
Abstract
Germline mutations in MBD4, which, like MUTYH and NTHL1, encodes a glycosylase of the DNA based excision repair system, cause an autosomal recessive syndrome characterised by increased risk of acute myeloid leukaemia, gastrointestinal polyposis, colorectal cancer (CRC) and, to a lesser extent, uveal melanoma and schwannomas. To better define the phenotypic spectrum and tumour molecular features associated with biallelic MBD4-associated cancer predisposition, and study if heterozygous variants are associated with gastrointestinal tumour predisposition, we evaluated germline MBD4 status in 728 patients with CRC, polyposis, and other suggestive phenotypes (TCGA and in-house cohorts). Eight CRC patients carried rare homozygous or heterozygous germline variants in MBD4. The information gathered on mode of inheritance, variant nature, functional effect of the variant, and tumour mutational characteristics suggested that none of the patients included in the study had an MBD4-associated hereditary syndrome and that the heterozygous variants identified were not associated with the disease.
Similar content being viewed by others


Germline biallelic pathogenic variants in the genes encoding the DNA base excision repair (BER) glycosylases MUTYH, NTHL1 and MBD4 cause recessive hereditary cancer syndromes characterised by the presence of gastrointestinal adenomatous polyposis and increased risk of colorectal cancer (CRC) and other tumour types [1,2,3]. The associated syndromes differ in prevalence, tumour spectrum, and type of mutations accumulated in the tumours, consequence of the glycosylase-specific defective DNA repair (Table 1).
The MBD4-associated neoplasia syndrome (MANS) is a rare recessive condition. Considering the reported cases (11 carriers in 8 families) [2, 4,5,6], the MANS-associated phenotype is characterised by acute myeloid leukaemia (AML) or myelodysplastic syndrome (MDS) [7/11 carriers; mean age at diagnosis 35 (range: 30–49)]; colorectal polyposis [9/11 carriers] diagnosed in adulthood (in their 30s), with variable expressivity, and associated with increased CRC risk [2/11 carriers]; schwannomas [3/11 carriers]; and uveal melanoma (UVM) [2/11 carriers] (Table S1). Other phenotypes observed in biallelic carriers include papillary thyroid cancer, lymphoma, meningioma, breast cancer, ovarian germ cell tumours, and other benign lesions including upper gastrointestinal polyps, making this a multi-tumour syndrome resembling the NTHL1 tumour syndrome [7] but with different extracolonic tumour spectrum. Based on reported phenotypes, clinical surveillance in MANS should focus on the high AML risk and gastrointestinal manifestations [2]. Due to recurrence of UVM and schwannomas, routine ophthalmological (eye) exams [8] and monitoring of schwannomas may be recommended. Based on response of MBD4-deficient UVMs [9,10,11], MANS tumours may be considered good candidates for immune checkpoint inhibitor-based therapies.
Sequencing data from MANS-associated colorectal adenomas revealed a somatic mutational spectrum enriched in CpG>TpG mutations, consequence of the failure to repair G:T mismatches resulting from deamination of 5’-methylcytosine. This generates a mutational signature, SBS96, that largely resembles SBS1 [2, 12], present in tumours with either inherited or acquired MBD4 mutations.
Heterozygous germline MBD4 pathogenic variants are associated with genetic predisposition to UVM. In these cases, the associated tumours show loss of the wildtype MBD4 allele and, like in MANS, elevated mutation burden enriched in CpG>TpG mutations (signature SBS96) [9, 10, 13]. Palles et al., identified four individuals heterozygous for loss-of-function MBD4 variants among 1,611 patients affected with ≥10 colorectal adenomas, familial and/or early onset CRC, or CRC in combination with other tumours. Adenomas developed by the heterozygotes did not show the typical mutational spectrum of MBD4-deficient tumours [2]. Nevertheless, these results were not conclusive of an association of heterozygous pathogenic variants with increased CRC risk or lack of it.
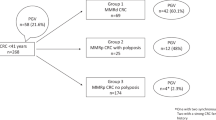
To expand the knowledge about MANS and the role of heterozygous MBD4 variants in CRC and polyposis predisposition, we studied the germline mutational status of MBD4 in patients affected with CRC (n = 543; data source: TCGA), adenomatous polyposis (n = 177; source: Hereditary Cancer Program, Catalan Institute of Oncology, IDIBELL), or tumours that fit the syndrome’s phenotypic spectrum (n = 8; source: Hereditary Cancer Program, Catalan Institute of Oncology, IDIBELL). Somatic analysis of the TCGA CRC cohort had been performed in two pan-cancer analyses that considered the identification of loss-of-function MBD4 variants and/or MBD4-deficient tumours [4, 9]. Sequencing of MBD4 coding exons and exon-intron boundaries was performed in the in-house cohorts with direct automated (Sanger) sequencing. Variants with a population minor allele frequency (MAF) <0.5% were considered. Description of patients and methods is included in Supplementary Materials and Methods. Review of MANS patients reported in the literature and re-analysis of the sequencing data available from five MBD4-deficient tumours, including three MANS-associated adenomas [3, 5,6,7] and two TCGA cancers [9], were performed.
No loss-of-function germline MBD4 variants were detected in any of the 728 patients studied. No rare (MAFgnomAD_non-cancer <0.5%) variants were identified in either the 177 adenomatous polyposis patients studied or the eight probands with MANS-suspected phenotypes, confirming the extremely low frequency of MBD4 pathogenic variants as cause of polyposis and the rarity of MANS. Eight of the 543 TCGA CRC patients analysed harboured rare germline missense variants in MBD4: one in homozygosis and the other seven in heterozygosis. Only two of the five variants identified, c.368C>T;p.(Ser123Leu) and c.1400A>G;p.(Asn467Ser), had REVEL [14] pathogenicity scores >0.500 (Table 2).
MBD4 c.181T>C;p.(Cys61Arg), predicted benign, was identified in homozygosis in a patient diagnosed with a sigmoid colon cancer at age 68 (TCGA-D5-6924), and in heterozygosity in a 69-year-old CRC patient (TCGA-AA-3685). Two additional homozygous individuals, one of them a >80-year-old cancer patient, were reported in the Genome Aggregation Database (gnomAD v.2.1.1). No effect of this variant on MBD4 glycosylase activity was observed in a previous study [15].
An Asian patient diagnosed with CRC at age 46 (TCGA-CA-6718) resulted heterozygous for a predicted pathogenic variant, c.368C>T;p.(Ser123Leu), relatively common in individuals of East Asian origin (MAF: 0.5%; source: gnomAD). Three patients, diagnosed with CRC in their 70 s (TCGA-AA-3869, TCGA-AA-3950, TCGA-DM-A28K), were heterozygous for c.1400A>G;p.(Asn467Ser) (REVEL: 0.540; MAFgnomAD_non-cancer: 0.17%; ~0.3% in Europeans). The presence of two homozygous individuals in the gnomAD v.2.1.1. non-cancer dataset, together with the experimental evidence showing no effect on MBD4 glycosylase activity [13], suggest a non-pathogenic nature for c.1400A>G. Two additional variants, predicted benign, were identified in the other two CRC patients (Table 2). A somatic MBD4 alteration (potential second hit) was detected in the CRC of the c.368C>T;p.Ser123Leu heterozygote but not in the other patients’ tumours.
MDB4-deficient tumours accumulate relatively high number of somatic mutations due to defective DNA repair, with an average tumour mutational burden (TMB) of ~10 mutations per Mb (mut/Mb) (range: 2.7–24.2) (Tables S1 and S2). Exome sequencing data was obtained from TCGA for the CRCs developed by the eight heterozygous or homozygous MBD4 variant carriers identified. The highest TMBs were identified in the CRCs developed by the c.368C>T;p.(Ser123Leu) and c.1160C>T;p.(S387L) heterozygotes (TCGA-CA-6718 ~ 100 mut/Mb; TCGA-D5-6530 24.4 mut/Mb). These tumours were deficient in polymerase proofreading and MMR, respectively, which caused the elevated TMB as showed the associated tumour mutational signatures (Table S3). The other six tumours had TMBs <5 mut/Mb (Table 2).
An agnostic analysis of tumour mutational signatures using FitMS through the web-based application Signal (all Cancer Reference Signatures, not selecting by tumour type) [16] was performed in eight TCGA CRCs belonging to the MBD4 variant carriers herein identified, five previously reported MBD4-deficient tumours (three MANS adenomas, one MBD4-deficient TCGA UVM, and one MBD4-deficient TCGA glioblastoma), and 43 randomly selected TCGA CRCs. While the five MBD4-deficient tumours reached SBS96 contributions of ~100% (range: 97–100%), all six polymerase proofreading-proficient and MMR-proficient CRCs from MBD4 variant carriers had SBS96 contributions of 32% to 69%; similar to the profiles of the 43 TCGA CRCs (Fig. S1 and Table S3). Due to the similarity between SBS96 and SBS1, the latter ubiquitously detected in tumours and associated with aging, we next analysed solely those two signatures. While the five MBD4-deficient tumours had 100% SBS96 contribution (0% SBS1), the eight TCGA CRCs developed by the MBD4 variant carriers preferentially harboured SBS1 over SBS96 (SBS1 contribution: 73-100%; SBS96 contribution: 0% in all except for one MMR-deficient CRC with 17% SBS96 contribution), confirming no MDB4 deficiency (Fig. S2 and Table S3).
Classification of the MBD4 variants identified in this study following the ACMG/AMP classification guidelines [17] and taking into consideration: presence of homozygous individuals in the gnomAD non-cancer dataset (BS2), in silico pathogenicity predictions (BP4, PP3), effect on MBD4 glycosylase activity (BS3, PS3), and major presence (or not) of tumour SBS96 (over SBS1) when the variant is in homozygosis or there is a clear somatic second hit in MDB4 (BP5, PP4), indicates that c.181T>C;p.Cys61Arg and c.1400A>G;p.Asn467Ser are benign, while the other four are variants of unknown significance (Table 2). Nonetheless, our findings, mostly supported by the tumour mutational spectra, indicate that none of the herein identified variants had an impact on CRC development.
Most germline MBD4 pathogenic variants reported, either biallelic or monoallelic, are loss-of-function variants scattered along the MBD4 sequence (Fig. 1). In-frame deletions and missense pathogenic variants occur at the C-terminal half of the protein, where the glycosylase ___domain for DNA repair is located. A relevant proportion of truncating variants also affect the protein C-terminal part, likely not inducing RNA nonsense-mediated decay but affecting the integrity or structural stability of the glycosylase ___domain, highlighting the relevance of this region in the associated cancer predisposition syndromes. An in vitro assay that evaluates MBD4 glycosylase activity [15, 18] may be used to characterise variants of unknown significance in this gene, particularly when tumour sequencing data are unavailable or non-informative.

Homozygous and compound heterozygous variants identified in MANS patients (Table S1) are represented in the upper part of the figure, while heterozygous variants associated with monoallelic cancer predisposition (Table S4) are represented in the bottom. Black dots represent truncating mutations, including nonsense, frameshift deletions or insertions and splice-site variants. Grey dots indicate missense variants and in-frame deletions or insertions. Position of the protein domains: Methyl-CpG binding ___domain (MBD), 80–149 amino acids; DNA glycosylase, 461–535 amino acids.
Due to the extreme rarity of MANS, as confirmed in our study, the inclusion of MBD4 in multi-gene panels designed to study cancer predisposition in the clinical practice will likely not increase the diagnostic yield. Nevertheless, if not included in the diagnostic gene panel, we recommend performing individual analysis of MBD4 when either phenotypic (patients with any combination of MDS/AML, polyposis, CRC, UVM and/or schwannomas) or tumour molecular characteristics (~100% SBS96 contribution) suggest an MBD4-related genetic aetiology. Major contribution of tumour SBS96 (~100%; >90%) may be used as pathogenicity-supporting evidence for variant interpretation.
In conclusion, our negative results confirm that MANS is an extremely infrequent syndrome and show that the few rare heterozygous (missense) MBD4 germline variants identified in our study are not driver carcinogenic events leading to tumour MBD4 deficiency.
Data availability
Data supporting the reported results may be found in the article, supplementary material and public repositories (TCGA, gnomAD).
References
Weren RD, Ligtenberg MJ, Geurts van Kessel A, De Voer RM, Hoogerbrugge N, Kuiper RP. NTHL1 and MUTYH polyposis syndromes: two sides of the same coin? J Pathol. 2018;244:135–42.
Palles C, West HD, Chew E, Galavotti S, Flensburg C, Grolleman JE, et al. Germline MBD4 deficiency causes a multi-tumor predisposition syndrome. Am J Hum Genet. 2022;109:953–60.
Krokan HE, Bjørås M. Base excision repair. Cold Spring Harb Perspect Biol. 2013;5:a012583.
Sanders MA, Chew E, Flensburg C, Zeilemaker A, Miller SE, Al Hinai AS, et al. MBD4 guards against methylation damage and germ line deficiency predisposes to clonal hematopoiesis and early-onset AML. Blood. 2018;132:1526–34.
Griffin BL, Majhail NS, Lesmana H. Germline Biallelic loss in MBD4 leading to early onset AML with hyper-mutator genomic signature. Blood. 2021;138:3385.
Blombery P, Ryland GL, Fox LC, Stark Z, Wall M, Jarmolowicz A, et al. Methyl-CpG binding ___domain 4, DNA glycosylase (MBD4)-associated neoplasia syndrome associated with a homozygous missense variant in MBD4: Expansion of an emerging phenotype. Br J Haematol. 2022;198:196–9.
Kuiper RP, Nielsen M, De Voer RM, Hoogerbrugge N. NTHL1 Tumor Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews ® [Internet]. Seattle (WA): University of Washington; 1983–2020.
Solnik M, Paduszyńska N, Czarnecka AM, Synoradzki KJ, Yousef YA, Chorągiewicz T, et al. Imaging of uveal melanoma-current standard and methods in development. Cancers (Basel). 2022;14:3147.
Rodrigues M, Mobuchon L, Houy A, Fiévet A, Gardrat S, Barnhill RL, et al. Outlier response to anti-PD1 in uveal melanoma reveals germline MBD4 mutations in hypermutated tumors. Nat Commun. 2018;9:1866.
Johansson PA, Stark A, Palmer JM, Bigby K, Brooks K, Rolfe O, et al. Prolonged stable disease in a uveal melanoma patient with germline MBD4 nonsense mutation treated with pembrolizumab and ipilimumab. Immunogenetics. 2019;71:433–6.
Saint-Ghislain M, Derrien AC, Geoffrois L, Gastaud L, Lesimple T, Negrier S, et al. MBD4 deficiency is predictive of response to immune checkpoint inhibitors in metastatic uveal melanoma patients. Eur J Cancer. 2022;173:105–12.
Degasperi A, Zou X, Dias Amarante T, Martinez-Martinez A, Koh GCC, Dias JML, et al. Substitution mutational signatures in whole-genome–sequenced cancers in the UK population. Science. 2022;376:abl9283.
Derrien AC, Rodrigues M, Eeckhoutte A, Dayot S, Houy A, Mobuchon L, et al. Germline MBD4 mutations and predisposition to uveal melanoma. J Natl Cancer Inst. 2021;113:80–7.
Pejaver V, Byrne AB, Feng BJ, Pagel KA, Mooney SD, Karchin R, et al. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am J Hum Genet. 2022;109:2163–77.
Sjolund AB, Senejani AG, Sweasy JB. MBD4 and TDG: multifaceted DNA glycosylases with ever expanding biological roles. Mutat Res. 2013;743-744:12–25.
Degasperi A, Amarante TD, Czarnecki J, Shooter S, Zou X, Glodzik D, et al. A practical framework and online tool for mutational signature analyses show inter-tissue variation and driver dependencies. Nat Cancer. 2020;1:249–63.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Tricarico R, Cortellino S, Riccio A, Jagmohan-Changur S, Van der Klift H, Wijnen J, et al. Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis. Oncotarget. 2015;6:42892–904.
Acknowledgements
We thank Adriana Costal and Laura Berrocal (Hereditary Cancer Program, Catalan Institute of Oncology) for technical support.
Funding
This research was funded by the Spanish Ministry of Science and Innovation (Agencia Estatal de Investigación), co-funded by FEDER funds a way to build Europe [SAF2016-80888-R (LV), PID2020-112595RB-I00 (LV), PID2019-111254RB-I00 (GC)]; Instituto de Salud Carlos III [CIBERONC CB16/12/00234]; Government of Catalonia [AGAUR 2021SGR01112, CERCA Programme for institutional support]; Scientific Foundation “Asociación Española Contra el Cáncer” [AECC Investigador (MT)]; Marie Skłodowska-Curie Individual Fellowship [Grant agreement No. 897064 (NG-A)].
Author information
Authors and Affiliations
Contributions
LV conceived and supervised the project, analysed and interpreted the obtained results, and wrote the manuscript. MT, NG-A and GA conceived and carried out the experiments. MT, NG-A, SG-M, JV-E and JMP obtained and analysed data. JB and GC provided samples, clinical information, and resources. All authors were involved in writing and/or critically reviewing the manuscript and had final approval of the submitted and published versions.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The study received the approval of the IDIBELL ethics committee (Reference number: PR247/15), and all patients studied signed an informed consent. The study was performed in accordance with the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Terradas, M., Gonzalez-Abuin, N., García-Mulero, S. et al. MBD4-associated neoplasia syndrome: screening of cases with suggestive phenotypes. Eur J Hum Genet 31, 1185–1189 (2023). https://doi.org/10.1038/s41431-023-01418-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-023-01418-5
This article is cited by
-
Genetics, genomics and clinical features of adenomatous polyposis
Familial Cancer (2025)
-
Genetic predisposition to polyposis syndromes
Clinical and Translational Oncology (2025)
-
Expanding what we know about rare genetic diseases
European Journal of Human Genetics (2023)
