
Abstract
Anomalous pulmonary venous return (APVR) frequently occurs with other congenital heart defects (CHDs) or extra-cardiac anomalies. While some genetic causes have been identified, the optimal approach to genetic testing in individuals with APVR remains uncertain, and the etiology of most cases of APVR is unclear. Here, we analyzed molecular data from 49 individuals to determine the diagnostic yield of clinical exome sequencing (ES) for non-isolated APVR. A definitive or probable diagnosis was made for 8 of those individuals yielding a diagnostic efficacy rate of 16.3%. We then analyzed molecular data from 62 individuals with APVR accrued from three databases to identify novel APVR genes. Based on data from this analysis, published case reports, mouse models, and/or similarity to known APVR genes as revealed by a machine learning algorithm, we identified 3 genes—EFTUD2, NAA15, and NKX2-1—for which there is sufficient evidence to support phenotypic expansion to include APVR. We also provide evidence that 3 recurrent copy number variants contribute to the development of APVR: proximal 1q21.1 microdeletions involving RBM8A and PDZK1, recurrent BP1-BP2 15q11.2 deletions, and central 22q11.2 deletions involving CRKL. Our results suggest that ES and chromosomal microarray analysis (or genome sequencing) should be considered for individuals with non-isolated APVR for whom a genetic etiology has not been identified, and that genetic testing to identify an independent genetic etiology of APVR is not warranted in individuals with EFTUD2-, NAA15-, and NKX2-1-related disorders.
Similar content being viewed by others


Introduction
Anomalous pulmonary venous return (APVR) is a spectrum of congenital heart defects (CHDs) that involves the direct or indirect drainage of some or all of the pulmonary veins into the right atrium rather than the left atrium [1, 2]. In cases of partial anomalous pulmonary venous return (PAPVR) at least one, but not all, of the pulmonary veins fails to connect to the left atrium. In cases of total anomalous pulmonary venous return (TAPVR), all of the pulmonary veins fail to connect to the left atrium. PAPVR and TAPVR can be further described as supra-cardiac, cardiac, infracardiac, and mixed based on their anomalous connections.
APVR affects approximately 1 in 10,000 newborns and accounts for up to 2.3% of all CHDs [3, 4]. TAPVR with additional CHDs is seen in approximately 23% of cases [5]. While PAPVR may rarely present as an isolated anomaly, it occurs more commonly with other CHDs, most often an atrial septal defect [1]. Individuals with TAPVR or PAPVR may also have non-cardiac anomalies. Up to 35% of individuals with APVR are described as having heterotaxy, which is defined by the abnormal lateralization or abnormal arrangement of organs in the chest and/or abdomen [6]. In this paper, we have defined non-isolated APVR as having APVR and at least one other CHD, a non-cardiac birth defect, or a neurodevelopmental disorder.
For a subset of non-isolated APVR cases, a gross chromosomal disorder, a microdeletion/microduplication, or a single gene disorder can be identified as the cause. Examples include Turner syndrome (45,X), cat eye syndrome (MIM# 115470) caused by a partial tetrasomy of 22q11, and cardiac-urogenital syndrome (MIM# 618280) caused by pathogenic heterozygous variants in MYRF. Identifying a molecular cause for APVR can aid in the development of individualized medical plans and provides the basis for more accurate risk estimations. However, uncertainty regarding the efficacy of clinical genetic testing in individuals with APVR may pose a barrier to genetic testing in some settings, and a lack of knowledge regarding the genes that cause APVR may lead to an extension of the diagnostic odyssey and unwarranted genetic testing for those in whom the causative variant has already been identified.
Here we use data from 49 individuals to determine the diagnostic efficacy of clinical exome sequencing (ES) and data from 62 individuals to identify phenotypic expansions involving APVR.
Subjects and methods
Database analysis and clinical review
From ~20,000 individuals referred to Baylor Genetics (BG) for ES, we identified 49 individuals with non-isolated APVR based on their test indications. Individuals who received a molecular diagnosis based on tests other than ES were excluded from our efficacy analysis with the exception of S10 whose diagnosis of thrombocytopenia-absent radius (TAR) syndrome (MIM# 274000) required identification of a copy number variant (CNV) and a single nucleotide variant (SNV) in the trans configuration.
Coded information was obtained on 39 individuals with APVR who carried small CNVs ( < 2.5 Mb and containing 1-20 protein-coding genes) from the Cytogenomics of Cardiovascular Malformations (CCVM) Consortium Registry [7].
De-identified data from 12 individuals with non-isolated APVR were identified from the DECIPHER database, 4 with SNVs and 8 with small CNVs ( < 2.5 Mb and containing 1-20 protein-coding genes) [8]. Each submitting center was contacted and approved the publication of their patient’s clinical and molecular data. S57 is also listed in the European Cytogeneticists Association Register of Unbalanced Chromosome Aberrations (ECARUCA) database [9].
Classification of SNVs
SNVs reported by BG or listed in the DECIPHER database were reanalyzed and classified as pathogenic, likely pathogenic, variants of uncertain significance (VUS), likely benign, or benign based on the 2015 American College of Medical Genetics and Genomics (ACMG) standards for variant interpretation using currently available data [10].
Determination of diagnostic certainty
The molecular and clinical data of each case were reviewed to determine if a definitive, probable, or provisional diagnosis could be made based on criteria outlined by Scott et al. [11]. Assuming that the phenotypic data provided in the indication were suggestive of a putative diagnosis, and the inheritance pattern was consistent, a definitive diagnosis was made if the individual carried a pathogenic variant(s) in a causative gene. A probable diagnosis was made if the individual carried 1) a likely pathogenic variant in an autosomal dominant gene or an X-linked gene in a male, 2) a pathogenic variant and a likely pathogenic variant or VUS in trans, or two likely pathogenic variants in trans, in an autosomal recessive gene, or 3) a mosaic pathogenic or likely pathogenic variant in an autosomal dominant gene or an X-linked gene in a male. A provisional diagnosis was made if the individual carried 1) only a single pathogenic variant in a autosomal recessive gene, 2) two VUSs in trans or a likely pathogenic variant and a VUS in trans in an autosomal recessive gene, 3) a heterozygous pathogenic variant in an X-linked gene in a female, or 4) only a VUS(s) in a causative gene.
When determining the diagnostic efficacy of clinical ES, we used the highest diagnostic certainty associated with the SNVs identified in each subject.
Classification of CNVs
CNVs were reanalyzed and classified as pathogenic, likely pathogenic, variants of uncertain significance (VUS), likely benign, benign, or susceptibility locus based on the 2020 American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen) technical standards for the interpretation and reporting of constitutional copy-number variants [12]. Single gene deletions were called according to the 2015 ACMG standards for the variant interpretation using currently available data [10].
All CNVs coordinates reported in this manuscript are defined based on hg38.
Literature and database searches
We searched the OMIM database (https://www.omim.org/), the Mouse Genome Informatics database (MGI; http://www.informatics.jax.org/), and the literature for reports in which each candidate gene, their associated genetic disorder(s), or their mouse homologs were associated with the development of APVR or other heart defects [13, 14].
Machine learning
We used a previously published machine learning algorithm to rank the similarity of all RefSeq genes to a training set of 35 genes known to cause APVR in humans and the human homologs of genes that cause APVR in mice—ACVR2B, ANKRD1, ANKS6, B9D2, BMPR2, CFAP53, CFC1, CHTOP, CITED2, DHCR24, DHCR7, DNAH5, FOXC2, GDF1, IFT88, MAPKAPK5, MED13L, MEGF8, MMP21, MYRF, NODAL, NTF3, NUP188, PDGFRA, PDZK1, PITX2, RPGRIP1L, SLC20A2, SMAD2, TBX1, TBX5, TMEM260, WBP11, WT1, ZIC3—based on data from Gene Ontology (GO), the Mouse Genome Database (MGI), the Protein Interaction Network Analysis (PINA) platform, the GeneAtlas expression distribution, and transcription factor binding and epigenetic histone modifications data from NIH Roadmap Epigenomics Mapping Consortium [15, 16].
Leave-one-out cross-validation studies were used to test the performance of our machine learning algorithm [15, 16]. The algorithm’s effectiveness is represented by the area between the receiver operating characteristic (ROC) style curves in Fig. 1A and the diagonal line that represents random chance results. This demonstrated that our scoring algorithm is able to identify known APVR genes more effectively than random chance.

A The machine learning algorithm was trained using 35 genes known to cause APVR in humans and the human homologs of genes that cause APVR in mice. Receiver operating characteristic (ROC) style curves were generated based on a leave-one-out validation study analysis performed for each knowledge source: Gene Ontology (GO), Mouse Genome Database (MGI), Protein Interaction Network Analysis (PINA), GeneAtlas expression distribution (Exp), and transcription factor binding (TF) and epigenetic histone modifications data (Epi) from NIH Roadmap Epigenomics Mapping Consortium. The black curve represents an omnibus score whose positive deviation indicates that our algorithm can identify genes in the training set more effectively than chance (diagonal line). After validation, ARMs-specific pathogenicity scores were calculated for all RefSeq genes. B Box plot showing the algorithmically generated APVR-specific pathogenicity scores for APVR training genes.
After validating the algorithm, APVR-specific pathogenicity scores were generated for all RefSeq genes based on the percentile rank of an omnibus score produced using fit data from all knowledge sources. By definition, the APVR-specific pathogenicity scores for all RefSeq genes range from 0 to 100%, with a median score of 50%. In contrast, the APVR-specific pathogenicity scores of the 35 genes in the training set had a range of 52.3%–100% with a median score of 98.2%, with CFAP53 (52.3%), B9D2 (56.9%), MYRF (58.7%) WBP11 (59.5%), and MED13L (60.8%) being outliers (Fig. 1B).
Statistical analyses
Two-tailed Fisher’s exact tests were performed using a 2×2 contingency table calculator available through GraphPad QuickCalcs (https://www.graphpad.com/quickcalcs/contingency1). P-values of < 0.05 were considered significant. Box plots were generated using the Alcula.com Statistical Calculator: Box Plot program (http://www.alcula.com/calculators/statistics/box-plot/).
Results
Diagnostic efficacy of clinical ES
We searched a database of ~20,000 individuals who were referred for clinical ES and identified 49 individuals with non-isolated APVR based on phenotypes included in their indications for testing. ES provided a definitive diagnosis for 7 individuals and a probable diagnosis for 1 individual yielding a molecular diagnostic efficacy rate of 16.3% (8/49). Provisional diagnoses were made in 3 additional cases. If included, the molecular diagnostic efficacy rate of ES for non-isolated APVR increased to 22.4% (11/49). Molecular and clinical data for all subjects in which a definitive, probable, or provisional diagnosis was made (S1-S11) are shown in Supplemental Table S1.
Of the 49 individuals with non-isolated APVR, 15 (30.6%) were also described as having heterotaxy. For the heterotaxy sub-cohort, the ES diagnostic yield was 20% (3/15) when only individuals with a definitive or probable diagnosis were considered, or 26.7% (4/15) when individuals with a provisional diagnosis were included. These diagnostic yields were not significantly higher than the ES diagnostic yield for individuals not described as having heterotaxy: 14.7% (5/34), p = 0.6869, and 20.5% (7/34), p = 0.7165, respectively.
Of the 49 individuals with non-isolated APVR, 29 (59.2%) were described as having TAPVR, 15 (30.6%) were described as having PAPVR, and 5 (10.2%) had unspecified APVR. No statistically significant differences were found between the diagnostic rates among individuals with TAPVR vs. PAPVR (3/29, 10.3% vs. 4/15, 26.7%; p = 0.2067) even when provisional diagnoses were included (5/29, 17.2% vs. 4/15, 26.7%; p = 0.4640). Similarly, no difference was found in the diagnostic rates between individuals with TAPVR with heterotaxy and those without heterotaxy (2/12, 16.7% vs 1/17, 5.9%; p = 0.5534), even when provisional diagnoses were included (3/12, 25% vs. 2/17, 11/7%; p = 0.6221). The same was true for the diagnostic rates between individuals with PAPVR with heterotaxy and those without heterotaxy (1/1, 100% vs. 3/14, 21.4%; p = 0.2667). Last, no difference was found between the diagnostic yields of the TAPVR and PAPVR sub-cohorts with heterotaxy (2/12, 16.7% vs 1/1, 100%; p = 0.2308) or without heterotaxy (1/17, 5.9% vs. 3/14, 21.4%; p = 0.3041), even when provisional diagnoses were included (3/12, 25% vs. 1/1, 100%; p = 0.2308 and 2/17, 11.8% vs. 3/14, 21.4%; p = 0.6358).
Phenotypic expansions involving APVR
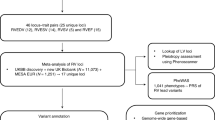
To identify phenotypic expansions involving APVR, we analyzed the molecular data from 62 individuals with non-isolated APVR identified from the BG clinical database (S1-S11), the CCVM Consortium Registry (S12-S50), and the DECIPHER database (S51-S62) (Fig. 2). The SNVs and CNVs reported in these individuals are summarized in Supplemental Table S1 and Supplemental Table S2, respectively. Within this combined cohort, only S6 carried an SNV in a gene clearly associated with APVR in humans—a de novo likely pathogenic c.3118 A > G, p.(R1040G) variant in MYRF. This subject was previously published, and MYRF has clearly been associated with scimitar syndrome, a rare variant of PAPVR (MIM# 618280) [17].

A diagram showing the workflow by which phenotypic expansions involving APVR were identified.
The other genes affected by SNVs or CNVs in these individuals were considered candidate APVR genes. For genes affected by microdeletions, we focused on genes that are predicted to have high loss-of-function intolerance (pLI ≥ 0.8 in gnomAD) since they are the most likely to be able to cause APVR when deleted [18]. When all genes with high loss-of-function intolerance in a deleted region were not previously associated with CHD development, we then focused on genes previously associated with CHDs. For genes affected by copy number gains, we focused on genes with high triplosensitivity (pTriplo ≥ 0.8 in DECIPHER) and those predicted to have high loss-of-function intolerance (pLI ≥ 0.8 in gnomAD) whose function may have been disrupted by the duplications based on the presence of one or more breakpoints within the gene [18, 19].
This analysis revealed three genes for which there is sufficient evidence to support a phenotypic expansion including APVR—EFTUD2 (S2, S51, S52, and S59), NKX2-1 (S8), and NAA15 (S7)—based on having been previously associated with APVR in published case reports and/or in mouse models and having a high APVR-specific pathogenicity score ( ≥ 80%). Data about these genes are summarized in Table 1. Data about genes for which there is insufficient evidence to support a phenotypic expansion including APVR but have previously been clearly associated with the development of other forms of CHDs are summarized in Table 2.
CNVs associated with APVR and CHD
We also identified three recurrently deleted or duplicated chromosomal regions in our cohort that have been previously associated with APVR as a typical or atypical cardiac finding. S17 carried a heterozygous 7q11.23 deletion that includes the Williams-Beuren syndrome (MIM# 194050) critical region [20]. S23-S26 and S58 carried heterozygous breakpoint 1 and 2 (BP1-BP2) 15q11.2 deletions (MIM# 615656), and S57 carries a slightly larger deletion that also includes GOLGA6L1. S46 carried a 22q11.1q11.21 duplication that contains the critical region for cat eye syndrome (MIM# 115470). There was insufficient evidence to implicate a specific gene in any of these regions as causative.
To meet criteria for phenotypic expansion, a CNV has to have been previously associated with APVR in published case reports and must contain at least one gene with a high APVR-specific pathogenicity score ( ≥ 80%) that has been previously associated with APVR or other CHD in published case reports or in a mouse model. This analysis revealed two CNVs that met these criteria (Table 3). The first is a 384.4 kb deletion of chromosome 1q21.1 containing RBM8A and PDZK1 carried by S12 (Fig. 3A). S10, who has molecularly confirmed TAR syndrome (MIM# 27400), also carries a 1q21.1 deletion of an unspecified size in trans with a pathogenic c.-21G>A RBM8A variant. The second is a 1.05 Mb deletion of 22q11.21 containing CRKL associated with LCR22-B to LCR22-D central 22q11.2 deletion syndrome carried by S31 (Fig. 3B).

A S10 and S12 carried deletions affecting a region of chromosome 1q21.1. The recurrent deletions associated with TAR syndrome (MIM# 274000) occur between BP2 and BP3 low copy repeat (LCR) regions (shown). S10 has molecularly confirmed TAR syndrome caused by a pathogenic RBM8A sequence in trans with a 1q21.1 deletion of an unspecified size (not shown). S12 had TAPVR and heterotaxy and carried a 384.4 kb deletion. Liu et al. described an individual who had a complex CHD with laterality defects and heterotaxy who carried a deletion that included PDZK1 but not RBM8A [37]. The smallest region of overlap is shown by dashed vertical lines. B S31 had PAPVR and carried a 1.05 Mb central 22q11.21 deletion shown. A mother and her son were previously reported to both have TAPVR and central 22q11.21 deletions [40]. The smallest region of overlap includes CRKL and is shown by dashed vertical lines. C 13 had TAPVR and carried a 555.4 kb deletion of chromosome 2q31.3q32.1. Lalani et al. previously reported an individual with APVR and 625 kb deletion [20]. The smallest region of overlap is shown by dashed vertical lines. Currently there is insufficient evidence to support an association between this region and the development of APVR.
Data from CNVs for which there is currently insufficient evidence to support a phenotypic expansion including APVR but that have previously been clearly associated with the development of CHDs are summarized in Table 4. Among these CNVs, only a 555.4 kb deletion of chromosome 2q31.3q32.1 (chr2:181,915,576-182,470,985) carried by S13 has specifically been associated with APVR (Fig. 3C). This deletion contains 3 protein coding genes ITPRID2, PPP1R1C, and PDE1A. One individual with a 625 kb deletion (chr2:181,573,843-182,199,076) including CERKL, NEUROD1, ITPRID2, PPP1R1C, and PDE1A, and APVR was previously reported [21]. PDE1A has been suggested as the most likely candidate gene in the deleted region for CHD development due to its calmodulin dependence, expression in the heart, and roles in regulating cardiac hypertrophy in animal models [21, 22]. However, we note that PDE1A has a pLI score of 0.01, LOEUF score of 0.53, and an APVR-specific pathogenicity score of 23.1%. Neither ITPRID2 (pLI = 0, LOEUF = 0.55, APVR-specific pathogenicity score = 50.1%) nor PPP1R1C (pLI = 0, LOEUF = 1.74, APVR-specific pathogenicity score = 29.3%) appear to be compelling candidate genes. Hence, if deletion of this region is associated with the development of APVR, it is likely that it is due to the deletion of two or more genes or the disruption of undefined regulator elements.
Discussion
To inform genetic testing workflows and improve the interpretation of genetic testing results for individuals with APVR, it is critical to determine the efficacy of ES in this population and to identify novel APVR genes. With this in mind, we used data from 49 individuals to determine the diagnostic efficacy of clinical ES and data from 62 individuals to identify phenotypic expansions involving APVR.
Diagnostic yield of clinical ES for non-isolated APVR
The diagnostic yield of ES in critically ill newborns with isolated and non-isolated CHD is relatively low at 7.9% and 15.7%, respectively [23]. Similarly, we found that molecular diagnostic yield of ES in individuals with non-isolated APVR was relatively low: 16.3% (8/49) when considering only definitive and probable diagnoses and 22.4% (11/49) when provisional diagnoses were included.
The molecular diagnostic yield in individuals with APVR and heterotaxy was higher than those who did not have heterotaxy, 20% (3/15) vs. 14.7% (5/34), but was not significantly different (p = 0.6869). Liu et al. identified a similar trend, where the diagnostic yield of a 485-gene panel for cardiac malformations was 13.7% (7/51) in fetuses with complex CHDs and heterotaxy and 7% (2/28) for those with complex CHDs without heterotaxy [24]. These data suggest that identifiable genetic factors may play a greater role in the development of APVR and other CHDs in individuals with heterotaxy than in those without heterotaxy, but additional studies will be needed before this can be confirmed.
Although our data provide clear evidence that ES can be utilized to identify a molecular diagnosis in a significant percentage of individuals with non-isolated APVR, our results are limited by the retrospective and de-identified nature of this study. Since the individuals in this study were ascertained from a clinical ES database, our results may also be affected by a referral bias in which a distinct subpopulation of individuals with non-isolated APVR are more likely to be referred for clinical ES. Prospective, multi-institution or clinic-based studies may confirm our findings. Such studies may also allow comparisons between the yields of ES and other genetic tests (i.e., gene panels, chromosomal microarray analysis (CMA), or whole genome sequencing) in individuals with isolated and non-isolated APVR.
We also recognize that limited sample size may have made it impossible to identify differences in the diagnostic yields of TAPVR and PAPVR subcohorts. Such difference may become apparent when larger APVR cohorts are analyzed or in future metanalyses.
Phenotypic expansions involving APVR
APVR associated with EFTUD2 variants
Heterozygous pathogenic variants in EFTUD2 are associated with mandibulofacial dysostosis with microcephaly (MFDM), Guion-Almeida type (MIM# 610536). MFDM is characterized by microcephaly, developmental delay, speech delay and distinctive facial features including midface and malar hypoplasia, micrognathia, microtia, dysplastic ears, and preauricular skin tags. Other features may include cleft palate, choanal atresia, facial asymmetry, cardiac anomalies, short stature, vertebral anomalies, and epilepsy. In our cohort, S2 and S51 had TAPVR and, respectively, carried a de novo pathogenic nonsense variant and a maternally inherited pathogenic frameshift variant predicted to result in nonsense-mediated mRNA decay (NMD) or truncation. S51’s mother had abnormal pinna morphology and a wide nasal bridge. S52 had TAPVR and carried a de novo missense variant classified as likely pathogenic. S59 had PAPVR and carried a de novo pathogenic deletion of exons 7-10 of EFTUD2. Two siblings with TAPVR who were heterozygous for a c.944delG, p.Ser315fs frameshift variant in EFTUD2, predicted to result in loss of function, have been previously described [25]. The identification of 6 individuals with MFDM and APVR combined with EFTUD2’s high APVR-specific pathogenicity score (80.8%) leads us to conclude that individuals who carry deleterious variants in EFTUD2 can present with APVR as part of MFDM.
APVR associated with NAA15 variants
Heterozygous pathogenic variants in NAA15 are associated with intellectual developmental disorder, autosomal dominant 50, with behavioral abnormalities (MIM# 617787). Most individuals reported with heterozygous pathogenic variants in NAA15 have varying levels of neurodevelopmental delays, intellectual disabilities, and/or autism spectrum disorder, but other documented features include dysmorphic facies without a consistent pattern, poor growth, seizures, feeding difficulties, and less commonly, cardiac defects [26]. In our cohort, S7 carried a maternally inherited pathogenic frameshift variant predicted to result in truncation or NMD and had scimitar anomaly as part of a more complex heart defect. This individual was previously reported by Fick et al. [27]. A 7-year-old male with heterotaxy syndrome and a complex heart defect including TAPVR who carried a c.1009_1012delGAAA, p.Lys335Lysfs*6 pathogenic frameshift variant in NAA15 had been previously reported [26, 28, 29]. This, combined with NAA15’s high APVR-specific pathogenicity score (81.2%), leads us to conclude that individuals with pathogenic variants in NAA15 can present with APVR in addition to other features.
APVR associated with NKX2-1 variants
Heterozygous pathogenic variants in NKX2-1 are associated with choreoathetosis, hypothyroidism, and neonatal respiratory distress (MIM# 610978) and hereditary benign chorea (MIM# 118700). While loss of NKX2-1 function has been reported to result in a predominantly neurologic, endocrine, and pulmonary phenotypes—including infantile onset of choreoathetosis, hypothyroidism, neonatal respiratory distress, and recurrent pulmonary infections—other phenotypes including CHDs have been reported [30]. In our cohort, S8 had APVR and carried a nonsense pathogenic variant in NKX2-1 that was not inherited from her mother. An individual with an NKX2-1 frameshift mutation and TAPVR has previously been reported [31]. NKX2-1 also has a high APVR-specific pathogenicity score (87.2%), suggesting that individuals with deleterious variants affecting NKX2-1 can also present with APVR.
APVR associated with 15q11.2 deletions
Monoallelic deletions of the 15q11.2 region between BP1-BP2 that include NIPA1, NIPA2, CYFIP1, and TUBGCP5 have been associated with low penetrance neurodevelopmental disorders, schizophrenia, epilepsy, learning disabilities, and CHDs (MIM# 615656). We identified 6 individuals in our cohort (S23-S26, S57, and S58) who carried monoallelic deletions including the 15q11.2 BP1-BP2 region. 15q11.2 deletions have been previously published in association with APVR in a case-control study [32], in a familial case report describing 2 siblings with TAPVR [33], and in a case series that identified 1 individual with APVR and the deletion [34]. Based on the pLI and LOEUF values from gnomAD and APVR-specific pathogenicity scores for NIPA1 (pLI = 0.01, LOEUF = 1.04, 32.8%,), NIPA2 (pLI = 0.92, LOEUF = 0.38, 37.7%), CYFIP1 (pLI = 0.97, LOEUF = 0.3, 72.9%), and TUBGCP5 (pLI = 0, LOEUF = 0.67, 33.3%), CYFIP1 would appear to be the most likely gene to contribute to the association with APVR. We note that CYFIP1 was independently identified as a CHD candidate gene by Meerschaut et al. based on their analysis of CNVs in individuals with CHD [35].
APVR associated with proximal 1q21.1 microdeletions
TAR syndrome is most commonly caused by a combination of a 1q21 microdeletion that includes RBM8A in trans with one of two noncoding SNVs in RBM8A and has been associated with the development of a variety of CHDs [36]. However, S10, who has molecularly confirmed TAR syndrome based on the identification of a pathogenic RBM8A sequence variant in trans with a 1q21.1 deletion of an unspecified size, is the first to be described as having TAPVR. S10 was also noted to have heterotaxy.
By themselves, heterozygous proximal 1q21.1 microdeletions that include the TAR syndrome region between BP2 and BP3 are associated with developmental delay, dysmorphic features, failure to thrive, skeletal anomalies, and occasionally, CHDs [37]. S12 had TAPVR and heterotaxy and carried a 384.4 kb deletion (chr1:145,635,432-146,019,857) of this proximal 1q21.1 region which included RBM8A, HFE2 (HJV), TXNIP, POLR3GL, ANKRD34A, LIX1L, PEX11B, ITGA10, ANKRD35, PIAS3, NUDT17, POLR3C, RNF115, CD160, PDZK1, and GPR89A (Fig. 3A).
It is possible that heterozygous loss of RBM8A function contributes to the development of TAPVR (pLI = 0.57, LOEUF = 0.56, APVR-specific pathogenicity score = 68.6%) in some individuals. Alternatively, haploinsufficiency of other genes in this region of chromosome 1q21.1 may be required. PDZK1 is another 1q21.1 gene deleted in these patients. Liu et al. described an individual with a 302.5 kb 1q21.1 deletion (chr1:145,601,946-145,809,957; chr1:147,952,586-147,997,136) including RNF115, CD160, PDZK1, GPR89A, and GPR89C with heterotaxy and dextrocardia, single atrium, partial atrioventricular canal, and persistent left superior vena cava [38]. While this individual did not have APVR, the presence of a complex CHD with laterality defects suggests that this CNV may increase the likelihood of other laterality-associated CHDs, including APVR. With a pLI score of 0 (LOEUF = 1.71), it is unlikely that a monoallelic deletion of PDZK1 is sufficient to cause TAPVR. However, PDZK1 has an APVR-specific pathogenicity score of 81.1% and data from Deciphering the Mechanisms of Developmental Disorders consortium indicate that Pdzk1-knockout mice have abnormal cardiac morphology including APVR [39]. Hence, monoallelic deletions of PDZK1 may also be contributing to the development of TAPVR, heterotaxy, or other forms of CHD associated with 1q21.1 deletions alone or in the setting of TAR syndrome.
APVR associated with central 22q11.21 deletions
Central 22q11.2 deletions span the region between low copy repeats blocks LCR22-B to LCR22-D and do not include TBX1. These deletions are associated with renal and urinary tract malformations, developmental delays, cognitive impairments, behavioral problems, and to a lesser extent than the common 3 Mb 22q11.2 deletion, CHDs [40]. In our cohort, S31 had PAPVR and a 1.05 Mb central 22q11.21 deletion (chr22:20,354,589-21,405,291) (Fig. 3B). A mother and her son were previously reported to both have central 22q11.21 deletions (chr22:20,319,470-21,108,064) and TAPVR [41]. The mother in this case report also carried the 15q11.2 deletion previously described; however, her son did not inherit that deletion.
CRKL is located in the deleted region and has previously been proposed as a candidate gene for heart defects [41,42,43]. CRKL has a pLI score of 0.45 and a LOEUF score of 0.64 in gnomAD and an APVR-specific pathogenicity score of 80.7%. Crkl-/- mice exhibit defects in multiple cardiac and cranial neural crest derivatives including the aortic arch arteries, cardiac outflow tract, cranial ganglia, thymus, parathyroid glands, and craniofacial structures [44]. Currently, there is insufficient evidence to conclude that haploinsufficiency of CRKL is sufficient to cause APVR but it may be contributing to the development of APVR and other forms of CHD seen in individuals with central 22q11.2 deletions.
Two other genes in this region have high pLI scores and high APVR-specific pathogenicity scores: MED15 (pLI =1, LOEUF = 0.24, 62.5%) and SCARF2 (pLI = 1, LOEUF = 0.19, 90.7%). Haploinsufficiency of these genes are not currently known to be associated with the development of cardiac defects in humans or mice.
Conclusion
Based on our results, we conclude that clinical ES should be considered for individuals with non-isolated APVR for whom a molecular diagnosis has not been detected by other genetic testing. Our data suggest that EFTUD2, NAA15, and NKX2-1 play a role in the development of APVR. If a clearly pathogenic variant in one of these gene is identified, additional testing aimed at identifying an independent cause for the APVR may not be warranted. We additionally conclude that CMA is also a valuable diagnostic test that can identify a causative CNVs in a subset of individuals with non-isolated APVR including proximal 1q21.1 microdeletions (RBM8A, PDZK1), recurrent BP1-BP2 15q11.2 deletions, and central 22q11.2 deletions (CRKL). Genome sequencing is becoming more available on a clinical basis and is a reasonable alternative to obtaining ES and CMA in individuals with non-isolated APVR. As additional cases are reported, it is likely that a subset of the candidate genes and genomic regions for which there is currently insufficient evidence to support an association with APVR will ultimately be implicated in the development of this form of CHD.
Data availability
The data generated during this study can be found within the published article and its supplementary files. All previously unpublished variants from the BG clinical database and the CCVM database have been submitted to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/; SNVs = SCV003915701 - SCV003915719; CNVs = SCV003919100 - SCV003919143).
References
Douglas YL, Jongbloed MR, Deruiter MC, Gittenberger-de Groot AC. Normal and abnormal development of pulmonary veins: state of the art and correlation with clinical entities. Int J Cardiol. 2011;147:13–24.
Verma AK, Sethi S, Kohli N. Partial anomalous pulmonary venous connection: state-of-the-art review with assessment using a multidetector computed tomography angiography. Pol J Radio. 2022;87:e549–e56.
Bjornard K, Riehle-Colarusso T, Gilboa SM, Correa A. Patterns in the prevalence of congenital heart defects, metropolitan Atlanta, 1978 to 2005. Birth Defects Res A Clin Mol Teratol. 2013;97:87–94.
Liu Y, Chen S, Zuhlke L, Black GC, Choy MK, Li N, et al. Global birth prevalence of congenital heart defects 1970-2017: updated systematic review and meta-analysis of 260 studies. Int J Epidemiol. 2019;48:455–63.
St Louis JD, Harvey BA, Menk JS, Raghuveer G, O’Brien JE Jr, Bryant R 3rd, et al. Repair of “simple” total anomalous pulmonary venous connection: a review from the Pediatric Cardiac Care Consortium. Ann Thorac Surg. 2012;94:133–7. discussion 137-8
Spigel ZA, Edmunds EE, Caldarone CA, Hickey EJ, Binsalamah ZM, Heinle JS. Total anomalous pulmonary venous connection: Influence of heterotaxy and venous obstruction on outcomes. J Thorac Cardiovasc Surg. 2022;163:387–95.e3.
Hinton RB, McBride KL, Bleyl SB, Bowles NE, Border WL, Garg V, et al. Rationale for the cytogenomics of cardiovascular malformations consortium: a phenotype intensive registry based approach. J Cardiovasc Dev Dis. 2015;2:76–92.
Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, et al. DECIPHER: Database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Hum Genet. 2009;84:524–33.
Vulto-van Silfhout AT, van Ravenswaaij CM, Hehir-Kwa JY, Verwiel ET, Dirks R, van Vooren S, et al. An update on ECARUCA, the European Cytogeneticists Association Register of Unbalanced Chromosome Aberrations. Eur J Med Genet. 2013;56:471–4.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Scott TM, Campbell IM, Hernandez-Garcia A, Lalani SR, Liu P, Shaw CA, et al. Clinical exome sequencing data reveal high diagnostic yields for congenital diaphragmatic hernia plus (CDH+) and new phenotypic expansions involving CDH. J Med Genet. 2021;59:270–8.
Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. 2020;22:245–57.
Blake JA, Bult CJ, Eppig JT, Kadin JA, Richardson JE. Mouse Genome Database G. The Mouse Genome Database: integration of and access to knowledge about the laboratory mouse. Nucleic Acids Res. 2014;42:D810–7.
McKusick VA. Mendelian Inheritance in Man and its online version, OMIM. Am J Hum Genet. 2007;80:588–604.
Campbell IM, Rao M, Arredondo SD, Lalani SR, Xia Z, Kang SH, et al. Fusion of large-scale genomic knowledge and frequency data computationally prioritizes variants in epilepsy. PLoS Genet. 2013;9:e1003797.
Callaway DA, Campbell IM, Stover SR, Hernandez-Garcia A, Jhangiani SN, Punetha J, et al. Prioritization of candidate genes for congenital diaphragmatic hernia in a critical region on chromosome 4p16 using a machine-learning algorithm. J Pediatr Genet. 2018;7:164–73.
Rossetti LZ, Glinton K, Yuan B, Liu P, Pillai N, Mizerik E, et al. Review of the phenotypic spectrum associated with haploinsufficiency of MYRF. Am J Med Genet A 2019;179:1376–82.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581:434–43.
Collins RL, Glessner JT, Porcu E, Lepamets M, Brandon R, Lauricella C, et al. A cross-disorder dosage sensitivity map of the human genome. Cell 2022;185:3041–55.e25.
Zou Y, Wu J, Wang G, Zhou C, Dong N. Infracardiac total anomalous pulmonary venous return in a patient with Williams syndrome: A case report. Med (Baltim). 2019;98:e16276.
Lalani SR, Shaw C, Wang X, Patel A, Patterson LW, Kolodziejska K, et al. Rare DNA copy number variants in cardiovascular malformations with extracardiac abnormalities. Eur J Hum Genet. 2013;21:173–81.
Miller CL, Oikawa M, Cai Y, Wojtovich AP, Nagel DJ, Xu X, et al. Role of Ca2+/calmodulin-stimulated cyclic nucleotide phosphodiesterase 1 in mediating cardiomyocyte hypertrophy. Circ Res. 2009;105:956–64.
Meng L, Pammi M, Saronwala A, Magoulas P, Ghazi AR, Vetrini F, et al. Use of exome sequencing for infants in intensive care units: ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr. 2017;171:e173438.
Liu H, Giguet-Valard AG, Simonet T, Szenker-Ravi E, Lambert L, Vincent-Delorme C, et al. Next-generation sequencing in a series of 80 fetuses with complex cardiac malformations and/or heterotaxy. Hum Mutat. 2020;41:2167–78.
McDermott JH, Study DD, Clayton-Smith J. Sibling recurrence of total anomalous pulmonary venous drainage. Eur J Med Genet. 2017;60:265–7.
Cheng H, Dharmadhikari AV, Varland S, Ma N, Domingo D, Kleyner R, et al. Truncating variants in NAA15 are associated with variable levels of intellectual disability, autism spectrum disorder, and congenital anomalies. Am J Hum Genet. 2018;102:985–94.
Fick TA, Scott DA, Lupo PJ, Weigand J, Morris SA. The frequency and efficacy of genetic testing in individuals with scimitar syndrome. Cardiol Young-. 2022;32:550–7.
Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 2015;350:1262–6.
Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013;498:220–3.
Thorwarth A, Schnittert-Hubener S, Schrumpf P, Muller I, Jyrch S, Dame C, et al. Comprehensive genotyping and clinical characterisation reveal 27 novel NKX2-1 mutations and expand the phenotypic spectrum. J Med Genet. 2014;51:375–87.
Hayasaka I, Cho K, Akimoto T, Ikeda M, Uzuki Y, Yamada M, et al. Genetic basis for childhood interstitial lung disease among Japanese infants and children. Pediatr Res. 2018;83:477–83.
Li X, Shi G, Li Y, Zhang X, Xiang Y, Wang T, et al. 15q11.2 deletion is enriched in patients with total anomalous pulmonary venous connection. Journal of medical genetics. 2020.
Kuroda Y, Ohashi I, Naruto T, Ida K, Enomoto Y, Saito T, et al. Familial total anomalous pulmonary venous return with 15q11.2 (BP1-BP2) microdeletion. J Hum Genet. 2018;63:1185–8.
Vanlerberghe C, Petit F, Malan V, Vincent-Delorme C, Bouquillon S, Boute O, et al. 15q11.2 microdeletion (BP1-BP2) and developmental delay, behaviour issues, epilepsy and congenital heart disease: a series of 52 patients. Eur J Med Genet. 2015;58:140–7.
Meerschaut I, Vergult S, Dheedene A, Menten B, De Groote K, De, et al. A reassessment of copy number variations in congenital heart defects: picturing the whole genome. Genes (Basel). 2021;12:1048.
Greenhalgh KL, Howell RT, Bottani A, Ancliff PJ, Brunner HG, Verschuuren-Bemelmans CC, et al. Thrombocytopenia-absent radius syndrome: a clinical genetic study. J Med Genet. 2002;39:876–81.
Rosenfeld JA, Traylor RN, Schaefer GB, McPherson EW, Ballif BC, Klopocki E, et al. Proximal microdeletions and microduplications of 1q21.1 contribute to variable abnormal phenotypes. Eur J Hum. Gene 2012;20:754–61.
Liu C, Cao R, Xu Y, Li T, Li F, Chen S, et al. Rare copy number variants analysis identifies novel candidate genes in heterotaxy syndrome patients with congenital heart defects. Genome Med. 2018;10:40.
Mohun T, Adams DJ, Baldock R, Bhattacharya S, Copp AJ, Hemberger M, et al. Deciphering the mechanisms of developmental disorders (DMDD): a new programme for phenotyping embryonic lethal mice. Dis Model Mech. 2013;6:562–6.
Rump P, de Leeuw N, van Essen AJ, Verschuuren-Bemelmans CC, Veenstra-Knol HE, Swinkels ME, et al. Central 22q11.2 deletions. Am J Med Genet A 2014;164A:2707–23.
Faurschou S, Lildballe DL, Maroun LL, Helvind M, Rasmussen M. Total anomalous pulmonary venous connection in mother and son with a Central 22q11.2 microdeletion. Case Rep. Genet. 2021;2021:5539855.
Breckpot J, Thienpont B, Bauters M, Tranchevent LC, Gewillig M, Allegaert K, et al. Congenital heart defects in a novel recurrent 22q11.2 deletion harboring the genes CRKL and MAPK1. Am J Med Genet A 2012;158A:574–80.
Racedo SE, McDonald-McGinn DM, Chung JH, Goldmuntz E, Zackai E, Emanuel BS, et al. Mouse and human CRKL is dosage sensitive for cardiac outflow tract formation. Am J Hum Genet. 2015;96:235–44.
Guris DL, Fantes J, Tara D, Druker BJ, Imamoto A. Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat Genet. 2001;27:293–8.
Lehalle D, Gordon CT, Oufadem M, Goudefroye G, Boutaud L, Alessandri JL, et al. Delineation of EFTUD2 haploinsufficiency-related phenotypes through a series of 36 patients. Hum Mutat. 2014;35:478–85.
Panizzi JR, Becker-Heck A, Castleman VH, Al-Mutairi DA, Liu Y, Loges NT, et al. CCDC103 mutations cause primary ciliary dyskinesia by disrupting assembly of ciliary dynein arms. Nat Genet. 2012;44:714–9.
Postma AV, van Engelen K, van de Meerakker J, Rahman T, Probst S, Baars MJ, et al. Mutations in the sarcomere gene MYH7 in Ebstein anomaly. Circ Cardiovasc Genet. 2011;4:43–50.
Chirita Emandi A, Dobrescu AI, Doros G, Hyon C, Miclea D, Popoiu C, et al. A novel 3q29 deletion in association with developmental delay and heart malformation-case report with literature review. Front Pediatr. 2019;7:270.
Tomita-Mitchell A, Mahnke DK, Struble CA, Tuffnell ME, Stamm KD, Hidestrand M, et al. Human gene copy number spectra analysis in congenital heart malformations. Physiol Genomics. 2012;44:518–41.
Nagamani SC, Erez A, Bader P, Lalani SR, Scott DA, Scaglia F, et al. Phenotypic manifestations of copy number variation in chromosome 16p13.11. Eur J Hum Genet. 2011;19:280–6.
Monteiro RAC, de Freitas ML, Vianna GS, de Oliveira VT, Pietra RX, Ferreira LCA, et al. Major contribution of genomic copy number variation in syndromic congenital heart disease: the use of MLPA as the first genetic test. Mol Syndromol. 2017;8:227–35.
Acknowledgements
This study makes use of data generated by the DECIPHER community. A full list of centers who contributed to the generation of the data is available from https://deciphergenomics.org/about/stats and via email from [email protected]. We note that those who carried out the original analysis and collection of the DECIPHER data bear no responsibility for the further analysis or interpretation of the data.
Funding
This work was supported, in part, by National Institutes of Health/Eunice Kennedy Shriver National Institute of Child Health and Human Development grant R01HD098458 to DAS. The CCVM Registry is supported in part by American Heart Association Transformational Award AHA 19TPA34850054 (SMW). Funding for the DECIPHER project was provided by Wellcome.
Author information
Authors and Affiliations
Contributions
DAS conceived the study. EAH wrote the first draft of the manuscript. CAS and PNL were responsible for the machine learning. XZ and NO were responsible for providing updated SNV and CNV variant interpretations, respectively, based on ACMG criteria. IV, ILHD, SJ, JC-S, MJP, JJL, MG, JB, AK, ES, UK, TB, TYT, RA, KN, GBF, AB, WSK-F, JR, LRH, BJL, GCG, KLM, SMW, and SRL obtained and provided clinical and molecular data. EAH and DAS analyzed clinical and molecular data. All authors reviewed, edited, and approved the final draft.
Corresponding author
Ethics declarations
Competing interests
The Department of Molecular & Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing completed at Baylor Genetics.
Ethical approval
This study was approved by the institutional review board of Baylor College of Medicine (protocol H-47546) and was conducted in accordance with the ethical standards of this institution’s committee on human research and international standards.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Huth, E.A., Zhao, X., Owen, N. et al. Clinical exome sequencing efficacy and phenotypic expansions involving anomalous pulmonary venous return. Eur J Hum Genet 31, 1430–1439 (2023). https://doi.org/10.1038/s41431-023-01451-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-023-01451-4
This article is cited by
-
The Role of Genetic Testing in Congenital Heart Disease
Current Treatment Options in Cardiovascular Medicine (2025)
-
Ambivalence and regret in genome sequencing
European Journal of Human Genetics (2023)
