
Abstract
Musculoskeletal disorders, including osteoarthritis, rheumatoid arthritis, osteoporosis, bone fracture, intervertebral disc degeneration, tendinopathy, and myopathy, are prevalent conditions that profoundly impact quality of life and place substantial economic burdens on healthcare systems. Traditional bulk transcriptomics, genomics, proteomics, and metabolomics have played a pivotal role in uncovering disease-associated alterations at the population level. However, these approaches are inherently limited in their ability to resolve cellular heterogeneity or to capture the spatial organization of cells within tissues, thus hindering a comprehensive understanding of the complex cellular and molecular mechanisms underlying these diseases. To address these limitations, advanced single-cell and spatial omics techniques have emerged in recent years, offering unparalleled resolution for investigating cellular diversity, tissue microenvironments, and biomolecular interactions within musculoskeletal tissues. These cutting-edge techniques enable the detailed mapping of the molecular landscapes in diseased tissues, providing transformative insights into pathophysiological processes at both the single-cell and spatial levels. This review presents a comprehensive overview of the latest omics technologies as applied to musculoskeletal research, with a particular focus on their potential to revolutionize our understanding of disease mechanisms. Additionally, we explore the power of multi-omics integration in identifying novel therapeutic targets and highlight key challenges that must be overcome to successfully translate these advancements into clinical applications.
Similar content being viewed by others
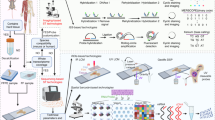

Introduction
The musculoskeletal system in vertebrates is essential for movement and the protection of internal organs; however, it is highly susceptible to debilitating diseases, including osteoarthritis (OA), rheumatoid arthritis (RA), osteoporosis (OP), bone fractures, intervertebral disc degeneration (IDD), tendon injuries and tendinopathy, as well as myopathy and muscle wasting (sarcopenia, cachexia), among others.1,2,3,4 These disorders cause pain, stiffness, and inflammation, leading to significant disability, with their prevalence exacerbating with aging and obesity.5,6,7,8 Despite the limited availability of effective treatments, substantial research efforts have been made in recent years to map the “omics” landscape (e.g., RNA sequencing, genomics, proteomics, and metabolomics, etc.) of different musculoskeletal cells in conditions of health and disease, as well as the interactions among cells and different signaling pathways. While single-cell technologies have advanced, bulk omics remain indispensable, particularly in proteomics and metabolomic,9,10,11,12,13,14 where technical challenges have constrained single-cell resolution. These approaches continue to provide critical insights into the regulatory mechanisms underlying musculoskeletal pathologies.
Bulk omics methods, despite their contributions, provide only population-averaged gene and protein expression data, failing to capture cellular heterogeneity and intercellular interactions. This limitation obscures the complexity of musculoskeletal tissues, hindering the identification of distinct cellular lineages and disease-associated signaling pathways. To overcome these challenges, recent advances in single-cell and spatial omics technologies have emerged as essential complementary approaches.15,16,17,18,19,20
Recently, advances in single-cell sequencing and spatial sequencing technologies have revolutionized musculoskeletal research and fundamentally enhanced our understanding of these complex diseases. Techniques such as single-cell RNA sequencing (scRNA-seq), single-nucleus RNA sequencing (snRNA-seq), single-cell assay for transposase-accessible chromatin with sequencing (scATAC-seq), and single-cell proteomics/metabolomics have ushered in a new era of precision in biological research.15,16,21,22,23 These methodologies (development timeline shown in Fig. 1) address the inherent limitations of traditional approaches by enabling the exploration of cellular heterogeneity and the complexities of tissues at an unprecedented resolution.24,25,26,27,28 For instance, scRNA-seq, pioneered by Tang et al., allows for transcriptome profiling at the individual cell level, revealing distinct cell populations, developmental trajectories, and new cellular states, thereby profoundly impacting the study of musculoskeletal diseases.24

The timeline of technological developments in exploring musculoskeletal diseases spans multiple biological levels, including transcriptomics, epigenomics, proteomics, and metabolomics
Moreover, advances in spatial omics techniques, such as spatial RNA sequencing, spatial proteomics, and spatial metabolomics,21,29,30,31,32 are pivotal in capturing the spatial arrangement and interactions of cells within tissues. Spatial transcriptomics, introduced by Ståhl et al. in 2016, has revolutionized gene expression analysis by visualizing it within tissue sections, enhancing our comprehension of gene activity in context.29 Recent innovations like single-cell deep visual proteomics32 and advanced mass spectrometry (MS) imaging31,33 have further propelled the capabilities of spatial proteomics and metabolomics, enabling detailed mapping of protein and metabolite distributions. These spatial omics techniques have garnered increasing interest in musculoskeletal research, underlining the critical importance of understanding cellular spatial organization in unraveling the complex mechanisms behind these diseases.
In this review, we will provide a comprehensive overview of the current landscape of advanced multi-omics and cutting-edge single-cell omics techniques in musculoskeletal research, emphasizing their transformative potential in enhancing our understanding of and management strategies for these challenging conditions. We will explore the latest methodologies applied to musculoskeletal tissues, examine key cellular and molecular mechanisms enabled by these technologies, and discuss the prospects for multi-omics applications that integrate various datasets to provide an understanding of disease mechanisms and the identification of novel therapeutic targets. Additionally, we will discuss the challenges in omics research and explore opportunities that exist for translating these compelling findings into impactful clinical applications.
Current omics technologies in articular cartilage
Single-cell RNA-seq and spatial transcriptomics in articular cartilage
Cartilage is a specialized connective tissue that covers the ends of diarthrosis joints and is crucial for joint function. However, its limited self-repair capacity makes it vulnerable to degeneration.8,34,35 OA, the most common joint disease, is characterized by progressive cartilage breakdown, synovial inflammation, and subchondral bone remodeling, leading to pain and mobility loss.8,35 In contrast, RA, an autoimmune disease, drives chronic inflammation, cartilage destruction, and bone erosion due to immune system dysregulation.34 Chondrocytes, the resident cells of articular cartilage, exhibit functional and molecular heterogeneity across its layered structure, with distinct subpopulations playing roles in maintaining joint homeostasis or driving degenerative pathologies. Traditional bulk omics approaches obscure this cellular diversity, masking critical disease-driving subpopulations and their pathogenic interactions. Recent advances in scRNA-seq have overcome these limitations by enabling high-resolution profiling of individual chondrocyte states, uncovering novel subtypes and communication networks that drive cartilage degeneration.36 Studies utilizing scRNA-seq have characterized chondrocytes from human OA and RA cartilage, identifying distinct subpopulations, including novel phenotypes with unique gene expression profiles and functions.37,38 These discoveries provide critical insights into the pathogenesis of OA and RA, paving the way for targeted therapies, early diagnosis, and improved treatment strategies.23
The pioneering application of scRNA-seq in human OA was first reported in 2018, where researchers identified seven distinct molecular subpopulations of chondrocytes within articular cartilage.37 This landmark study not only highlighted the diversity of chondrocyte states but also illuminated their potential roles in disease progression. Subsequent analyses have further refined these findings, revealing specific chondrocyte subpopulations—termed effector (EC), regulatory (RegC), and homeostatic (HomC)—each characterized by unique expression profiles and implicated in different facets of OA pathology.37 Notably, scRNA-seq has identified gene signatures associated with OA susceptibility, such as GLIS3, TGFB1, TNC, and WWP2. Moreover, one of the earlier studies demonstrating scRNA-seq’s utility in animal models showcased that many cell clusters identified in humans also exist in mouse knee cartilage.39 Further investigations using scRNA-seq have mapped several chondrocyte subpopulations in OA, including homeostatic, hypertrophic, prehypertrophic, regulatory, fibro-chondrocytes, prefibrocartilage, reparative, pre-inflammatory, and inflammatory chondrocytes, each with distinct molecular signatures.23,40 The discovery of inflammatory chondrocytes and their activation of the MIF-CD74 pathway sheds light on inflammatory processes in OA.40 Additionally, scRNA-seq has highlighted a senescent pathogenic cell cluster within cartilage, with regulators like FAP and ZEB1 emerging as novel therapeutic targets.41 This technology has also unveiled a chondrocyte subpopulation associated with anti-senescence and OA progression, identifying master regulator proteins such as NDRG2, TSPYL2, JMJD6, and HMGB2 as potential therapeutic avenues.42 Furthermore, scRNA-seq results of human OA cartilage identified seven distinct chondrocyte clusters, and demonstrated the upregulated SPP1 and downregulated VISFATIN signaling in OA.43 Notably, studies have also revealed alterations in cell–cell communication among chondrocyte subtypes, mediated by signaling pathways like PTN, VISFATIN, SPP1, and TGF-β, which may play crucial roles in OA progression.43
Beyond the characterization of chondrocyte subpopulations, scRNA-seq has been instrumental in exploring the involvement of pyroptosis-related genes in OA. Researchers have identified prognostic factors such as CASP6, NOD1, and PYCARD, with hub genes linked to pyroptosis implicated in the notch and oxidative phosphorylation pathways. These findings not only enhance our understanding of OA mechanisms but also present potential biomarkers for diagnosis and prognosis.44 Additionally, the identification of a ferroptotic chondrocyte cluster and the role of TRPV1 as an anti-ferroptotic target45,46 suggest innovative therapeutic possibilities; activation of TRPV1 has been shown to mitigate OA progression by enhancing GPX4 expression.46 Fu et al. also used scRNA-seq technology to discover changes in the expression distribution of seven distinct cell clusters and target genes in human articular cartilage, such as TNFRSF1B, GRN, and YWHAE.47 Moreover, their recent study revealed that Nav1.7 (SCN9A) is upregulated in OA articular cartilage tissue compared to normal articular cartilage, and it regulates chondrocyte biology, particularly chondrocyte metabolism. Targeted blocking or knockout of Nav1.7 has the potential to protect joint cartilage from degeneration and slow OA progression, thereby alleviating OA-associated pain.48
Integrative analyses combining miRNA profiles in cartilage-derived extracellular vesicles with scRNA-seq data in knee OA have yielded fresh insights into cartilage injury and the pathogenesis of knee OA, potentially enhancing diagnostic and therapeutic strategies. By identifying differentially expressed miRNAs and their target genes, researchers have pinpointed significant enrichment in pathways such as MAPK signaling, focal adhesion, and FoxO signaling.49 In another study, scRNA-seq identified three chondrocyte subsets: C1 involved in collagen turnover, synovial-like C2, and middle C3 maintaining cartilage matrix. And miR-17 suppresses HIF-1α to sustain homeostasis. Its loss reduces C1/C2, disrupts collagen balance, and elevates OA-associated enzymes (MMP3, MMP13 and ADAMTS5), while restoration via GDF-5 induction reverses degeneration.50 Furthermore, scRNA-seq has informed studies investigating chondrocyte regeneration mechanisms in OA, demonstrating that microtubule stabilization can effectively promote cartilage regeneration by inhibiting YAP activity—a promising therapeutic target for OA and cartilage injury.51
In the context of RA, scRNA-seq has been applied to explore the effects of mechanical loading on chondrocytes. By comparing cartilage from weight-bearing and non-weight-bearing regions of the femur in RA patients, researchers have identified two novel immune-related chondrocyte subtypes, suggesting that mechanical loading may influence chondrocyte function and contribute to the development of knee RA. This study highlights the contrasting functions of macrophage-like chondrocytes (MCs) in different regions and provides insight into the role of immune and mechanical factors in chondrocyte behavior and disease progression.38
In conclusion, scRNA-seq has provided unprecedented insights into the cellular and molecular mechanisms underlying OA and RA, identifying novel chondrocyte subpopulations and signaling pathways that may serve as therapeutic targets (Fig. 2a). However, scRNA-seq alone lacks spatial context, a limitation addressed by spatial transcriptomics, which maps transcriptional activities within intact tissue architecture to identify microenvironment-specific drivers of disease progression [reviewed in ref. 52]. For example, a recent multi-omics study integrating scRNA-seq and spatial transcriptomics in human OA knee identified 11 chondrocyte populations, including previously unrecognized pre-inflammatory and inflammatory subsets, marked by MIF-CD74 signaling activation.40 The study also revealed that prehypertrophic/hypertrophic chondrocytes localize to the articular surface, while prefibrocartilage chondrocytes distinguish inflammatory and non-inflammatory OA subtypes. These findings underscore how spatial context enhances molecular classification and uncovers localized disease mechanisms. By integrating cellular heterogeneity with tissue architecture, these advanced approaches refine our understanding of OA and RA pathogenesis, enabling the development of targeted therapies and improving clinical outcomes.

Application and pipeline of single-cell RNA-seq, single-cell ATAC-seq, and proteomics in OA and RA Cartilage. a Singel-cell RNA-seq. b Single-cell ATAC-seq. c Proteomics
Single-cell ATAC-seq and whole-genome bisulfite sequencing in articular cartilage
In addition to genomic and transcriptomic profiling, recent epigenetic advancements have begun to dissect the complex molecular underpinnings of joint disorders, offering new insights into its pathogenesis and potential therapeutic targets. ATAC-seq technology is crucial for mapping accessible chromatin regions in articular cartilage and joint-associated cells, uncovering transcriptional regulatory networks critical in OA and RA pathogenesis.53 This tool identifies active regulatory elements, such as enhancers and promoters, that control gene expression linked to inflammatory responses and tissue remodeling. scATAC-seq extends these capabilities by enabling chromatin accessibility analysis at the single-cell level, allowing researchers to explore cellular heterogeneity in joint tissues—a key factor in disease progression and treatment response. A groundbreaking study produced the first whole-genome chromosome conformation map (Hi-C) of primary human chondrocytes, integrating three-dimensional genomics with genetic association and epigenetic data. This integration has identified novel candidate effector genes for OA, such as SPRY4, PAPPA, and SLC44A2, revealing that OA-associated genetic variants often reside within chromatin loop anchors, indicating their regulatory roles in gene expression through enhancer-promoter interactions.54 Concurrently, ATAC-seq analyses have identified altered enhancers and target genes in articular cartilage of OA patients, revealing pathways involved in ossification and mesenchymal stem cell differentiation. This finding highlights the importance of direct chromatin profiling in clinical tissues for comprehensive epigenetic insights into disease mechanisms.55 Furthermore, Creb5 has emerged as a crucial regulator of Prg4/lubricin expression in the superficial zone articular chondrocytes, essential for joint lubrication and arthritis protection. Its role has been confirmed through chromatin immunoprecipitation and ATAC-seq analyses.56 The evolutionary history of the human knee, shaped by adaptations to bipedalism, influences OA risk, with epigenetic profiling revealing both ancient selection and recent constraints on knee regulatory elements that overlap with risk variants. This may lead to adult pathology, exemplified by the causal enhancer variant at the GDF5-UQCC1 locus, which influences knee shape and susceptibility of OA.57 While scATAC-seq has provided valuable snapshots of cell–cell variability in chromatin organization, gathering data from hundreds to thousands of single cells in parallel and highlighting distinct epigenetic profiles across chondrocyte subpopulations and other joint-resident cells, it has unfortunately not yet been used on chondrocytes (Fig. 2b).
DNA methylation, as a key mechanism for altering chromatin accessibility, has drawn extensive attention in studies of OA and RA.58,59,60 John Loughlin’s group has significantly advanced this field by identifying differentially methylated regions (DMRs) in genes such as COLGALT2, TMEM129, and WWP2, which play crucial roles in OA progression and present new therapeutic targets.60,61,62,63,64 Additional findings indicate that genes such as TBX4, ZBTB16, TRAF1, CTGF and CX3CL1 undergo methylation changes in OA that affect transcriptional regulation and chondrocyte function, linking these alterations to developmental and inflammatory signaling pathways.65,66 Researchers have also demonstrated stage-specific methylation changes and age-related overlaps in OA pathology, providing potential therapeutic avenues.67,68,69
Despite these advancements, studies on DNA methylation have primarily relied on beadchip microarray-based technologies, which assess only a limited subset of the genome. In a study by Shen et al., the impact of Dnmt3b ablation in articular chondrocytes was investigated using whole-genome bisulfite sequencing (WGBS).59 Chondrocytes from Dnmt3b f/f mice, transfected with either Adeno-GFP or Adeno-Cre, showed no significant differences in global methylation levels, yet identified 4 271 DMRs, predominantly in introns and intergenic regions (44% hypomethylated and 56% hypermethylated).59 Notably, DMR-associated genes significantly overlapped with differentially expressed genes (DEGs) related to osteoblasts and chondrocytes. Among the 104 genes exhibiting both expression and DNA methylation changes, two-thirds were hypomethylated, including key cartilage-related OA genes such as Ucma, Bmp4 and Igf1. Enriched transcription factor binding sites in DMRs were associated with chondrogenesis (Sox9 and Sox4), inflammation (Nfact2), and energy metabolism (Fkhr, Foxc1 and Foxk1)59 Moreover, a recent study by Wu et al. investigated the role of DNA methylation in antler chondrogenesis by comparing whole-genome DNA methylation between precartilage and cartilage in deer antlers.70 Despite similar overall methylation levels at CpG and non-CpG sites, WGBS analysis revealed 140 784 DMRs and 3 941 DMR-related genes between the two tissues. These DMR-related genes were enriched in pathways critical to chondrogenesis, including insulin receptor binding, PI3K/AKT signaling, Hippo signaling, and glycosaminoglycan biosynthesis. Key genes such as CD44, IGF1, RUNX1 and COL2A1 were closely linked to antler chondrogenesis, highlighting the regulatory role of DNA methylation in cartilage formation and regeneration.70 These findings provide valuable insights into the epigenetic mechanisms underpinning tissue regeneration and cartilage development, with implications for understanding cartilage-related diseases (Fig. 3).

Application of advanced epigenomics, transcriptomics, proteomics, and metabolomics in cartilage
The ongoing development and application of scATAC-seq hold great promise for refining our understanding of OA and RA at the cellular level. Integrating WGBS with other omics technologies—such as genomics, transcriptomics, proteomics, and metabolomics—allows researchers to comprehensively analyze the epigenetic landscape of joint diseases.71,72 This integration can pave the way for the development of personalized treatment approaches.
Proteomics in articular cartilage
The application of proteomics technology in the study of articular cartilage, particularly in the context of OA and RA, represents a significant advancement in understanding the pathobiology of these diseases. Proteomics, which involves the large-scale study of proteins, their structures, and functions, has been instrumental in identifying biomarkers and elucidating the molecular mechanisms underlying cartilage degeneration and disease progression. In articular cartilage research, proteomics mainly focuses on four components: articular cartilage tissue, cartilage explants, articular chondrocytes, and chondrocyte secretions.
Articular cartilage tissue proteomics has proven challenging due to the tissue’s complex and rigid structure. Initial evaluations of OA cartilage through tandem MS identified over 100 different proteins, including proteolytic fragments of link protein, aggrecan, and fibronectin.73 However, recent advancements have allowed for the comparative proteomic analysis of articular cartilage from OA patients and normal donors. One of the first studies identified 814 distinct proteins, with 59 proteins showing different levels between OA and non-OA cartilage.74 Bioinformatics characterization revealed many of these proteins are involved in regulating extracellular matrix (ECM) turnover, including matrix metalloproteinases (MMPs) and their inhibitors (TIMPs). Two-dimensional electrophoresis followed by a linear ion trap mass spectrometer with a Fourier Transform Ion Cyclotron Resonance mass spectrometer (LTQ-FT/MS) can effectively characterize the proteome of cartilage without in vitro culturing, which could obfuscate physiological differences.75 A total of 1 436 ± 49 or 1 472 ± 7 protein spots were resolved from normal and OA cartilage extractions, respectively,75 highlighting the potential of proteomics in identifying biomarkers and mediators of OA.
Cartilage explants offer another avenue for proteomic analysis. This approach involves culturing sections of cartilage tissue and analyzing the proteins secreted into the culture medium. Although it requires large amounts of starting material and primarily detects secreted components, it provides valuable insights into the proteins involved in cartilage catabolism and the secretory response of chondrocytes to various stimuli. The first comparative proteomic analysis of cartilage explants from OA and non-OA cartilage was conducted by Hermansson et al.,76 identifying proteins categorized as matrix proteins (COMP, different types of collagens, CILP1, FMOD, FN, SPARC, chitinase 3-like 2 (YKL-39), and chitinase 3-like 1 (YKL-40)) and matrix proteinases with their inhibitors, further confirming OA-associated ECM remodeling.76 Additionally, regulatory molecules, such as connective tissue growth factor, cytokine-like protein C-17, and inhibin βA, were found to be differentially expressed.76 Furthermore, Peffers et al. used a distinct approach to analyze the proteomic profile of IL-1β-treated OA cartilage explants, identifying a total of 252 proteins using QconCAT technology.77 This comparative analysis and absolute quantification of proteins involved in molecular pathways enhance our understanding of OA pathogenesis.
Proteomic analyses of articular chondrocytes have also contributed significantly to our understanding of cartilage biology. The initial proteomic investigation of osteoarthritic chondrocytes utilized 2D gel electrophoresis, while subsequent studies have primarily employed nanoscale LC-MS/MS, enabling shotgun proteomic analyses.78 One study using 2D gel electrophoresis and MALDI-TOF MS identified 93 proteins from normal chondrocytes involved in cytoskeleton and cellular organization, energy pathways, lipid metabolism, and stress response.78 A comparative proteomic analysis uncovered 9 proteins with increased expression and 9 proteins with decreased levels in OA chondrocytes relative to normal chondrocytes, with bioinformatics analysis facilitating their categorization into subgroups based on cellular function.79 iTRAQ-based proteomics has revealed 76 proteins with differing expression levels between OA patients and controls, with LECT2, BAALC, and PRDX6 identified as potential novel biomarkers, suggesting their combined use with conventional markers could enhance the assessment of OA severity and prognosis.80 Additionally, proteomics methods have been used to study protein interactions in chondrocytes, leading to discoveries such as complex with functional downstream effects from progranulin/TNFR2 signaling.47,81
The chondrocyte secretome is another critical area of research. Proteomic analysis of the secretome can identify changes in the secretory profile of chondrocytes in OA, providing insights into the disease’s pathogenesis and potential therapeutic targets. For instance, proteomic analysis of conditioned media from OA and normal chondrocytes treated with IL-1β, with or without nicotine, unveiled alterations in the profile of chondrocyte-secreted proteins. Notably, nicotine influenced nineteen proteins within the articular inflammation model, including cytokines (IL-6 and IL-8) and ECM remodeling factors (ACAN, MMP-2, MMP-3, MMP-1 and PGS2).82 Recently, Fu et al. also utilized proteomics to identify key downstream effector molecules responsible for Nav1.7 blockade-mediated regulation of chondrocyte metabolism, such as pro-anabolic HSP70 and anti-catabolic midkine.48 Blockade of Nav1.7 increases the secretion of HSP70 and midkine in articular chondrocytes, and elevated secretion of these molecules affects all chondrocytes and other joint cells through both autocrine and paracrine effects, thus regulating OA progression.48 These analyses can reveal proteins involved in inflammatory and metabolic pathways, which are crucial for understanding the multifactorial nature of OA.83
In conclusion, proteomics technology has significantly advanced our understanding of the complex molecular processes occurring in articular cartilage during both health and disease (Fig. 2c). The ongoing application of proteomics across these various areas is likely to lead to the identification of novel biomarkers and therapeutic targets for OA and RA, ultimately enhancing our comprehension of these debilitating conditions.84,85
Metabolomics in articular cartilage
Due to the avascular, aneural, and alymphatic nature of cartilage, chondrocytes primarily rely on anaerobic glycolysis for ATP production, supporting crucial processes such as the synthesis of ECM components like collagen and proteoglycans.86,87,88,89 While glucose metabolism is fundamental, glutamine also plays a crucial role in chondrocyte metabolism.90 As the most abundant amino acid in circulation, glutamine serves not only as an energy substrate but also as a precursor for biosynthetic processes. It aids in nucleotide and amino acid synthesis while supporting the tricarboxylic acid (TCA) cycle through its conversion to α-ketoglutarate.90,91 Studies indicate that the deletion of GLUT1 significantly impairs both chondrocyte proliferation and ECM synthesis, prompting a metabolic shift toward glutamine utilization.92,93 Beyond glucose and glutamine, lipids, including fatty acids and cholesterols, are now also recognized as important players in the regulation of chondrocyte function and cartilage integrity.86,88,94,95 Proper regulation of multiple metabolic pathways, such as glucose, glutamine, and lipid metabolism, ensures that chondrocytes have the necessary energy and biosynthetic resources to support cartilage homeostasis. Disruptions in this metabolic balance can lead to elevated oxidative stress, altered energy production, and inflammation, accelerating chondrocyte hypertrophy and degradation of the ECM.14,88,96,97,98,99
Chondrocyte metabolomics offers a powerful approach to explore the metabolic alterations that contribute to the onset and progression of OA.97,100,101,102,103,104,105 Studies employing carbon-13 glucose isotopes with LC/MS have demonstrated increased glycolysis and decreased TCA cycle activity, indicating a shift toward anaerobic energy production.86,88,96,97 Metabolic profiling has identified over 1 010 metabolite features significantly altered in OA cartilage, particularly in lipid, mitochondrial, and amino acid metabolism.101 Additionally, chondrocytes, being highly mechanosensitive, undergo significant metabolic changes in response to mechanical loading. Cyclical compression of human chondrocytes reveals that substrate stiffness and loading duration modulate key metabolic pathways, including those involving purines, glutamine, and glycolysis.106 Reduced mechanical loading, such as in simulated microgravity, results in alterations in protein synthesis, energy metabolism, and oxidative catabolism,107 while injury further increased carbohydrate and amino acid metabolism.108
While bulk metabolomics studies have provided important insights into global metabolic shifts associated with cartilage health, mechanical loading, injury, and OA progression, they do not fully capture the complexity of individual chondrocyte behaviors within the cartilage matrix. Chondrocytes exist in a heterogeneous microenvironment,109,110 with distinct subpopulations potentially exhibiting unique responses to mechanical stress and metabolic alterations. These differential cellular responses may significantly contribute to OA pathogenesis at the single-cell level; thus, single-cell metabolomics offers the opportunity to detect these cellular variations and identify specific metabolic alterations in individual chondrocytes, leading to a more precise understanding of metabolic dysfunctions driving OA progression. However, the application of single-cell metabolomics in chondrocyte studies remains in its early stages. Although this technique has been extensively utilized in cancer research,111,112 its use in cartilage biology is limited by several technical challenges. Chief among these is the inherently low metabolic activity of chondrocytes, particularly in mature cartilage. Chondrocytes reside within a dense ECM and function in a hypoxic environment, leading to lower metabolic turnover than proliferative cells like those in cancer. This reduced activity can result in metabolite concentrations falling below the detection thresholds of current analytical methods, limiting the sensitivity of available techniques. Advancements in extraction methods and detection technologies will be crucial for accurately profiling low-abundance metabolites in individual chondrocytes.
While single-cell metabolomics offers insights into metabolic heterogeneity at the cellular level, spatial metabolomics provides the added advantage of preserving the spatial context of cartilage tissue architecture. This approach allows for precise mapping of metabolic alterations within the cartilage matrix, capturing metabolic dynamics in their native environment. It could effectively identify region-specific metabolic changes, particularly in areas subjected to varying mechanical stress, inflammation, or tissue degeneration during OA progression (as shown in Fig. 3). Cillero-Pastor et al.113 were the first to employ time-of-flight secondary ion mass spectrometry (TOF-SIMS) to investigate the spatial metabolomics of healthy and OA human cartilage, revealing significant molecular alterations linked to OA pathology. Their TOF-SIMS analysis uncovered a distinct lipidomic signature in OA cartilage, highlighted by the accumulation of cholesterol, oleic acid, and other fatty acids predominantly localized in the superficial layer, contrasting with the uniform lipid distribution seen in healthy cartilage. Additionally, OA cartilage exhibited localized deposits of calcium and phosphate ions around chondrocytes, indicating dysregulated ion homeostasis. In a related study, Eveque-Mourroux et al. 114 explored the lipidomic and proteomic differences in OA cartilage between patients with and without type 2 diabetes mellitus (T2DM) using a combination of label-free proteomics and matrix-assisted laser desorption/ionization mass spectrometry imaging (MALDI-MSI). They found distinct lipid and protein profiles between the two groups, with phosphatidylcholine (PC) and sphingomyelin (SM) more prevalent in OA without T2DM patients, while lysolipids dominate in OA with T2DM patients. Furthermore, significant differences in lipid distribution were observed between the superficial and deep layers of cartilage. MALDI-MSI provided spatially resolved lipid analysis, revealing the localization of phosphatidylcholine (PC) and sphingomyelin (SM) in the superficial cartilage, while lysolipids were predominantly concentrated in the deeper layers, particularly in OA patients with T2DM. These molecular and spatial differences underscore the effectiveness of TOF-SIMS and MALDI-MSI in studying cartilage spatial metabolomics, offering critical insights into OA pathology and the metabolic alterations driving the disease.
In conclusion, understanding the molecular heterogeneity in OA cartilage is essential for developing more targeted therapeutic approaches, as these insights are crucial for recognizing metabolic reprogramming during OA progression. Integrating spatial and single-cell metabolomics is vital for obtaining a comprehensive view of OA pathology at both the cellular and tissue levels. By combining these approaches, we can gain deeper insights into how localized metabolic dysfunctions contribute to OA pathogenesis, ultimately identifying spatially distinct metabolic targets for therapeutic intervention. Therefore, targeting these dysregulated metabolic pathways in chondrocytes presents a promising strategy for restoring cellular homeostasis, reducing inflammation, and slowing cartilage degeneration. These single-cell omics techniques also have important implications in understanding cartilage development in processes such as the chondrogenic differentiation of pluripotent stem.115 Such advancements could significantly enhance the development of more effective treatments for OA.
Current omics technologies in synovium tissues
Single-cell RNA-seq and spatial transcriptomics in synovium
The synovium is a specialized connective tissue that lines the inner surface of the capsules of synovial, or diarthrodial joints. The healthy synovium contains two main types of cells within the synovial membrane lining: Type A synoviocytes (macrophage-like synoviocytes) and Type B synoviocytes (fibroblast-like synoviocytes, FLS). Type A synoviocytes perform phagocytic activities similar to macrophages, removing debris and waste products from the synovial fluid while maintaining joint homeostasis. Type B synoviocytes are primarily involved in the production of synovial fluid, including secreting hyaluronic acid, other lubricating substances, and various proteins and enzymes that are essential for the maintenance and repair of the synovial membrane and joint cartilage. Importantly, the synovium also contains numerous other immune cell types, including macrophages, B cells, T cells, and dendritic cells, which contribute to joint health, injury response, and a balanced environment within the joint.
In arthritis, including RA and OA, the synovium undergoes significant changes characterized by inflammation, cellular infiltration, and thickening (hyperplasia), a condition known as synovitis. Nearly all forms of arthritis are characterized by significant changes in the number and phenotype of immune cells. However, the significant complexity and often low prevalence of many of these cell types have made it difficult in the past to identify their specific roles in physiologic or pathologic synovial function. In particular, traditional molecular and histologic methods have proven difficult in identifying interactions among cell types within the synovium, as well as with other joint tissues such as the articular cartilage or bone.
In this regard, recent advances in scRNA-seq, snRNA-seq, and MS have uncovered novel synovial cell identities and therapeutic targets, enabling detailed characterization of individual musculoskeletal cells in both health and disease.116 However, obtaining healthy synovial tissue, particularly from individuals without joint disease, remains a significant challenge. As a result, early single-cell studies of the synovium primarily analyzed tissue from RA patients using methods such as Cel-Seq2 (plate-based), droplet-based, or microfluidic (e.g., 10x Genomics).
A critical milestone in single-cell analysis focused on developing the methodologies for single-cell isolations from cryopreserved synovial tissues.117 Researchers collected and cryopreserved human synovial samples, optimizing parameters for mechanical and enzymatic dissociation to ensure high cell viability while preserving surface proteins critical for downstream analyses, such as cell sorting, mass cytometry, and RNA-seq. These refined protocols yielded high frequencies of viable cells with minimal transcriptome variability, preserving key surface markers essential for accurate cell profiling. Mass cytometry, employing a 35-marker panel, identified diverse cell phenotypes, while RNA-seq provided robust transcriptomic profiles, capturing expression data for over 1 000 genes per cell. This foundational work established a foundational pipeline for analyzing cryopreserved synovial specimens.
Building on this groundwork, subsequent investigations leveraged droplet-based microfluidics to characterize the synovium at single-cell resolution. Investigators performed single-cell transcriptome profiling of disaggregated synovial tissue from five RA patients.118 In this early study, 20 387 single cells were sequenced, revealing 13 transcriptomically distinct clusters, including 10 immune populations that broadly expressed PTPRC (CD45) and three fibroblast populations, expressing uniform high levels of COL1A2. Within immune cells, clear markers of known subtypes, including canonical macrophage markers (MARCO), T cell (CD3), and B-cell (MS4A1) markers, were found. These encompassed an unsupervised draft atlas of the autoimmune infiltrate contributing to disease biology. Notably, this study also identified a previously uncharacterized fibroblast subpopulation within the synovium.
The importance of FLS in inflammatory arthritis extends beyond RA to juvenile idiopathic arthritis (JIA), an autoimmune disease wherein the synovium is highly affected and appears to play a pathologic role. FLS in JIA are known to be heterogeneous, and subpopulations of FLS can show aggressive phenotypes associated with invasive and destructive disease activity. To investigate JIA FLS heterogeneity and gene expression in JIA, scRNA-seq was used to profile cells from multiple JIA subtypes.119 FLS were found to be heterogeneous and exhibited characteristics of fibroblasts, chondrocytes, and smooth muscle cells. Among these, the chondrocyte-like subpopulation was the predominant cell type, with its percentage increasing with disease severity. Despite overlapping subpopulations across subtypes, the chondrocyte-like cells had unique genetic fingerprints that distinguished between JIA subtypes. The study found biologically relevant differences in gene expression between JIA subtypes that supported a critical role for FLS in pathogenesis, and that gene expression profiles within the chondrocyte-like subpopulation could distinguish among subtypes.119 Besides, the pathological role of synovial fibroblasts was further explored through studies of NOTCH3 signaling. Using scRNA-seq and synovial tissue organoids, they demonstrated that NOTCH3 signaling drives both transcriptional and spatial gradients in fibroblasts originating from vascular endothelial cells outward. In active RA, synovial fibroblasts exhibited marked upregulation of NOTCH3 and NOTCH target genes, indicating that endothelium-derived NOTCH signaling regulates the positional identity of fibroblasts. This stromal crosstalk pathway underlies inflammation and pathology in inflammatory arthritis.120
In addition to exploring fibroblast-driven inflammation, recent scRNA-seq applications have provided insights into the mechanisms of joint pain in RA. While RA pain is often assumed to be directly linked to synovial inflammation, simple measures of inflammation do not correlate with pain severity in patients. A recent study identified an 815-gene expression module associated with pain in synovial biopsy samples from patients with established RA who exhibited limited synovial inflammation at the time of arthroplasty.121 Further scRNA-seq analyses revealed that the majority of these 815 genes were expressed by lining layer synovial fibroblasts. In these studies, receptor-ligand interaction analysis predicted crosstalk between human lining layer fibroblasts and human dorsal root ganglion neurons expressing calcitonin gene-related peptide (CGRP+). These findings suggest that synovial lining fibroblasts express pain-associated genes that promote the growth of pain-sensing neurons into hypertrophic synovial regions in RA.121
While most studies have focused on the role of scRNA-seq in identifying subpopulations of synovial cells that are integral in RA pathology, few studies have attempted to identify cells involved in RA remission. Using integrated scRNA-seq data and CellChat to analyze cell–cell communication, the EGF signaling pathway was identified for its potential role in this regard.122 Eleven clusters of synovial cells, including fibroblasts, T cells, macrophages, and B cells, were found, with extensive communication networks involving EGF signaling between fibroblasts and synovial macrophages. HBEGF was highly expressed in a fibroblast subset, downregulated in RA, and upregulated after treatment. HBEGF+ fibroblasts were defined by high HBEGF expression and related genes like CLIC5, PDGFD, BDH2 and ENPP1, which were downregulated in RA and elevated after therapy. These findings provide an example of the potential application of single-cell methods to uncover pathways involved in both disease progression and remission.122
As for OA synovium, scRNA-seq has been used to evaluate the effect of obesity on OA synovium. Given that obesity is one of the primary risk factors for OA in both weight-bearing (e.g., hip, knee, foot) and non-weight-bearing (e.g., hand) joints. However, the mechanisms by which obesity mediates OA synovium remains poorly understood. To address it, synovial tissues from the hand, hip, knee, and foot joints were collected from OA patients classified as either obese (BMI > 30) or normal weight (BMI 18.5–24.9). Fibroblasts from these tissues were analyzed using proteomic panels along with bulk and single-cell RNA-seq.123 These analyses revealed that the inflammatory profile of OA synovial fibroblasts is independently affected by obesity, joint loading, and anatomical site, with significant heterogeneity between obese and normal weight patients. scRNA-seq identified four molecular endotypes, including obesity-specific subsets defined by an inflammatory endotype associated with immune cell regulation, fibroblast activation, and inflammatory signaling, characterized by upregulated CXCL12, CFD and CHI3L1 expression. Fibroblast subsets in obese patients were spatially localized in the sublining and lining layers of the synovium and were distinguished by differential expression of the transcriptional regulators MYC and FOS. These findings underscore the impact of obesity on altering the inflammatory profile of synovial fibroblasts in both load-bearing and non-load-bearing joints, and provide potential targets for new OA therapies.123
Further elucidation of fibroblast heterogeneity in OA synovium has come from studies using flow cytometry to isolate fibroblast subsets based on the surface markers CD39 and CD55.124 Subsequent scRNA-seq, MS, and proteomic profiling revealed that CD39+CD55− fibroblasts expressed high levels of myogenic markers, such as CNN1, IGFBP7, MYH11, and TPM1, compared to CD39−CD55+ fibroblasts. KEGG analysis of upregulated DEGs in CD39+CD55− fibroblasts identified the Apelin and cGMP-PKC-signaling pathways as potential contributors to pain. LC/MS analysis showed significantly higher levels of proteins encoded by myogenic marker genes, including CNN1, IGFBP7, and MYH11, in CD39+CD55− fibroblasts. In contrast, CD39−CD55+ fibroblasts highly expressed PRG4 genes and proteins, with upregulated DEGs enriched for pro-inflammatory pathways. Of particular interest was that the proportion of CD39+CD55− fibroblasts in synovium significantly correlated with both resting and active pain levels, but not joint space width, in OA patients.124
Comparative studies between RA and OA synovial tissues have expanded our understanding of unique cellular phenotypes and states associated with each condition. Analysis of 51 synovial tissue samples from patients with RA or OA identified 18 unique cell populations, including T cells, B cells, monocytes, and fibroblasts.125 Combining mass cytometry and transcriptomics revealed cell states expanded in RA synovia, such as THY1(CD90)+HLA-DRAhi sublining fibroblasts, IL1B+ pro-inflammatory monocytes, ITGAX+TBX21+ autoimmune-associated B cells, as well as PDCD+ peripheral helper T (T(PH)) cells and follicular helper T [T(FH)] cells. Distinct subsets of CD8+ T cells with GZMK+, GZMB+, and GNLY+ phenotypes were also characterized. Using mass cytometry, inflammatory mediators were identified and mapped to their source cell populations, such as IL6 expression to THY1+HLA-DRAhi fibroblasts and IL1B production to pro-inflammatory monocytes. This integrated approach provides a unique workflow for identifying populations as potential mediators of RA or OA pathogenesis.125
Beyond scRNA-seq, researchers have also utilized snRNA-seq to generate a comprehensive transcriptomic profile of synovial cells in the subacromial synovium from young and aged individuals. By delineating aging-related transcriptomic changes across different cell types and their associated regulatory networks, the researchers identified two subsets of stromal cells in the human synovium: lining cells and sublining cells. In aged stromal cells, genes associated with angiogenesis and fibrosis were upregulated, while genes related to cell adhesion and cartilage development were downregulated. Additionally, specific cell–cell communications in the aged synovium mirrored aging-related inflammation and tissue remodeling, such as vascular hyperplasia and tissue fibrosis. One of the major findings was the identification of forkhead box O1 (FOXO1) as a significant regulon for aging DEGs in synovial stromal cells. FOXO1 was found to be downregulated in both lining and sublining cell populations of the aged synovium. These data indicate that FOXO1 plays an important role in the regulation of human synovial aging.126
While these studies provided initial “atlases” of synovial cell populations, rodent models of RA or OA have provided deeper mechanistic insights into specific populations that drive the pathogenesis of these diseases. One of the first scRNA-seq studies of mouse RA used the serum-transfer (K/BxN) model with transgenic mouse models to identify distinct subsets of fibroblasts responsible for mediating either inflammation or tissue damage in arthritis.127 Deletion of fibroblast activation protein-α (FAPα)+ fibroblasts suppressed both inflammation and bone erosions in mouse models of resolving and persistent arthritis. Single-cell transcriptional analysis identified two distinct fibroblast subsets within the FAPα+ population: FAPα+THY1+ immune effector fibroblasts located in the synovial sublining, and FAPα+THY1− destructive fibroblasts restricted to the synovial lining layer. When adoptively transferred into the joint, FAPα+THY1− fibroblasts selectively mediated bone and cartilage damage with little effect on inflammation, whereas the transfer of FAPα+THY1+ fibroblasts resulted in a more severe and persistent inflammatory arthritis, with minimal effect on bone and cartilage. The findings demonstrate how single-cell analysis can be used to identify distinct fibroblast subsets with non-overlapping functions in the synovium.127
Building on this, the role of macrophages in regulating synovial function has been elucidated using fate mapping and scRNA-seq in the K/BxN mouse model.128 This work identified dynamic membrane-like structures, consisting of a distinct population of CX3CR1+ tissue-resident macrophages, that form an internal immunological barrier at the synovial lining that physically secludes the joint. These barrier-forming macrophages exhibit features typical of epithelial cells and maintain their numbers through a pool of locally proliferating CX3CR1− mononuclear cells embedded into the synovial tissue. Unlike recruited monocyte-derived macrophages, which actively contribute to joint inflammation, these epithelial-like CX3CR1+ lining macrophages restrict the inflammatory reaction by providing a tight-junction-mediated shield for intra-articular structures. These findings revealed functional diversity of macrophages in synovial homeostasis and inflammation.128
Expanding on the heterogeneity of macrophage populations, recent studies have explored the breadth of the distribution of macrophages in a mouse model of injury- and obesity-induced OA, using scRNA-seq combined with lineage-tracing to study the origin and phenotype of sorted myeloid subtypes in the mouse synovium.129 scRNA-seq results revealed that joint injury and obesity differentially and synergistically alter the architectural, cellular, and molecular profiles of the synovial capsule. Joints showed the presence of multiple populations of macrophages that uniquely expressed Ccr2, H-2Aa and Lyve1, as well as established macrophage genes including Csf1r, Adgre1, and Itgam. Four macrophage populations, Ccr2+MHCII− cells, Ccr2+MHCII+ cells, Lyve1+MHCII+ cells, and Lyve1+MHCII− cells, displayed unique transcriptomic profiles. Fewer patrolling monocytes were observed in obese mice, whereas there was a significantly higher influx of pro-inflammatory monocyte-derived macrophages in the first 3 days after joint injury in obese compared to control (lean) mice. Joint injury and obesity also changes in other immune cell subtypes, including T, B, mast cells, and neutrophils, as well as local synovial fluid cytokines associated with injury and obesity.129
Finally, the role of synovial cell populations has also been investigated in the context of post-traumatic osteoarthritis (PTOA). Several studies have shown that the synovium is altered following joint injury and in post-traumatic arthritis,130 yet the roles of specific cell types in this process remain to be determined. To study the effects of joint injury on the synovium, mice were subjected to non-invasive anterior cruciate ligament rupture as a model, and scRNA-seq was performed on synovial cell populations. Seven distinct functional subsets of synovial fibroblasts were uncovered in healthy and injured synovium, and their temporal dynamics in early and established PTOA were defined. Wnt/β-catenin signaling was found to be overactive in PTOA synovium, and trajectory analyses predicted that Prg4(hi) lining fibroblasts arise from a pool of Dpp4+ mesenchymal progenitors in the synovium, with SOX5 identified as a potential regulator of this emergence. This study shows that synovial fibroblasts assume distinct functional identities during PTOA in mice, and Prg4hi lining fibroblasts secrete R-spondin 2, which may drive pathological joint crosstalk after injury.
Advancing beyond single-cell transcriptomics, spatial transcriptomics has provided valuable insights into the spatial organization and interactions of synovial cells within the joint microenvironment. By linking transcriptional profiles to the physical locations of cells, this approach offers a clearer understanding of cell localization and crosstalk in RA and OA synovium. When integrated with scRNA-seq, spatial transcriptomics reveals additional complexities in synovial inflammation and tissue remodeling, particularly in macrophage phenotypes. One recent study has focused on the spatial distribution and functional states of macrophages in RA and OA.131 Using scRNA-seq coupled with spatial transcriptomics, researchers identified three distinct macrophage clusters: M0-like MARCO+ Mϕ1, M2-like CSF1R+ Mϕ2, and M1-like PLAUR+ Mϕ3. Mϕ1 was broadly found in the synovium, whereas Mϕ2 and Mϕ3 were sparse. Trajectory analysis showed that Mϕ1 existed at the start of the differentiation trajectory. HOXB6, STAT1, and NFKB2 were specific TFs for Mϕ1, Mϕ2, and Mϕ3 under RA conditions, respectively. Compared to OA, all three macrophage clusters upregulated CXCL2, CXCL1, IL1B, TNFAIP3, ICAM1, CXCL3, PLAU, CCL4L2, CCL4, and TNF within the NF-kappa B signaling pathway.131 This study identifies macrophage subsets with different polarized states and their molecular signatures, providing a more precise understanding of these cell subtypes in RA and OA.
In addition to studying macrophages, scRNA-seq and spatial transcriptomics have been used to investigate other immune cell populations, such as B cells, by employing pre-sorting techniques like flow cytometry to increase sequencing resolution. While the role of B cells in established RA is well recognized, their presence and function in joint tissue at the onset of the disease remain unclear. Synovial biopsies from untreated patients at the time of RA diagnosis were sorted for B cells and then underwent RNA sequencing, and paired tissue pieces were subjected to spatial transcriptomics.132 Of note was the presence of both local B-cell maturation via T cell help and plasma cell survival niches with a strong CXCL12-CXCR4 axis. Immunoglobulin sequence analyses revealed clonality between the memory B and plasma cell pools, further supporting local maturation of the B cells. These findings suggest that plasma cell niches are not a consequence of chronic inflammation but are already present at the time of RA diagnosis.132
In conclusion, the synovium is a complex tissue consisting of multiple cell types. Advances in single-cell techniques have shown that the cellular makeup and phenotype of the synovium can be altered drastically with joint injury, OA, or RA. Specific changes occur in various cell populations, such as fibroblasts, macrophages, or other immune cells, highlighting their roles in driving inflammation, joint damage, and pain. Moreover, advanced omic techniques as well as bioinformatic analyses have provided insight into the intricate crosstalk and interaction action among the cells in the synovium, as well as with other cells in the joint and potentially, systemically. Most recently, scRNA-seq studies have extended to synovial fluid, which could offer a non-invasive way to study immune activation and inflammatory pathways in joint disease.133 Additionally, scRNA-seq and snRNA-seq have recently been applied to the infrapatellar fat pad (IFP) in both mouse and human. These studies revealed that the synovium and IFP share a population of common mesenchymal progenitors.134,135 Among these, Dpp4+ mesenchymal progenitors were identified as the source of synovial lining fibroblasts, adipocytes, and myofibroblasts, highlighting the integration of these tissues as a functional unit.134 In OA, this functional unit undergoes profound changes, characterized by fibrosis and cartilage degeneration driven by biglycan-positive fibroblasts and Apolipoprotein E signaling.135 All these investigations offer a promising strategy to elucidate the exact origin and recruitment of these cells to the synovium, synovial fluid, and IFFP in future research. By identifying these mechanisms, novel therapeutic targets could be developed to modulate immune and inflammatory responses more effectively and to improve treatments for joint diseases (Fig. 4).

Application of advanced epigenomics, transcriptomics, proteomics, and metabolomics in synovium
Whole-genome bisulfite sequencing in synovium
RA also involves epigenetic modifications, notably DNA methylation, which influences disease mechanism.136 In RA, abnormal DNA methylation patterns in synovial fibroblasts and immune cells contribute to chronic inflammation and autoimmune responses. These methylation changes can promote the activation of inflammatory pathways and immune cell infiltration, which are characteristic of RA pathology.136 WGBS have also been applied to identify specific DNA methylation patterns and methylated positions associated with the disease. In contrast to OA studies that primarily focus on cartilage, RA studies tend to concentrate on synovial tissues and fibroblasts from RA patients. This focus arises from the nature of RA as a chronic autoimmune disease primarily targeting the synovial lining of joints, leading to inflammation, synovial hyperplasia, and subsequent cartilage and bone destruction.137 A study by Ham et al. focusing on synoviocytes found that while the overall DNA methylation patterns of RA and OA patients were broadly similar, specific epigenetic variations were observed in RA. Notably, they identified 523 low-methylated regions that were uniquely associated with RA, suggesting distinct regulatory mechanisms at play in this autoimmune condition.138 Another study led by Ai et al. used synoviocytes isolated from RA and OA to identify specific epigenetic regions that distinguish RA cells from OA cells. Their analysis revealed that the majority of these DMRs were concentrated within active enhancers and promoters.139 Moreover, Li Yim et al. exploited WGBS and RNA sequencing to investigate DNA methylation and gene expression changes in synovial tissues from early arthralgia and established RA patients, and they indicated that differences in DNA methylation and gene expression were associated with disease severity, as measured by the swollen joint count 66. Patients with a swollen joint count 66 of 9 or higher showed distinct activation of immune response and dysregulation of cell adhesion pathways at both the transcript and methylation levels.140
Taken together, in the context of RA, WGBS offers critical insights into the epigenetic regulation of genes involved in immune response, inflammation, and synovial tissue remodeling. WGBS can help elucidate how methylation changes contribute to the activation of pro-inflammatory pathways and immune cell infiltration. By mapping the methylome of synovial tissue or immune cells, epigenetic markers could be identified. This information is invaluable for developing epigenetic-based therapies that modulate immune responses or mitigate synovial inflammation. Future research combining WGBS with other omics technologies will offer a deeper understanding of the molecular mechanisms driving RA.141,142
Proteomic and metabolomic techniques in synovium
In addition to these recent studies the transcriptome and epigenomics of the synovium, the advent of new proteomic, lipidomic, and metabolomic techniques based on MS have led to an improved understanding of the metabolic profile of synovial tissues in health and disease. These techniques provide a more detailed snapshot of the functional state of the synovium by providing data at the protein level, including lipids and metabolites, and their potential interactions. While the majority of this work to date has been performed on serum or synovial fluid, several studies have focused on the metabolic dysfunction of the synovial tissue itself in order to identify potential biomarkers as well as disease-related processes in the context of RA or OA [reviewed in refs. 143,144]. In both conditions, the synovium has been implicated as a critical mediator of the disease process as well as a potential mediator of joint pain.144,145
Analyses of the RA synovium have shown specific changes in synovial matrix proteins with various forms of arthritis. For example, high-resolution MS studies have been used to identify a novel citrullination site on fibronectin in the synovium,146 suggesting critical post-translational modifications of arginine. While the specific effects of this phenomenon remain to be determined, identification of new metabolic or protein modifications such as these has the potential to uncover new biomarkers or therapeutic targets for arthritis.
With a focus on OA, Rocha et al. used MALDI-MSI to examine the lipidomic profile of the osteoarthritic synovium and to compare it with healthy synovium and other forms of inflammatory arthropathies as RA and psoriatic arthritis.147 Their findings revealed complex lipidomic profiles that differed between OA and control samples, with osteoarthritic synovium exhibiting elevated levels of phosphatidylcholines, fatty acids, and lysophosphatidic acids and lower levels of lysophosphatidylcholines compared to control tissues. However, osteoarthritic tissue showed lower amounts of phosphatidylethanolamine-based plasmalogens as compared to tissues from inflammatory arthritis. In other studies, lipidomic studies of osteoarthritic synovium showed a significant upregulation in most differential lipids, particularly triacylglycerides and long-chain unsaturated fatty acids, in comparison to a treatment group that was intra-articularly with chitosan as a therapeutic.148
Clearly, significant additional work is needed in this area to identify changes in the proteomic and metabolomic profile of synovial tissues. While several ongoing studies have targeted the synovial fluid or serum using multi-omics strategies in this regard,149 many questions remain about specific matrix and cellular changes within the synovial tissue itself. A further understanding of these changes will provide new insights into cell–cell and cell-matrix interactions in the synovium that may drive joint disease.
Current omics technologies in bone compartments
Transcriptomics in mouse and human bone marrow cells
Omics technologies have also transformed the understanding of bone biology, particularly in bone fracture healing and OP. Bone marrow cells attract significant interest in current omics studies due to their critical role in bone homeostasis, hematopoiesis, and the repair processes following injury. To fulfill its dual functions of mechanical support and blood production, bone marrow is primarily made of two lineages of cells. On the one hand, mesenchymal stem and progenitor cells (MSPCs, also termed skeletal stem and progenitor cells, SSPCs) give rise to bone-forming osteoblasts/osteocytes, which produce mineralized bone matrix for mechanically supporting the body, and marrow adipocytes, whose function is still under extensive debate. On the other hand, hematopoietic stem and progenitor cells (HSPCs) give rise to all of the different types of mature blood cells inside the bone marrow and in the peripheral blood. While hematopoietic cells constitute the vast majority of bone marrow cells (>98%), the rare mesenchymal cells are also critical for blood production as they provide a niche for hematopoiesis. The recent emergence of single-cell omics techniques has generated an unprecedented opportunity to investigate cellular heterogeneity of these two essential bone marrow cell lineages and examine their crosstalk (as shown in Fig. 5).

Application of advanced transcriptomics and metabolomics in bone cells and bony callus
Since 2019, many studies have performed scRNA-seq of mouse bone marrow mesenchymal lineage cells using a variety of cell labeling and sorting strategies. Initial studies examined postnatal mice, mainly at young and adult ages. The first study by the Scadden group sorted non-hematopoietic (Ter119-Cd71-Lin-) cells from the long bones of young mice.150 The second study by the Aifantis group profiled gene expression in various bone marrow HSC niches, including cells labeled by Lepr-Cre (mesenchymal stromal niche), Cdh5-Cre (vascular niche), and Col2.3-Cre (osteoblast niche).151 Following those, the Qin group analyzed mesenchymal cells labeled by Col2-Cre from mice at 1 (young), 3 (adult), and 16 (aging) months of age;152 the Ayturk group examined long bone endocortical cells from PBS or Sclerostin-neutralizing antibody treated mice;153 the Hass group analyzed total bone marrow cells, followed by progressive depletion of abundant cell types or enrichment of rare populations;154 the Ono group analyzed CXCL12-abundant reticular (CAR) cells, a major component of bone marrow Mesenchymal progenitor cells (MPCs), using Cxcl12GFPhigh cells from Cxcl12-CreER Td Cxcl12-GFP mice;155 the Long group examined Gli1+ mesenchymal cells in mice with or without parathyroid hormone treatment.156 Interestingly, regardless of mouse ages, reporters, and sorting strategies, all datasets from these studies contain a large cell cluster (or more clusters) that highly and specifically express bona fide adipocyte markers, including Cebpa, Pparg, Adipoq, and Lpl, as well as hematopoietic regulatory factors, such as Cxcl12, Kitl, Il7, Angpt1, etc. This cell cluster was given many names, such as MALP (marrow adipogenic lineage precursor),152,156 LepR-MPC (due to its high expression of Lepr),150 Adipo-CAR, Osteo-CAR,154,157 etc. However, when datasets from different groups are merged and examined, it is clear that they are the same mesenchymal subpopulation expressing hematopoietic supportive factors at the highest level. Using Adipoq-Cre to label these cells, conditional knockout mice have demonstrated the critical role of MALPs in suppressing bone formation, promoting bone resorption, and maintaining hematopoiesis.158 scRNA-seq analysis revealed that bone marrow injuries, such as radiation and chemotherapy, appear to expand these cells and convert them into myelofibrolasts.151,159 Furthermore, in myelofibrosis (MF), a type of bone marrow disease, the so-called “adipogenic mesenchymal stromal cells,” which are similar to MALPs, were identified by scRNA-seq as the fibrosis-driving cells.160
Two recent studies examining fetal mouse bone marrow revealed a very different pattern of mesenchymal subpopulations.161,162 Fetal bone marrow is mostly constituted of mesenchymal progenitors, such as Pdgfra+Sca1+ (PαS) cells, which are only a small portion of adult bone marrow. Strikingly, it completely lacks a MALP-like cell cluster, which leads to an overall low expression of hematopoietic supportive factors in fetal bone marrow. Based on scRNA-seq datasets and reporter mouse analysis, MALPs emerge right after birth in mice (P0). These observations are consistent with the current view that HSPCs colonize bone marrow postnatally.
scRNA-seq of human bone marrow mesenchymal cells has also been reported by various groups in the past several years. Most of these studies analyzed bone marrow aspirate or biopsy from adult and aged patients and generated a relatively homogenous cell clustering pattern. Similar to mouse bone marrow, the major cell cluster(s) identified in those studies highly and specifically express adipogenic markers, Lepr, as well as hematopoietic regulatory factors.163,164,165,166 In MF patients, scRNA-seq analysis discovered a loss of hematopoietic supportive factors and significant upregulation of ECM genes.160 By enzymatically digesting femoral head trabecular bone, Bandypadhyay et al. generated a comprehensive bone marrow cell atlas including mesenchymal cells with much more heterogeneity. A total of six mesenchymal subpopulations were identified, including Fibro- MPC, APOD+ MPC, Osteo- MPC, Osteoblast, Adipo- MPC, and THY1+ MPC.167 While Fibro- MPC is likely to be the most primitive MSPCs, Adipo- MPC and THY1+ MPC express adipogenic markers and a high level of hematopoietic regulatory factors and thus resemble MALPs in mice.
Similar to mouse, scRNA-seq suggested that human fetal bone marrow contains much more MSPCs than adult or aged human bone marrow. One study divided mesenchymal lineage cells into osteochondral precursors, osteoblast precursors, early osteoblasts, Adipo- CARs, and chondrocytes.168 Another study found 7 mesenchymal clusters, including 4 osteolineage cells, a cluster of cycling cells, and a cluster of mesenchymal cells with no obvious differentiation markers.169 Similar to mouse datasets, this fetal bone marrow dataset does not have MALP counterparts.
Applying spatial transcriptomics to bone marrow presents significant challenges due to the low resolution of current technologies and the close proximity of bone marrow cells. Dr. Tower’s group was the first to demonstrate the feasibility of using spatial transcriptomics to characterize the cellular components of the stem cell niche and identify cell–cell interactions.170 To address the resolution limitations, a single-cell proteomic imaging approach has been successfully applied to human bone marrow.167 By employing Co-Detection by Indexing (CODEX), over 1.2 million bone marrow cells were spatially profiled using a 53-antibody panel. Integrating scRNA-seq and CODEX data revealed a hyperoxygenated arterio-endosteal neighborhood for early myelopoiesis, and an adipocytic localization for early HSPCs.
Future technology improvement in either increasing the resolution of whole transcriptome sequencing-based spatial transcriptomics or increasing the number of genes in imaging-based spatial transcriptomics will generate a more detailed cell mapping of bone marrow mesenchymal and hematopoietic cells and provide more crosstalk information between or among these two types of cells.
Transcriptomics in bony callus
Bone fracture healing involves a complex process characterized by callus formation, a temporary structure of cartilage and bone that stabilizes the fracture site and supports new bone growth.171,172,173,174,175 This intricate process is tightly regulated by growth factors, cytokines, diverse cell lineages, and signaling pathways.171,172,173 scRNA-seq has been instrumental in elucidating the cellular populations and gene expression profiles involved in callus formation, uncovering heterogeneity among mesenchymal stem cells (MSCs) and their differentiation into chondroblasts and osteoblasts176,177,178,179,180,181,182,183,184 (as shown in Fig. 5). Additionally, scRNA-seq analyses of calluses have provided deeper insights into the cellular dynamics and molecular mechanisms of bone repair.
Bone healing declines with aging, but the underlying mechanisms and rejuvenation strategies remain under active investigation. Thomas H. Ambrosi et al. explored age-related declines in bone healing using scRNA-seq.176 In comparing calluses from young (2-month-old) and aged (24-month-old) mice 10 days post-fracture, they observed higher clonal activity and diversity of skeletal stem cells (SSCs) in younger mice.176 Eight SSC subpopulations were identified, with aging impairing differentiation into osteogenic and chondrogenic lineages and shifting the bone marrow niche toward a pro-inflammatory state. However, aged mice treated with BMP2 and low-dose aCSF1 exhibited enhanced osteochondrogenic gene expression and increased bone-forming SSC fractions, leading to improved regeneration.176
Further studies have highlighted aging-related changes in SSPCs. Lin et al. identified three major SSPC clusters (osteogenic, proliferative, and adipogenic) in young and aged calluses.177,178 Aging increased inflammatory osteogenic cells, characterized by IRF and NF-κB activation, reducing osteogenic potential.177 The same group found senescence-associated gene signatures enriched in aged callus clusters, suggesting inflammation and senescence as contributors to impaired fracture repair.178 Zou et al. identified pro-senescent macrophage-derived factors, such as grancalcin (GCA), that impair SSPC function in aging. GCA’s receptor, PlexinB2, was found highly expressed in SSPCs, influencing stemness and differentiation.179
Inflammatory and immune components in the bone marrow microenvironment also affect callus formation.180 Zhang et al. identified B-cell dynamics during fracture healing, noting reduced B-cell populations in aged fractures. B-cell numbers peaked during callus remodeling, highlighting their temporal role in repair.180 Another scRNA-seq study by Avin et al. obtained canal tissues from human femoral nonunion patients and autologous bone graft, and identified 23 cell clusters, with higher proportions of monocytes, CD14+ dendritic cells, and lower proportions of T cells, myelocytes and promyelocytes in nonunion samples.182 Additional studies have probed signaling pathways in fracture repair. Xiao et al. examined RA-induced fracture nonunion, finding that inflammation disrupted progenitor proliferation and differentiation, thereby impairing fracture healing. Their scRNA-seq analysis revealed that the Rbpjk/Notch pathway was upregulated due to reduced DNA methylation, further exacerbating the repair deficits175,181,185,186 under systemic inflammatory conditions. Serowoky et al. demonstrated that Sonic Hedgehog (Shh) signaling in periosteal SSPCs is essential for soft callus formation. Their scRNA-seq analyses of Sox9+ lineage knockout mice revealed a reduction in Cxcl12-expressing cells, underscoring the therapeutic potential of Shh for large-scale skeletal injuries.183
Spatial transcriptomics has further advanced fracture healing research.154,170,187 Jiang et al. applied this technique to study metastatic breast cancer’s effects on fracture healing, revealing significant downregulation of essential bone matrix genes and compromised repair. By localizing gene expression within the three-dimensional architecture of bone tissues, the study provided unparalleled insights into spatial genetic changes during fracture repair.188 Using the Visium CytAssist spatial transcriptomics platform, researchers successfully mapped genes associated with hard callus (e.g., Dmp1 and Sost) and soft callus (e.g., Acan and Col2a1) while preserving the spatial integrity of the tissue. Notably, the presence of MDA-MB-231 metastatic breast cancer cells corresponded to significant downregulation of regulators essential for bone matrix and structural integrity, potentially contributing to fragility in pathological fractures.188 Dr. Tower utilized the spatial transcriptomics approach in murine and human fracture callus tissue, identifying a spatially restricted activation of the BMP pathway. This localized activation was observed in both a murine model and human patients with neurofibromatosis type 1 loss of function.189
Collectively, these studies underscore the transformative potential of single-cell and spatial omics in unraveling the cellular and molecular dynamics of fracture healing. Future research integrating these cutting-edge techniques may provide comprehensive insights into bone regeneration, particularly in aging and inflammatory contexts, paving the way for targeted therapies to enhance repair.
Metabolomics in osteoblasts/osteocytes
Osteoblast differentiation is typically associated with broad transcriptional and proteomic changes. In addition, it has become more appreciated that osteoblasts rewire their metabolism to facilitate the increased energetic and biosynthetic demands that are associated with bone formation.190,191,192 Metabolic rewiring produces distinct metabolites that are essential at different stages of differentiation and regulate distinct cellular processes. For example, a-ketoglutarate (a-KG) is essential for SSC proliferation,193 glutathione (GSH) regulates cell viability and protects RUNX2 from oxidative damage to promote osteoblast differentiation,90,194,195,196 while amino acids are required for bone matrix production.194,197,198,199,200,201 Identifying and quantifying the metabolomic changes that occur during normal osteoblast differentiation, and during fracture repair and in disease, is essential to understand, diagnose and treat human bone disease. However, untargeted metabolomic analysis is very challenging because of the difficulty of detecting and characterizing all metabolites in a cell.
Initial metabolic studies in cultured cells elucidated many important nutrients and metabolic pathways utilized during osteoblast differentiation. Many of these studies utilized candidate approaches to study the metabolism of individual nutrients (e.g., glucose or glutamine). With advancements in metabolomics, technologies like nuclear magnetic resonance (NMR) and MS have been applied to broadly interrogate cellular metabolism in cultured cells as well as in isolated bone tissue. NMR is a rapid, nondestructive, reproducible technique that provides important structural information and has been used to characterize metabolic changes in MC-3T3 cells,202 human osteoblasts,203 and adipose-derived mesenchymal stem cells.204 However, the low sensitivity and spectral resolution have limited the implementation of NMR in the bone field. Compared to NMR, MS has several advantages, including increased sensitivity and specificity, and has played a central role in metabolomic and stable isotope tracing studies in both bone and other tissues. Over the last two decades, advances in liquid chromatography (LC) and gas chromatography (GC) have enabled the quantitative analysis of several hundred metabolites in either a targeted or non-targeted manner (as shown in Fig. 5).
Several studies have used LC/MS data to quantify metabolite abundance in several systems including primary bone marrow stromal cells (BMSC), primary calvarial osteoblasts, immortalized osteoblast and bone marrow-derived cell lines,193,196,205,206,207 whole bones from wild type,208 aged209 or diabetic mice,210 or serum from either humans211,212 or rodents.213 These studies have shaped our understanding of how the metabolome changes during osteoblast differentiation and in disease. For example, the abundance of several amino acids, the antioxidant GSH, and glycolytic metabolites like lactate are increased in mature osteoblasts.205 This is consistent with several studies demonstrating that mature osteoblasts increase amino acid uptake and synthesis, GSH synthesis, and use glycolysis for ATP production.194,196,197,201,205,214,215 Another study described distinct metabolomic profiles in adipogenic and osteogenic BMSC clones.207 This suggests that metabolism, in addition to changing during differentiation, likely plays an instructive role in governing cell-fate decision.207 Consistent with this, lipid availability can alter chondrocyte differentiation.216 More recent studies have begun to apply LC/MS to evaluate the metabolomic footprint in whole bone and bone marrow. These studies have described sexually dimorphic metabolite profiles in mice, with increased amino acid, purine, and energy metabolism in males and increased lipid metabolism in females.208 Studies in humans found significant associations between circulating amino acids and risk for low bone mineral density.217 Moreover, Liang et al. explored the multi-omics (RNA-seq, scRNA-seq, proteomics) to map osteokines involved in regulating bone and systemic health.218 By analyzing bone, osteoblasts, marrow, and serum, they identified 375 osteokines influenced by aging and mechanical stress. Osteoblasts were key producers, with senescent cells secreting fatty-acid-binding protein 3 to drive vascular smooth muscle cell aging, linking bone and vascular health.218 Future studies comparing metabolome profiles in bones from mouse models of human bone disease will be important to understand how metabolism is affected. Likewise, harnessing the power of mouse genetics to inhibit signaling pathways like WNT, PTH, or BMP will be important to elucidate the metabolomic effects of bone anabolic agents.
While steady-state snapshots of metabolite profiles provide essential information about cellular metabolism, such snapshots do not directly reflect enzymatic or pathway activity. Instead, stable isotope tracing can be used to monitor the metabolic fate of substrates containing heavy isotopes, typically 15N, 13C, 2H, or 18O, through a technique known as stable isotope tracing.219,220 GC/MS is used primarily for stable isotope tracing studies, although LC/MS can also be used. GC/MS is the favorite for these studies because many amino acids and metabolites in glycolysis and the TCA cycle are directly amenable. Stable isotope tracing has been particularly fruitful for tracing the flux of glucose and several different amino acids during osteoblast differentiation in cultured cells193,194,197,198,221 and more recently in whole bones in vivo.210,222 These studies have yielded interesting insights into how the metabolism of individual nutrients changes during differentiation and disease. For example, tracing studies using either 13C or 15N labeled glutamine found that glutamine broadly contributes to several amino acids, TCA intermediates like citrate and a-KG, GSH, and the purine metabolite uridine monophosphate.193,194,197,221 Conversely, asparagine contributed only to non-essential amino acid synthesis, whereas proline was not metabolized and is directly incorporated into nascent protein in osteoblasts.197,198 By comparison, glucose is mainly converted to lactate in osteoblasts, although it also contributes to the malate-aspartate shuttle and the TCA cycle.223 Consistent with this, in vivo tracing found that glucose is primarily traced to lactate with lesser enrichment in the TCA cycle in cortical bone.222 Follow-up studies in a mouse model of type 2 diabetes found reduced labeling of pyruvate, glutamate, and glutamine, indicative of suppressed glucose metabolism.210 These studies highlight the unique metabolism of different nutrients in osteoblasts and how metabolic flux is altered in disease. It is important to note that while the in vivo studies are inherently more challenging than the in vitro studies, they offer a critical look at the metabolic activity of bone cells in their native environment. The recent progress in this area is encouraging.
Although metabolomic analysis by LC/MS remains the gold standard in metabolite analysis, cell metabolism is both cell-type and microenvironment dependent. Specific techniques have been optimized to allow for cell sorting, enriching for individual cell types while preserving the metabolic signature during processing.224 However, these analysis techniques erase the spatial context of these cells within their local microenvironment. Previous works have shown that cell metabolism is directly responsive to external signals from both the microenvironment, such as hypoxia,225 and from cell–cell signaling, such as cytokine signaling from macrophages during inflammation,226 underscoring the importance of spatial registration of cell metabolism within tissue.
Over the last several years, spatial transcriptomics has evolved as a powerful tool to visualize the transcriptional landscape within the native tissue environment.29 Spatial transcriptomics makes use of tissue sections rather than dissociated cells, meaning that metabolic changes associated with prolonged exposure to enzymatic digestion at elevated temperatures can be bypassed.227 Though primarily used for soft tissue, owing to the ease of sample preparation, the use of spatial transcriptomics has been used in several musculoskeletal tissues, including the skeletal muscle,228 calvaria,229 digit tip,230 femur,170 and tendon,231,232 as well as human fetal limbs.233 In addition to these published works, several studies are currently available as preprints, suggesting that the application of spatial transcriptomics to musculoskeletal tissue will continue to expand.234,235 Within the mouse femur, transcripts related to glycolysis, oxidative phosphorylation, and fatty acid metabolism were found to be differentially regulated within the bone marrow relative to the proximity to blood vessels or bone surfaces.170 Within the regenerating digit tip, spatial transcriptomics was used to identify elevated glucose metabolism pathways as a result of aging.230 While several limitations currently exist in terms of sample processing,236 as well as the limited spatial resolution and/or sensitivity, these studies suggest the feasibility of using spatial transcriptomics for the unbiased assessment of tissue metabolism within its native context.
Spatial transcriptomics serves only as a proxy for measuring cell metabolism, using transcripts to estimate the activity of key metabolic enzymes.222 Ultimately, the direct, rather than indirect, interrogation of metabolites is critical in understanding the current metabolic status of cells and tissues. Techniques89 such as MALDI-MSI allows the visualization of metabolites within the spatial context of native tissue. Spatial metabolomics is still not yet widely utilized in musculoskeletal research, limited by the same tissue processing, resolution, and sensitivity obstacles presented by spatial transcriptomics. However, early studies in bone237 and muscle tissue238 show the feasibility of using spatial metabolomics to assess changes in energy production and phosphate homeostasis, suggesting that these technological challenges can be overcome. These spatial platforms will together provide key tools needed to unravel cellular metabolism in sync with transcriptomics and offer insight on the spatial relationship between cell metabolism and tissue fate and function in bone.
Current omics technologies in the intervertebral disc
Transcriptomics in intervertebral disc
The intervertebral disc (IVD) consists of three functionally distinct regions: the shock-absorbing nucleus pulposus (NP), the collagen-rich annulus fibrosus (AF) for structural support; and cartilaginous end plates (CEP) enabling nutrient exchange.239 This intricate architecture balances spinal flexibility with mechanical resilience, yet the IVD’s avascular nature and limited regenerative capacity make it susceptible to IDD, a progressive process characterized by ECM breakdown, cellular dysfunction, and inflammatory signaling, ultimately leading to structural failure and chronic pain.240,241 Emerging technologies such as scRNA-seq have begun to unravel these complexities. The earliest reported application of scRNA-seq for the IVD was in 2020 to assess the differentiation state of notochordal cells derived from human embryonic stem cells.242 Since then, the usage of the technique in published studies has rapidly increased in various model systems. These scRNA-seq studies discovered unreported IVD cell populations and biological processes during disease and aging (as shown in Fig. 6).

Application of advanced transcriptomics, proteomics, and metabolomics in intervertebral disc
scRNA-seq is an incredibly powerful tool to help identify the diverse cell populations and their complex intracellular processes within the IVD: AF, NP, CEP, exogenous tissues (e.g., nerves and vasculature), as well as interactions between immune cells and mesenchymal stem cells.243 Early application of scRNA-seq in the IVD identified gene markers and their relative expression levels for distinct IVD cell populations. For example, FOXM1 and KDM4E were identified in healthy human lumbar IVDs as unique transcription factors that regulate protein–protein interactions for the AF and NP.244 Another study of human NP, AF, and CEP tissues from patients within a variety of spinal disorders identified regulatory, homeostatic, and effector populations of chondrocytes with distinct functions for IVD homeostasis. Notably, they also identified new subtypes of NP-progenitor cells that are PDGFRA and PROC positive.245,246,247
In animal models, scRNA-seq has expanded our understanding of species-specific IVD cell markers. Findings in Sprague-Dawley rats uncovered more specific, unique NP cell markers such as Krt7, Prrg4, and Akap12; and novel markers to distinguish between inner AF (iAF): Bpifa2f, Mmp3, and IL11, and outer AF (oAF): Fibin, Myoc, and Igfbp5248 with wide functions from matrix synthesis to antibacterial response. Another study in the bovine tail identified distinct NP populations expressing CP, S100B, H2AC18, SNORC, CRELD2, PDIA4, DNAJC3, CHCHD7 and RCN2; further analysis identified a notochord populations distinct from the NP clusters that expressed KRT8, ATP6V1G3, C1QTNF3, CD55 and SPP1, in addition to iAF and oAF markers.249 Combining bulk and single-cell RNA sequencing identified stem cells from the NP and AF populations that expressed LGALS3, CD24, and CD44, notochordal-like cells expressing CD24, and fibroblast-like cells expressing SPARC in the NP and AF populations.250 Though these studies are done in different species, they identify many overlapping markers in similar populations, as well as transcriptional markers that exist uniquely in their respective biological context.
Given that IVD degeneration is a major contributor to back pain, a leading disability worldwide.251 scRNA-seq has facilitated the identification of gene expression changes in IVD cells associated with degeneration and damage. Analysis of iAF and NP tissues in degenerate IVDs identified a list of predictive degeneration genes: MT2A, UPP1, PRG4, MT1G, IGFBP3, and SOD2.252 Another study in the human lumbar CEPs identified FOXF2, PRRX2, S100A2, ID4, ENO1, TST (associated with anabolic, metabolic, development, and cell cycling) in non-degenerated CEP cells; while degenerated cells were enriched in genes that regulate chondrocyte hypertrophy and calcification such as BMPR1B, FMN1, NPR3, and CEMIP.245,247
Trajectory inference analyses have further demonstrated how the lineage patterns of IVD progenitors are altered with degeneration. For instance, in human scoliotic discectomy tissues, the MCAM+ AF progenitor trajectory to iAF shifted towards hypertrophic and pro-inflammatory AF trajectories, which expressed hypoxia, apoptosis, ossification, and inflammation genes, were more enriched in mild and severely degenerated IVDs. Similarly, CD24+/MKI67+ NP progenitors were enriched towards the EffectorNP1 cluster in normal IVDs but changed trajectories to EffectorNP3 in severely degenerated IVDs. During degeneration, subtypes of B cells, Cd8+ T cells, neutrophils, mast cells, dendritic cells, and monocytes/macrophages in the IVDs had a significantly different percentage of representation in degenerated IVDs.253
Studies utilizing injury models to mimic IVD degeneration have further revealed the dynamic participation of immune cells. Injury models are common to invoke degeneration,254 and a rapid degenerative process stimulates the diverse participation of immune cells.255,256 In Sprague-Dawley rats, injured IVDs contained 1.5 times more myeloid cells with abundant pro-inflammatory M1 macrophages. Additionally, the lymphoid populations expand dramatically by 4.6x, consisting of B-cell subtypes and T cells. Ngf+ and Ngfr+ (p75NTR) in the outer AF cells suggest that these cells upregulate angiogenesis, and can sensitize nociceptive neurons that lead to hyperalgesia.257,258 Another study found a role for a local NP cell subtype that can promote discogenic back pain and prime towards a painful hyperalgesia response.259 Other studies have found specific upregulation of anabolic, metabolic, and inflammatory processes.260,261,262 These studies bridge the mechanistic understanding between IVD degeneration and the clinically prominent problem of low back pain.
scRNA-seq has also offered new insight into the utilization of various stem cells to replenish IVD cell populations by enabling the evaluation of the transcriptomic transitions resulting from treatment conditions. For example, human embryonic stem cells exposed to Fibroblast Growth Factor treatment (concomitant with inhibition of BMP and retinoic acid signaling) appear to yield NP cells that recapitulate the transcriptomic signatures and cellular diversity of authentic human NP cells.242 Implanting chondrocyte-like NP cells from induced human pluripotent stem cells into the nuclectomized IVD space of nude rats prevented degeneration and preserved mechanical properties; and the scRNA-seq revealed that the implanted cells gave rise to distinct cell lineages with BNIP3L- and HIF-1α-signaling.263 Lineage-tracing and fate-mapping studies have further expanded our understanding of IVD development. Analyses of embryonic tissues from human and mouse embryos at multiple gestational milestones during IVD development identified critical markers, Sox10, CTSK, Spp1, and Krt15, that are key to the transition to developing notochord and nucleus pulposus.264 These notochordal cells largely differentiate into nucleus pulposus cells from neonatal stages into adulthood, but some notochordal cells preserve their identity and function into adulthood.265
Overall, scRNA-seq has advanced IVD biology and enabled the profiling of transcriptional changes occurring with growth, aging, and disease. In addition to these new insights, the support of the scientific community to deposit datasets also enhances opportunities to combine datasets or conduct new analyses on existing datasets. Studies that have reanalyzed previously deposited scRNA-seq data deposited on GEO have uncovered new findings from publicly available datasets such as: linking infiltrating monocytes and macrophages in the pathogenesis of IVD degeneration,266,267 identifying AF resident Grem1+ progenitor and Lum+ vasculogenesis promoting cell populations,268 and elucidating the effect of HSP90 fibrotic NP populations associated with pathogenic angiogenesis.269 The reanalysis of datasets from different types of sequencing modalities, such as high-throughput sequencing and microarray studies in combination with scRNA-seq, offers a powerful approach to identify disease markers from these large datasets. A study with this analysis found three DEGs, GREM1, LRPPRC, and SLC39A4, significantly regulated from six GEO deposited datasets and four key microRNAs that act as upstream regulators of target genes.270
While scRNA-seq reveals cell-specific transcriptomic changes, spatial transcriptomics preserves the locational specificity of the cell transcriptome and provides context on how the cells function in their tissue-specific environment. Examining the progressive localization of NP-progenitor gene markers (Cd24a, Thy1, Sox9, Pdgfra, Fgfr3, Uts2r, Lepr, Tie2, Ctsk) revealed that postnatal progenitor cells are predominantly located in the outer, peripheral regions of the NP and differentiate towards the center. Additionally, Ctsk-positive NP-progenitor cells had the capacity to generate the entire NP structure as the IVD tissue matures.271 These findings highlight the critical importance of anatomical ___location for the interpretation of tissue repair and function.
Despite their advantages, scRNA-seq and spatial transcriptomics present challenges, particularly in the context of IVD research. The IVD is a connective tissue with low cell numbers, and thus multiple spinal levels or animals may need to be pooled to achieve sufficient cell numbers for scRNA-seq experiments. Harsh or lengthy digestion protocols to render IVD tissue into a single-cell suspension can also cause cell loss for fragile cell populations, stimulate cell stress pathways, and create variability in results. scRNA-seq does not provide locational information of identified cell types, and this technique must be paired with immunostaining or histology to correlate gene expression with cluster identification or alternative “omics” techniques such as spatial RNA transcriptomics must be used. Spatial transcriptomics has a low RNA transcript capture efficiency, there is a lack of single-cell resolution, and great care must be taken to prevent RNA degradation on sample sections and mRNA diffusion during permeability steps.271,272 scRNA-seq data can be noisier than bulk-RNA sequencing or microarray analysis due to a variety of technical and biological factors, and extensive bioinformatic analyses of the raw data is required.273
Proteomics in the intervertebral disc
By leveraging established techniques of MS and modern informatics approaches, proteomics provides comprehensive protein, peptide, and other small molecules from a single preparation. Studies dating back to 2009 have identified pertinent ECM and tissue-specific proteins critical for IVD function by using MS.274 High-resolution tandem MS modalities have been utilized in the IVD field to characterize distinct portions of the proteome, such as the cellular content, matrisome, and secretome. Proteomics is a salient technique that continues to uncover the biological functions important for IVD health as measured via proteomic changes during IVD development, growth, and degeneration (as shown in Fig. 6).
Early applications of utilizing proteomics in the field were focused on constructing a complete proteomic characterization of the IVD. A study from 2015 identified 1 360 proteins from healthy, wild-type CD1 mice were categorized into either intracellular and plasma membrane, organelle, macromolecular complex, or extracellular region, with catalytic activity being the most common molecular function based on PANTHER gene ontology analyses.275 There are distinct proteomic profiles of the NP and AF in healthy mice IVDs, where the NP largely contained lysosomal and gap junction proteins, while oxidative stress-specific proteins were in the AF. The finding permitted the formation of the “PRIMUS” dataset: Proteomic Resource for IVD of MUS musculus. Cross-species comparisons of IVD proteomic profiles between rodent and human samples determined that the AF is most similar between the two than the NP; however, mice are still an appropriate model system to study the IVD.276 Proteomic analysis of human IVD tissue has also better informed the field of the types of patient samples that should be considered biologically “healthy” and used as experimental controls. Human IVD tissue derived from cadaver, scoliotic, and spinal trauma patients is often used as “healthy” controls in studies, but the proteomic profiles of the samples can differ from those of truly healthy tissue. Comparative analyses between IVD samples collected from scoliosis and lumbar fracture patients and healthy IVD tissue brain-dead organ donors found that inflammatory, oxidative stress, and degenerative pathways are elevated in the former.277,278 These studies highlight the importance of understanding the proteomic profiles of IVD across species and biological states to better inform experimentation.
IVD tissue damage changes the RNA transcriptomic profiles, but the proteomic profile is highly altered as well. The first study to do a comparative analysis between human AF cells isolated from control IVD tissue from scoliotic patients and degenerated IVD tissue from herniations found distinct results where three proteins were decreased with degeneration: HSPA8Q, G6PD, and protocadherin-23 and seven proteins were increased: GNAI2, TMEM51, LPPR2, FAR1, PSMC5, ADORA3, and SOD2.279 Another study analyzing control and degenerated IVD tissues showed aging decrease available IVD proteins and increases SLRPS and fibril-forming molecules that promote fibrosis.280 In addition to degeneration and aging, mechanical loading can also alter the proteome of the IVD. Low compressive forces applied to non-degenerated human NP cells had beneficial outcomes on protein production, where pro-survival and ECM homeostasis proteins are upregulated,281,282 but high compressive forces dysregulated NP senescence, apoptosis, and ECM catabolism protein production. These observations led to the discovery of potential protective bioregulators, such as MIF, that aid in NP cell survival under high compression.283 There are also interactions between the genome that culminate in the proteome—SM/J mice (poor healers) have an ectopic expression of prehypertrophic chondrocyte protein markers in the outer NP region, including PPR, IHH, and PTCH.284
Beyond the tissue-level proteome, analyses of the IVD secretome can determine how the expression levels of chemokines, growth factors, and ECM proteins change with IVD degeneration. Proteomic analysis on collected media from cultured degenerative bovine IVD cells showed an increase in inflammatory cytokine production that correlated with increased chemokine production, CCL5 and CXCL6, which have a chemotactic effect on MSCs.285 Increased IVD degeneration leads to ECM degradation, where changes in the levels of fragmented ECM components can be measured in the secretome. NP cells isolated from non-chondrodystrophic and degenerative, chondrodystrophic canine IVDs had an increase in fragmented core proteins from SLRPS decorin, BIGLYCAN, and higher levels of other ECM proteins such as FIBRONECTIN and FIBROMODULIN.286 Additionally, the cross-species analysis of notochordal/NP cultured medium from human, canine, and porcine IVD samples to identify secreted factors that could be targets for regenerative therapies found 60 secreted proteins shared amongst the models that were mainly ECM or organelle-derived or membrane-bound proteins. The NP cultured medium had the capacity to stimulate regenerative effects, such as increased ECM deposition, in the NP chondrocyte-like cells that replace healthy NP cells with disease.287 These studies highlight the value of analyzing the IVD secretome to better identify regenerative factors or immunomodulatory and ECM changes.
Assessing the biological pathways that are enriched in fetal tissue and dysregulated in degenerative tissue by comparison could pinpoint targetable factors to stimulate IVD tissue repair. During development, the proteomic profile of fetal IVDs changes as the tissue develops and progresses into skeletal maturity. Proteomic analysis of human fetal IVD samples found ten clusters of proteins grouped based on biological function, such as ribosomal biogenesis, matrilin and thrombospondin, collagens, SLRPS, and others.288 Further analysis of how the human IVD proteome changes in fetal tissue compared to healthy and degenerate adult tissue samples identified Collagens 1 and 2 as those with anabolic potential and plausible protein targets for tissue engineering strategies, while Collagen X was identified as a marker of IVD degeneration.289 Aging also affects the proteome that can prompt IVD degeneration. An analysis of the matrisome from fetal, young, and aged bovine NP tissue using STRING to assess protein–protein interactions found a high degree of connectivity and large protein networks for protein groups such as GAG binding, polysaccharide and carbohydrate binding, collagen fibril organization, skeletal system and cartilage development, and glycolysis. Collagen XI was shown to be overexpressed in the fetal NP matrisome, while Prolargin increases with age.290 Reanalysis of this proteomic dataset to focus on the NP cellulome instead found aged NPs overexpressed several proteins such as Chitinase‐3‐like protein 1, Clusterin, Endoplasmin, Alpha‐2‐HS‐glycoprotein, Lysozyme C, Melanoma‐derived growth regulatory protein, Protein disulfide‐isomerase A3, Ribonuclease 4, and Tumor necrosis factor receptor superfamily member 11B compared to young NPs.291
There have been no IVD studies to date that have performed true spatial proteomics, which is defined as the localization of proteins at the subcellular level.292 There has been a study that has constructed a spatiotemporal proteomic atlas of the IVD. This technique differs from spatial profiling techniques since the targets are not identified in situ, like spatial transcriptomics, but many IVD regions are dissected and analyzed separately, and the anatomical identification of locational changes in the proteome is reconstructed to form a proteomic map. There is only one study that has constructed a spatiotemporal proteomic atlas of the IVD thus far and aimed to profile spatiotemporal changes of the proteomic profile of the different anatomical locations of young and aged IVDs. The findings from this study created the DIPPER database, which functions to provide a comprehensive proteomic resource to study the spatial proteome of young, aged, and degenerative human IVDs. This study performed a thorough isolation of eleven distinct anatomical regions of the IVD, where seven segments were isolated from the central left-to-right lateral axis and five segments were isolated from the central anteroposterior axis. These segments correspond to four areas from the oAF, two from the iAF, one from the central NP, and four from the IAF/NP boundary. This is the first study to provide higher spatial resolution of the proteomic difference between IVD regions and how the proteomic profile of those regions changes between young and aged IVDs.293
The DIPPER database represents a significant milestone in spatial proteomics, offering an unprecedented resource for understanding proteomic variations across IVD anatomical regions and age groups. It has inspired other proteomics studies that have reanalyzed its publicly available data to uncover new findings. The application of an MS analysis technique called Peptide Location Fingerprinting (PLF) on the DIPPER dataset identified age-associated protein structural differences and tissue region differences in many ECM proteins. These findings highlight the potential of PLF in identifying potential degenerative biomarkers.294 A follow-up study using PLF to further analyze the DIPPER database by conducting a comparative study of the aged human IVD proteome from the DIPPER database to human arterial atherosclerosis and aged mouse lung datasets to determine common cross-species, age-associated differences in protein structures conserved between mouse and human IVDs. The IVD, lung, and arteries contain ECM-rich tissues, which this study found that ECM proteins are the largest class of protein dysregulated with aging, independent of tissue, and there are similarities in the types of ECM proteins most affected in IVD, lung, and arterial tissues.295
Metabolomics in intervertebral disc
Metabolomics, a methodology that identifies low molecular weight biomolecules in tissues, has been less utilized in the IVD field than scRNA-seq or proteomics, but has still provided valuable insights into biological changes in response to IVD disease or degeneration.296 Similar to proteomics, MS methodologies are often used, but other tools such as NMR or Raman spectroscopy can also be utilized to interrogate metabolome changes. Since proteomics is one of the oldest “omics” techniques employed in the IVD field, and both proteomics and metabolomics have many similarities in experimental procedures, a protocol for the parallel analysis of mouse IVD tissues for metabolomics and proteomics via MS was established in 2020 in an effort to standardize the usage of these techniques.297
Since then, studies have utilized metabolomics to assess the metabolomic and lipidomic changes of the IVD due to degeneration and profile how the metabolome of quiescent IVD stem cells supports cell survival and functionality by reducing cell energy requirements via downregulating metabolism (as shown in Fig. 6).298 More specifically, the detection of changes to IVD metabolites as degeneration progresses is a comprehensive way to uncover potential drivers of ECM degradation, a prominent feature of IVD degeneration. By using NMR and MS metabolomics methodologies, metabolites associated with IVD degeneration progression were measured in IVD tissues at different stages of degeneration in tissues collected from 6-month-old Sprague-Dawley rats inflicted with an IVD needle puncture and IVD tissue from discectomy patients experiencing degeneration or acute injury. Differential expression of metabolites between healthy and degenerated tissues focused on amino acid metabolism and sugar metabolism, since carbohydrate and protein metabolism were the main processes identified. Divergences in metabolic pathways before and after degeneration were associated with three biological activities: carbohydrate utilization, antioxidant pathways, and Gly-Ser-Thr metabolism.299,300 In addition to metabolomics, the IVD lipidome is an important area of study since lipids can act as signaling molecules in cellular processes such as proliferation, survival, apoptosis, and metabolism.301 Francisco et al. were the first to characterize IVD cellular lipidomics and identify the types of lipids that are affected by IVD pathology. Triacylglycerols, diacylglycerol, fatty acids, phosphatidylcholine, lysophosphatidylinositols, and sphingomyelin were all decreased, while bile acids and ceramides were increased. Interestingly, there were divergences in the metabolic profiles due to sex, where male patients had a decreased metabolic profile and female an increased profile in degenerated IVDs. The metabolomic profile changes of IVDs with degeneration could indicate that metabolite dysregulation is promoting senescence, autophagy alteration, or potentially cell death, which exacerbates tissue pathology.302
The etiology of IVD degeneration is still unclear, but a controversial hypothesis concerning degeneration stimuli is the role of subclinical bacterial infections in IVD degeneration. Bacterial colonization has been detected in human IVD samples, but bacterial presence could simply be the result of contamination during tissue extraction.303 To determine if bacterial-specific metabolites could be detected in human IVD samples and if there were changes in metabolite expression with IVD degeneration, Rajasekaran et al. identified 39 bacterial-specific metabolites via used untargeted metabolite profiling. Primary bacterial metabolites associated with processes such as bacterial growth, survival and quorum sensing were found in healthy and degenerative IVDs. These findings could indicate a role for infection-mediated pathology during IVD degeneration; however, additional studies need to corroborate these findings.304
Overall, proteomics and metabolomics are unbiased methodologies capable of measuring protein, metabolite, and lipid expression of the complete IVD proteome or of the matrisome, cellulome, or secretome individually. These techniques provide more direct measurements of biological responses via proteins, biomolecules, or lipid expression measurements than via RNA transcriptomics, which quantifies biological changes more indirectly via mRNA transcripts.305 The limitations are similar to scRNA-seq, where many IVDs may need to be pooled to accumulate enough tissue for analyses. Since the current iterations of spatial proteomics do not measure protein content in situ, homogenization of the tissue is required and can cause variability in protein identification.
Current omics technologies in the tendon
Tendons are viscoelastic tissues that facilitate efficient movement between muscle and bone. The complex hierarchical organization of collagen fibers allows tendons to withstand large forces via extensive stretching before returning to normal length. Resident tendon fibroblasts, or tenocytes, are aligned along the collagen fibers and are the primary cells that coordinate ECM formation and degradation. As tendons are mechanosensitive tissues, aberrant amounts of load (e.g., under- and overuse) can cause onset of tendinopathy, a broad term used to describe the complex pathology characterized by pain and limited mobility. Underuse is devoid of the mechanical loading that is necessary to activate and maintain the homeostatic function of tenocytes. Conversely, overuse from repetitive mechanical loading creates micro-injuries that the tendon is unable to repair, entering a degenerative cycle.306 Tendinopathic tissues from under- and overuse are commonly characterized by evidence of degeneration: alterations in tendon size, cell density and morphology, ECM disorganization, and severe reductions in mechanical function.306,307 Tendinopathy affects 1%–2% of adults,308,309 and because tendons are critical for day-to-day movement, tendinopathy can drastically impact patient’s quality of life. Clinical presentation of tendinopathy often occurs too late for preventative measures to be effective; as a result, there are currently no current biological treatments to remedy tendinopathy. Current therapeutics include non-steroidal anti-inflammatory drugs, massage, and exercise-based therapies, but these strategies merely alleviate painful symptoms and do not effectively regenerate the damaged tissue and restore function. Further complicating this, because the architecture of the tendon is compromised during tendinopathy, tendons are at risk for rupture and often require surgical intervention. In addition to degeneration of the native tendon structure, which can lead to bulk tissue rupture, tendons are also very prone to acute traumatic injuries due in part to their superficial locations (e.g., palmar and dorsal sides of the hands and feet). The fibrotic nature through which tendons heal provides an initial structural framework between the native tendon ends at the injury site. However, this provisional tissue and the subsequent mature scar tissue lack the mechanical properties of the native tissue,310 resulting in limitations in tissue function and an increase in re-injury.
Given the complex etiology of tendinopathy and response to acute tendon injuries, identification of tenable therapeutic targets has been a massive challenge, and there are currently no consensus biologics to restore tendon structure-function. A key barrier to the identification of translational strategies has been the limited definition of the cell and molecular environment underlying tendon pathology and repair, as most studies have primarily focused on the characterization of histological, morphological, and functional properties. Moreover, the tendon cell environment has historically not been well-defined, in part due to reliance on low-throughput approaches such as single-marker lineage-tracing and reporter animal models. While providing critical information on cell origins, lineage contributions, and cell-fate decisions, lineage-tracing has limited scalability and is constrained by the availability of cell-type and tissue-specific drivers. However, recent adoption of high-throughput sequencing approaches has facilitated a clear appreciation of the heterogeneity of the tendon cell environment in human tendinopathy,311,312,313 mouse tendon healing,314,315 zebrafish tendon regeneration316 and in spatially distinct regions of healthy human hamstring,317 rat patellar tendon,318 and human tendinopathy.313 Within the last decade, high-throughput “omics” studies have expedited the tendon field’s understanding of the cellular landscape during homeostasis, aging, tendinopathy, and injury. Here, we review the transformative impact of “omics” technologies on the tendon field and outline key gaps that remain to be addressed.
Transcriptomics in tendon
Traditional sequencing techniques such as bulk-RNA-seq have suggested that different anatomical tendons are transcriptionally distinct,319 and that there are region-specific transcriptomic signatures in different regions of the same tendon.320 Bulk-RNA sequencing indicated spatial variation between anatomical tendons, but does not give us any information about the potential diversity of cell populations within these regions. Additionally, lineage-tracing and reporter constructs of select tenocyte markers (e.g., Scleraxis, S100a4, etc.) define some degree of tendon cell heterogeneity, however, the molecular definition of these populations is limited to a few genes.321 However, robust transcriptomic analyses such as scRNA-seq and snRNA-seq reveals an unprecedented understanding of the heterogeneity within uninjured tendons (as shown in Fig. 7). For example, snRNA-seq analysis of healthy human hamstring identified twelve total populations: two fibroblast populations, three skeletal muscle populations, two endothelial cells, satellite cells, adipocytes, immune cells, mural cells, and nerve cells.317 Specifically, two fibroblast populations were suggested to play a major role in maintaining tendon homeostasis, given upregulations in ECM production and organization genes.317 Moreover, in various healthy human tendons, five subpopulations of tenocytes were identified by scRNA-seq: Two populations that co-express microfibril genes, a fibro–adipogenic progenitor population, a TPPP3/PRG4+ chondrogenic group, and ITGA7+ smooth muscle-mesenchymal cells.311 These findings support the previously unrecognized presence of multiple tenocyte clusters, but additional work is needed to define the potential distinct functional roles of these populations and to understand the relationship between tenocyte subpopulations between different studies.

Application of single-cell RNA-seq and spatial transcriptomics in Tendon
Despite the advances brought by scRNA-seq, a significant limitation lies in its ability to provide spatial information on the ___location of different subpopulations. Spatial transcriptomics is emerging as an important tool for understanding the ___location of different molecular programs within the tendon during homeostasis,317 pathogenesis,313 and healing314 (as shown in Fig. 7). However, currently commercially available spatial sequencing technologies lack consistent single-cell resolution. As such, integrating spatial sequencing and scRNA sequencing datasets will provide a comprehensive understanding of the cellular diversity and how different populations coordinate their functions in a spatially dependent manner. Moreover, discerning how these populations change in their distribution and expression across anatomical tendons will be important to delineate their tendon-specific function.
In the context of tendon aging, these advanced technologies offer insights into the mechanisms driving age-related changes. Tendon aging represents a critical area of study, as it is associated with impairments in homeostasis, accelerated tendinopathy, and failed healing.322 Specifically, aged tendons demonstrate disordered ECM organization, tissue degeneration, and severe reductions in cellularity and mechanical properties.323 Bulk-RNA-seq analysis of aged uninjured human Achilles (~69 years average age) reveals that they are transcriptionally distinct from their young counterparts (~19 years average age).324 DEGs in aged tendons represent pathways related to dysregulation of cellular function and maintenance, cellular growth and proliferation, and cellular development.324 While this study established dynamic alterations in the aged tendon transcriptome, gene expression is averaged out over the entire tissue, obscuring novel cell populations that are responsible for driving these age-related impairments. Defining how cell populations change in proportion and molecular profile during aging via “omics” can motivate potential therapeutics. Indeed, comparative scRNA-seq datasets of young (6 months) and aged (21 months) mouse flexor tendons indicated that aged tendons lose a significant proportion of tenocytes associated with “ECM biosynthesis.” As loss of ECM homeostasis is a hallmark of tendon aging, preventing loss of this population is a promising strategy to retain tendon function and highlights the power of leveraging “omics” to find novel potential targets to temper the effects of aging on tendon health. Furthermore, scRNA-seq analysis of young and old superficial digital flexor tendons established age-specific alterations in the interfascicular matrix (IFM) cell clusters, with differential expression related to senescence, dysregulated proteostasis, and inflammation. Given the IFM’s sensitivity to aging and declines in stiffness,325,326 future studies focusing on proteoglycans and other IFM-specific components during aging325 may provide key insights into age-related degeneration. In addition, incorporation of spatial sequencing will be critical to delineate the IFM’s contributions to the mechanical and structural deficits seen during aging. As anatomical tendons are transcriptionally distinct from each other,319 and have transcriptionally unique spatial regions,320 characterizing each tendon and their spatial compartments during aging is a rich area for future investigation. Comprehensively defining the aging tendon cellular landscape over the course of the lifespan with multi-omics approaches can pinpoint the timepoint and mechanisms responsible for the switch in age-related deficits, such as cellularity and ECM breakdown.
Moving from aging to disease states, tendinopathy presents a unique challenge for researchers. Tendinopathy, a common and debilitating condition, is characterized histologically by disorganized ECM, hypercellularity, inflammation, and altered tenocyte morphology in the samples from animal models and clinical samples of tendinopathy.327,328 These studies have provided a strong foundation to our characterization of tendinopathy, but do not investigate the underlying cellular and molecular mechanisms. scRNA-seq studies have identified a variable number of tenocyte subpopulations, with some of this diversity accounted for when considering differences between tendons and species.232,311,312,329 However, understanding the relationship and potential functional and molecular overlap between subpopulations, how this may different between anatomically distinct tendons, and combining a clear understanding cellular heterogeneity with spatial information relative to different tissue features represents key action items that will facilitate leveraging these powerful datasets to identify central regulations of tendinopathy pathogenesis and novel therapeutic targets.
The challenges of tendinopathy extend into the broader issue of tendon healing, where insufficient or inappropriate tenocyte functions are thought to be a primary driver of impaired tendon healing.330 As such, efforts to understand tendon injuries have typically employed lineage-tracing of tenocyte markers to track their contributions during healing.321,331,332 While lineage-tracing of tenocytes during healing allows definition of the spatial localization of different populations, these approaches are limited to a few target genes, and additional definition of these populations is limited. Multiplex immunofluorescence profiling can provide some additional resolution on the molecular diversity of these populations,321 although this approach is somewhat limited by the technical constraints of traditional fluorescence microscopy. However, the combination of lineage-tracing approaches with high-throughput omics is a powerful approach to further identify cell-fate and differentiation dynamics.333 For example, combined lineage-tracing and scRNA-seq have identified both pro-tenogenic and pro-fibrotic roles for epitenon cells (thin layer of cells on the surface of tendons) during mouse flexor tendon healing.315 Additionally, pseudotime trajectory analysis from spatial transcriptomics of mouse flexor tendon using a Scleraxis-lineage trace demonstrates tenocyte trifurcation into (1) tenogenic, (2) fibroblastic/fibrotic, and (3) reactive tissue sites, with the reactive tissue primarily enriched for Scleraxis-lineage cells.314 Collectively, combining lineage-tracing and “omics” studies provide tremendous power to identify markers of specialized populations, define regulators of cell-fate decisions, and can inform generation of animal novel models to functionally interrogate different populations in space and time, which will facilitate a more informed understanding of the healing process and therapeutic candidate identification.
Another recent study further reveals the importance of sensory innervation in tendon healing and repair, using spatial transcriptomics and scRNA-seq.231 Spatial transcriptomics identified distinct regions of the injured tendon, each exhibiting unique transcriptional responses, such as neurovascular ingrowth and increased nerve growth factor (NGF) expression.231 Complementary scRNA-seq analysis delineated key cell clusters, including mesenchymal and immune cells, with distinct regenerative roles.231 Importantly, sensory nerves, through NGF signaling, regulate mesenchymal cell behaviors and inflammatory responses, emphasizing their critical role in tendon regeneration. Disruption of sensory innervation impairs repair by dysregulating key signaling pathways.
Overall, the tendon field is at the precipice of an “omics” renaissance in which integrated multi-omics can be a critical step in the identification and validation of new therapeutic drug targets. While there is a rapidly expanding body of information regarding the transcriptomic and/or proteomic regulation of tendon homeostasis, pathology, and repair, there remains a limited definition of the epigenetic and metabolomic states of tendon cells across these different states. Moreover, integration across multi-omics data modalities will provide a more holistic understanding of how the tendon responds, adapts/maladapts to insults, including aging, aberrant loading, and systemic co-morbidities, or to acute injury.334 This comprehensive approach will pave the way for targeted therapies to improve tendon health and function across the lifespan.
Current omics technologies in muscle
Skeletal muscle is the prime mover of the musculoskeletal system, uniquely contributing to movement through coordinated force generation in thousands of large, multinucleated myofibers.335 While myofibers constitute ~80% of muscle volume, muscle-resident mononuclear cells also support efficient contraction (Fig. 8). Dependence of muscle contraction on the myofiber is intuitive, as this is the cell responsible for force generation. Likewise, myofiber contraction is clearly dependent on the nervous system for excitation and the vascular system for the delivery of oxygen and metabolites.335 However, the role of the other, diverse, mononuclear cells in muscle physiology has only begun to be elucidated. The satellite cell is the best studied of these as it has been recognized as the primary muscle stem/progenitor cell since the 1960s.336 Satellite cells have also long been noted to rely on other cells residing or active in their microenvironment for support during development and regeneration.337 In the early days of flow cytometry, many of these were classified together as “other” (e.g., side population cells) of unknown composition and function.338 However, much work over the past 20 years has defined substantial heterogeneity in these mononuclear cells which includes fibro/adipogenic progenitors (FAPs), pericytes, tissue-resident immune cells and other precursor cells with myogenic potential (e.g., PW1+ cells and TWIST2+ cells). The exact role of each cell type is an area of intense investigation. With the advancement of large-scale unbiased (omics) technologies to the single-cell level, we are now uncovering cellular diversity within mononuclear and myofiber type classifications. Further overlaying this information on temporal or disease trajectories is poised to dramatically increase our understanding of cell–cell signaling in homeostasis and how it changes in response to stimuli. Unique features of skeletal muscle structure and function have required specialized approaches to maximize the insight gained from each experimental question, which, when leveraged appropriately, can be integrated into our understanding of the musculoskeletal system as a whole. This part of the review will focus on the unique challenges in applying omics tools to skeletal muscle and insights being gained on the frontier of these approaches.

Application of single-cell RNA-seq, single-nucleus RNA-seq, spatial transcriptomics, and metabolomics in muscle
Single-cell RNA-seq in muscle
Over the past 20 years, bulk-RNA sequencing has provided incredible insight into the muscle transcriptomic response to stimuli such as injury, exercise, aging, and disease.339,340,341,342,343,344,345 However, because the vast majority of nuclei in muscle are myonuclei serving muscle fibers, transcriptional changes in mononuclear cells including immune cells, FAPs, and satellite cells can be overwhelmed in bulk sequencing.346 Thus, one of the most significant advancements in RNA sequencing technologies for skeletal muscle has been the ability to capture the transcriptome of single cells by scRNA-seq, unveiling previously undiscovered subpopulations of the cells described above, each with unique transcriptional responses to stimuli and with specialized roles in homeostasis and disease (as shown in Fig. 8).
scRNA-seq has revealed a surprising heterogeneity in satellite cells within muscle.347,348,349,350 For example, 17 subpopulations of satellite cells were recently identified in uninjured human muscle.351 One of these, positive for the surface marker Cav1, was found to have a resistance to activation, very high engraftment capacity, and enhanced ability to differentiate into myogenic cells and to repopulate the satellite cell niche. In this way, scRNA-seq could be leveraged to identify novel cell sub-phenotypes that can be prospectively isolated and utilized in cell-based therapies. In addition to profiling homeostasis, scRNA-seq has been useful in characterizing cell population dynamics during development and regeneration.352,353,354 For instance, scRNA-seq in a murine toxin-injury model described a pseudotemporal mapping of satellite cell transcription during regeneration, marked by a progressive increase in mitochondrial genes as satellite cells terminally differentiated into myoblasts.350 Outside of satellite cells, temporal dynamics and heterogeneity of FAPs during muscle regeneration have also been elucidated by scRNA-seq.355,356,357,358,359 Early (0.5–3 days post toxin-induced injury) response genes in murine FAPs suggest significant contribution to cytokine interactions and immune cell recruitment to muscle,359 whereas later (10 days post injury) response genes point to differentiation into myofibroblasts and involvement in ECM remodeling.358 These studies have uncovered a dichotomous role for FAPs in muscle regeneration where they play a critical signaling role in early-stage regeneration but then can contribute to the pathological features of fibrosis and intramuscular adipocytes at later stages. This type of information can be utilized as an atlas of regeneration to reference disease models against and uncover stages at which regeneration becomes compromised.360 Atlases of development and aging are being similarly utilized.361,362,363 In the development sphere, scRNA-seq studies have identified novel mitochondrial-rich and myocyte-like subpopulations of FAPs in neonatal porcine muscle that may promote or directly contribute to myogenesis.352 In the aging sphere, scRNA-seq has demonstrated alterations in the response of aged satellite cells to activation cues364,365 characterized by a lag in satellite cell differentiation resulting in further disturbances in satellite cell fusion and maturation, cumulatively leading to a diminished regeneration response.365 While these examples highlight satellite cells and FAPs, scRNA-seq has also provided insight into the temporal and transcriptional dynamics of immune populations,366 vascular associated cells367 and cells of the nervous system.368 Over the past 6–8 years, this technique has enabled a significant stride forward in understanding mononuclear cell dynamics in muscle.
Single-nucleus RNA-seq in muscle
The primary limitation of scRNA-seq for skeletal muscle is the inability to transcriptionally profile myonuclei as myofibers are too large for the typical droplet-based cell isolation technique. snRNA-seq circumvents this issue by isolating and profiling nuclei from lysed cells. While there is a loss of cytoplasmic transcriptional information with the use of snRNA-seq, this technique is believed to still capture the majority of transcriptional activity369 and allows transcripts to be mapped to nuclei of origin in multinucleated cells, which is impossible in traditional scRNA-seq. Using this technique, substantial myonuclear heterogeneity has been uncovered, with subpopulations of myonuclei specializing by subcellular ___location and temporal response to stimuli (as shown in Fig. 8).
Interpreting the physiological significance of snRNA-seq data can be more challenging than scRNA-seq since validation of cell types with surface markers is not possible, and information about spatial drivers of heterogeneity is lost. Because of this, snRNA-seq is most powerful when combined with other approaches. Combining snRNA-seq with single-molecule fluorescence in situ hybridization (smRNA-FISH) in adult mouse muscle, unique markers of myonuclei associated with the neuromuscular junction and myotendinous junction were spatially validated.370 This enabled previously uncharacterized genes involved in neuromuscular junction maintenance to be uncovered.370 Using a similar strategy, a recent study found a substantial lack of neuromuscular junction myonuclei in mice with muscular dystrophy (mdx), which reflects the disruption of neuromuscular junction morphology that others have observed in this mouse.371 Further combining snRNA-seq with single-nucleus transposase-accessible chromatin using sequencing (snATAC-seq) identified regulators of regional transcriptional programs, such as collagens in the myotendinous junction.372 The bulk of snRNA-seq studies have focused on transcriptional heterogeneity in myonuclei, but it is important to note that other muscle-resident cells may also be excluded from scRNA-seq, such as intramuscular adipocytes and macrophage clusters, and may be washed out by the sheer number of myonuclei in snRNA-seq. A spatial approach may be useful in identifying changes in cells not captured by scRNA-seq or snRNA-seq and offers the additional benefit of spatial mapping and selection.
Spatial transcriptomics in muscle
Skeletal muscle pathology is highly heterogeneous and frequently features sporadic necrosis, localized fibrosis, and isolated pockets of intramuscular adipocytes. Conventional RNA-FISH has been used to spatially map a few genes at a time, providing additional insight into myonuclei and satellite cell specialization.373,374,375 However, with the advent of spatial transcriptomics, combining full coverage of the transcriptome with complete spatial information is poised to dramatically increase our understanding of cell–cell signaling in aging and disease (as shown in Fig. 8).
An early spatial transcriptomics study in muscle captured transitional progenitor states during toxin-injury induced skeletal muscle regeneration, in which FAPs were the cell type most likely to interact with myogenic cells, primarily through a paracrine signaling mechanism, further supporting previous evidence for a crucial role for FAPs in muscle regeneration.360 In the same injury model, another group using spatial transcriptomics identified a transient, “acute” senescence-like state in macrophages and FAPs local to the area of muscle necrosis.376 Others have utilized spatial transcriptomics to understand the local transcriptional response to more translationally relevant muscle injury.228,377,378,379 Insights into cell–cell signaling in mouse models of volumetric muscle loss and muscular dystrophy revealed that there is an enrichment in Trem2+ pro-fibrotic macrophages that colocalize with mesenchymal-derived cells, driving fibrotic progression within the injury area and inhibiting the influx of satellite cell-mediated regeneration.228,378 Moreover, the inhibition of fibrotic signaling within this model reduced Trem2+ macrophage activity and increased satellite cell homing to the injury site, attenuating fibrosis and improving regeneration, providing insights into therapeutic options to mitigate these effects.378 While spatial transcriptomics applied to skeletal muscle is still in its infancy, it has tremendous potential to shed light on the micro- and macro-environmental drivers of the transcriptional heterogeneity identified in scRNA-seq and snRNA-seq studies described above. For a detailed review of the current and future applications of transcriptomics to skeletal muscle research, see ref. 380
Proteomic/Metabolomics approaches in muscle
While transcriptomics can provide a wealth of data at the gene expression level, gene changes frequently do not track with protein expression or functional changes. An example of this in muscle is Myf5, a key myogenic determination gene, which shows no difference in transcription levels between quiescent and activated satellite cells but a high difference at the protein level, as Myf5 mRNA is sequestered by miR-31, preventing its translation until activation.381 Bulk proteomics and metabolomics by LC/MS have been extensively used in skeletal muscle to better understand protein and metabolite dynamics in myogenesis, myofiber type specification, and the effects of disuse atrophy, muscular dystrophy, neuromuscular disorders, sarcopenia, and exercise.382,383,384,385,386,387 However, this approach cannot differentiate between individual cellular contributors to the bulk proteome/metabolome, and much of the field today is leveraging new technology for a more sensitive and spatial approach to better appreciate the heterogeneity across muscle structure and pathology.
A major advance for skeletal muscle has been the characterization of the proteome of individual myofibers, which was impossible even 10 years ago due to the limited sensitivity for low-abundance proteins, which includes most non-sarcomeric proteins in skeletal muscle.388,389,390,391 Like scRNA-seq for mononuclear cells, single myofiber proteomics has highlighted distinct subpopulations of myofibers within the traditional classifications. Surprising differences in protein abundance in some subpopulations were also noted, including in mitochondrial enzymes, proteins involved in lipid metabolism, and cytoskeletal proteins.388,390,391 Though less utilized in muscle, single-cell proteomics can also characterize mononuclear cell populations identified by antibody labeling via cytometry by time-of-flight (CyTOF).348,392 However, like scRNA-seq, these strategies also lose all spatial information. Spatial approaches applying LC/MS to laser capture microdissected tissue regions are poised to provide more insight into localized pathology.
However, application of spatial proteomics to skeletal muscle has lagged its use in other tissue types, in part due to the ability to capture myofibers manually and apply traditional LC/MS as discussed above. A few studies have utilized this method to characterize isolated regions of pathology in myopathy393 and rotator cuff injury,394 revealing important features of the degenerative microenvironment of muscle. Recently, spatial metabolomics revealed a new subtype of muscle fiber, deemed type IIb mito-high, that was enriched for fatty acid oxidative metabolism and observed to express a superfast isoform of myosin heavy chain, Myh13, previously only found in extraocular muscles (as shown in Fig. 8).238 As the field of spatial proteomics/metabolomics in skeletal muscle emerges, current work is establishing a bedrock on which future studies can build, with some early studies beginning to detail the proteomics landscape in myopathies. Future work will also evolve in parallel with MS technology as detection sensitivity increases and substantially more targets can be captured. For a detailed review of the current and future applications of proteomics to skeletal muscle research, see ref. 395
Each of the technologies discussed has advanced a piece of muscle research: scRNA-seq has dramatically expanded our understanding of mononuclear cell population heterogeneity, snRNA-seq has defined myonuclear transcriptional heterogeneity within a myofiber, spatial transcriptomics has tied some of this heterogeneity to tissue structure and pathology and single myofiber and spatial proteomics/metabolomics has refined our definitions of myofiber types. In theory, the overlay of all of this information would provide a clear picture of a process (e.g., regeneration) so that we could therapeutically target breakdown in the process (e.g., in muscular dystrophy) to improve muscle health and function. In practice, this will require many more years of data interpretation, dissemination, and validation.
Conclusion and prospective
The musculoskeletal system is highly susceptible to degenerative and inflammatory disorders, which arise from intricate cellular and molecular interactions. Recent advancements in single-cell and spatial omics technologies have revolutionized the field by providing unprecedented resolution into cellular heterogeneity and tissue microenvironments. scRNA-seq has identified disease-specific cell states and lineage trajectories in musculoskeletal disorders. However, due to challenges in dissociating certain musculoskeletal tissues, snRNA-seq has emerged as a powerful alternative, enabling transcriptomic profiling of dense, matrix-rich tissues by analyzing nuclear transcripts without requiring complete cell dissociation. Beyond transcriptomics, scATAC-seq and WGBS complement these approaches by mapping chromatin accessibility and DNA methylation landscapes, respectively, revealing epigenetic drivers of disease mechanisms. Despite their transformative potential, these techniques must be integrated with other omics workflows to connect transcriptional and epigenetic dynamics to functional outcomes. Advances in proteomics and metabolomics offer deeper insights into functional cellular states and metabolic alterations. However, single-cell proteomics and metabolomics face challenges in detecting low-abundance proteins and metabolites, resolving post-translational modifications, and addressing technical variability. Their application in musculoskeletal research remains in its early stages, necessitating further methodological refinements to enhance resolution, reproducibility, and biological interpretability.
Spatial omics techniques bridge the gap between cellular resolution and tissue architecture, enabling the precise mapping of molecular gradients in musculoskeletal tissues. However, each modality presents distinct technical and analytical challenges. Spatial transcriptomics is constrained by the trade-off between resolution and transcript coverage, reduced sensitivity for low-abundance genes, and the complexity of data integration. Spatial proteomics faces limitations in antibody availability, particularly for musculoskeletal-specific targets, as well as challenges in achieving comprehensive protein coverage and detecting post-translational modifications. Similarly, spatial metabolomics encounters obstacles related to metabolite stability, restricted spatial resolution, and the complexity of spectral interpretation. A major challenge across all spatial omics platforms is the integration of multimodal datasets, as differences in spatial resolution, molecular stability, and biological timescales complicate computational alignment. Additionally, musculoskeletal tissues, particularly calcified or mineralized structures, pose difficulties for optimal sectioning and permeabilization, further limiting data acquisition. Innovations in imaging MS and AI-driven deconvolution algorithms are addressing these challenges by enhancing sensitivity, improving data integration, and enabling high-resolution mapping of post-translational modifications and metabolic flux. However, barriers related to cost, standardization, and protocol optimization for musculoskeletal tissues remain critical for broader adoption and clinical translation.
Future priorities include multi-omics integration to construct comprehensive cellular atlases, the development of AI-driven computational tools to decode high-dimensional datasets, and the validation of biomarkers in diverse patient cohorts. By harmonizing bulk, single-cell, and spatial approaches, researchers can unravel the multiscale complexity of musculoskeletal diseases, spanning systemic dysregulation to spatially organized cellular communication within discrete tissue niches. These advancements hold immense potential for precision therapies, ultimately aiming to restore tissue function and improve patient outcomes in musculoskeletal disorders.
References
Felsenthal, N. & Zelzer, E. Mechanical regulation of musculoskeletal system development. Development 144, 4271–4283 (2017).
Frontera, W. R. Physiologic changes of the musculoskeletal system with aging: a brief review. Phys. Med. Rehabil. Clin. N. Am. 28, 705–711 (2017).
Mitchell, T. The Musculoskeletal System (Master Books, 2015).
Shen, J., Li, S. & Chen, D. TGF-beta signaling and the development of osteoarthritis. Bone Res. 2, 14002 (2014).
El-Tallawy, S. N. et al. Management of musculoskeletal pain: an update with emphasis on chronic musculoskeletal pain. Pain. Ther. 10, 181–209 (2021).
Mease, P. J. Inflammatory musculoskeletal disease: identification and assessment. J. Rheumatol. 38, 557–561 (2011).
Abdelhamid, R. E. & Sluka, K. A. ASICs mediate pain and inflammation in musculoskeletal diseases. Physiology 30, 449–459 (2015).
Li, X. et al. Mitigation of articular cartilage degeneration and subchondral bone sclerosis in osteoarthritis progression using low-intensity ultrasound stimulation. Ultrasound Med. Biol. 45, 148–159 (2019).
Mobasheri, A. Applications of proteomics to osteoarthritis, a musculoskeletal disease characterized by aging. Front. Physiol. 2, 108 (2011).
Nielson, C. M., Jacobs, J. M. & Orwoll, E. S. Proteomic studies of bone and skeletal health outcomes. Bone 126, 18–26 (2019).
Pauper, M. et al. Proteomic profiling towards a better understanding of genetic based muscular diseases: the current picture and a look to the future. Biomolecules 15, 130 (2025).
Swank, K. R. et al. Metabolomic profiling in the characterization of degenerative bone and joint diseases. Metabolites 10, 223 (2020).
Kumar, A. et al. Metabolomic analysis of primary human skeletal muscle cells during myogenic progression. Sci. Rep. 10, 11824 (2020).
Wei, G. et al. Risk of metabolic abnormalities in osteoarthritis: a new perspective to understand its pathological mechanisms. Bone Res. 11, 63 (2023).
Wu, S., Ohba, S. & Matsushita, Y. Single-cell RNA-sequencing reveals the skeletal cellular dynamics in bone repair and osteoporosis. Int. J. Mol. Sci. 24, 9814 (2023).
Wang, T. et al. Single-cell RNA sequencing in orthopedic research. Bone Res. 11, 10 (2023).
Wang, S. et al. The evolution of single-cell RNA sequencing technology and application: progress and perspectives. Int. J. Mol. Sci. 24, 2943 (2023).
Arias-Hidalgo, C. et al. Single-cell proteomics: the critical role of nanotechnology. Int. J. Mol. Sci. 23, 6707 (2022).
Ahmad, R. & Budnik, B. A review of the current state of single-cell proteomics and future perspective. Anal. Bioanal. Chem. 415, 6889–6899 (2023).
Zhang, Z. et al. When cancer drug resistance meets metabolomics (bulk, single-cell and/or spatial): progress, potential, and perspective. Front. Oncol. 12, 1054233 (2022).
Feng, S., Li, J., Tian, J., Lu, S. & Zhao, Y. Application of single-cell and spatial omics in musculoskeletal disorder research. Int. J. Mol. Sci. 24, 2271 (2023).
Rai, M. F. et al. Single cell omics for musculoskeletal research. Curr. Osteoporos. Rep. 19, 131–140 (2021).
Rai, M. F. et al. Three decades of advancements in osteoarthritis research: insights from transcriptomic, proteomic, and metabolomic studies. Osteoarthr. Cartil. 32, 385–397 (2024).
Tang, F. et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 (2009).
Buenrostro, J. D. et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015).
Bendall, S. C. et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 332, 687–696 (2011).
Hughes, A. J. et al. Single-cell western blotting. Nat. Methods 11, 749–U794 (2014).
Kennedy, R. T., Stclaire, R. L., White, J. G. & Jorgenson, J. W. Chemical-analysis of single neurons by open tubular liquid-chromatography. Mikrochim. Acta 2, 37–45 (1987).
Stahl, P. L. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016).
Rappsilber, J., Siniossoglou, S., Hurt, E. C. & Mann, M. A generic strategy to analyze the spatial organization of multi-protein complexes by cross-linking and mass spectrometry. Anal. Chem. 72, 267–275 (2000).
Hu, T. et al. Single-cell spatial metabolomics with cell-type specific protein profiling for tissue systems biology. Nat. Commun. 14, 8260 (2023).
Rosenberger, F. A. et al. Spatial single-cell mass spectrometry defines zonation of the hepatocyte proteome. Nat. Methods 20, 1530–1536 (2023).
Planque, M., Igelmann, S., Ferreira Campos, A. M. & Fendt, S. M. Spatial metabolomics principles and application to cancer research. Curr. Opin. Chem. Biol. 76, 102362 (2023).
Pap, T. & Korb-Pap, A. Cartilage damage in osteoarthritis and rheumatoid arthritis-two unequal siblings. Nat. Rev. Rheumatol. 11, 606–615 (2015).
Tong, L. et al. Current understanding of osteoarthritis pathogenesis and relevant new approaches. Bone Res. 10, 60 (2022).
Chen, Y. et al. A high-resolution route map reveals distinct stages of chondrocyte dedifferentiation for cartilage regeneration. Bone Res. 10, 38 (2022).
Ji, Q. et al. Single-cell RNA-seq analysis reveals the progression of human osteoarthritis. Ann. Rheum. Dis. 78, 100–110 (2019).
Yan, M. et al. Single-cell RNA sequencing reveals distinct chondrocyte states in femoral cartilage under weight-bearing load in Rheumatoid arthritis. Front. Immunol. 14, 1247355 (2023).
Sebastian, A. et al. Single-cell RNA-seq reveals transcriptomic heterogeneity and post-traumatic osteoarthritis-associated early molecular changes in mouse articular chondrocytes. Cells 10, 1462 (2021).
Fan, Y. et al. Unveiling inflammatory and prehypertrophic cell populations as key contributors to knee cartilage degeneration in osteoarthritis using multi-omics data integration. Ann. Rheum. Dis. 83, 926–944 (2024).
Swahn, H. et al. Senescent cell population with ZEB1 transcription factor as its main regulator promotes osteoarthritis in cartilage and meniscus. Ann. Rheum. Dis. 82, 403–415 (2023).
Guang, Z., Min, Z., Jun-Tan, L., Tian-Xu, D. & Xiang, G. Single-cell protein activity analysis reveals a novel subpopulation of chondrocytes and the corresponding key master regulator proteins associated with anti-senescence and OA progression. Front. Immunol. 14, 1077003 (2023).
Kang, X., Zhang, K., Wang, Y., Zhao, Y. & Lu, Y. Single-cell RNA sequencing analysis of human chondrocytes reveals cell-cell communication alterations mediated by interactive signaling pathways in osteoarthritis. Front. Cell Dev. Biol. 11, 1099287 (2023).
Chen, Y., Zhang, Y., Ge, Y. & Ren, H. Integrated single-cell and bulk RNA sequencing analysis identified pyroptosis-related signature for diagnosis and prognosis in osteoarthritis. Sci. Rep. 13, 17757 (2023).
Li, H. et al. Combining single-cell RNA sequencing and population-based studies reveals hand osteoarthritis-associated chondrocyte subpopulations and pathways. Bone Res. 11, 58 (2023).
Lv, Z. et al. Single cell RNA-seq analysis identifies ferroptotic chondrocyte cluster and reveals TRPV1 as an anti-ferroptotic target in osteoarthritis. EBioMedicine 84, 104258 (2022).
Fu, W. et al. 14-3-3 epsilon is an intracellular component of TNFR2 receptor complex and its activation protects against osteoarthritis. Ann. Rheum. Dis. 80, 1615–1627 (2021).
Fu, W. et al. Na(v)1.7 as a chondrocyte regulator and therapeutic target for osteoarthritis. Nature 625, 557–565 (2024).
Ning, Y. et al. Integrative analysis of miRNA in cartilage-derived extracellular vesicles and single-cell RNA-seq profiles in knee osteoarthritis. Arch. Biochem. Biophys. 748, 109785 (2023).
Zhang, Y. et al. Dual functions of microRNA-17 in maintaining cartilage homeostasis and protection against osteoarthritis. Nat. Commun. 13, 2447 (2022).
Li, J. et al. Microtubule stabilization targeting regenerative chondrocyte cluster for cartilage regeneration. Theranostics 13, 3480–3496 (2023).
Fan, X. et al. Spatial analysis of the osteoarthritis microenvironment: techniques, insights, and applications. Bone Res. 12, 7 (2024).
Lareau, C. A. et al. Mitochondrial single-cell ATAC-seq for high-throughput multi-omic detection of mitochondrial genotypes and chromatin accessibility. Nat. Protoc. 18, 1416–1440 (2023).
Bittner, N. et al. Primary osteoarthritis chondrocyte map of chromatin conformation reveals novel candidate effector genes. Ann. Rheum. Dis. 83, 1048–1059 (2024).
Liu, Y. et al. Chromatin accessibility landscape of articular knee cartilage reveals aberrant enhancer regulation in osteoarthritis. Sci. Rep. 8, 15499 (2018).
Zhang, C. H. et al. Creb5 establishes the competence for Prg4 expression in articular cartilage. Commun. Biol. 4, 332 (2021).
Richard, D. et al. Evolutionary selection and constraint on human knee chondrocyte regulation impacts osteoarthritis risk. Cell 181, 362–381.e328 (2020).
Shen, J. et al. Inhibition of 4-aminobutyrate aminotransferase protects against injury-induced osteoarthritis in mice. JCI Insight 4, e128568 (2019).
Shen, J. et al. DNA methyltransferase 3b regulates articular cartilage homeostasis by altering metabolism. JCI Insight 2, e93612 (2017).
Loughlin, J. & Reynard, L. N. Osteoarthritis: epigenetics of articular cartilage in knee and hip OA. Nat. Rev. Rheumatol. 11, 6–7 (2015).
Rushton, M. D. et al. Characterization of the cartilage DNA methylome in knee and hip osteoarthritis. Arthritis Rheumatol. 66, 2450–2460 (2014).
Kehayova, Y. S., Wilkinson, J. M., Rice, S. J. & Loughlin, J. Mediation of the same epigenetic and transcriptional effect by independent osteoarthritis risk-conferring alleles on a shared target gene, COLGALT2. Arthritis Rheumatol. 75, 910–922 (2023).
Roberts, J. B. et al. Specific isoforms of the ubiquitin ligase gene WWP2 are targets of osteoarthritis genetic risk via a differentially methylated DNA sequence. Arthritis Res. Ther. 26, 78 (2024).
Brumwell, A. et al. Identification of TMEM129, encoding a ubiquitin-protein ligase, as an effector gene of osteoarthritis genetic risk. Arthritis Res. Ther. 24, 189 (2022).
Zhao, L., Wang, Q., Zhang, C. & Huang, C. Genome-wide DNA methylation analysis of articular chondrocytes identifies TRAF1, CTGF, and CX3CL1 genes as hypomethylated in osteoarthritis. Clin. Rheumatol. 36, 2335–2342 (2017).
Alvarez-Garcia, O. et al. Increased DNA methylation and reduced expression of transcription factors in human osteoarthritis cartilage. Arthritis Rheumatol. 68, 1876–1886 (2016).
Zhang, Y. et al. Genome-wide DNA methylation profile implicates potential cartilage regeneration at the late stage of knee osteoarthritis. Osteoarthr. Cartil. 24, 835–843 (2016).
Kreitmaier, P. et al. An epigenome-wide view of osteoarthritis in primary tissues. Am. J. Hum. Genet. 109, 1255–1271 (2022).
Izda, V. et al. A pilot analysis of genome-wide DNA methylation patterns in mouse cartilage reveals overlapping epigenetic signatures of aging and osteoarthritis. ACR Open Rheumatol. 4, 1004–1012 (2022).
Wu, J., Yang, F., Wu, X., Liu, X. & Zheng, D. Comparison of genome-wide DNA methylation patterns between antler precartilage and cartilage. Mol. Genet. Genom. 298, 343–352 (2023).
Li, J. et al. Multi-omics molecular biomarkers and database of osteoarthritis. Database 2022, baac052 (2022).
Wei, Y. et al. Progress in multi-omics studies of osteoarthritis. Biomark. Res. 13, 26 (2025).
Garcia, B. A. et al. Protein profile of osteoarthritic human articular cartilage using tandem mass spectrometry. Rapid Commun. Mass Spectrom. 20, 2999–3006 (2006).
Wu, J. et al. Comparative proteomic characterization of articular cartilage tissue from normal donors and patients with osteoarthritis. Arthritis Rheum. 56, 3675–3684 (2007).
Guo, D. et al. Proteomic analysis of human articular cartilage: identification of differentially expressed proteins in knee osteoarthritis. Jt. Bone Spine 75, 439–444 (2008).
Hermansson, M. et al. Proteomic analysis of articular cartilage shows increased type II collagen synthesis in osteoarthritis and expression of inhibin betaA (activin A), a regulatory molecule for chondrocytes. J. Biol. Chem. 279, 43514–43521 (2004).
Peffers, M. J., Beynon, R. J. & Clegg, P. D. Absolute quantification of selected proteins in the human osteoarthritic secretome. Int. J. Mol. Sci. 14, 20658–20681 (2013).
Ruiz-Romero, C., Lopez-Armada, M. J. & Blanco, F. J. Proteomic characterization of human normal articular chondrocytes: a novel tool for the study of osteoarthritis and other rheumatic diseases. Proteomics 5, 3048–3059 (2005).
Ruiz-Romero, C. et al. Proteomic analysis of human osteoarthritic chondrocytes reveals protein changes in stress and glycolysis. Proteomics 8, 495–507 (2008).
Ikeda, D., Ageta, H., Tsuchida, K. & Yamada, H. iTRAQ-based proteomics reveals novel biomarkers of osteoarthritis. Biomarkers 18, 565–572 (2013).
Fu, W. et al. TNFR2/14-3-3epsilon signaling complex instructs macrophage plasticity in inflammation and autoimmunity. J. Clin. Investig. 131, e144016 (2021).
Lourido, L. et al. Secretome analysis of human articular chondrocytes unravels catabolic effects of nicotine on the joint. Proteom. Clin. Appl. 10, 671–680 (2016).
Hsueh, M. F., Onnerfjord, P. & Kraus, V. B. Biomarkers and proteomic analysis of osteoarthritis. Matrix Biol. 39, 56–66 (2014).
Sanchez, C. et al. Chondrocyte secretome: a source of novel insights and exploratory biomarkers of osteoarthritis. Osteoarthr. Cartil. 25, 1199–1209 (2017).
Trachana, V., Mourmoura, E., Papathanasiou, I. & Tsezou, A. Understanding the role of chondrocytes in osteoarthritis: utilizing proteomics. Expert Rev. Proteom. 16, 201–213 (2019).
Zheng, L., Zhang, Z., Sheng, P. & Mobasheri, A. The role of metabolism in chondrocyte dysfunction and the progression of osteoarthritis. Ageing Res. Rev. 66, 101249 (2021).
Wu, X., Fan, X., Crawford, R., Xiao, Y. & Prasadam, I. The metabolic landscape in osteoarthritis. Aging Dis. 13, 1166–1182 (2022).
Adam, M. S., Zhuang, H., Ren, X., Zhang, Y. & Zhou, P. The metabolic characteristics and changes of chondrocytes in vivo and in vitro in osteoarthritis. Front. Endocrinol. 15, 1393550 (2024).
Arai, Y. et al. Cartilage homeostasis under physioxia. Int. J. Mol. Sci. 25, 9398 (2024).
Stegen, S. et al. Glutamine metabolism controls chondrocyte identity and function. Dev. Cell 53, 530–544 e538 (2020).
Arra, M. et al. Glutamine metabolism modulates chondrocyte inflammatory response. eLife 11, e80725 (2022).
Lee, S. Y., Abel, E. D. & Long, F. Glucose metabolism induced by Bmp signaling is essential for murine skeletal development. Nat. Commun. 9, 4831 (2018).
Wang, C. et al. Deletion of Glut1 in early postnatal cartilage reprograms chondrocytes toward enhanced glutamine oxidation. Bone Res. 9, 38 (2021).
Tabbaa, S. M. et al. Elevated lipid metabolites in stored clinical OCA media correlate with chondrocyte death. Am. J. Sports Med. 52, 2119–2128 (2024).
Tang, R. et al. Gene therapy for fat-1 prevents obesity-induced metabolic dysfunction, cellular senescence, and osteoarthritis. Proc. Natl. Acad. Sci. USA 121, e2402954121 (2024).
Defois, A. et al. Osteoarthritic chondrocytes undergo a glycolysis-related metabolic switch upon exposure to IL-1b or TNF. Cell Commun. Signal. 21, 137 (2023).
Wu, X. et al. Dysregulated energy metabolism impairs chondrocyte function in osteoarthritis. Osteoarthr. Cartil. 31, 613–626 (2023).
Tan, C., Li, L., Han, J., Xu, K. & Liu, X. A new strategy for osteoarthritis therapy: inhibition of glycolysis. Front. Pharmacol. 13, 1057229 (2022).
Pi, P. et al. The role of targeting glucose metabolism in chondrocytes in the pathogenesis and therapeutic mechanisms of osteoarthritis: a narrative review. Front. Endocrinol. 15, 1319827 (2024).
Jaggard, M. K. J. et al. A systematic review of the small molecule studies of osteoarthritis using nuclear magnetic resonance and mass spectroscopy. Osteoarthr. Cartil. 27, 560–570 (2019).
Welhaven, H. D. et al. Metabolomic profiles and pathways in osteoarthritic human cartilage: a comparative analysis with healthy cartilage. Metabolites 14, 183 (2024).
Brahmachary, P., Erdogan, E., Myers, E. & June, R. K. Metabolomic profiling and characterization of a novel 3D culture system for studying chondrocyte mechanotransduction. bioRxiv (2024).
Batushansky, A., Lopes, E. B. P., Zhu, S., Humphries, K. M. & Griffin, T. M. GC-MS method for metabolic profiling of mouse femoral head articular cartilage reveals distinct effects of tissue culture and development. Osteoarthr. Cartil. 27, 1361–1371 (2019).
Zhai, G., Randell, E. W. & Rahman, P. Metabolomics of osteoarthritis: emerging novel markers and their potential clinical utility. Rheumatology 57, 2087–2095 (2018).
Borel, M. et al. Longitudinal profiling of articular cartilage degradation in osteoarthritis by high-resolution magic angle spinning 1H NMR spectroscopy: experimental study in the meniscectomized guinea pig model. J. Proteome Res. 8, 2594–2600 (2009).
McCutchen, C. N., Zignego, D. L. & June, R. K. Metabolic responses induced by compression of chondrocytes in variable-stiffness microenvironments. J. Biomech. 64, 49–58 (2017).
Bergstrom, A. R. et al. Metabolic profiles of encapsulated chondrocytes exposed to short-term simulated microgravity. bioRxiv (2024).
Southan, J., McHugh, E., Walker, H. & Ismail, H. M. Metabolic signature of articular cartilage following mechanical injury: an integrated transcriptomics and metabolomics analysis. Front. Mol. Biosci. 7, 592905 (2020).
Hu, X. et al. Identification of cellular heterogeneity and immunogenicity of chondrocytes via single-cell RNA sequencing technique in human osteoarthritis. Front. Pharmacol. 13, 1004766 (2022).
Xu, W. et al. Engineered biomechanical microenvironment of articular chondrocytes based on heterogeneous GelMA hydrogel composites and dynamic mechanical compression. Biomater. Adv. 153, 213567 (2023).
Zhang, C., Le Devedec, S. E., Ali, A. & Hankemeier, T. Single-cell metabolomics by mass spectrometry: ready for primetime? Curr. Opin. Biotechnol. 82, 102963 (2023).
Wei, D., Xu, M., Wang, Z. & Tong, J. The development of single-cell metabolism and its role in studying cancer emergent properties. Front. Oncol. 11, 814085 (2021).
Cillero-Pastor, B., Eijkel, G., Kiss, A., Blanco, F. J. & Heeren, R. M. Time-of-flight secondary ion mass spectrometry-based molecular distribution distinguishing healthy and osteoarthritic human cartilage. Anal. Chem. 84, 8909–8916 (2012).
Eveque-Mourroux, M. R. et al. Heterogeneity of lipid and protein cartilage profiles associated with human osteoarthritis with or without type 2 diabetes mellitus. J. Proteome Res. 20, 2973–2982 (2021).
Wu, C. L. et al. Single cell transcriptomic analysis of human pluripotent stem cell chondrogenesis. Nat. Commun. 12, 362 (2021).
Baldwin, M. et al. A roadmap for delivering a human musculoskeletal cell atlas. Nat. Rev. Rheumatol. 19, 738–752 (2023).
Donlin, L. T. et al. Methods for high-dimensional analysis of cells dissociated from cryopreserved synovial tissue. Arthritis Res. Ther. 20, 139 (2018).
Stephenson, W. et al. Single-cell RNA-seq of rheumatoid arthritis synovial tissue using low-cost microfluidic instrumentation. Nat. Commun. 9, 791 (2018).
Simonds, M. M., Sullivan, K. E. & Brescia, A. C. Single-cell analysis reveals heterogeneity of juvenile idiopathic arthritis fibroblast-like synoviocytes with implications for disease subtype. Arthritis Res. Ther. 24, 225 (2022).
Wei, K. et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature 582, 259–264 (2020).
Bai, Z. et al. Synovial fibroblast gene expression is associated with sensory nerve growth and pain in rheumatoid arthritis. Sci. Transl. Med. 16, eadk3506 (2024).
Chen, N., Fan, B., He, Z., Yu, X. & Wang, J. Identification of HBEGF+ fibroblasts in the remission of rheumatoid arthritis by integrating single-cell RNA sequencing datasets and bulk RNA sequencing datasets. Arthritis Res. Ther. 24, 215 (2022).
Wijesinghe, S. N. et al. Obesity defined molecular endotypes in the synovium of patients with osteoarthritis provides a rationale for therapeutic targeting of fibroblast subsets. Clin. Transl. Med. 13, e1232 (2023).
Tsuchiya, M. et al. CD39+CD55- Fb subset exhibits myofibroblast-like phenotype and is associated with pain in osteoarthritis of the knee. Biomedicines 11, 3047 (2023).
Zhang, F. et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat. Immunol. 20, 928–942 (2019).
Liu, F. et al. Identification of FOXO1 as a geroprotector in human synovium through single-nucleus transcriptomic profiling. Protein Cell 15, 441–459 (2024).
Croft, A. P. et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 570, 246–251 (2019).
Culemann, S. et al. Locally renewing resident synovial macrophages provide a protective barrier for the joint. Nature 572, 670–675 (2019).
Harasymowicz, N. et al. Injury and obesity differentially and synergistically induce dysregulation of synovial immune cells in osteoarthritis. Ann. Rheum. Dis. S0003-4967, 00813-1 (2024).
Wu, C. L. et al. Conditional macrophage depletion increases inflammation and does not inhibit the development of osteoarthritis in obese macrophage Fas-induced apoptosis-transgenic mice. Arthritis Rheumatol. 69, 1772–1783 (2017).
Li, X. et al. A single-cell RNA-sequencing analysis of distinct subsets of synovial macrophages in rheumatoid arthritis. DNA Cell Biol. 42, 212–222 (2023).
Hardt, U. et al. Integrated single cell and spatial transcriptomics reveal autoreactive differentiated B cells in joints of early rheumatoid arthritis. Sci. Rep. 12, 11876 (2022).
Durham, L. E. et al. Substantive similarities between synovial fluid and synovial tissue T cells in inflammatory arthritis via single-cell RNA and T cell receptor sequencing. Arthritis Rheumatol. 76, 1594–1601 (2024).
Li, J. et al. Synovium and infrapatellar fat pad share common mesenchymal progenitors and undergo coordinated changes in osteoarthritis. J. Bone Min. Res. 39, 161–176 (2024).
Tang, S. et al. Single-cell atlas of human infrapatellar fat pad and synovium implicates APOE signaling in osteoarthritis pathology. Sci. Transl. Med. 16, eadf4590 (2024).
Ciechomska, M., Roszkowski, L. & Maslinski, W. DNA methylation as a future therapeutic and diagnostic target in rheumatoid arthritis. Cells 8, 953 (2019).
Taylor, P. C., Moore, A., Vasilescu, R., Alvir, J. & Tarallo, M. A structured literature review of the burden of illness and unmet needs in patients with rheumatoid arthritis: a current perspective. Rheumatol. Int. 36, 685–695 (2016).
Ham, S. et al. Epigenetic analysis in rheumatoid arthritis synoviocytes. Exp. Mol. Med. 51, 1–13 (2019).
Ai, R. et al. Comprehensive epigenetic landscape of rheumatoid arthritis fibroblast-like synoviocytes. Nat. Commun. 9, 1921 (2018).
Li Yim, A. Y. F. et al. Novel insights into rheumatoid arthritis through characterization of concordant changes in DNA methylation and gene expression in synovial biopsies of patients with differing numbers of swollen joints. Front. Immunol. 12, 651475 (2021).
Jiang, F. et al. A landscape of gene expression regulation for synovium in arthritis. Nat. Commun. 15, 1409 (2024).
Tsuchiya, H., Ota, M. & Fujio, K. Multiomics landscape of synovial fibroblasts in rheumatoid arthritis. Inflamm. Regen. 41, 7 (2021).
Gong, X. et al. An overview of multi-omics technologies in rheumatoid arthritis: applications in biomarker and pathway discovery. Front. Immunol. 15, 1381272 (2024).
Falconer, J. et al. Review: synovial cell metabolism and chronic inflammation in rheumatoid arthritis. Arthritis Rheumatol. 70, 984–999 (2018).
Scanzello, C. R. & Goldring, S. R. The role of synovitis in osteoarthritis pathogenesis. Bone 51, 249–257 (2012).
Hermansson, M. et al. MS analysis of rheumatoid arthritic synovial tissue identifies specific citrullination sites on fibrinogen. Proteom. Clin. Appl. 4, 511–518 (2010).
Rocha, B. et al. Identification of a distinct lipidomic profile in the osteoarthritic synovial membrane by mass spectrometry imaging. Osteoarthr. Cartil. 29, 750–761 (2021).
Su, Q. H. et al. Course-based intra-articular injection of medical chitosan mitigates excessive deposition of triacylglycerides in the synovial tissue of the knee osteoarthritis. J. Chin. Med. Assoc. 87, 870–877 (2024).
Ge, M. et al. Multi-omics analysis of synovial tissue and fluid reveals differentially expressed proteins and metabolites in osteoarthritis. J. Transl. Med. 23, 285 (2025).
Baryawno, N. et al. A cellular taxonomy of the bone marrow stroma in homeostasis and leukemia. Cell 177, 1915–1932.e1916 (2019).
Tikhonova, A. N. et al. The bone marrow microenvironment at single-cell resolution. Nature 569, 222–228 (2019).
Zhong, L. et al. Single cell transcriptomics identifies a unique adipose lineage cell population that regulates bone marrow environment. eLife 9, e54695 (2020).
Ayturk, U. M. et al. Single-cell RNA sequencing of calvarial and long-bone endocortical cells. J. Bone Miner. Res. 35, 1981–1991 (2020).
Baccin, C. et al. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat. Cell Biol. 22, 38–48 (2020).
Matsushita, Y. et al. A Wnt-mediated transformation of the bone marrow stromal cell identity orchestrates skeletal regeneration. Nat. Commun. 11, 332 (2020).
Shi, Y. et al. Gli1+ progenitors mediate bone anabolic function of teriparatide via Hh and Igf signaling. Cell Rep. 36, 109542 (2021).
Matsushita, Y., Chu, A. K. Y., Ono, W., Welch, J. D. & Ono, N. Intercellular interactions of an adipogenic CXCL12-expressing stromal cell subset in murine bone marrow. J. Bone Miner. Res. 36, 1145–1158 (2021).
Zhong, L., Yao, L., Seale, P. & Qin, L. Marrow adipogenic lineage precursor: a new cellular component of marrow adipose tissue. Best. Pr. Res. Clin. Endocrinol. Metab. 35, 101518 (2021).
Zhong, L. et al. Transient expansion and myofibroblast conversion of adipogenic lineage precursors mediate bone marrow repair after radiation. JCI Insight 7, e150323 (2022).
Leimkuhler, N. B. et al. Heterogeneous bone-marrow stromal progenitors drive myelofibrosis via a druggable alarmin axis. Cell Stem Cell 28, 637–652.e638 (2021).
Hall, T. D. et al. Murine fetal bone marrow does not support functional hematopoietic stem and progenitor cells until birth. Nat. Commun. 13, 5403 (2022).
Liu, Y. et al. A specialized bone marrow microenvironment for fetal haematopoiesis. Nat. Commun. 13, 1327 (2022).
de Jong, M. M. E. et al. The multiple myeloma microenvironment is defined by an inflammatory stromal cell landscape. Nat. Immunol. 22, 769–780 (2021).
Li, H. et al. Identification of phenotypically, functionally, and anatomically distinct stromal niche populations in human bone marrow based on single-cell RNA sequencing. eLife 12, e81656 (2023).
Wang, Z. et al. Single-cell transcriptome atlas of human mesenchymal stem cells exploring cellular heterogeneity. Clin. Transl. Med. 11, e650 (2021).
Wang, Z. et al. Single-cell RNA sequencing deconvolutes the in vivo heterogeneity of human bone marrow-derived mesenchymal stem cells. Int. J. Biol. Sci. 17, 4192–4206 (2021).
Bandyopadhyay, S. et al. Mapping the cellular biogeography of human bone marrow niches using single-cell transcriptomics and proteomic imaging. Cell 2, 00408–00402 (2024).
Jardine, L. et al. Blood and immune development in human fetal bone marrow and Down syndrome. Nature 598, 327–331 (2021).
Zhang, P. et al. Characterization of mesenchymal stem cells in human fetal bone marrow by single-cell transcriptomic and functional analysis. Signal Transduct. Target. Ther. 8, 126 (2023).
Xiao, X. et al. Spatial transcriptomic interrogation of the murine bone marrow signaling landscape. Bone Res. 11, 59 (2023).
Bigham-Sadegh, A. & Oryan, A. Basic concepts regarding fracture healing and the current options and future directions in managing bone fractures. Int. Wound J. 12, 238–247 (2015).
Sheen, J. R., Mabrouk, A. & Garla, V. V. Fracture healing overview. in StatPearls (StatPearls Publishing, 2024).
Wildemann, B. et al. Non-union bone fractures. Nat. Rev. Dis. Prim. 7, 57 (2021).
Salichos, L., Thayavally, R., Kloen, P. & Hadjiargyrou, M. Human nonunion tissues display differential gene expression in comparison to physiological fracture callus. Bone 183, 117091 (2024).
Wang, C., Abu-Amer, Y., O’Keefe, R. J. & Shen, J. Loss of Dnmt3b in chondrocytes leads to delayed endochondral ossification and fracture repair. J. Bone Miner. Res. 33, 283–297 (2018).
Ambrosi, T. H. et al. Aged skeletal stem cells generate an inflammatory degenerative niche. Nature 597, 256–262 (2021).
Lin, X. et al. Aged callus skeletal stem/progenitor cells contain an inflammatory osteogenic population with increased IRF and NF-kappaB pathways and reduced osteogenic potential. Front. Mol. Biosci. 9, 806528 (2022).
Liu, J. et al. Molecular signatures distinguish senescent cells from inflammatory cells in aged mouse callus stromal cells. Front. Endocrinol. 14, 1090049 (2023).
Zou, N. Y. et al. Age-related secretion of grancalcin by macrophages induces skeletal stem/progenitor cell senescence during fracture healing. Bone Res. 12, 6 (2024).
Zhang, H. et al. Single-cell RNA sequencing reveals B cells are important regulators in fracture healing. Front. Endocrinol. 12, 666140 (2021).
Xiao, D. et al. DNA methylation-mediated Rbpjk suppression protects against fracture nonunion caused by systemic inflammation. J. Clin. Investig. 134, e168558 (2023).
Avin, K. G. et al. Single-cell RNAseq provides insight into altered immune cell populations in human fracture nonunions. J. Orthop. Res. 41, 1060–1069 (2023).
Serowoky, M. A. et al. A murine model of large-scale bone regeneration reveals a selective requirement for Sonic Hedgehog. NPJ Regen. Med. 7, 30 (2022).
Yao, L. et al. Activin A marks a novel progenitor cell population during fracture healing and reveals a therapeutic strategy. eLife 12, e89822 (2023).
Xu, T., Wang, C., Shen, J., Tong, P. & O’Keefe, R. Ablation of Dnmt3b in chondrocytes suppresses cell maturation during embryonic development. J. Cell Biochem. 119, 5852–5863 (2018).
Wang, C. et al. Targeting angiogenesis for fracture nonunion treatment in inflammatory disease. Bone Res. 9, 29 (2021).
Mancinelli, L., Schoedel, K. E., Weiss, K. R. & Intini, G. Methods to enable spatial transcriptomics of bone tissues. J. Vis. Exp. 3 (2024).
Jiang, W., Caruana, D. L., Back, J. & Lee, F. Y. Unique spatial transcriptomic profiling of the murine femoral fracture callus: a preliminary report. Cells 13, 522 (2024).
Rios, J. J. et al. Spatial transcriptomics implicates impaired BMP signaling in NF1 fracture pseudarthrosis in murine and patient tissues. JCI Insight 9, 522 (2024).
Shen, L., Hu, G. & Karner, C. M. Bioenergetic metabolism in osteoblast differentiation. Curr. Osteoporos. Rep. 20, 53–64 (2022).
Stegen, S. & Carmeliet, G. Metabolic regulation of skeletal cell fate and function. Nat. Rev. Endocrinol. 20, 399–413 (2024).
van Gastel, N. & Carmeliet, G. Metabolic regulation of skeletal cell fate and function in physiology and disease. Nat. Metab. 3, 11–20 (2021).
Yu, Y. et al. Glutamine metabolism regulates proliferation and lineage allocation in skeletal stem cells. Cell Metab. 29, 966–978.e964 (2019).
Stegen, S. et al. Glutamine metabolism in osteoprogenitors is required for bone mass accrual and PTH-induced bone anabolism in male mice. J. Bone Miner. Res. 36, 604–616 (2021).
Stegen, S. et al. HIF-1alpha promotes glutamine-mediated redox homeostasis and glycogen-dependent bioenergetics to support postimplantation bone cell survival. Cell Metab. 23, 265–279 (2016).
Hu, G. et al. Glutathione limits RUNX2 oxidation and degradation to regulate bone formation. JCI Insight 8, e166888 (2023).
Sharma, D., Yu, Y., Shen, L., Zhang, G. F. & Karner, C. M. SLC1A5 provides glutamine and asparagine necessary for bone development in mice. eLife 10, e71595 (2021).
Shen, L. et al. SLC38A2 provides proline to fulfill unique synthetic demands arising during osteoblast differentiation and bone formation. eLife 11, e76963 (2022).
Shen, L., Yu, Y. & Karner, C. M. SLC38A2 provides proline and alanine to regulate postnatal bone mass accrual in mice. Front. Physiol. 13, 992679 (2022).
Elefteriou, F. et al. ATF4 mediation of NF1 functions in osteoblast reveals a nutritional basis for congenital skeletal dysplasiae. Cell Metab. 4, 441–451 (2006).
Rached, M. T. et al. FoxO1 is a positive regulator of bone formation by favoring protein synthesis and resistance to oxidative stress in osteoblasts. Cell Metab. 11, 147–160 (2010).
Bian, Xue-Feng, L, J.-F., Sun, Jia-Ming & Zhang, Hui 1HNMR metabolomics of MC3T3-E mouse osteoblast proliferation and alkaline phosphatase content by deer antler peptide amine. Chin. J. Anal. Chem. 49, 103–109 (2021).
Araujo, R. et al. NMR metabolomics to study the metabolic response of human osteoblasts to non-poled and poled poly (L-lactic) acid. Magn. Reson. Chem. 57, 919–933 (2019).
Bispo, D. S. C. et al. NMR metabolomics assessment of osteogenic differentiation of adipose-tissue-derived mesenchymal stem cells. J. Proteome Res. 21, 654–670 (2022).
Misra, B. B., Jayapalan, S., Richards, A. K., Helderman, R. C. M. & Rendina-Ruedy, E. Untargeted metabolomics in primary murine bone marrow stromal cells reveals distinct profile throughout osteoblast differentiation. Metabolomics 17, 86 (2021).
Helderman, R. C. et al. Loss of function of lysosomal acid lipase (LAL) profoundly impacts osteoblastogenesis and increases fracture risk in humans. Bone 148, 115946 (2021).
Tencerova, M. et al. Metabolic programming determines the lineage-differentiation fate of murine bone marrow stromal progenitor cells. Bone Res. 7, 35 (2019).
Welhaven, H. D. et al. The cortical bone metabolome of C57BL/6J mice is sexually dimorphic. JBMR 6, e10654 (2022).
Nam, M. et al. Metabolic alterations in the bone tissues of aged osteoporotic mice. Sci. Rep. 8, 8127 (2018).
Song, F. et al. Osteoblast-intrinsic defect in glucose metabolism impairs bone formation in type II diabetic male mice. eLife 12, e85714 (2023).
Zhang, X. et al. Metabolomics insights into osteoporosis through association with bone mineral density. J. Bone Miner. Res. 36, 729–738 (2021).
Mei, Z. et al. Association between the metabolome and bone mineral density in a Chinese population. EBioMedicine 62, 103111 (2020).
Hu, Y., Zhang, X. & Shan, Y. LC-MS-based plasma metabolomics reveals metabolic variations in ovariectomy-induced osteoporosis in female Wistar rats. RSC Adv. 8, 24932–24941 (2018).
Guntur, A. R. et al. Osteoblast-like MC3T3-E1 cells prefer glycolysis for ATP production but adipocyte-like 3T3-L1 cells prefer oxidative phosphorylation. J. Bone Min. Res. 33, 1052–1065 (2018).
Esen, E. et al. WNT-LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast differentiation. Cell Metab. 17, 745–755 (2013).
van Gastel, N. et al. Lipid availability determines fate of skeletal progenitor cells via SOX9. Nature 579, 111–117 (2020).
Su, Y. et al. Circulating amino acids are associated with bone mineral density decline and ten-year major osteoporotic fracture risk in older community-dwelling adults. Bone 129, 115082 (2019).
Liang, W. et al. An integrated multi-omics analysis reveals osteokines involved in global regulation. Cell Metab. 36, 1144–1163 e1147 (2024).
Violante, S., Berisa, M., Thomas, T. H. & Cross, J. R. Stable isotope tracers for metabolic pathway analysis. Methods Mol. Biol. 1978, 269–283 (2019).
Jang, C., Chen, L. & Rabinowitz, J. D. Metabolomics and isotope tracing. Cell 173, 822–837 (2018).
Karner, C. M., Esen, E., Okunade, A. L., Patterson, B. W. & Long, F. Increased glutamine catabolism mediates bone anabolism in response to WNT signaling. J. Clin. Investig. 125, 551–562 (2015).
Lee, W. C., Ji, X., Nissim, I. & Long, F. Malic enzyme couples mitochondria with aerobic glycolysis in osteoblasts. Cell Rep. 32, 108108 (2020).
Zhang, Y. et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol. 20, 1181–1192 (2018).
DeVilbiss, A. W. et al. Metabolomic profiling of rare cell populations isolated by flow cytometry from tissues. eLife 10, e61980 (2021).
Semenza, G. L. Hypoxia-inducible factors in physiology and medicine. Cell 148, 399–408 (2012).
Walsh, M. C. et al. Osteoimmunology: interplay between the immune system and bone metabolism. Annu. Rev. Immunol. 24, 33–63 (2006).
Badur, M. G., Zhang, H. & Metallo, C. M. Enzymatic passaging of human embryonic stem cells alters central carbon metabolism and glycan abundance. Biotechnol. J. 10, 1600–1611 (2015).
Larouche, J. A., Wallace, E. C., Spence, B. D., Buras, E. & Aguilar, C. A. Spatiotemporal mapping of immune and stem cell dysregulation after volumetric muscle loss. JCI Insight 8, e162835 (2023).
Tower, R. J. et al. Spatial transcriptomics reveals a role for sensory nerves in preserving cranial suture patency through modulation of BMP/TGF-beta signaling. Proc. Natl. Acad. Sci. USA 118, e2103087118 (2021).
Tower, R. J. et al. Spatial transcriptomics reveals metabolic changes underly age-dependent declines in digit regeneration. eLife 11, e71542 (2022).
Cherief, M. et al. TrkA-mediated sensory innervation of injured mouse tendon supports tendon sheath progenitor cell expansion and tendon repair. Sci. Transl. Med. 15, eade4619 (2023).
Fu, W., Yang, R. & Li, J. Single-cell and spatial transcriptomics reveal changes in cell heterogeneity during progression of human tendinopathy. BMC Biol. 21, 132 (2023).
Zhang, B. et al. A human embryonic limb cell atlas resolved in space and time. Nature 635, 668–678 (2023).
Mathavan, N. et al. Spatial transcriptomics in mechanomics: new horizons in exploring the mechanoregulation of bone regeneration. bioRxiv (2024).
Bian, F. et al. Spatial transcriptomics reveals the requirement of ADGRG6 in maintaining chondrocyte homeostasis in mouse growth plates. bioRxiv (2023).
Wehrle, E., Gunther, D., Mathavan, N., Singh, A. & Muller, R. Protocol for preparing formalin-fixed paraffin-embedded musculoskeletal tissue samples from mice for spatial transcriptomics. STAR Protoc. 5, 102986 (2024).
Buck, A., Prade, V. M., Kunzke, T., Erben, R. G. & Walch, A. Spatial metabolomics reveals upregulation of several pyrophosphate-producing pathways in cortical bone of Hyp mice. JCI Insight 7, e162138 (2022).
Luo, L. et al. Spatial metabolomics reveals skeletal myofiber subtypes. Sci. Adv. 9, eadd0455 (2023).
Chan, W. C., Sze, K. L., Samartzis, D., Leung, V. Y. & Chan, D. Structure and biology of the intervertebral disk in health and disease. Orthop. Clin. North Am. 42, 447–464 (2011).
Walk, R. et al. The progression of neurovascular features and chemokine signatures of the intervertebral disc with degeneration. bioRxiv (2024).
Lintz, M., Walk, R. E., Tang, S. Y. & Bonassar, L. J. The degenerative impact of hyperglycemia on the structure and mechanics of developing murine intervertebral discs. JOR Spine 5, e1191 (2022).
Diaz-Hernandez, M. E. et al. Derivation of notochordal cells from human embryonic stem cells reveals unique regulatory networks by single cell-transcriptomics. J. Cell Physiol. 235, 5241–5255 (2020).
Li, Z. et al. Single-cell RNA sequencing reveals the difference in human normal and degenerative nucleus pulposus tissue profiles and cellular interactions. Front. Cell Dev. Biol. 10, 910626 (2022).
Fernandes, L. M. et al. Single-cell RNA-seq identifies unique transcriptional landscapes of human nucleus pulposus and annulus fibrosus cells. Sci. Rep. 10, 15263 (2020).
Gan, Y. et al. Spatially defined single-cell transcriptional profiling characterizes diverse chondrocyte subtypes and nucleus pulposus progenitors in human intervertebral discs. Bone Res. 9, 37 (2021).
Han, S. et al. Single-cell RNA sequencing of the nucleus pulposus reveals chondrocyte differentiation and regulation in intervertebral disc degeneration. Front. Cell Dev. Biol. 10, 824771 (2022).
Kuchynsky, K. et al. Transcriptional profiling of human cartilage endplate cells identifies novel genes and cell clusters underlying degenerated and non-degenerated phenotypes. Arthritis Res. Ther. 26, 12 (2024).
Wang, J. et al. Novel biomarkers of intervertebral disc cells and evidence of stem cells in the intervertebral disc. Osteoarthr. Cartil. 29, 389–401 (2021).
Panebianco, C. J., Dave, A., Charytonowicz, D., Sebra, R. & Iatridis, J. C. Single-cell RNA-sequencing atlas of bovine caudal intervertebral discs: discovery of heterogeneous cell populations with distinct roles in homeostasis. FASEB J. 35, e21919 (2021).
Calió, M., Gantenbein, B., Egli, M., Poveda, L. & Ille, F. The cellular composition of bovine coccygeal intervertebral discs: a comprehensive single-cell RNAseq analysis. Int. J. Mol. Sci. 22, 4917 (2021).
Disease, G. B. D., Injury, I. & Prevalence, C. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392, 1789–1858 (2018).
Cherif, H. et al. Single-cell RNA-seq analysis of cells from degenerating and non-degenerating intervertebral discs from the same individual reveals new biomarkers for intervertebral disc degeneration. Int. J. Mol. Sci. 23, 3993 (2022).
Wang, D. et al. Single-cell transcriptomics reveals heterogeneity and intercellular crosstalk in human intervertebral disc degeneration. iScience 26, 106692 (2023).
Walk, R. E., Moon, H. J., Tang, S. Y. & Gupta, M. C. Contrast-enhanced microCT evaluation of degeneration following partial and full width injuries to the mouse lumbar intervertebral disc. Sci. Rep. 12, 15555 (2022).
Clayton, S. W., Walk, R. E., Mpofu, L., Easson, G. W. D. & Tang, S. Y. Analysis of infiltrating immune cells following intervertebral disc injury reveals recruitment of gamma-delta (gammadelta) T cells in female mice. bioRxiv (2024).
Ling, Z. et al. Single-cell RNA-seq analysis reveals macrophage involved in the progression of human intervertebral disc degeneration. Front. Cell Dev. Biol. 9, 833420 (2021).
Rohanifar, M. et al. Single cell RNA-sequence analyses reveal uniquely expressed genes and heterogeneous immune cell involvement in the rat model of intervertebral disc degeneration. Appl. Sci. 12, 8244 (2022).
Swahn, H. et al. Shared and compartment-specific processes in nucleus pulposus and annulus fibrosus during intervertebral disc degeneration. Adv. Sci. 11, e2309032 (2024).
Jiang, W. et al. Intervertebral disc human nucleus pulposus cells associated with back pain trigger neurite outgrowth in vitro and pain behaviors in rats. Sci. Transl. Med. 15, eadg7020 (2023).
Shao, T. et al. The role of immunocyte infiltration regulatory network based on hdWGCNA and single-cell bioinformatics analysis in intervertebral disc degeneration. Inflammation 47, 1987–1999 (2024).
Wang, P., Li, Z. & Ye, D. Single-cell RNA-seq analysis reveals the Wnt/Ca(2+) signaling pathway with inflammation, apoptosis in nucleus pulposus degeneration. BMC Musculoskelet. Disord. 25, 321 (2024).
Zhang, Y. et al. Single-cell RNA-seq analysis identifies unique chondrocyte subsets and reveals involvement of ferroptosis in human intervertebral disc degeneration. Osteoarthr. Cartil. 29, 1324–1334 (2021).
Kamatani, T. et al. Human iPS cell-derived cartilaginous tissue spatially and functionally replaces nucleus pulposus. Biomaterials 284, 121491 (2022).
Zhou, T. et al. Spatiotemporal characterization of human early intervertebral disc formation at single-cell resolution. Adv. Sci. 10, e2206296 (2023).
Jiang, W. et al. Single-cell atlas unveils cellular heterogeneity and novel markers in human neonatal and adult intervertebral discs. iScience 25, 104504 (2022).
Wang, L. et al. Revealing the immune infiltration landscape and identifying diagnostic biomarkers for lumbar disc herniation. Front. Immunol. 12, 666355 (2021).
Li, W. et al. Deciphering the sequential changes of monocytes/macrophages in the progression of IDD with longitudinal approach using single-cell transcriptome. Front. Immunol. 14, 1090637 (2023).
Sun, H., Wang, H., Zhang, W., Mao, H. & Li, B. Single-cell RNA sequencing reveals resident progenitor and vascularization-associated cell subpopulations in rat annulus fibrosus. J. Orthop. Transl. 38, 256–267 (2023).
Zhang, S. et al. Inhibiting heat shock protein 90 attenuates nucleus pulposus fibrosis and pathologic angiogenesis induced by macrophages via down-regulating cell migration-inducing protein. Am. J. Pathol. 193, 960–976 (2023).
Zhang, Z., Huo, J., Ji, X., Wei, L. & Zhang, J. GREM1, LRPPRC and SLC39A4 as potential biomarkers of intervertebral disc degeneration: a bioinformatics analysis based on multiple microarray and single-cell sequencing data. BMC Musculoskelet. Disord. 24, 729 (2023).
Chen, Y. et al. Characterization of the nucleus pulposus progenitor cells via spatial transcriptomics. Adv. Sci. 11, e2303752 (2024).
Williams, C. G., Lee, H. J., Asatsuma, T., Vento-Tormo, R. & Haque, A. An introduction to spatial transcriptomics for biomedical research. Genome Med. 14, 68 (2022).
Chen, G., Ning, B. & Shi, T. Single-cell RNA-seq technologies and related computational data analysis. Front. Genet. 10, 317 (2019).
Haglund, L., Ouellet, J. & Roughley, P. Variation in chondroadherin abundance and fragmentation in the human scoliotic disc. Spine 34, 1513–1518 (2009).
McCann, M. R. et al. Proteomic signature of the murine intervertebral disc. PLoS ONE 10, e0117807 (2015).
Kudelko, M. et al. PRIMUS: comprehensive proteomics of mouse intervertebral discs that inform novel biology and relevance to human disease modelling. Matrix Biol. 12, 100082 (2021).
Rajasekaran, S. et al. Proteomic signatures of healthy intervertebral discs from organ donors: a comparison with previous studies on discs from scoliosis, animals, and trauma. Neurospine 17, 426–442 (2020).
Rajasekaran, S. et al. Can scoliotic discs be controls for molecular studies in intervertebral disc research? Insights from proteomics. Glob. Spine J. 12, 598–609 (2022).
Ye, D. et al. Comparative and quantitative proteomic analysis of normal and degenerated human annulus fibrosus cells. Clin. Exp. Pharmacol. Physiol. 42, 530–536 (2015).
Yee, A. et al. Fibrotic-like changes in degenerate human intervertebral discs revealed by quantitative proteomic analysis. Osteoarthr. Cartil. 24, 503–513 (2016).
Easson, G. W. D. et al. Modulation of TRPV4 protects against degeneration induced by sustained loading and promotes matrix synthesis in the intervertebral disc. FASEB J. 37, e22714 (2023).
Easson, G. W. D., Savadipour, A., Gonzalez, C., Guilak, F. & Tang, S. Y. TRPV4 differentially controls inflammatory cytokine networks during static and dynamic compression of the intervertebral disc. JOR Spine 6, e1282 (2023).
Wang, Y. et al. Deficiency of MIF accentuates overloaded compression-induced nucleus pulposus cell oxidative damage via depressing mitophagy. Oxid. Med. Cell. Longev. 2021, 6192498 (2021).
Zhang, Y. et al. Early onset of disc degeneration in SM/J mice is associated with changes in ion transport systems and fibrotic events. Matrix Biol. 70, 123–139 (2018).
Pattappa, G. et al. CCL5/RANTES is a key chemoattractant released by degenerative intervertebral discs in organ culture. Eur. Cell Mater. 27, 124–136 (2014). discussion 136.
Erwin, W. M. et al. The biological basis of degenerative disc disease: proteomic and biomechanical analysis of the canine intervertebral disc. Arthritis Res. Ther. 17, 240 (2015).
Bach, F. C. et al. Soluble and pelletable factors in porcine, canine and human notochordal cell-conditioned medium: implications for IVD regeneration. Eur. Cell Mater. 32, 163–180 (2016).
Rajasekaran, S. et al. Proteomic signature of nucleus pulposus in fetal intervertebral disc. Asian Spine J. 14, 409–420 (2020).
Rajasekaran, S. et al. Novel biomarkers of health and degeneration in human intervertebral discs: in-depth proteomic analysis of collagen framework of fetal, healthy, scoliotic, degenerate, and herniated discs. Asian Spine J. 17, 17–29 (2023).
Caldeira, J. et al. Matrisome profiling during intervertebral disc development and ageing. Sci. Rep. 7, 11629 (2017).
Molinos, M. et al. Alterations of bovine nucleus pulposus cells with aging. Aging Cell 22, e13873 (2023).
Lundberg, E. & Borner, G. H. H. Spatial proteomics: a powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 20, 285–302 (2019).
Tam, V. et al. DIPPER, a spatiotemporal proteomics atlas of human intervertebral discs for exploring ageing and degeneration dynamics. eLife 9, e64940 (2020).
Eckersley, A. et al. Peptide ___location fingerprinting reveals tissue region-specific differences in protein structures in an ageing human organ. Int. J. Mol. Sci. 22, 10408 (2021).
Eckersley, A. et al. Peptide ___location fingerprinting identifies species- and tissue-conserved structural remodelling of proteins as a consequence of ageing and disease. Matrix Biol. 114, 108–137 (2022).
Dai, X. & Shen, L. Advances and trends in omics technology development. Front. Med. 9, 911861 (2022).
Veras, M. A. et al. Protocol for parallel proteomic and metabolomic analysis of mouse intervertebral disc tissues. JOR Spine 3, e1099 (2020).
Chen, Q. et al. Quiescence preconditioned nucleus pulposus stem cells alleviate intervertebral disc degeneration by enhancing cell survival via adaptive metabolism pattern in rats. Front. Bioeng. Biotechnol. 11, 1073238 (2023).
Wu, X. et al. Glycine-serine-threonine metabolic axis delays intervertebral disc degeneration through antioxidant effects: an imaging and metabonomics study. Oxid. Med. Cell. Longev. 2021, 5579736 (2021).
Guo, Z. et al. The role of IL-6 and TMEM100 in lumbar discogenic pain and the mechanism of the glycine-serine-threonine metabolic axis: a metabolomic and molecular biology study. J. Pain. Res. 16, 437–461 (2023).
Wymann, M. P. & Schneiter, R. Lipid signalling in disease. Nat. Rev. Mol. Cell Biol. 9, 162–176 (2008).
Francisco, V. et al. Metabolomic signature and molecular profile of normal and degenerated human intervertebral disc cells. Spine J. 23, 1549–1562 (2023).
Granville Smith, I. et al. Evidence for infection in intervertebral disc degeneration: a systematic review. Eur. Spine J. 31, 414–430 (2022).
Rajasekaran, S. et al. Bacteria in human lumbar discs - subclinical infection or contamination? Metabolomic evidence for colonization, multiplication, and cell-cell cross-talk of bacteria. Spine J. 23, 163–177 (2023).
Boonen, K. et al. Beyond genes: re-identifiability of proteomic data and its implications for personalized medicine. Genes 10, 682 (2019).
Williamson, P. M. et al. Tendinopathy and tendon material response to load: What we can learn from small animal studies. Acta Biomater. 134, 43–56 (2021).
Roffino, S. et al. Negative impact of disuse and unloading on tendon enthesis structure and function. Life Sci. Space Res. 29, 46–52 (2021).
Riel, H., Lindstrom, C. F., Rathleff, M. S., Jensen, M. B. & Olesen, J. L. Prevalence and incidence rate of lower-extremity tendinopathies in a Danish general practice: a registry-based study. BMC Musculoskelet. Disord. 20, 239 (2019).
Linsell, L. et al. Prevalence and incidence of adults consulting for shoulder conditions in UK primary care; patterns of diagnosis and referral. Rheumatology 45, 215–221 (2006).
Nichols, A. E. C., Best, K. T. & Loiselle, A. E. The cellular basis of fibrotic tendon healing: challenges and opportunities. Transl. Res. 209, 156–168 (2019).
Kendal, A. R. et al. Multi-omic single cell analysis resolves novel stromal cell populations in healthy and diseased human tendon. Sci. Rep. 10, 13939 (2020).
Still, C. et al. Single-cell transcriptomic profiling reveals distinct mechanical responses between normal and diseased tendon progenitor cells. Cell Rep. Med. 2, 100343 (2021).
Akbar, M. et al. Single cell and spatial transcriptomics in human tendon disease indicate dysregulated immune homeostasis. Ann. Rheum. Dis. 80, 1494–1497 (2021).
Ackerman, J. E. et al. Defining the spatial-molecular map of fibrotic tendon healing and the drivers of Scleraxis-lineage cell fate and function. Cell Rep. 41, 111706 (2022).
Nichols, A. E. C., Wagner, N. W., Ketonis, C. & Loiselle, A. E. Epitenon-derived cells comprise a distinct progenitor population that contributes to both tendon fibrosis and regeneration following acute injury. bioRxiv (2023).
Rajan, A. et al. Single-cell analysis reveals distinct fibroblast plasticity during tenocyte regeneration in zebrafish. Sci. Adv. 9, eadi5771 (2023).
Mimpen, J. Y. et al. Single nucleus and spatial transcriptomic profiling of healthy human hamstring tendon. FASEB J. 38, e23629 (2024).
Steffen, D., Mienaltowski, M. & Baar, K. Spatial gene expression in the adult rat patellar tendon. Matrix Biol. 19-20, 100138 (2023).
Disser, N. P. et al. Widespread diversity in the transcriptomes of functionally divergent limb tendons. J. Physiol. 598, 1537–1550 (2020).
Disser, N. P. et al. Achilles tendons display region-specific transcriptomic signatures associated with distinct mechanical properties. Am. J. Sports Med. 50, 3866–3874 (2022).
Best, K. T. & Loiselle, A. E. Scleraxis lineage cells contribute to organized bridging tissue during tendon healing and identify a subpopulation of resident tendon cells. FASEB J. 33, 8578–8587 (2019).
Korcari, A., Przybelski, S. J., Gingery, A. & Loiselle, A. E. Impact of aging on tendon homeostasis, tendinopathy development, and impaired healing. Connect Tissue Res. 64, 1–13 (2023).
Svensson, R. B., Heinemeier, K. M., Couppe, C., Kjaer, M. & Magnusson, S. P. Effect of aging and exercise on the tendon. J. Appl Physiol. 121, 1237–1246 (2016).
Peffers, M. J. et al. Transcriptome analysis of ageing in uninjured human Achilles tendon. Arthritis Res. Ther. 17, 33 (2015).
Zamboulis, D. E. et al. The interfascicular matrix of energy storing tendons houses heterogenous cell populations disproportionately affected by aging. Aging Dis. 15, 295–310 (2024).
Patel, D. et al. Structure-function specialisation of the interfascicular matrix in the human Achilles tendon. Acta Biomater. 131, 381–390 (2021).
Soslowsky, L. J. et al. Neer Award 1999: overuse activity injures the supraspinatus tendon in an animal model: a histologic and biomechanical study. J. Shoulder Elb. Surg. 9, 79–84 (2000).
Malmgaard-Clausen, N. M., Kjaer, M. & Dakin, S. G. Pathological tendon histology in early and chronic human patellar tendinopathy. Transl. Sports Med. 2022, 2799665 (2022).
Guo, J. et al. Integrative single-cell RNA and ATAC sequencing reveals that the FOXO1-PRDX2-TNF axis regulates tendinopathy. Front. Immunol. 14, 1092778 (2023).
Best, K. T. et al. Scleraxis-lineage cell depletion improves tendon healing and disrupts adult tendon homeostasis. eLife 10, e62203 (2021).
Howell, K. et al. Novel model of tendon regeneration reveals distinct cell mechanisms underlying regenerative and fibrotic tendon healing. Sci. Rep. 7, 45238 (2017).
Sakabe, T. et al. Transcription factor scleraxis vitally contributes to progenitor lineage direction in wound healing of adult tendon in mice. J. Biol. Chem. 293, 5766–5780 (2018).
Wagner, D. E. & Klein, A. M. Lineage tracing meets single-cell omics: opportunities and challenges. Nat. Rev. Genet. 21, 410–427 (2020).
Zhang, X. et al. Multi-omics analysis of human tendon adhesion reveals that ACKR1-regulated macrophage migration is involved in regeneration. Bone Res. 12, 27 (2024).
Mukund, K. & Subramaniam, S. Skeletal muscle: a review of molecular structure and function, in health and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 12, e1462 (2020).
Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 9, 493–495 (1961).
Bentzinger, C. F., Wang, Y. X., Dumont, N. A. & Rudnicki, M. A. Cellular dynamics in the muscle satellite cell niche. EMBO Rep. 14, 1062–1072 (2013).
Asakura, A. Stem cells in adult skeletal muscle. Trends Cardiovasc. Med. 13, 123–128 (2003).
McFarland, A. J., Ray, P. R., Bhai, S., Levine, B. D. & Price, T. J. RNA sequencing on muscle biopsy from a 5-week bed rest study reveals the effect of exercise and potential interactions with dorsal root ganglion neurons. Physiol. Rep. 10, e15176 (2022).
Zhang, X. et al. Characterization of cellular senescence in aging skeletal muscle. Nat. Aging 2, 601–615 (2022).
Wang, L., Zhou, Y., Wang, Y. & Shan, T. Integrative cross-species analysis reveals conserved and unique signatures in fatty skeletal muscles. Sci. Data 11, 290 (2024).
Tumasian, R. A. et al. Skeletal muscle transcriptome in healthy aging. Nat. Commun. 12, 2014 (2021).
Southerland, K. W. et al. Skeletal muscle regeneration failure in ischemic-damaged limbs is associated with pro-inflammatory macrophages and premature differentiation of satellite cells. Genome Med. 15, 95 (2023).
Pillon, N. J. et al. Transcriptomic profiling of skeletal muscle adaptations to exercise and inactivity. Nat. Commun. 11, 470 (2020).
Nieves-Rodriguez, S. et al. Transcriptomic analysis of paired healthy human skeletal muscles to identify modulators of disease severity in DMD. Front. Genet. 14, 1216066 (2023).
Cai, C., Yue, Y. & Yue, B. Single-cell RNA sequencing in skeletal muscle developmental biology. Biomed. Pharmacother. 162, 114631 (2023).
De Micheli, A. J., Spector, J. A., Elemento, O. & Cosgrove, B. D. A reference single-cell transcriptomic atlas of human skeletal muscle tissue reveals bifurcated muscle stem cell populations. Skelet. Muscle 10, 19 (2020).
Giordani, L. et al. High-dimensional single-cell cartography reveals novel skeletal muscle-resident cell populations. Mol. Cell 74, 609–621.e606 (2019).
Cho, D. S. & Doles, J. D. Single cell transcriptome analysis of muscle satellite cells reveals widespread transcriptional heterogeneity. Gene 636, 54–63 (2017).
Dell’Orso, S. et al. Single cell analysis of adult mouse skeletal muscle stem cells in homeostatic and regenerative conditions. Development 146, dev174177 (2019).
Barruet, E. et al. Functionally heterogeneous human satellite cells identified by single cell RNA sequencing. eLife 9, e51576 (2020).
Xu, D. et al. Single-cell RNA-sequencing provides insight into skeletal muscle evolution during the selection of muscle characteristics. Adv. Sci. 10, e2305080 (2023).
De Micheli, A. J. et al. Single-cell analysis of the muscle stem cell hierarchy identifies heterotypic communication signals involved in skeletal muscle regeneration. Cell Rep. 30, 3583–3595.e3585 (2020).
Qin, T. et al. Single-cell RNA-seq reveals novel mitochondria-related musculoskeletal cell populations during adult axolotl limb regeneration process. Cell Death Differ. 28, 1110–1125 (2021).
Uapinyoying, P. et al. Single-cell transcriptomic analysis of the identity and function of fibro/adipogenic progenitors in healthy and dystrophic muscle. iScience 26, 107479 (2023).
Fitzgerald, G. et al. MME+ fibro-adipogenic progenitors are the dominant adipogenic population during fatty infiltration in human skeletal muscle. Commun. Biol. 6, 111 (2023).
Negroni, E. et al. Muscle fibro-adipogenic progenitors from a single-cell perspective: focus on their “virtual” secretome. Front. Cell Dev. Biol. 10, 952041 (2022).
Oprescu, S. N., Yue, F., Qiu, J., Brito, L. F. & Kuang, S. Temporal dynamics and heterogeneity of cell populations during skeletal muscle regeneration. iScience 23, 100993 (2020).
Ma, L. et al. Single-cell RNA-seq reveals novel interaction between muscle satellite cells and fibro-adipogenic progenitors mediated with FGF7 signalling. J. Cachexia Sarcopenia Muscle 15, 1388–1403 (2024).
McKellar, D. W. et al. Large-scale integration of single-cell transcriptomic data captures transitional progenitor states in mouse skeletal muscle regeneration. Commun. Biol. 4, 1280 (2021).
Kedlian, V. R. et al. Human skeletal muscle aging atlas. Nat. Aging 4, 727–744 (2024).
Voisin, S. et al. Meta-analysis of genome-wide DNA methylation and integrative omics of age in human skeletal muscle. J. Cachexia Sarcopenia Muscle 12, 1064–1078 (2021).
Dos Santos, M. et al. Opposing gene regulatory programs governing myofiber development and maturation revealed at single nucleus resolution. Nat. Commun. 14, 4333 (2023).
Kimmel, J. C., Hwang, A. B., Scaramozza, A., Marshall, W. F. & Brack, A. S. Aging induces aberrant state transition kinetics in murine muscle stem cells. Development 147, dev183855 (2020).
Kurland, J. V. et al. Aging disrupts gene expression timing during muscle regeneration. Stem Cell Rep. 18, 1325–1339 (2023).
Krasniewski, L. K. et al. Single-cell analysis of skeletal muscle macrophages reveals age-associated functional subpopulations. eLife 11, e77974 (2022).
Cameron, A. et al. Identification of underexplored mesenchymal and vascular-related cell populations in human skeletal muscle. Am. J. Physiol. Cell Physiol. 323, C1586–C1600 (2022).
Proietti, D. et al. Activation of skeletal muscle-resident glial cells upon nerve injury. JCI Insight 6, e143469 (2021).
Barthelson, R. A., Lambert, G. M., Vanier, C., Lynch, R. M. & Galbraith, D. W. Comparison of the contributions of the nuclear and cytoplasmic compartments to global gene expression in human cells. BMC Genom. 8, 340 (2007).
Petrany, M. J. et al. Single-nucleus RNA-seq identifies transcriptional heterogeneity in multinucleated skeletal myofibers. Nat. Commun. 11, 6374 (2020).
Kim, M. et al. Single-nucleus transcriptomics reveals functional compartmentalization in syncytial skeletal muscle cells. Nat. Commun. 11, 6375 (2020).
Dos Santos, M. et al. Single-nucleus RNA-seq and FISH identify coordinated transcriptional activity in mammalian myofibers. Nat. Commun. 11, 5102 (2020).
Chau, J. et al. Relationship of DUX4 and target gene expression in FSHD myocytes. Hum. Mutat. 42, 421–433 (2021).
Solovyeva, E. M. et al. New insights into molecular changes in skeletal muscle aging and disease: differential alternative splicing and senescence. Mech. Ageing Dev. 197, 111510 (2021).
Kann, A. P. & Krauss, R. S. Multiplexed RNAscope and immunofluorescence on whole-mount skeletal myofibers and their associated stem cells. Development 146, dev179259 (2019).
Young, L. V. et al. Muscle injury induces a transient senescence-like state that is required for myofiber growth during muscle regeneration. FASEB J. 36, e22587 (2022).
Stec, M. J. et al. A cellular and molecular spatial atlas of dystrophic muscle. Proc. Natl. Acad. Sci. USA 120, e2221249120 (2023).
Coulis, G. et al. Single-cell and spatial transcriptomics identify a macrophage population associated with skeletal muscle fibrosis. Sci. Adv. 9, eadd9984 (2023).
Ruoss, S. et al. Spatial transcriptomics tools allow for regional exploration of heterogeneous muscle pathology in the pre-clinical rabbit model of rotator cuff tear. J. Orthop. Surg. Res. 17, 440 (2022).
Williams, K., Yokomori, K. & Mortazavi, A. Heterogeneous skeletal muscle cell and nucleus populations identified by single-cell and single-nucleus resolution transcriptome assays. Front. Genet. 13, 835099 (2022).
Crist, C. G., Montarras, D. & Buckingham, M. Muscle satellite cells are primed for myogenesis but maintain quiescence with sequestration of Myf5 mRNA targeted by microRNA-31 in mRNP granules. Cell Stem Cell 11, 118–126 (2012).
Hostrup, M. et al. High-intensity interval training remodels the proteome and acetylome of human skeletal muscle. eLife 11, e69802 (2022).
Mucha, O. et al. Proteome profiling of the dystrophic. Biomolecules 13, 1648 (2023).
Szczerbinski, L. et al. Metabolomic profile of skeletal muscle and its change under a mixed-mode exercise intervention in progressively dysglycemic subjects. Front. Endocrinol. 12, 778442 (2021).
Deshmukh, A. S. et al. Deep muscle-proteomic analysis of freeze-dried human muscle biopsies reveals fiber type-specific adaptations to exercise training. Nat. Commun. 12, 304 (2021).
Hunt, L. C. et al. Integrated genomic and proteomic analyses identify stimulus-dependent molecular changes associated with distinct modes of skeletal muscle atrophy. Cell Rep. 37, 109971 (2021).
Ubaida-Mohien, C. et al. Discovery proteomics in aging human skeletal muscle finds change in spliceosome, immunity, proteostasis and mitochondria. eLife 8, e49874 (2019).
Kallabis, S. et al. High-throughput proteomics fiber typing (ProFiT) for comprehensive characterization of single skeletal muscle fibers. Skelet. Muscle 10, 7 (2020).
Melby, J. A. et al. High sensitivity top-down proteomics captures single muscle cell heterogeneity in large proteoforms. Proc. Natl. Acad. Sci. USA 120, e2222081120 (2023).
Murgia, M. et al. Protein profile of fiber types in human skeletal muscle: a single-fiber proteomics study. Skelet. Muscle 11, 24 (2021).
Murgia, M. et al. Single muscle fiber proteomics reveals unexpected mitochondrial specialization. EMBO Rep. 16, 387–395 (2015).
Borok, M. et al. Progressive and coordinated mobilization of the skeletal muscle niche throughout tissue repair revealed by single-cell proteomic analysis. Cells 10, 744 (2021).
Maerkens, A. et al. New insights into the protein aggregation pathology in myotilinopathy by combined proteomic and immunolocalization analyses. Acta Neuropathol. Commun. 4, 8 (2016).
Olie, C. S. et al. The metabolic landscape in chronic rotator cuff tear reveals tissue-region-specific signatures. J. Cachexia Sarcopenia Muscle 13, 532–543 (2022).
Dowling, P., Swandulla, D. & Ohlendieck, K. Mass spectrometry-based proteomic technology and its application to study skeletal muscle cell biology. Cells 12, 2560 (2023).
Acknowledgements
This work was supported by two DoD grants (HT94252310534 to R.J.T. and HT94252310519 to C.M.K.) and the following NIH/NIAMS grants: R01 grants (AR078035 and AR076900 to C.L.; AG069401 and AG067698 to L.Q.; AI186118, HD112474, and HD107034 to R.J.T.; AR076325 and AR071967 to C.M.K.; AR080902 and AR072999 to F.G.; AR074441 and AR077678 to S.Y.T.; AR082667 and AR077527 to A.E.L.; AR083900, AR075860 and AR077616 to J.S.), R21 grants (AR077226 to J.S.; AR083217 to A.E.L.; AR081517 to S.Y.T.), a T32 grant (HD007434 to D.R.K.) and P30 center grants (AR074992 and AR073752).
Author information
Authors and Affiliations
Contributions
Manuscript preparation contributions: cartilage—X.L., L.F., R.Z., C.L., C.W., and J.S.; synovium—F.G.; bone marrow—L.Y. and L.Q.; bone callus—X.L., L.F., and J.S.; bone cells—R.J.T. and C.M.K.; spine—S.W.C. and S.Y.T.; tendon—S.M. and A.E.L.; muscle—D.R.K. and G.A.M. J.S. designed the overall structure of the review and finalized the manuscript. C.L., C.W., F.G., L.Q., R.J.T., C.M.K., S.Y.T., A.E.L., G.A.M., and J.S. led their respective sections of the review.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, X., Fang, L., Zhou, R. et al. Current cutting-edge omics techniques on musculoskeletal tissues and diseases. Bone Res 13, 59 (2025). https://doi.org/10.1038/s41413-025-00442-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41413-025-00442-z
