). To test if EZH2/CELF2-regulated alternative splicing is directly contributing to their role as oncogene/tumour-suppressor, we examined the role of one of the top genes showing consistent differential splicing upon EZH2 inhibition and CELF2 over-expression: CDC14B (Fig. 5C), a protein kinase that has been implicated in mitotic survival [32]. While the perl script reported increased levels of one specific isoform (ex12-16), closer inspection of the aligned reads indicated the presence of several isoforms (Supplementary Fig. 10A), and analysis of exon-junction spanning reads revealed that EZH2 inhibition and CELF2 expression caused a general shift from exon 13 containing isoforms to exon 13 skipping isoforms (Fig. 5D). Interestingly, exon 13 has been shown to contain a pre-mature stop codon [33], and when we specifically targeted this isoform with DsiRNAs (Dicer-Substrate Short Interfering RNAs), we detected decreased cell viability and colony formation ability of K562 cells. This effect was not seen when we targeted one of the isoforms without exon 13 (Fig. 5E, Supplementary Fig. 10B).

A Differential splicing events in K562 cells after CELF2 over-expression determined with our in-house perl script. Plot on the left gives differential splice indices (ΔSI) of annotated isoforms (ISO), skipped exons (SE), and retained introns (RI), table on the right summarizes up- and down-regulated events, as well as the number of associated genes. B Heatmaps presenting ΔSI values at differential slicing events found after EZH2 inhibition, together with the corresponding ΔSI values observed after CELF2 over-expression. Red: up, blue: down. The black square highlights splicing events significantly up- or down-regulated in both scenarios. C The CDC14B isoform ex12-16 is increased upon EZH2 inhibitor treatment or CELF2 overexpression (perl script). D Analysis of exon-junction covering reads reveals that EZH2 inhibition or CELF2 expression leads to a shift from CDC14B isoforms containing exon 3 with a premature stop codon (shown as inverted red triangle), to isoforms skipping exon 13. Left panel: overview of CDC14B isoforms distinguishable by diagnostic exon-junction covering reads (shown as lines underneath the boxes that represent exons). Right panel: black numbers: reads covering the relevant diagnostic junctions were counted, and are given as ratio between treated samples (EZH2 inhibitor, pL-CELF2) over corresponding controls (DMSO, pL). Note that treatment leads to a relative enrichment of isoforms lacking exon 13 (see orange numbers, showing their increased presence relative to exon 13 containing isoforms). E DsiRNA (Dicer-Substrate Short Interfering RNA) mediated knock-down of exon 13 containing isoforms (ex13), but not the exon 13 skipping isoform ex12-15, leads to reduced cell viability and colony formation ability in K562 cells. * p value < 0.05 (t test, paired, 2-tailed). F Volcano plot showing differential gene-expression after CELF2 expression in K562 cells. G Gene set enrichment analysis of top ranked “Hallmark” genes upon CELF2 over-expression.
Next, we analysed gene expression changes caused by CELF2 over-expressing in K562. In agreement with its predominant role as splicing regulator we found comparatively less genes to be transcriptionally de-regulated (Fig. 5F). However, transcriptional changes were targeting important pathways in CML biology (Fig. 5G). We note that the up-regulation of apoptosis has been proposed previously as a mechanism of EZH2 inhibitor action in CML [10], and interferon alpha has been in clinical practice to treat CML before the arrival of TKIs [34]. MYC has been reported to be important at various stages of CML development, during transformation as well as to overcome TKI resistance [35,36,37,38,39]. All three pathways responded in a similar fashion to EZH2 inhibitor treatment and CELF2 over-expression, but the clearest correlation was found for the repression of MYC target genes (Supplementary Fig. 11A, B). A reduced MYC signature was also found in EPZ-6438 treated KCL22 and AR230-s cell lines (Supplementary Fig. 11C). Interestingly, while MYC transcript levels were not changed with either treatment, we did observe a reduction in protein levels, pointing towards post-translational regulation (Supplementary Fig. 11D–F). Using the translational inhibitor cycloheximide (CHX) followed by Western blot experiments we monitored potential changes to the half-life of MYC, without observing obvious alterations. However, when we examined the phosphorylation status of Thr58 and Ser62, two sites important for the regulation of MYC protein stability [40], we noticed specific hyper-phosphorylation of one of the residues, Thr58 (Supplementary Fig. 11G, H). In order to directly link Thr58 phosphorylation changes to EZH2/CELF2 activity, we sought to identify splicing or expression changes in key genes known to regulate Thr58 phosphorylation, but none of the genes tested (GSK3, FBW7, EYA3, PPP2R2A, PPP2R5A, PPP2CA, TESPA1, SENP1, AXIN1, PIN1, TRIB3, UBE3B, USP28) showed consistent deregulation, pointing towards a more indirect regulation of MYC phosphorylation.
The splicing landscape in patient derived CML samples
To examine the relevance of our findings to CML patients, we extended our study to CML patient derived samples. We obtained seven chronic phase CML patient samples after diagnosis (6 bone marrow, 1 peripheral blood). In addition, we also obtained CD34+ HSCs from the bone marrow of healthy donors (HDs) as controls (Table 1). First, we performed RNA-seq of CD34+ cells. Gene expression analysis revealed major differences between CML and HD, leading to a clear separation based on principal component analysis (Supplementary Fig. 12A, B). Using the pizzly fusion gene detection programme, we found independent validation for the presence of the BCR::ABL1 fusion transcript in 5/7 CML patient samples, but not in HD samples (Supplementary Fig. 12C). We also saw the enrichment of gene-sets for “BCR::ABL1 signalling” and “CML”, albeit mostly not reaching statistical significance, potentially due to the low number of genes contained in the gene-sets (Supplementary Fig. 12D).
Next, we aimed to establish the splicing landscape in CML. We employed the in-house perl script, yielding 759 significant differential splicing events (409 ISOs, 15 SEs, and 335 RIs) in 600 unique genes (Fig. 6A). Surprisingly, we noticed a consistent reduction of ISOs, SEs, and RIs, indicating a general increase in splicing efficacy in CML. We also found a high relative abundance of decreased intron retention events. Associated transcripts were found to display increased mRNA levels, in line with reduced nonsense mediated decay (Supplementary Fig. 13). To complement our analysis, we analysed the same datasets using rMATS, leading to the identification of 10,783 differential splicing events (|ΔSI | > 0.05) in 4798 unique genes. Consistent with the perl script approach we found an overall strong shift towards increased splicing efficacy (for SE, RI, A5SS, and A3SS, Supplementary Fig. 14A, B). Overlapping the DSGs identified by both methods, we found that 377/576 (65.5%) of the perl targets were also present in the pool of differentially spliced rMATS target genes (Supplementary Fig. 14C). GSEA analysis of DSGs revealed that genes involved in “mRNA-processing”, “mRNA-splicing”, “mRNA-binding” as well as “Mitosis” were enriched in targets found by either method (Supplementary Fig. 14D). We also searched for RNA binding motifs enriched at differentially spliced SEs (rMATS), and strikingly the same GU-rich CELF motif identified earlier showed increased association (Supplementary Fig. 14E). Importantly, in contrast to conditions with increased CELF2, CML samples showed enrichment of the motif in up-regulated SEs.

A Alternative splicing events in CD34+ cells from CML patients exhibiting differential splicing compared to CD34+ from healthy donors (HD) determined with our in-house perl script. Plot on the left gives differential splice indices (ΔSI) of annotated isoforms (ISO), skipped exons (SE), and retained introns (RI), table on the right summarizes up- and down-regulated events, as well as the number of associated genes. B Percentage of differentially expressed genes from the GO categories “RNA-splicing” and “Development” in CD34 + CML patient samples compared to CD34+ control samples. Up-regulated genes are presented in red, down-regulated genes in blue. C Bar-graph showing the differential expression of selected splicing genes in CML patients. The blot encompassed genes previously identified as EZH2 targets in K562 cells. Red: up-regulated, blue: down-regulated. D Basal expression levels of genes from (E) in CD34+ control samples determined by RNA-seq. E Normalized read counts of CELF2 in CD34+ healthy donor (HD) and CML chronic phase (CP) patient samples. F H3K27me3 levels at the CELF2 promoter in CML (pink) and non-CML (grey) haematopoietic stem/progenitor cells (HSCs/HPCs) as determined by ChIP-seq. Figure was generated using the dataset PRJEB8291. Tracks represent normalized read counts and the same scale is used for all samples. BM: samples derived from bone marrow, PB: derived from peripheral blood, CP: chronic phase (G) CELF2 promoter H3K27me3 levels in non-CML (black, grey) and CML CD34+ cells in chronic phase (CP, pink) or blast phase (BP, green). Data was derived from the previously published ChIP-seq dataset PRJNA777969, tracks are shown using the same scale. BM: samples derived from bone marrow, PB: derived from peripheral blood.
CELF2 in CML patient samples
Based on our cell line derived data and other previously published datasets, we hypothesized that splicing de-regulation in CML is partially caused by EZH2 mediated transcriptional de-regulation of splicing factors, including CELF2. Indeed, 41% of genes within the GO category “RNA splicing” were de-regulated in CML, comparable to other GO categories such as “Development” (Fig. 6B). Interestingly, when we focused on splicing factors directly regulated by EZH2 in K562 cells, we found that eight out of eleven were down-regulated, and transcripts were present at low abundance (Fig. 6C, D), pointing towards a generally repressed state. CELF2, was included in the set of down-regulated genes, but the difference between CML and healthy donors did not reach statistical significance (Fig. 6E, p = 0.0934). However, when we expanded our analysis to three other expression datasets containing CD34 + CML and normal cells [10, 15, 41], all of them exhibiting reduced CELF2 levels in CML, with two reaching statistical significance (Supplementary Fig. 15). We also examined H3K27me3 levels at the CELF2 promoter in ChIP-seq datasets generated from CML patient samples, and noticed increased signals in CML compared to non-CML (Fig. 6F, G). This suggests that CELF2 is indeed suppressed by EZH2 in CML patients.
To understand its contribution to splicing and gene-regulation patterns in CML, we transduced three BM-derived CD34+ patient samples with lentiviral constructs encoding CELF2, leading to moderate over-expression. As shown in Fig. 7A we detected 578 differential splicing events in 527 genes. Of note, over-expression led to the partial reversal of disease-associated splicing changes (Fig. 7B, Supplementary Fig. 16, Supplementary Table 3). Focusing on gene-expression, we observed few changes, in line with its primary role in splicing. However, when we tested the enrichment of de-regulated pathways, the down-regulation of the MYC target gene signature was still clearly detectable (Fig. 7C). This trend was also observed upon analysis of expression data of CP CD34+ samples treated with the EZH2 inhibitor GSK343, derived from a previously published dataset (Supplementary Fig. 17) [10]. Finally, reduced colony formation ability was observed upon CELF2 over-expression as well as EZH2 inhibitor treatment, corroborating our findings from K562 cells (Fig. 7D).

A Differential splice index values (ΔSI) for ISO, SE, and RI splice events found differentially spliced upon CELF2 over-expression in CML patient samples (left chart). The table on the right summarized the number of de-regulated events and associated genes. B Heatmaps presenting ΔSI values at differential splicing events found after CELF2 over-expression in CML patient samples, together with the corresponding ΔSI values observed in CML versus HD samples. Red: up, blue: down. The black square highlights splicing events with opposing regulation patterns. C Enrichment plots of the “cMyc gene set V1” in CML patients (left blot) and after CELF2 over-expression in CML patient samples (right blot). In between, a heatmap represents the log2 fold-change expression values of genes in the “cMyc gene set V1”. D Colony forming ability of CML samples was assessed after treatment with the EZH2 inhibitor GSK126, or CELF2 over-expression. CP: chronic phase, BM: samples derived from bone marrow, PB: derived from peripheral blood. E Correlation between EZH2 and CELF gene family members in 9 selected TCGA datasets. Correlation coefficients associated with p values < 0.05 are plotted in red (positive correlation) or blue (negative correlation). BRCA breast cancer, COAD colon adenocarcinoma, GBM glioblastoma, LUAD lung adenocarcinoma, LUSC lung squamous cell carcinoma, STAD stomach adenocarcinoma, TGCT testicular germ cell tumour, THYM thymoma, UCEC uterine corpus endometrial carcinoma.
EZH2 mediated splicing factor regulation in other cancers
Given the current interest in EZH2 inhibitors as potential treatment options for a growing number of cancer types, we wanted to put our findings into a broader context. Therefore, we analysed the impact of EZH2 inhibition on splicing factors expression in a variety of cancer cell lines. Although treatment conditions differed between the sample-sets, we observed that while splicing genes tend to be less enriched than developmental genes, they are de-regulated upon treatment with EZH2 inhibitors (Supplementary Fig. 18A), consistent with our findings in CML. Interestingly, several splicing factors, including CELF2, were found up-regulated in more than one dataset, suggesting a common mode of regulation across different tissues (Supplementary Fig. 18B). Finally, we examined the correlation between splicing gene- and EZH2-expression in TCGA patient sample datasets from a total of 33 cancer types. We found that correlation tends to be conserved across most cancer types (Supplementary Fig. 19). CELF2, together with other CELF-protein family members exhibited mostly negative correlation with EZH2, in line with direct regulation and repression by EZH2 (Fig. 7E).
Discussion
Aberrant splicing is a common feature of many cancers; however, the underlying mechanism varies in a context-dependent manner. While splicing changes are frequently caused by the mutation of SRs in haematological malignancies, altered expression levels and copy number changes are more prominent in solid tumours [4, 7]. CML as a primarily clonal disease driven by the BCR::ABL1 oncogene, is usually associated with few mutations, instead exhibiting a profound rewiring of transcriptional networks [42]. Indeed, our analysis of CML and control samples confirmed wide-spread transcriptional changes, including the mis-expression of numerous SRs, well beyond the factors previously reported [13,14,15]. This was accompanied by the disruption of normal splicing patterns, and the alternative splicing of hundreds of genes. Interestingly, CML samples exhibited a marked reduction in splicing across most event types. This runs contrary to the general trend of increased alternative splicing in many (but not all) cancers [6, 43,44,45]. Previously published work on alternative splicing in CML led to inconsistent findings [46, 47], possibly due to sample variability connected to the use of whole blood (PBMCs) as source for data generation. In this study we specifically enriched our samples for CD34+ expressing cells, mostly eliminating the effect of other cell types obscuring the CML splicing signature. Our data suggests that the transcriptional de-regulation of SRs is a major factor contributing to mis-regulation of altered splicing efficacy in CML.
The polycomb protein EZH2 is an important regulator of transcription, and it is up-regulated in CML [10, 11]. The combined use of EZH2 inhibitors together with TKIs has been suggested to target LSCs which remain refractory to TKI treatment alone. However, the current understanding of the mechanism of action underlying the beneficial effect of EZH2 inhibition is limited. Transcriptional regulation of genes important for apoptosis and cell cycle progression has been suggested as possible mechanisms [10]. Indeed, our findings using EZH2 inhibitors support such a model, as we confirmed the role of EZH2 as important transcriptional regulator in CML cell lines, with thousands of targets, including apoptotic genes. In this study we add another group of relevant targets, splicing regulators, and show that EZH2 elicits part of its effect on CML cells through their regulation. Treating CML cell lines with EZH2 inhibitors caused differential splicing of hundreds of genes, and the up-regulation of splicing factors repressed by H3K27me3. The relevance of individual EZH2 mediated alternative splicing events was illustrated by our finding that CDC14B alternative splicing modulates cell growth, potentially due to its antagonistic role to the pro-mitotic factor CDK1 [32].
Over-expression of several targets and analysis of binding motifs marked CELF2 as candidate to mediate EZH2 dependent effects on splicing and cell growth. Indeed, CELF2 over-expression in cell lines or patient samples pheno-copied some of the effects observed after EZH2 inhibition, including reduced growth, altered splicing and a shift in gene expression patterns. This clearly shows that CELF2 acts as a tumour-suppressor in CML. Of note, a recent publication also implicates CELF2 in leukaemic stem cell maintenance [30]. EZH2 inhibition seems a promising avenue to increase CELF2 levels, and work in breast cancer cells suggests that DNA-de-methylating agents might be a feasible alternative [26]. Another therapeutic approach that has been recently put forward is the use of spliceosome inhibitors [14]. This is interesting, especially in the light of our finding that SR gene expression and splicing patterns in CML cells are in a severely dis-balanced state, potentially explaining their increased vulnerability to further perturbation of this pathway.
An additional finding of our study is the identification of MYC signalling as one of the important downstream targets of EZH2 and CELF2. MYC plays a crucial role at different stages of CML, including initiation, drug response and resistance [35,36,37,38,39]. Here we show that elevating CELF2 levels via EZH2 inhibition reduces MYC signalling, hampering CML cell growth. Importantly this effect seems to be BCR::ABL1 independent. While the exact mechanism remains unclear, our findings point to the regulation of MYC protein stability, mediated via the modulation of Thr58 phosphorylation.
Analysis of datasets from inhibitor treated cancer cell lines, together with correlation analysis between EZH2 and splicing gene expression in patient samples, suggests that the role of EZH2 as a splicing modulator might not be restricted to CML, but that it is a common mechanism across different cancer types. Specifically, CELF-family genes emerge as potential relevant downstream targets that warrant further studies in the future.
Finally, our work also highlights the possibility that other transcriptional activators and repressors might act as modulators of cellular splicing, through their ability to influence the expression of multiple downstream targets, including SRs. Their de-regulation might be a contributing factor to the wide-spread splicing mis-regulation that is observed across many cancers, even in the absence of splicing factor mutations.
Data availability
The datasets generated during the current study are available in the European Genome-Phenome Archive: PRJEB51336, PRJEB51550, PRJEB72776, PRJEB72797-8. Datasets used for analysis published elsewhere are listed in Supplementary Table 2.
References
Brown SJ, Stoilov P, Xing Y. Chromatin and epigenetic regulation of pre-mRNA processing. Hum Mol Genet. 2012;21:R90–96.
Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science. 2010;327:996–1000.
Tellier M, Maudlin I, Murphy S. Transcription and splicing: a two-way street. Wiley Interdiscip Rev RNA. 2020;11:e1593.
Bradley RK, Anczukow O. RNA splicing dysregulation and the hallmarks of cancer. Nat Rev Cancer. 2023;23:135–55.
Cherry S, Lynch KW. Alternative splicing and cancer: insights, opportunities, and challenges from an expanding view of the transcriptome. Genes Dev. 2020;34:1005–16.
Kahles A, Lehmann KV, Toussaint NC, Huser M, Stark SG, Sachsenberg T, et al. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell. 2018;34:211–24.e216.
Sveen A, Kilpinen S, Ruusulehto A, Lothe RA, Skotheim RI. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene. 2016;35:2413–27.
Eich ML, Athar M, Ferguson JE 3rd, Varambally S. EZH2-targeted therapies in cancer: hype or a reality. Cancer Res. 2020;80:5449–58.
Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22:128–34.
Scott MT, Korfi K, Saffrey P, Hopcroft LE, Kinstrie R, Pellicano F, et al. Epigenetic reprogramming sensitizes CML stem cells to combined EZH2 and tyrosine kinase inhibition. Cancer Discov. 2016;6:1248–57.
Xie H, Peng C, Huang J, Li BE, Kim W, Smith EC, et al. Chronic myelogenous leukemia- initiating cells require polycomb group protein EZH2. Cancer Discov. 2016;6:1237–47.
Ng JJ, Ong ST. Therapy resistance and disease progression in CML: mechanistic links and therapeutic strategies. Curr Hematol Malig Rep. 2022;17:181–97.
Jin Y, Zhou J, Xu F, Jin B, Cui L, Wang Y, et al. Targeting methyltransferase PRMT5 eliminates leukemia stem cells in chronic myelogenous leukemia. J Clin Invest. 2016;126:3961–80.
Lebecque B, Bourgne C, Munje C, Berger J, Tassin T, Cony-Makhoul P, et al. The spliceosome: a new therapeutic target in chronic myeloid leukaemia. Cancers (Basel). 2022;14:4695.
Sinnakannu JR, Lee KL, Cheng S, Li J, Yu M, Tan SP, et al. SRSF1 mediates cytokine-induced impaired imatinib sensitivity in chronic myeloid leukemia. Leukemia. 2020;34:1787–98.
An O, Tan KT, Li Y, Li J, Wu CS, Zhang B, et al. CSI NGS portal: an online platform for automated NGS data analysis and sharing. Int J Mol Sci. 2020;21:3828.
Sherman BT, Hao M, Qiu J, Jiao X, Baseler MW, Lane HC, et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022;50:W216–W221.
Madan V, Kanojia D, Li J, Okamoto R, Sato-Otsubo A, Kohlmann A, et al. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat Commun. 2015;6:6042.
Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, et al. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci USA. 2014;111:E5593–5601.
Park JW, Jung S, Rouchka EC, Tseng YT, Xing Y. rMAPS: RNA map analysis and plotting server for alternative exon regulation. Nucleic Acids Res. 2016;44:W333–338.
Mehmood A, Laiho A, Venalainen MS, McGlinchey AJ, Wang N, Elo LL. Systematic evaluation of differential splicing tools for RNA-seq studies. Brief Bioinform. 2020;21:2052–65.
Ladd AN, Charlet N, Cooper TA. The CELF family of RNA binding proteins is implicated in cell-specific and developmentally regulated alternative splicing. Mol Cell Biol. 2001;21:1285–96.
Mallory MJ, Allon SJ, Qiu J, Gazzara MR, Tapescu I, Martinez NM, et al. Induced transcription and stability of CELF2 mRNA drives widespread alternative splicing during T-cell signaling. Proc Natl Acad Sci USA. 2015;112:E2139–2148.
Chatrikhi R, Mallory MJ, Gazzara MR, Agosto LM, Zhu WS, Litterman AJ, et al. RNA binding protein CELF2 regulates signal-induced alternative polyadenylation by competing with enhancers of the polyadenylation machinery. Cell Rep. 2019;28:2795–806.e2793.
de la Rosa J, Weber J, Friedrich MJ, Li Y, Rad L, Ponstingl H, et al. A single-copy Sleeping Beauty transposon mutagenesis screen identifies new PTEN-cooperating tumor suppressor genes. Nat Genet. 2017;49:730–41.
Pique L, Martinez de Paz A, Pineyro D, Martinez-Cardus A, Castro de Moura M, Llinas-Arias P, et al. Epigenetic inactivation of the splicing RNA-binding protein CELF2 in human breast cancer. Oncogene. 2019;38:7106–12.
Fan Z, Zhang Z, Piao C, Liu Z, Wang Z, Kong C. Genome-wide analyses of prognostic and therapeutic alternative splicing signatures in bladder urothelial carcinoma. Front Oncol. 2021;11:626858.
Ramalingam S, Ramamoorthy P, Subramaniam D, Anant S. Reduced expression of RNA binding protein CELF2, a putative tumor suppressor gene in colon cancer. Immunogastroenterology. 2012;1:27–33.
Sah B, Singh J, Shen Y, Goldfarb N, Samie FH, Geskin LJ et al. Loss of CELF2 promotes skin tumorigenesis and increases drug resistance. Int J Dermatol. 2025;64:101–10
Guo T, Wang Y, Sun X, Hou S, Lan Y, Yuan S, et al. Loss of RNA-binding protein CELF2 promotes acute leukemia development via FAT10-mTORC1. Oncogene. 2024;43:1476–87.
Teo WW, Cao X, Wu CS, Tan HK, Zhou Q, Gao C, et al. Non-coding RNA LEVER sequestration of PRC2 can mediate long range gene regulation. Commun Biol. 2022;5:343.
Dietachmayr M, Rathakrishnan A, Karpiuk O, von Zweydorf F, Engleitner T, Fernandez-Saiz V, et al. Antagonistic activities of CDC14B and CDK1 on USP9X regulate WT1-dependent mitotic transcription and survival. Nat Commun. 2020;11:1268.
Muys BR, Shrestha RL, Anastasakis DG, Pongor L, Li XL, Grammatikakis I, et al. Matrin3 regulates mitotic spindle dynamics by controlling alternative splicing of CDC14B. Cell Rep. 2023;42:112260.
Talpaz M, Mercer J, Hehlmann R. The interferon-alpha revival in CML. Ann Hematol. 2015;94:S195–207.
Albajar M, Gomez-Casares MT, Llorca J, Mauleon I, Vaque JP, Acosta JC, et al. MYC in chronic myeloid leukemia: induction of aberrant DNA synthesis and association with poor response to imatinib. Mol Cancer Res. 2011;9:564–76.
Krishnan V, Schmidt F, Nawaz Z, Venkatesh PN, Lee KL, Ren X, et al. A single-cell atlas identifies pretreatment features of primary imatinib resistance in chronic myeloid leukemia. Blood. 2023;141:2738–55.
Sawyers CL, Callahan W, Witte ON. Dominant negative MYC blocks transformation by ABL oncogenes. Cell. 1992;70:901–10.
Winter GE, Rix U, Carlson SM, Gleixner KV, Grebien F, Gridling M, et al. Systems-pharmacology dissection of a drug synergy in imatinib-resistant CML. Nat Chem Biol. 2012;8:905–12.
Peter B, Eisenwort G, Sadovnik I, Bauer K, Willmann M, Rulicke T, et al. BRD4 degradation blocks expression of MYC and multiple forms of stem cell resistance in Ph(+) chronic myeloid leukemia. Am J Hematol. 2022;97:1215–25.
Thomas LR, Tansey WP. Proteolytic control of the oncoprotein transcription factor Myc. Adv Cancer Res. 2011;110:77–106.
Gerber JM, Gucwa JL, Esopi D, Gurel M, Haffner MC, Vala M, et al. Genome-wide comparison of the transcriptomes of highly enriched normal and chronic myeloid leukemia stem and progenitor cell populations. Oncotarget. 2013;4:715–28.
Krishnan V, Kim DDH, Hughes TP, Branford S, Ong ST. Integrating genetic and epigenetic factors in chronic myeloid leukemia risk assessment: toward gene expression-based biomarkers. Haematologica. 2022;107:358–70.
Dvinge H, Bradley RK. Widespread intron retention diversifies most cancer transcriptomes. Genome Med. 2015;7:45
Shah JS, Milevskiy MJG, Petrova V, Au AYM, Wong JJL, Visvader JE, et al. Towards resolution of the intron retention paradox in breast cancer. Breast Cancer Res. 2022;24:100.
Zhang D, Hu Q, Liu X, Ji Y, Chao HP, Liu Y, et al. Intron retention is a hallmark and spliceosome represents a therapeutic vulnerability in aggressive prostate cancer. Nat Commun. 2020;11:2089.
Li SQ, Liu J, Zhang J, Wang XL, Chen D, Wang Y, et al. Transcriptome profiling reveals the high incidence of hnRNPA1 exon 8 inclusion in chronic myeloid leukemia. J Adv Res. 2020;24:301–10.
Schmitz U, Shah JS, Dhungel BP, Monteuuis G, Luu PL, Petrova V, et al. Widespread aberrant alternative splicing despite molecular remission in chronic myeloid leukaemia patients. Cancers (Basel). 2020;12:3738
Acknowledgements
We thank Prof. Ong Sin Tiong, DUKE-NUS, Singapore, for the kind gift of TF1 cells, Dr. Juan Felipe Ortiz Quinonez, CSI Singapore, for help with bioinformatic analysis, Dr. Daniel G. Tenen, CSI Singapore, for sharing the K562 EZH2 CRISPR/Cas9 knock-out cell line and the Ministry of Education for funding support (MOE-T2EP30220-0013).
Author information
Authors and Affiliations
Contributions
Performed experiments: RB, JY, YTH, BL, HK, LQZ, CG; analysed data: RB, YL, HY; provided material: GMO, JWY, LPK, LKT, EC, WWT; wrote manuscript: RB; conceptualized study: RB, WJC; supervised study: WJC.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The research was approved by the institutional review board (National University Hospital, DSRB# 2015/00804; DSRB#: 2007/00173) and all human participants gave written informed consent. All methods were performed in accordance with the relevant guidelines and regulations.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Brunmeir, R., Ying, L., Yan, J. et al. EZH2 modulates mRNA splicing and exerts part of its oncogenic function through repression of splicing factors in CML. Leukemia 39, 650–662 (2025). https://doi.org/10.1038/s41375-024-02509-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-024-02509-y
