
Abstract
Safe and effective vaccines against COVID-19 for children and adolescents are needed. This international multicenter, randomized, double-blind, placebo-controlled, phase III clinical trial assessed the efficacy, immunogenicity, and safety of CoronaVac® in children and adolescents (NCT04992260). The study was carried out in Chile, South Africa, Malaysia, and the Philippines. The enrollment ran from September 10, 2021 to March 25, 2022. For efficacy assessment, the median follow-up duration from 14 days after the second dose was 169 days. A total of 11,349 subjects were enrolled. Two 3-μg injections of CoronaVac® or placebo were given 28 days apart. The primary endpoint was the efficacy of the CoronaVac®. The secondary endpoints were the immunogenicity and safety. The vaccine efficacy was 21.02% (95% CI: 1.65, 36.67). The level of neutralizing antibody in the vaccine group was significantly higher than that in the placebo group (GMT: 390.80 vs. 62.20, P <0.0001). Most adverse reactions were mild or moderate. All the severe adverse events were determined to be unrelated to the investigational products. In conclusion, in the Omicron-dominate period, a two-dose schedule of 3 μg CoronaVac® was found to be safe and immunogenic, and showed potential against symptomatic COVID-19 in healthy children and adolescents.
Similar content being viewed by others
Introduction
The COVID-19 pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has led to more than 774 million confirmed cases and 7.03 million deaths worldwide up to February 11, 2024. (https://covid19.who.int) Children and adolescents were also at risk of COVID-19. Worldwide, over 8.8 million children under 14 years old suffered from SARS-CoV-2 infection from December 30, 2019 to October 25, 20211. Although infected children and adolescents cases are mainly mild or asymptomatic compared with adults2, severe diseases, hospitalization, and multisystem inflammatory syndrome in children (MIS-C) can occur, which may require intensive care3,4,5. Apart from the effects on physical health, being infected by SARS-CoV-2 can interrupt education and have negative impacts on social and emotional development and mental health6,7,8. Besides, adolescents play an important role in SARS-CoV-2 transmission9,10. Thus, safe and effective vaccines against COVID-19 for children and adolescents are needed to help prevent disease and contribute to herd immunity.
However, limited clinical evidence supported the efficacy of COVID-19 vaccines in persons under 18 years old, and their immunization schedule remained to be further explored. COVID-19 messenger RNA vaccines, mRNA-1273 (Moderna), showed an efficacy of 36.8% and 50.6% in recipients aged 2–5 years and 6–23 months by a two-dose injection schedule in phase III clinical trials during the Omicron-dominant phase11. In the phase II–III trial of recombinant subunit adjuvanted protein vaccines, NVX-CoV2373 (Novavax), for 2–17 years in India, the geometric mean titer ratio compared to those in adults were 1.33 and 1.93 for neutralizing antibodies in adolescents and children, respectively12. The vaccine efficacy against the delta variant for 12–17-year-olds was approximately 80%13. However, more international multicenter evidence is still needed to support the efficacy of NVX-CoV2373 in persons under 18, especially those younger than two years old.
CoronaVac® is an inactivated COVID-19 vaccine developed by Sinovac Life Sciences Co., Ltd. Three domestic phase I/II clinical trials proved the immunogenicity and safety of CoronaVac®14,15,16, and international phase III trials in adults further supported its efficacy and safety17,18. Thus, CoronaVac® was approved for conditional marketing in China on February 5, 2021 (approval number: 2021S00156, 2021S00157) and was approved by the World Health Organization for emergency use in adults on June 01, 202119. Furthermore, results from a phase I/II study supported the safety and immunogenicity of CoronaVac® administered as two-dose injection (day 0 and day 28) in healthy persons aged 3–17 years16, and supported the use of 3 μg dose with a two-dose immunization schedule for further studies in children and adolescents. Based on that, this global multicenter, randomized, double-blind, phase III clinical trial was launched in September 2021 at sites in Chile, South Africa, Malaysia, and the Philippines to assess the efficacy, immunogenicity, and safety of CoronaVac® in healthy children and adolescents aged 6 months–17 years. Here, we present the main findings of this study.
Results
Clinical trial profile
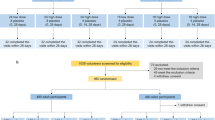
The enrollment ran from September 10, 2021, to March 25, 2022, followed for 12 months after the second dose. Due to the outbreak of the Omicron variant, which started in November 2021, the number of PCR-confirmed symptomatic COVID-19 cases quickly accumulated to the statistical requirement (at least 93 cases) in the interim analysis on March 10, 2022 (Table S1). A total of 11,349 subjects were enrolled in this study. Among them, 11,343 received the first dose of either the CoronaVac® vaccine (n = 6071) or placebo (n = 5272), and 11,037 received their second dose. For the efficacy analysis, 9678 subjects were included in the per-protocol set (E-PPS). For efficacy evaluation, the median duration of follow-up from 14 days after the second dose was 169 days (range, 2–310) in the vaccine group and 169 days (range, 3–310) in the placebo group. A total of 1404 subjects randomized were also assigned to the immunogenicity subgroup, of whom 1123 were included in the per-protocol set for immunogenicity (I-PPS). In addition, 2941 subjects randomized in either arm were also assigned to the safety subgroup (Fig. 1).

*Other: Vaccine group participants without corresponding control population. During the study period, CoronaVac® was approved for EUA (Emergency Use Authorization) in Chile for a population aged 3–17 years. Hence, participants aged 3–17 years in Chile (343 aged 6–17 years and 416 aged 3–5 years) were unblinded and administrated CoronaVac®. The data of these subjects were excluded from the efficacy analysis#. One participant, who was enrolled in the placebo group at the beginning of the study, had the first dose of placebo. However, this participant received CoronaVac® in the second injection. Thus, after the second dose, this participant was moved to the vaccine group in the safety analysis.
Efficacy analysis
The efficacy analysis was conducted based on the E-PPS. A total of 9678 subjects were finally included, with 4841 (50.02%) in the vaccine group and 4837 (49.98%) in the placebo group. Among them, 51.19% were male, 53.08% were Asian. The proportion of participants aged 6–35 months, 3–5 years, 6–11 years and 12–17 years were 1.97%, 15.01%, 39.13% and 43.88%, respectively. The overall median age was 11.20 years old. There were no significant differences in demographic and other characteristics between the vaccine and placebo groups at baseline (Table 1). The demographic and other characteristics at baseline were similar in each country’s vaccine and placebo groups (Table S2–Table S5). Among those enrolled, 4737 subjects (48.95%) had immunological evidence of SARS-CoV-2 infection at baseline based on the positive result of SARS-CoV-2 antigen or IgG/IgM rapid test, despite having reported no active infection by history of record.
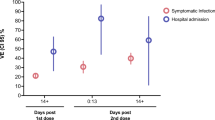
In the efficacy analysis of COVID-19 cases at least 14 days after the second dose vaccination, 339 confirmed cases were reported, including 152 cases in the vaccine group (6.59/100 person-years) and 187 cases in the placebo group (8.24/100 person-years). The vaccine efficacy against PCR-confirmed symptomatic COVID-19 cases was 21.02% with 95% confidence interval (95% CI) of 1.65 and 36.67 (Table 2). PCR-confirmed symptomatic COVID-19 cases accrual by study time was presented in Fig. 2.

PCR-confirmed symptomatic COVID-19 cases accrual by study time was presented. The inset shows the same data on an enlarged axis.
The efficacy of vaccine against COVID-19 cases with different severity, including hospitalization, severe, and death cases, was analyzed accordingly. Among 9678 subjects, seven hospitalization cases were reported, including one in the vaccine group (0.04/100 person-years) and six in the placebo group (0.26/100 person-years). The estimated efficacy against COVID-19 hospitalization was 84.19% (95% CI: −30.34, 99.66). No severe case was reported in the vaccine group, but two were reported in the placebo group, where the efficacy against severe COVID-19 was 100% (95% CI: −424.27, 100.00). No death occurred during the study period. Above all, due to the limited cases, the results were not powered to prove the efficacy against hospitalization and severe cases.
In addition, the vaccine efficacy against PCR-confirmed symptomatic COVID-19 cases in the population without immunological evidence of past SARS-CoV-2 infection was 26.72% (95% CI: 5.33, 43.42) (Table 2).
Respiratory tract specimens of PCR-confirmed symptomatic COVID-19 cases were sequenced to detect the variants of the circulating virus. In the E-PPS (interim analysis), a total of 87 respiratory tract specimens were sequenced, all of which were the Omicron variant, including 33 BA.1 subvariant, 50 BA.2 subvariant, and four unspecified. The vaccine efficacy against Omicron BA.1 and BA.2 was 50.40% (95%CI: −6.72, 78.28) and 22.20% (95% CI: −41.00, 57.58), respectively (Table S6).
Immunogenicity analysis
A total of 1123 participants were included in the I-PPS. The level of neutralizing antibody against the SARS-CoV-2 ancestral strain were measured and analyzed. Results showed that 749 (66.70%) participants were seropositive before the vaccination. Twenty-eight days after the second dose of vaccination, the seroconversion rates, geometric mean titers (GMT), and geometric mean fold increase (GMI) in the vaccine group were significantly higher than those in the placebo group (P < 0.0001), with the seroconversion rates being 70.10% (95% CI: 66.30, 73.70) vs. 23.29% (95% CI: 19.69, 27.20), GMT: 390.80 (95% CI: 353.90, 431.54) vs. 62.20 (95% CI: 51.11, 75.69), GMI: 11.22 (95% CI: 9.43, 13.35) vs. 1.34 (95% CI: 1.18, 1.52). Participants with seropositive neutralizing antibodies at baseline had a higher level of GMT than those with seronegative. The neutralizing antibody GMT of participants aged 6–35 months was 757.79 (95% CI: 582.20, 986.33) after two vaccination doses, while that in participants aged 3–17 years was 348.63 (95% CI: 314.11, 386.95) (Table 3). The overview of neutralizing antibodies between the vaccine and placebo groups in 3–17 years in each country was shown in Table S7. Participants in Chile were unblinded and administrated CoronaVac® because of national policy changes (CoronaVac® was approved for EUA). Satisfactory immunogenicity of the vaccine was observed in the other three countries. Twenty-eight days after the second dose of vaccination, seroconversion rates, GMT, and GMI in the vaccine group were significantly higher than those in the placebo group (all P < 0.01).
Immunogenicity analysis stratified by baseline immunological evidence of infection were shown in Table S8. Twenty-eight days after the second dose of vaccination, seroconversion rates, GMT, and GMI in the vaccine group were significantly higher than those in the placebo group (all P < 0.01). Participants with immunological evidence before vaccination had a higher level of GMT. These findings were consistent with the results of stratified analysis based on baseline neutralizing antibody levels.
Safety evaluation
In the safety subgroup, the onset for most adverse reactions (ARs) was 0–7 days. The ARs were mainly in Grade 1, with an incidence rate of 32.35% (582/1799) and 21.14% (241/1140) in the vaccine group and the placebo group, respectively. The incidence rate of solicited ARs in the vaccine group was significantly higher than that in the placebo group (P < 0.0001), especially the local ones, including vaccination site pain (P < 0.0001), vaccination site erythema (P < 0.0001), and vaccination site induration (P = 0.0016). For the participants aged 6–35 months, the overall incidence rate of ARs within 28 days after vaccination was 31.68% (64/202). The most common ARs within 28 days after each vaccination dose were fever, diarrhea, and vaccination site pain (Table 4). For the participants aged 3–17 years, the overall incidence rate of ARs within 28 days after vaccination was 30.91% (846/2737). The most common AR within 28 days after each vaccination dose was vaccination site pain (Table 4).
Safety evaluation among all participants were also performed. The results among all participants and the safety subgroup were comparable. Among all age groups (6–35 months, 3–5 years, 6–11 years, 12–17 years), the onset of most ARs was 0–7 days. Most ARs were in Grade 1, with an incidence rate of <30% in the vaccine group and <20% in the placebo group. (Table S9)
As for severe adverse events (SAEs), a total of 182 events were reported among 151 children and adolescents aged 6 months–17 years (74 SAEs in the vaccine group and 77 SAEs in the placebo group). All the SAEs were determined to be unrelated to the investigational products.
Discussion
To our knowledge, this is the first international multicenter clinical trial of inactivated COVID-19 vaccine among healthy children aged 6 months–17 years. Our results indicated that two doses of CoronaVac® administered in a 28-day apart schedule were safe and immunogenic, and showed potential efficacy against symptomatic COVID-19 in healthy children and adolescents aged 6 months–17 years during a period when Omicron was the predominant variant.
In this study, the efficacy of two-dose CoronaVac® in children and adolescents against PCR-confirmed symptomatic COVID-19 was only 21.02%. The efficacy was lower than those observed in previous CoronaVac® trials involving older age cohorts17,20,21, most likely due to the prevalence of the Omicron variant. Same findings were mentioned in the systematic reviews that primary immunization with COVID-19 vaccines would provide limited efficacy against the Omicron variant22,23. A limited efficacy of the COVID-19 vaccine was also observed in trials using other vaccines for children or adolescents during the emergence of the Omicron variant. For example, the mRNA-1273 vaccine efficacy against COVID-19 was 36.8% among 2–5 year-old and 50.5% among 6–23 month-old11. This phenomenon was highly related to the immune evasion of the Omicron variant22. In addition, our results showed that the vaccine efficacy varied in different Omicron subvariants (BA.1 and BA.2). A higher efficacy against Omicron BA.1 (50.40%) than against Omicron BA.2 (22.20%) was observed, suggesting relatively better cross-protection between our prototype and Omicron BA.1. Besides, previously exposed participants showed a lower level of efficacy (−0.85% vs. 26.72%) in this study. About 50% of the participants in our study had been previously exposed, thus the efficacy of CoronaVac® might be slightly underestimated.
Limited numbers of hospitalizations and severe cases, and no deaths were observed in our study because of the characteristics of Omicron infection24,25. Only seven hospitalization cases were reported (one in the vaccine group and six in the placebo group), while two severe cases were reported in the placebo group (none for the vaccine group). Thus, this trial was not powered to prove the efficacy against hospitalization and severe cases. In this study, CoronaVac® showed a trend to protect against hospitalization and severe cases, though without statistical significance. Nevertheless, real-world studies in Chile and Brazil during the Omicron-dominant period provided evidence for the effectiveness of CoronaVac® against hospitalization and severe cases26,27. A real-world study in children aged 3–5 years in Chile also showed that the effectiveness of CoronaVac® against hospitalization and intensive care unit admission was 64.6% and 69.0%, respectively26. On the other hand, a real-world study in children aged 6–11 years in Brazil showed that the effectiveness of CoronaVac® against hospitalization was 59.2%27. Those above real-world evidence indirectly verified our findings in this study that the CoronaVac® vaccine appears potential efficacious and effective against severe disease and hospitalization against COVID-19 in children and teenagers amidst the mild presentation of the Omicron variant. However, it still needs further evaluation in larger sample size and longer period to draw a precise conclusion.
Among all the age groups, participants with seronegative or seropositive neutralizing antibodies at baseline elicited a high antibody level after receiving two doses of CoronaVac®. This suggested that CoronaVac® had good immunogenicity in eliciting priming and anamnestic immune responses. Participants with seropositive neutralizing antibodies at baseline had a higher level of GMT than those with seronegative, which reflected the effect of a booster dose, the necessity of which has been increasingly recognized and explored in other studies23,28. Generally, vaccination with CoronaVac® for children and adolescents could achieve good immunogenicity. This study also proved the safety of CoronaVac® in children and adolescents, especially those aged 6–35 months.
There were several limitations. Firstly, due to a lack of enough prediction of the variants of the SARS-CoV-2 virus, we only considered about primary immunization without boosters when we designed this trial. The study coincided with the emergence of the Omicron variant; limited efficacy of vaccination against symptomatic infection was observed because of immune evasion of the Omicron variant. Further studies should be carried out to evaluate the efficacy of booster doses of CoronaVac®. Secondly, we designed the study such that the participants in the placebo or vaccine group at a ratio of 1:1. However, because of national policy changes in Chile during recruitment, the actual number of participants in the vaccine group exceeded that in the placebo group. These subjects were excluded from the efficacy analysis. Thirdly, even though all samples of PCR-confirmed symptomatic COVID-19 cases were sent to perform the whole viral genome sequencing, some samples did not accomplish the test after interim analysis. However, based on the epidemiological data29, we believe that the results of our study demonstrated efficacy against the Omicron variant in children and adolescents.
In conclusion, in the Omicron-dominate period, a two-dose schedule of 3 μg CoronaVac® was immunogenic and safe in healthy children and adolescents aged from 6 months–17 years, and showed potential efficacy against symptomatic COVID-19 disease. According to this study, children and adolescents, with or without immunological evidence of SARS-CoV-2 infection, might benefit from the CoronaVac® vaccination without severe adverse reactions. Thus, we provided a potential strategy for vaccinating this young population using CoronaVac®. In the meantime, developing vaccines for different variants should be considered to provide targeted protection.
Methods
The trial was conducted following the principles of the Declaration of Helsinki and the International Conference on Harmonization (ICH) tripartite guidelines for good clinical practice. The study protocol was approved by the national regulatory authorities of each site in all participating countries before it started (see Supplementary Note 6). This trial is registered with ClinicalTrials.gov (NCT04992260).
Study design and participants
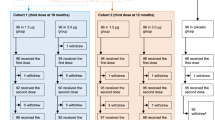
This multicenter, randomized, double-blind, placebo-controlled, phase III clinical trial evaluated the efficacy, immunogenicity, and safety of two doses (day 0 and day 28) of CoronaVac® in healthy children and adolescents aged from 6 months to 17 years. Participants were enrolled from Chile, South Africa, Malaysia, and the Philippines, with 37 sites. The enrollment ran from September 10, 2021, to March 25, 2022.
Eligible participants were healthy children and adolescents aged from 6 months to 17 years (participants aged 6–35 months were only recruited from South Africa), without a history of confirmed SARS-CoV-2 infection on PCR test, had no previous receipt of other COVID-19 vaccines, had no unstable or severe underlying medical conditions, had no allergy to vaccines or vaccine/placebo ingredients. The guardians must agree and sign the informed consent voluntarily. In addition, children and teenagers above seven years of age also needed to provide written assent to participate in the study. A complete list of inclusion/exclusion criteria was shown in the supplements (Supplementary Note 1).
Randomization and blinding
Central randomization was implemented for this study. The participants were randomly assigned to the vaccine or placebo groups according to a 1:1 ratio. The randomization was balanced by using randomly permuted blocks and stratified by country and age groups. Each site first selected the subjects in immunogenicity and safety subgroups according to participants’ willingness until the assigned number was reached. SAS software (version 9.4) was used to generate a random list, which was imported into the Interactive Web Response System (IWRS). Investigators entered the key information of the participants to obtain the random numbers when they were enrolled. Participants were assigned a treatment identification number to ensure adequate blinding. Before each vaccination, an authorized pharmacist logged into the IWRS and obtained the vaccine number. Participants and study staff remained blinded throughout the study.
Vaccine
CoronaVac® was derived from the novel coronavirus (strain CZ02) and then cultured in African green monkey kidney cell cultures (Vero Cell), followed by harvest, inactivation, concentration, purification, and aluminum hydroxide adsorption. The dose was 3 μg/0.5 ml. Aluminum hydroxide packaged in a pre-filled syringe was used as a placebo control. CoronaVac® and placebo were prepared and tested by Sinovac Life Sciences Co., Ltd. according to the requirements of Manufacturing and Quality Control Requirements of SARS-CoV-2 Vaccine (Vero Cell), Inactivated, then reviewed and retested by the National Institutes for Food and Drug Control (NIFDC), China.
Procedures
After determining eligibility and obtaining written informed consent and assent, if necessary, on day 0, respiratory tract samples were collected (nasopharyngeal swab, throat swab, or saliva) at baseline for rapid SARS-CoV-2 antigen and fingertip blood was collected for SARS-CoV-2 IgG/IgM rapid test (Assays were listed in Supplementary Note 7). Participants with a positive result of rapid antigen tests and/or SARS-CoV-2 IgG/IgM test (just positive result of IgG/IgM test in some sites) but no known history of symptomatic or confirmed SARS-CoV-2 infection using PCR were considered as having immunological evidence of SARS-CoV-2 infection. The latter could be included in the study. With or without immunological evidence of SARS-CoV-2 infection were used for stratified analysis. Two doses of CoronaVac® or placebo were administered to each participant in the deltoid of the upper arm via intramuscular according to a day 0 and 28 schedules.
Participants were contacted weekly by phone or text message for eight weeks as of the first dose and then at least every two weeks by phone or text messages until the end of the study to inquire whether they experienced any signs or symptoms that were consistent with COVID-19. The presence of any signs or symptoms of suspected COVID-19, including fever or chills, cough, nasal congestion or runny nose, muscle pain, etc. (see Supplementary Note 2), triggered the laboratory test by RT-PCR to confirm SARS-CoV-2 infection. For the PCR-confirmed symptomatic cases, the diagnosis date was defined as the date of the first qualifying symptom(s). To clarify the variants involved, whole viral genome sequencing was performed among the PCR-confirmed symptomatic COVID-19 cases in the qualified local laboratory. All COVID-19 cases were followed up until recovery.
Immunogenicity was assessed by neutralizing antibody levels against ancestral SARS-CoV-2. Each time, a 3 mL blood sample was collected from each participant in the immunogenicity subgroup at day 0 (pre-vaccination) and day 56 (28 days after the second dose), as well as six months and one year after the second vaccination. All the serum samples were sent to China and analyzed by the central laboratory of NIFDC using the micro-cytopathic test to measure the level of neutralizing antibody. Serum samples were inactivated and serially diluted with cell culture medium in two-fold steps. The diluted serum samples were incubated with equal volume of the live SARS-CoV-2 virus (GenBank: MT407649.1) suspension, with a 50% cell culture infective dose of 100 (100 CCID50) for 2 h at 36.5 °C. Vero cells (1.0–2.0 × 105 cells/mL) were then added to the serum-virus suspensions in microplates in duplicate and incubated at 36.5 °C for 5 days. Cytopathic effects were assessed under microscopes and the neutralizing antibody titer was calculated by the highest dilution that can protect 50% cells from 100 CCID50. The positive cutoff of the antibody titer was 1:8, the value < 1:8 was assigned to 1:4. According to WHO guideline (TRS No.927)30, seroconversion of antibody was defined as a change from seronegative at baseline to seropositive or a four-fold titer increase if the participant was seropositive at baseline in this study. More details about the method have been provided in the supplements (Supplementary Note 3).
For safety evaluation, all participants were observed in the study site for at least 30 min for immediate reactions after each vaccination. Local and systemic solicited adverse events (AEs) within seven days and unsolicited AEs within 28 days were monitored after each dose for participants in the safety subgroup by using Diary card. Safety information was also collected from participants that not in the safety subgroup via their active report or weekly/bi-weekly monitoring. Solicited local AEs (at the injection site) included pain, induration, swelling, erythema, rash, and pruritus; solicited systemic AEs included acute allergic reaction, skin and mucosa abnormality, diarrhea, anorexia, vomiting, nausea, muscle pain (non-injection site), headache, cough, fatigue, and fever. The reported AEs were graded according to the China National Medical Products Administration guideline31. The relationship between AEs and vaccination was established by the investigators. In addition, serious adverse events (SAEs) in all participants were monitored up to 12 months after complete vaccination.
Outcomes
In this clinical trial, the primary endpoint of efficacy was the incidence of symptomatic COVID-19 cases confirmed by RT-PCR 14 days after the second dose of vaccination. The secondary efficacy objectives were: (1) to evaluate the efficacy in preventing hospitalization, severe cases, or death caused by COVID-19 as of 14 days after the second dose; (2) to evaluate the efficacy in preventing symptomatic, RT-PCR-confirmed COVID-19 beginning 14 days after the second dose in participants without immunological evidence at baseline. Severe COVID-19 cases were defined according to the Chinese Diagnosis and Treatment Plan for COVID-19 (trial version 8), with one or more of high fever (>39.5 °C) that lasted for three days, tachypneic by age reference, poor saturation below 93%, and/or having assisted breathing, drowsiness, convulsion, poor feeding or feeding difficulties with dehydration32. Definitions related to COVID-19 are presented in Supplementary Note 2.
Immunogenicity objectives evaluated in participants in the immunogenicity subgroup included seropositivity rate, seroconversion rate, GMT, and GMI of neutralizing antibodies 28 days after the second dose of vaccination. For safety analysis, AEs and SAEs were collected.
Statistical analysis
Based on the incidence rate of COVID-19 in children and adolescents, to detect 50% vaccine efficacy, using a two-sided, type I error rate of 5%, a total of 93 PCR-confirmed symptomatic COVID-19 cases were required. Considering a 10% drop-out rate, approximately 14,000 participants would be needed and with 7000 participants in each arm (details of sample size calculation are displayed in Supplementary Note 4). In addition, 12% and 20% of all participants were enrolled in the immunogenicity and safety subgroups, respectively.
Efficacy evaluation was run on the per-protocol set (E-PPS) population, including the randomized eligible participants who completed the two-dose vaccination and had been followed up for case surveillance 14 days after the second vaccination dose. The participants who met any of the following conditions were excluded from E-PPS: (1) major protocol deviations or violations; (2) any conditions which may affect the evaluation of vaccine efficacy. The incidence density of confirmed COVID-19 cases in each group was calculated from (the number of confirmed COVID-19 cases/person-years at risk) × 100%. Stratification factors were disease severity and baseline status of infection (with or without immunological evidence of SARS-CoV-2 infection). The Poisson regression model was applied to estimate the vaccine efficacy and 95% confidence interval.
Immunogenicity was analyzed from the per-protocol set for immunogenicity (I-PPS), which included the randomized eligible participants who completed two doses of vaccination and had both valid immunogenicity data of pre-vaccination and 28 days after the second dose. Stratification factors were age, country, and baseline status of infection. The safety set (SS) included participants who received at least one dose of vaccine or placebo. Evaluation for solicited and unsolicited AEs was analyzed in the safety subgroup, stratified by age group. Safety evaluation for SAEs was run for all participants. The chi-square or Fisher exact tests were used for binary variables, and the Student’s t-test or ANOVA was used for continuous variables. The 95% CIs were derived from the Clopper-Pearson method for seropositivity and seroconversion rates. GMTs and 95% CIs were calculated based on a standard normal distribution of log-transformed GMT.
Details of the analysis sets above are displayed in the supplements (Supplementary Note 5). Statistical analysis was performed using SAS software (version 9.4). P values of less than 0.05 were considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data for this study are provided in the article or Supplementary Information. The original data are available under controlled conditions due to privacy laws pertaining to patient consent for data usage. Request for the original data in a de-identified format and solely for research purposes and the original study protocol can be directed to the corresponding authors.
References
WHO. Interim statement on COVID-19 vaccination for children and adolescents, 2021. World Health Organization, Geneva https://www.who.int/news/item/24-11-2021-interim-statement-on-covid-19-vaccination-for-children-and-adolescents.
Cui, X. et al. A systematic review and meta-analysis of children with coronavirus disease 2019 (COVID-19). J. Med Virol. 93, 1057–1069 (2021).
Ebina-Shibuya, R., Namkoong, H., Shibuya, Y. & Horita, N. Multisystem inflammatory syndrome in children (MIS-C) with COVID-19: insights from simultaneous familial Kawasaki disease cases. Int J. Infect. Dis. 97, 371–373 (2020).
Radia, T. et al. Multi-system inflammatory syndrome in children & adolescents (MIS-C): a systematic review of clinical features and presentation. Paediatr. Respir. Rev. 38, 51–57 (2021).
Feldstein, L. R. et al. Characteristics and outcomes of US children and adolescents with multisystem inflammatory syndrome in children (MIS-C) compared with severe acute COVID-19. JAMA 325, 1074–1087 (2021).
Engzell, P., Frey, A., Verhagen, M. D. Learning loss due to school closures during the COVID-19 pandemic. Proc. Natl Acad. Sci. USA https://doi.org/10.1073/pnas.2022376118 (2021).
López-Bueno, R. et al. Potential health-related behaviors for pre-school and school-aged children during COVID-19 lockdown: a narrative review. Prev. Med. 143, 106349 (2021).
McKune, S. L. et al. Psychosocial health of school-aged children during the initial COVID-19 safer-at-home school mandates in Florida: a cross-sectional study. BMC Public Health 21, 603 (2021).
Rumain, B., Schneiderman, M. & Geliebter, A. Prevalence of COVID-19 in adolescents and youth compared with older adults in states experiencing surges. PLoS ONE 16, e0242587 (2021).
Paul, L. A. et al. Association of age and pediatric household transmission of SARS-CoV-2 Infection. JAMA Pediatr. 175, 1151–1158 (2021).
Anderson, E. J. et al. Evaluation of mRNA-1273 Vaccine in Children 6 Months to 5 Years of Age. N. Engl. J. Med. 387, 1673–1687 (2022).
Gunale B., et al. Safety and immunogenicity of SARS-CoV-2 recombinant spike protein vaccine in children and adolescents in India: a phase 2-3 randomized clinical trial. JAMA Pediatr. https://doi.org/10.1001/jamapediatrics.2023.2552 (2023).
Áñez, G. et al. Safety, immunogenicity, and efficacy of the NVX-CoV2373 COVID-19 vaccine in adolescents: a randomized clinical trial. JAMA Netw. Open 6, e239135 (2023).
Zhang, Y. et al. Safety, tolerability, and immunogenicity of an inactivated SARS-CoV-2 vaccine in healthy adults aged 18-59 years: a randomised, double-blind, placebo-controlled, phase 1/2 clinical trial. Lancet Infect. Dis. 21, 181–192 (2021).
Wu, Z. et al. Safety, tolerability, and immunogenicity of an inactivated SARS-CoV-2 vaccine (CoronaVac) in healthy adults aged 60 years and older: a randomised, double-blind, placebo-controlled, phase 1/2 clinical trial. Lancet Infect. Dis. 21, 803–812 (2021).
Han, B. et al. Safety, tolerability, and immunogenicity of an inactivated SARS-CoV-2 vaccine (CoronaVac) in healthy children and adolescents: a double-blind, randomised, controlled, phase 1/2 clinical trial. Lancet Infect. Dis. 21, 1645–1653 (2021).
Tanriover, M. D. et al. Efficacy and safety of an inactivated whole-virion SARS-CoV-2 vaccine (CoronaVac): interim results of a double-blind, randomised, placebo-controlled, phase 3 trial in Turkey. Lancet 398, 213–222 (2021).
Palacios, R. et al. Double-blind, randomized, placebo-controlled phase III clinical trial to evaluate the efficacy and safety of treating healthcare professionals with the adsorbed COVID-19 (inactivated) vaccine manufactured by Sinovac - PROFISCOV: a structured summary of a study protocol for a randomised controlled trial. Trials 21, 853 (2020).
WHO. Prequalification of medical products (IVDs, medicines, vaccines and immunization devices, vector control), 2021. https://extranet.who.int/pqweb/vaccines/who-recommendation-sinovac-covid-19-vaccine-vero-cell-inactivated-CoronaVac.
Fadlyana, E. et al. A phase III, observer-blind, randomized, placebo-controlled study of the efficacy, safety, and immunogenicity of SARS-CoV-2 inactivated vaccine in healthy adults aged 18-59 years: an interim analysis in Indonesia. Vaccine 39, 6520–6528 (2021).
McMenamin, M. E. et al. Vaccine effectiveness of one, two, and three doses of BNT162b2 and CoronaVac against COVID-19 in Hong Kong: a population-based observational study. Lancet Infect. Dis. 22, 1435–1443 (2022).
Andrews, N. et al. Covid-19 vaccine effectiveness against the Omicron (B.1.1.529) variant. N. Engl. J. Med. 386, 1532–1546 (2022).
Chenchula, S., Karunakaran, P., Sharma, S. & Chavan, M. Current evidence on efficacy of COVID-19 booster dose vaccination against the Omicron variant: a systematic review. J. Med. Virol. 94, 2969–2976 (2022).
Li, M. et al. COVID-19 vaccine development: milestones, lessons and prospects. Signal Transduct. Target Ther. 7, 146 (2022).
Zhou, Y., Zhi, H. & Teng, Y. The outbreak of SARS-CoV-2 Omicron lineages, immune escape, and vaccine effectivity. J. Med. Virol. 95, e28138 (2023).
Jara, A. et al. Effectiveness of CoronaVac in children 3-5 years of age during the SARS-CoV-2 Omicron outbreak in Chile. Nat. Med. 28, 1377–1380 (2022).
Florentino, P. T. V. et al. Vaccine effectiveness of CoronaVac against COVID-19 among children in Brazil during the Omicron period. Nat. Commun. 13, 4756 (2022).
Muñoz, F. M. et al. Evaluation of BNT162b2 Covid-19 vaccine in children younger than 5 years of age. N. Engl. J. Med. 388, 621–634 (2023).
HQ WHO. Enhancing readiness for Omicron (B.1.1.529): technical brief and priority actions for Member States. https://www.who.int/ (2021).
Standardization WECoB. WHO Guidelines on Non-clinical Evaluation of Vaccines. Tech. Rep. Ser. 927 (WHO 2005).
Administration. CNMP. Guidelines for grading standards of adverse events in clinical trials of preventive vaccines (accessed 29 March 2020) https://www.nmpa.gov.cn/xxgk/ggtg/ypggtg/ypqtggtg/20191231111901460.html.
China NHCotPsRo. Diagnosis and treatment plan for COVID-19 (trial version 8). Chin. J. Clin. Infect. Dis. 13, 321–328 (2020).
Acknowledgements
We are grateful to all the participants and their guardians who volunteered for this study. We also thank all the study site personnel for their contributions. We especially acknowledge the study team members from 37 sites listed in Supplementary Note 6, for their contributions to this study. Thank you for the cooperation and support provided by Pontificia Universidad Católica de Chile and Numolux Group (Pty) Ltd.
Author information
Authors and Affiliations
Contributions
Q.X., Y.Y., X.M., H.Z., and G.Z contributed to the study design, data interpretation, clinical trial management, writing, or revising the manuscript. Z.J. did the data interpretation and statistical analysis. T.H.T. contributed ideas to the study conceptualization and design and worked as Malaysia’s national coordinating principal investigator. J.Y., K.W., and H.Y. conducted laboratory test of neutralizing antibodies. All the authors critically reviewed and approved the final manuscript as submitted for publication.
Corresponding authors
Ethics declarations
Competing interests
Q.X., Y.Y., and J.Y. are employees of Sinovac Life Sciences Co., Ltd. X.M., H.Z., and G.Z. are the employees of Sinovac Biotech Co., Ltd. All other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xin, Q., Wang, K., Toh, TH. et al. Efficacy, immunogenicity and safety of CoronaVac® in children and adolescents aged 6 months to 17 years: a multicenter, randomized, double-blind, placebo-controlled phase III clinical trial. Nat Commun 15, 6660 (2024). https://doi.org/10.1038/s41467-024-50802-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50802-2
