
Abstract
The quest for targeted therapies is critical in the battle against cancer. The RAS/MAP kinase pathway is frequently implicated in neoplasia, with ERK playing a crucial role as the most distal kinase in the RAS signaling cascade. Our previous research demonstrated that the interaction between ERK and MYD88, an adaptor protein in innate immunity, is crucial for RAS-dependent transformation and cancer cell survival. In this study, we examine the biological consequences of disrupting the ERK-MYD88 interaction through the ERK D-recruitment site (DRS), while preserving ERK’s kinase activity. Our results indicate that EI-52, a small-molecule benzimidazole targeting ERK-MYD88 interaction induces an HRI-mediated integrated stress response (ISR), resulting in immunogenic apoptosis specific to cancer cells. Additionally, EI-52 exhibits anti-tumor efficacy in patient-derived tumors and induces an anti-tumor T cell response in mice in vivo. These findings suggest that inhibiting the ERK-MYD88 interaction may be a promising therapeutic approach in cancer treatment.
Similar content being viewed by others


Introduction
In the course of carcinogenesis, cancer cells hijack and rewire cellular programs to acquire selective proliferation, survival, and adaptive advantages1. Indeed, the RAS/MAP kinase pathway is frequently over-activated in cancer, and since the effector proteins of this pathway (RAF, MEK, and ERK) are kinases, several small molecule kinase inhibitors have been developed and are used in the clinic. This approach has been demonstrated to be effective in shutting down the RAS signal to drive cells into an underactive state and restore control over tumor growth. However, clinical practice has revealed that blocking RAS signaling at the kinase level can be evaded by cell adaptation and leads to treatment resistance. This underscores the necessity for exploring alternative strategies.
We previously demonstrated that MYD88, an adapter protein involved in innate immunity, interacts via its D-___domain with the D-recruitment site (DRS) of the extracellular signal-regulated kinase proteins (ERK1 and ERK2, hereafter designated as ERK) and that this protein-protein interaction (PPI) is required for RAS-dependent cell transformation and cancer cell survival in vitro and in vivo2,3, suggesting that inhibiting this PPI could be an effective strategy for cancer therapy.
Here, we describe two families of small chemical molecules that inhibit ERK-MYD88 interaction, including the proof-of-concept compound, EI-52. We show that EI-52 induces rapid, immunogenic apoptosis specific to cancer cells in vitro and that it exerts an anti-tumoral activity in vivo via both the direct killing of tumor cells and the activation of an anti-tumoral T-cell response.
Results
Identification and characterization of EI-52, a small molecule that binds ERK1/2 and inhibits the ERK-MYD88 interaction
To identify molecules that disrupt the interaction between the MYD88 D-___domain and ERK DRS, we screened a library of about 66,000 small molecules in a homogeneous time-resolved fluorescence (HTRF) assay using a short peptide containing the MYD88 D-___domain and a recombinant ERK protein. Hits that inhibited the ERK-MYD88 interaction by more than 40% at 20 µM were retested and confirmed using newly synthesized lots (Supplementary table 1). At this stage, 75 compounds (hit rate 1.13%) belonging to 5 different chemical series were selected and ranked based on physicochemical parameters (solubility, logP, pKa) and structure-activity relationship. Finally, two distinct chemical families (benzimidazole and spiro scaffolds) with similar in vitro and cellular activities were chosen for further development. The proof-of-concept benzimidazole compound EI-52 (Fig. 1A) exhibits binding to both ERK1 and ERK2, as determined by fluorescence quenching (Fig. 1B), and inhibits ERK1 and ERK2 interaction with MYD88 by HTRF (Fig. 1C). The Kd values for ERK1 and ERK2 in both the fluorescence quenching and HTRF assays are in the same order of magnitude.

A Chemical structure of EI-52. B Binding of EI-52 to ERK1 and ERK2 by fluorescence quenching. Representative of two independent experiments. C ERK1/2-MYD88 interaction inhibition by HTRF. Mean ± SEM from three independent experiments. D EI-52 docking on ERK. Negatively charged residues are in red. Zone A includes the Common Docking (CD) ___domain (red dotted circle). MKP3 peptide in magenta. Key hydrophobic/aromatic residues in yellow. E MYD88-ERK1 interaction by BiFC and flow cytometry. Mean VENUS Fluorescence Intensity ± SEM at varying EI-52 concentrations (left panel) from three independent experiments, Tukey’s one-way ANOVA; 8 vs 0 h *p = 0.0446, 10- and 12 vs 0 h ****p < 0.0001 (95% confidence interval). F PLA of endogenous MYD88 and ERK in HCT116 cells treated with EI-52 (8 μM) or DMSO for 3 h using antibodies against MYD88 and ERK. The interactions are visualized as red dots and the nuclei are counterstained with DAPI (blue). The number of interactions per cell, quantified by ImageJ, is expressed as the fold difference relative to DMSO treatment. Mean ± SEM from three independent experiments, two-tailed Student’s t-test; *p = 0.0161 (95% confidence interval). G PLA in HeLa cells transfected with 6His-WT or 6His-D319N ERK2 and Flag-MYD88. Cells were treated with EI-52 (10 μM) or DMSO for 8 h using anti-His and -Flag. Detection was carried out as in 1 F. Mean ± SEM from three independent experiments, Tukey’s one-way ANOVA; *p = 0.0303 (95% confidence interval). H Cell recovery and cell death following EI-52 or ERK kinase inhibitors over 48 h at 8 μM. Mean ± SEM from three independent experiments, two-way Tukey’s ANOVA; Cell recovery at 48 h EI-52 vs DMSO *p = 0.0123, LY32149966 vs DMSO p = 0.0145; Death at 48 h EI-52 vs DMSO *p = 0.0456 (95% confidence interval). I Apoptosis (annexin V/PI) in HCT116 cells treated for 24 h with DMSO or EI-52 (8 μM) by flow cytometry. Mean percentage of annexin V+ cells ± SEM from three independent experiments, two-tailed Student’s t-test; **F = 0.0016 (95% confidence interval). Source data are provided as a Source Data file.
To determine the putative ligand binding site(s) of EI-52 on ERK, we used the P2Rank pocket prediction engine4 and identified several binding pockets, including the catalytic site (top predicted pocket not involved in EI-52 binding) and a relatively elongated groove (second best predicted pocket) that can be subdivided from an energetic and topological standpoint into two regions. These two sub-pockets will hereafter be called Zone A and Zone B (Fig. 1D). Several peptides have been co-crystallized with ERK and shown to bind to Zones A and B5,6, prompting us to focus our docking experiments on this binding groove. Zone A, which contains the DRS, displays several hot-spot residues, as estimated with pyDockEneRes7. This cavity contains mainly aromatic residues and three negatively charged residues (Fig. 1D). Contiguous to Zone A, Zone B makes a limited number of strong interactions with the co-crystallized peptides. It forms a small, partly hydrophobic cavity, which includes the ED site. Note that the catalytic site is located away (more than 10 Å) from Zones A and B. EI-52 was predicted to bind to Zone A, and the two main orientations with the highest binding scores and computed binding free energy are shown (Fig. 1D). While the binding scores for EI-52 from the three scoring functions used did not favor one pose over the other, the computed ∆G values favored pose 1. In both orientations, the dimethylaminophenyl group of EI-52 is predicted to interact with aspartic and glutamic acid residues (D316, D319, E79 in ERK2) of Zone A. This is consistent with the interactions described for ERK/D-___domain peptide co-crystals8,9. Interestingly, a representative compound from the spiro family (SP-26), was docked using the same protocol as EI-52, and the best ∆G value showed a binding pose similar to EI-52 pose 1 with SP-26 slightly protruding in the Zone B region (Supplementary Fig. 1A). The estimated binding free energy appears slightly more favorable for SP-26 (∆G = −32.7 kcal/mol) than to EI-52 (∆G = −29.9 kcal/mol).
The ability of EI-52 to inhibit ERK-MYD88 PPI was next investigated in cells, using two different approaches. First, a bimolecular fluorescence complementation (BiFC) assay was performed in HEK293T cells transfected with ERK and MYD88 tagged with the N- and C-terminal fragments of Venus protein, respectively. It showed that EI-52 inhibits ERK-MYD88 interaction in a dose-dependent manner in the micromolar range (Fig. 1E, Supplementary Fig. 2A). Using the same technique, we showed that the spiro compound SP-26 also inhibits ERK-MYD88 PPI in cells (Supplementary Fig. 1B). Then, using a proximity ligation assay (PLA), we showed that EI-52 inhibits endogenous ERK-MYD88 interaction in HCT116 cells (Fig. 1F, Supplementary Fig. 2B).
According to our predicted model (Fig. 1D), the aspartic acid at position 319 of ERK is important for EI-52 binding to ERK. To definitively establish target engagement in cellulo and confirm the in silico model, we conducted PLA in cells transfected with tagged MYD88 and ERK (WT or bearing the D319N mutation) constructs in the presence or absence of EI-52. Quantification revealed that while EI-52 strongly inhibited MYD88 interaction with WT ERK, it was unable to inhibit its interaction with D319N ERK (Fig. 1G, Supplementary Fig. 2C). This result confirms that aspartic acid in position 319 is indeed critical for EI-52 binding to ERK and inhibition of ERK-MYD88 PPI.
Taken together, these in vitro and in cellulo data conclusively demonstrate that EI-52 directly binds to the ERK DRS and inhibits ERK-MYD88 interaction.
Blocking ERK-MYD88 interaction with EI-52 induces cancer cell death
Having previously shown that the ERK-MYD88 PPI is key for cancer persistence3, we investigated whether treatment with EI-52 affects cancer cell survival. We treated the human colon cancer cells HCT116 (K-RasG13D) with EI-52 (8 µM) or ERK kinase inhibitors at the same concentration, and evaluated cell recovery and death over 48 h. We observed that EI-52 induced a rapid and robust cancer cell death (Fig. 1H). Importantly, the spiro compound SP-26, which was predicted to bind to the ERK DRS, also triggered cell death in HCT116 (Supplementary Fig. 1C). As expected, ERK kinase inhibitors only reduced cell recovery without inducing cell death (Fig. 1H). Flow cytometry analysis showed that HCT116 cells treated with EI-52 for 24 h exposed annexin V, indicating that EI-52-induced cell death is apoptotic (Fig. 1I, Supplementary Fig. 2D).
In addition, we evaluated the ability of dozens of EI-52 analogs to inhibit ERK-MYD88 interaction and induce cancer cell death. A limited structure-activity relationship (SAR) study showed that certain close analogs functionally behave like EI-52, while others do not inhibit MYD88-ERK interaction or induce HCT116 cell death. A representative sample of such analogs is shown in Supplementary Table 2. These results preclude the possibility that the biological activity of EI-52 is due to a non-specific effect of the benzimidazole chemical scaffold and form a basis for subsequent drug optimization efforts.
In light of these results, we extended the analysis to 301 cell lines from the OncopanelTM collection (Eurofins). Cell lines were tested with 10 concentrations of EI-52 in a two-fold dilution series starting from 30 μΜ. Cell viability was measured, and IC50 was calculated for each cell line. Out of 301 cell lines, 80% responded with an IC50 between 3.4 and 11 µM (intermediate sensitivity), 10% below 3.4 µM (high sensitivity), and 10% above 11 µM (low or no sensitivity) (Fig. 2A). This range of EI-52 concentrations required to kill cancer cells is consistent with the biological data described in Fig. 1, in which the effective concentrations of EI-52 ranged from 4–12 µM, depending on the type of experiment. Whereas the majority of the cancer cell lines tested showed high or intermediate sensitivity to the ERK-MYD88 PPI inhibitor EI-52 at the micromolar range, the response was heterogeneous within tumor types (Fig. 2B). Interestingly, comparing the EI-52 response pattern of these 301 human cancer cell lines to that of 63 anti-tumor reference compounds showed a distinctive pattern of sensitivity, different from that of kinase inhibitors and other classes of drugs (Fig. 2C), suggesting that EI-52 indeed represents a class of antitumor agents with a unique mechanism of action.

A Waterfall plot of IC50 values of 301 cell lines of the OncoPanel™ collection. Each bar represents one cell line, colored by relative drug response. B Classification of the response to EI-52 according to the origin of the cancer cell lines. Pie charts showing the distribution of cell lines classified as sensitive (red), relatively insensitive (blue), or intermediate (gray) within each cancer type represented in OncoPanel™. The number of cell lines for each tissue/tumor type is indicated in parentheses. C Dendrogram with EI-52 and 63 anti-tumor reference compounds clustered by drug sensitivity in 301 cell lines. Source data are provided as a Source Data file.
Cytosolic accumulation of phospho-ERK following EI-52 treatment is accompanied by an integrated stress response, leading to apoptotic death of cancer cells
To elucidate the mechanism of action of EI-52 and the consequences of ERK-MYD88 PPI inhibition on ERK signaling, we first ascertained that EI-52 does not indirectly interfere with ERK kinase activity. Using an in vitro kinase assay, we found that the catalytic activity of ERK remained intact following treatment with EI-52 at concentrations ranging from 1–100 µM, whereas it was lost upon treatment with increasing concentrations of the pan-kinase inhibitor K252a or the ERK kinase inhibitor FR18020410,11 (Fig. 3A). As expected, treatment with EI-52 induces cell death, resulting in an increased percentage of SubG0 cells (Fig. 3B left panel, Supplementary Fig. 3). However, focusing on the live cells, we found that EI-52 does not disrupt the cell cycle, consistent with the continued kinase activity of ERK (Fig. 3B right panel, Supplementary Fig. 3). Moreover, EI-52 did not affect ERK phosphorylation (Fig. 3C), consistent with the ability of MEK to interact with ERK independently of the DRS12. In addition, and in contrast to the kinase inhibitor U0126, treatment with EI-52 did not compromise the phosphorylation of the ERK substrate RSK (Fig. 3C), confirming in cellulo that ERK kinase activity is maintained during EI-52 treatment. Interestingly, the expression of the phosphatase DUSP5, which associates with ERK and controls its phosphorylation, was increased upon treatment of HeLa cells with EI-52 (Fig. 3C). DUSP5 was also increased in HCT116 cells harboring a K-Ras mutation that were treated with EI-52 and SP-26 (Supplementary Fig. 1D). Consistent with the crucial role of aspartic acid 319 in EI-52 binding to ERK and inhibiting ERK-MYD88 interaction (Fig. 1G), we observed that expression of DUSP5 is further increased in cells overexpressing WT ERK, but not in those transfected with the D319N ERK mutant (Fig. 3D). This result demonstrates that the biological effects of EI-52 depend on its ability to interact with ERK and block ERK-MYD88 interaction.

A In vitro ERK1 kinase assay with kinase inhibitors K252a or FR180204, or EI-52. Mean ± SEM from three independent experiments. B Cell cycle of HCT116 cells treated with DMSO, U0126 10 μM, paclitaxel 25 nM, or EI-52 8 μM for 24 h was measured by flow cytometry. Mean ± SEM of subG0 cells from three independent experiments, Dunnett’s one-way ANOVA; ****p < 0.0001 (95% confidence interval) (left panel). Percentage of viable cells across cell cycle phases (right panel). C Starved HeLa cells were treated for 8 h with DMSO or EI-52 8 μM, with or without U0126 10 μM. Where indicated, cells were activated for 8 min with EGF 100 ng/ml. ERK targets were analyzed by western blot. Representative data of three independent experiments. D HCT116 cells were transfected with empty vector, 6His-WT, or 6His-D319N ERK2, and treated with DMSO or EI-52 8 μM for 18 h. Representative of three independent DUSP5, ERK, and His western blots. E Phosphorylated-ERK and total ERK immunofluorescence after treatment of serum starved-HeLa cells with EI-52 8 μM for 6 h. Representative of three independent experiments. F Immunofluorescence staining of phosphorylated-ERK. Serum-starved HeLa cells were pretreated for 1 h with DMSO or EI-52 8 μM then activated 10 min with 20% serum. Representative of three independent experiments. G HCT116 cells were treated for the indicated times with DMSO or 8 μM of EI-52, and ISR markers were analyzed by western blot. Representative data of three independent experiments. H HCT116 cells were pre-incubated or not with ISRIB 400 nM for 1 h then treated with EI-52 8 μM for 16 h. Mean fold change in caspase 3/7 activity ± SEM from three independent experiments, Tukey’s one-way ANOVA; *p < 0.0233 (95% confidence interval). I HCT116 cells were transfected with siHRI, siPERK, siGCN2, siPKR, or siControl, then treated with EI-52 (8 μM) or DMSO. Expression of ATF4, HRI, PERK, CGN2, PKR, and tubulin was determined by western blot. Representative of three independent experiments. Source data are provided as a Source Data file.
Disruption of ERK interactions can alter subcellular protein distribution and function13,14. It was also reported that DUSP5 can paradoxically increase and prolong ERK cytoplasmic activity15. We therefore monitored phospho-ERK localization after exposure to EI-52. We found that after 6 h of treatment, ERK localization remained unperturbed, while almost all phospho-ERK had accumulated in the cytoplasm (Fig. 3E). However, phospho-ERK accumulation in the cytoplasm is not due to an inhibition of nuclear translocation since phospho-ERK goes to the nucleus following activation with serum, regardless of the presence or absence of EI-52 (Fig. 3F).
Phospho-ERK mislocalization was accompanied by increased eIF2α phosphorylation and ATF4 and CHOP protein levels (Fig. 3G), all of which characterize an ongoing integrated stress response (ISR), which has been reported to lead to apoptosis16. Indeed, inhibition of ERK-MYD88 PPI with EI-52 triggered the caspase 3/7-mediated apoptosis of HCT116 cells that was significantly reduced by co-treatment with ISRIB, an inhibitor of the key mediator of ISR, phospho-eIF2α (Fig. 3H). Significantly, the spiro compound SP-26 also induces an increase in ATF4 expression (Supplementary Fig. 1D), further confirming that the biological effects observed following treatment with EI-52 are not due to an off-target activity. Cellular stress is sensed by four specialized kinases, PERK, GCN2, HRI, and PKR, that converge on eIF2α phosphorylation17. Using specific siRNAs, we showed that only the HRI knockdown consistently impairs ATF4 accumulation, indicating a role for HRI in EI-52-triggered ISR (Fig. 3I).
Taken together, these results indicate that EI-52 is an inhibitor of ERK-MYD88 PPI which, without affecting ERK phosphorylation or kinase activity, results in phospho-ERK delocalization accompanied by an integrated stress response that leads to apoptosis of cancer cells.
To determine whether the effect of ERK-MYD88 PPI inhibition is specific to cancer cells, we compared EI-52 activity in the colorectal cell line HCT116 to that in untransformed human colonic epithelial cells (HCEC-hTERT)18. EI-52 treatment triggered the death of the cancer cell line but not of its untransformed counterpart at the same concentration (Fig. 4A). This result could be due to the differential ability of EI-52 to activate an ISR, illustrated by the induction of a substantial increase in ATF4 expression in cancer cells but not in non-transformed cells (Fig. 4B). Furthermore, primary HUVEC isolated from 3 different donors were not sensitive to EI-52 (Fig. 4C). Collectively, these data show that EI-52 induces an integrated stress response specifically in transformed cells, leading to cell death, further demonstrating that EI-52-induced cell death is not the result of non-specific toxicity.

A Cell death/confluence ratio was measured using the IncuCyte™ Kinetic Live Cell Imaging System following treatment of transformed (HCT116) or untransformed (HCEC) cell lines with EI-52 or DMSO for 24 h. Results are expressed as mean of ratio ± SEM from three independent experiments. Statistical analysis was performed using Bonferroni’s Two-way ANOVA; HCT116 DMSO vs EI-52 ****p < 0.0001 (95% confidence interval). B Transformed or untransformed cells were treated for the indicated times with DMSO or EI-52 8 μM, and the ISR marker ATF4 was analyzed by western blot. Representative data of three independent experiments are shown. C Caspase 3/7 activity in HCT116 cells or HUVEC (3 donors) was assessed after 16 h treatment with EI-52 8 μM or DMSO. Results are expressed as the mean fold activation of caspase 3/7 in EI-52-treated samples relative to DMSO-treated controls ± SEM. Statistical significance was assessed using a two-tailed one-sample t-test; **p = 0.0037 (95% confidence interval). Source data are provided as a Source Data file.
ERK-MYD88 PPI inhibitor EI-52 displays anti-tumor activity in mice and in patient-derived tumors
To investigate the anti-tumoral activity of EI-52 in vivo, we tested the compound in a syngeneic Lewis lung carcinoma (LLC) model. First, we established that EI-52 induces apoptosis of LLC cells in vitro (IC50 = 4 μM). Since EI-52 was not optimized for oral administration, a pharmacokinetic study was carried out for intraperitoneal (i.p.) administration. Bioavailability was 52.9% and the area under the curve (AUC) 1129 ng/ml/h (Supplementary Table 3), corresponding to 3μmol/L/h. These values are compatible with the IC50 determined in vitro; therefore, proof-of-concept studies could be conducted using i.p. administration in mice. LLC cells were implanted subcutaneously, and when tumors reached 100 mm3, mice were treated daily by intraperitoneal injection of EI-52, which resulted in a dose-dependent inhibition of tumor growth (Fig. 5A). More notably, EI-52 also displayed a protective effect in the aggressive Kras-LA2 spontaneous lung tumor model19. In this model, sporadic activation of K-RAS leads to lung tumors first detectable as small pleural nodules at 1 week of age. As shown in Fig. 5B, EI-52 treatment for 10 weeks induced a significant reduction of the tumor load in the lungs of treated mice. Anatomopathological analysis of liver, kidney, and spleen from mice treated for 10 weeks with EI-52 did not show signs of toxicity at the end of the protocol (Supplementary Fig. 4). Moreover, none of the mice treated with EI-52 died during the course of the study (n = 107). Finally, the toxicity of EI-52 was assessed in chicken embryonic chorioallantoic membranes (CAM), an alternative predictive model of acute toxicity for new drugs20. EI-52 treatment resulted in no embryo deaths or malformations (Table 1) in this highly sensitive model, further indicating that inhibition of ERK-MYD88 PPI may be safe in vivo.

A C57BL/6 mice (n = 9/group) injected subcutaneously with Lewis Lung Carcinoma cells were treated daily by intraperitoneal injection with vehicle (PBS, 40% PEG 400, 20% DMSO) or with 25 or 50 mg/kg of EI-52. Tumor volume was measured twice a week with an electronic caliper. B Five-week-old K-rasLA2 mice (WT n = 5, K-Ras mutant n = 7) were treated intraperitoneally 5 times a week for 10 weeks with 25 mg/kg of EI-52. Tumor load was evaluated on a Perkin Elmer Quantum FX microCT scan. Left panel: example of image reconstitution of lungs from 2 mice treated for 10 weeks with vehicle or with EI-52. Right panel: tumor load in the lungs of vehicle- or EI-52-treated mice at 10 weeks. Statistical analysis was performed using a two-tailed Mann-Withney test; *p = 0.0177 (95% confidence interval). C Patient-derived cancer tumoroids from colon (male, age 77) and lung (female, age 64) were treated for 48 h with DMSO or indicated concentrations of EI-52. Cell death (PI-positive) and apoptosis (caspase 3/7 cleavage) were observed by OPERA imaging. Shown are two of three independent experiments. D Thick (250 μm) sections of surgical pieces from head and neck cancer patients (3 females and 8 males, age 54–82) were cultured in optimized medium in presence of DMSO or EI-52 (8 μM). 24 h later, thin sections were sliced and stained for the apoptotic marker cleaved-PARP (left panel). IHC scoring allows a semi-quantitative analysis of apoptosis (right panel). Mean score ± SEM from 11 samples, statistical analysis was performed using a two-tailed Wilcoxon t-test; *p < 0.0469 (95% confidence interval). Source data are provided as a Source Data file.
We next investigated the effect of EI-52 in ex vivo human tumors using available surgical resection samples. Patient-derived tumor organoids (PDOs) were generated from fresh colorectal and lung tumor surgical samples and treated for 48 h at different doses of EI-52. Using propidium iodide uptake and caspase 3/7 activation, we demonstrated that of ERK-MYD88 PPI inhibition with EI-52 leads to apoptosis in both human colon and lung tumoroids (Fig. 5C). We then evaluated EI-52 efficacy in undissociated primary human cancers. We generated thick sections from head and neck cancer surgical pieces. Then, the tumor slices were cultured and treated with vehicle or EI-52 for 24 h. In these ex-vivo models, EI-52 treatment resulted in strong c-PARP staining that was almost exclusively restricted to tumor cells (Fig. 5D), consistent with the tumor specificity of EI-52-triggered apoptosis described above. Altogether, these results show that disrupting ERK-MYD88 PPI with EI-52 can kill human cancer cells ex vivo without apparent toxicity.
Inhibition of ERK-MYD88 PPI with EI-52 triggers immunogenic cancer cell death and an anti-tumoral T cell response
To gain insight into the effect of EI-52 on global transcriptional activity, gene expression was measured on Affymetrics arrays. Transcriptomic analysis of HCT116 cells treated for 16 h with EI-52 revealed upregulated transcription of NF-κB-dependent inflammatory genes (Fig. 6A), including chemokines (Fig. 6B), whose expression and secretion was confirmed at the RNA (Fig. 6C) and protein levels (Fig. 6D). In contrast, the MEK kinase inhibitor U0126 did not induce chemokine production (Fig. 6D).

A RNAseq GSEA plot and (B) top upregulated genes in HCT116 cells treated for 18 h with EI-52 (6 μM) or DMSO. C Chemokine mRNA from HCT116 cells treated for 16 h with DMSO or EI-52 8 μM. Mean ± SEM from three independent experiments, two-tailed one-sample t-test. CXCL8 **p = 0.0029; CXCL1 *p = 0.0312; CXCL2 *p = 0.0439 (95% confidence interval). D CXCL8, CXCL1, and CXCL2 in supernatants of HCT116 cells treated with DMSO, MEK inhibitor U0126 10 μM, or EI-52 8 μM for 24 h. Mean ± SEM from four (CXCL8) and three (CXCL1, CXCL2) independent experiments, unpaired two-tailed Student’s t-test; CXCL8 **p = 0.0073; CXCL1 *p = 0.0479; CXCL2 *p = 0.0325 (95% confidence interval) (E) NF-κB, CXCL1, and CXCL8 luciferase activity 16 h post-treatment with EI-52 8 μM or DMSO. Mean of treatment /DMSO ratio ± SEM from four (CXCL8) and three (CXCL1, CXCL2) independent experiments, unpaired two-tailed Student’s t-test; NF-κB *p = 0.0395; CXCL1 *p = 0.0165; CXCL8 *p = 0.0108 (95% confidence interval). F IL8 secretion in supernatant of HCT116 cells transfected for 24 h with control or p65 siRNA, then treated 16 h with DMSO or EI-52 8 μM. Mean ± SEM from three independent experiments, unpaired two-tailed Student’s t-test; ***p = 0.0002 (95% confidence interval) (G) Co-immunoprecipitation of ERK1-HA with p65-Flag after treatment of transfected HEK293T cells with EI-52 8 μM. Representative data of three independent experiments. H Extracellular ATP and HMGB1 in supernatants of HCT116 cells treated for 24 h with DMSO, U0126 10 μM, or EI-52 8 μM. Results are expressed as mean of treatment/DMSO ratio ± SEM from four (e-ATP) or three (HMGB1) independent experiments, two-tailed one sample t-test; e-ATP: U0126 *p = 0.0171; EI-52 **p = 0.0034 (95% confidence interval). HMGB1: EI-52 *p = 0.0157 (95% confidence interval). I Chemotaxis of migrating THP1 cells was evaluated in Boyden chamber containing supernatant of HCT116 cells treated 24 h with control, U0126 10 μM, or EI-52 8 μM. Mean ± SEM from three independent experiments, Dunnett’s one-way ANOVA; **EI-52 p = 0.0013 (95% confidence interval). Source data are provided as a Source Data file.
We therefore investigated the status of p65/RELA, one of the main NF-κB transcription factors. We treated HCT116 cells with EI-52 and found that it leads to the activation of NF-κB, CXCL1, and CXCL8 transcription (Fig. 6E). Knocking down p65 strongly inhibited IL-8 production in response to EI-52, indicating that NF-κB is required for its secretion (Fig. 6F). Interestingly, transfected p65 and ERK co-immunoprecipitated in HEK293T cells treated with EI-52 (Fig. 6G), indicating that interference with the ERK CD ___domain not only modifies the ERK protein complex by losing interactions but also by gaining other partners such as p65, with different downstream biological outcomes.
Production of chemokines by dying cancer cells has been reported to be an indication of an immunogenic cell death (ICD) program21,22. Indeed, EI-52-induced cell death was also accompanied by the release of damage-associated molecular patterns such as ATP, which has chemotactic activity, and HMGB1, an endogenous TLR4 ligand (Fig. 6H). Consistent with the ability of EI-52 to induce the production of the chemotactic mediators CXCL8 and ATP by dying cells, we showed that supernatant from EI-52-treated cells was able to attract THP1 macrophages (Fig. 6I). In contrast, U0126 did not induce either mediators of ICD or chemoattraction of macrophages by HCT116 cells (Fig. 6H, I), further highlighting the functional differences between inhibition of ERK kinase activity and ERK-MYD88 PPI.
To investigate ICD in vivo, we first quantified EI-52 in the tumors and verified whether it induces direct killing of murine tumor cells implanted subcutaneously in mice. Twenty-four hours after intraperitoneal administration, 240 – 440 ng/g of EI-52 were found in the tumors (Table 2), which exhibited strong PARP cleavage (Fig. 7A, B). These data demonstrate that systemic EI-52 administration leads to efficient tumor exposure to the compound and rapid apoptotic cancer cell death in vivo.

Following s.c. implantation of syngeneic CT26 cells, BALB/c mice were treated intraperitoneally for 24 h with vehicle or 50 mg/kg of EI-52 and tumors were collected. A Cleaved-PARP was observed by IHC staining, representative of four separate mice or (B) by western Blot of tumor cell lysates recovered from four mice per treatment. C Following s.c. implantation of CT26 cells, syngeneic wild-type and nude BALB/c mice (n = 10/group) were treated daily with vehicle or with 25 mg/kg of EI-52 intraperitoneally. Tumor volume was measured twice a week with an electronic caliper. Results are presented as Mean tumor volume ± SEM, Sidak’s two-way ANOVA, Balb/c day 14 Vehicle/vs EI-52 ****p < 0,0001 (95% confidence interval). D Following s.c. implantation of CT26 cells, mice (n = 10/group) were treated daily with vehicle or with 25 mg/kg of EI-52 intraperitoneally. At days 0, 3, and 5, mice received an intraperitoneal injection of 200 μg isotype control or anti-PD1 antibody. Tumor volume was measured twice a week with an electronic caliper. Results are presented as Mean tumor volume ± SEM, Tukey’s two-way ANOVA, day 14 Vehicle/isotype control vs EI-52/isotype control *p = 0.0115, EI-52/isotype control vs EI-52/anti-PD1 *p = 0.0335 (95% confidence interval). Source data are provided as a Source Data file.
Next, we investigated a possible T-cell contribution to the antitumor activity of EI-52. Murine CT26 colon cancer cells were implanted subcutaneously into syngeneic wild-type and nude BALB/c mice, which differ by the presence or absence of T cells, respectively. When tumors reached 100 mm3, mice were treated daily with a suboptimal dose of EI-52 (25 mg/kg) to limit direct killing and thus facilitate the detection of a possible contribution from T-cell immunity. Indeed, EI-52 treatment only weakly slowed the tumor growth in nude mice, whereas it had a much greater antitumor effect in T-cell competent mice. These results demonstrate that treatment with EI-52 triggers immunogenic cancer cell death and an anti-tumoral T cell response in vivo (Fig. 7C). Having shown a role for T cells in EI-52 tumor growth inhibition, we assessed the effect of combining EI-52 with an immune checkpoint antibody. Murine CT26 colon cancer cells were implanted subcutaneously into syngeneic mice and when tumors reached 100 mm3, mice received three injections of 200μg of anti-PD1 at days 0, 3, and 6 in combination with daily injections of EI-52 (25 mg/kg). As shown in Fig. 7D, the combination of anti-PD1 with EI-52 significantly improves anti-tumoral efficacy.
Discussion
The extracellular signal-regulated kinase proteins represent a protein kinase hub, interfaced to different cellular sub-routings regulating cell division and survival, cellular biomass and metabolism, as well as differentiation and tissue fate23.
While the rules that drive ERK signal across this functional diversity are not fully understood, some key elements regarding ERK signal integration have been identified. First, although ERK targets a vast catalog of proteins with more than five hundred direct substrates24, specific substrate repertoires are selected by at least two distinct interaction modes. Indeed, the specificity and fidelity of ERK-induced pathways rely on docking sites distant from the catalytic site, known as the D- and F-recruitment sites (DRS and FRS, respectively), which interact with conserved motifs, the D or F domains, harbored by the interacting proteins25. These interaction sites are also used by scaffold proteins to regulate the spatial distribution of ERK, decoupling its nuclear from its cytosolic functions. Finally, in addition to this spatial assignation, ERK-signaling outcome is influenced both in duration and frequency by a set of phosphatases. Together, these elements sustain the diversity and selectivity of ERK responses to extra- and intracellular cues.
For over two decades, researchers in the field of cancer have been striving to develop inhibitors for the MAPK enzymatic activity of the RAS pathway. These inhibitors have been found to impact cell proliferation by affecting the quantitative signaling output of ERK. While interruption of RAS signaling eventually affects cell survival and death, this process is typically slow, resulting from prolonged cell cycle arrest.
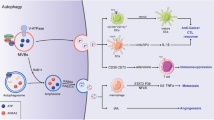
In this report, we present the discovery of two chemical families (spiro and benzimidazole) that target ERK through its DRS, leading to the loss of ERK-MYD88 interaction and perturbation of the ERK complex. This results in early immunogenic apoptosis and anti-tumoral T cell response in vivo. The detailed molecular consequences of ERK-MYD88 PPI inhibition with EI-52 on cancer cells need further investigation. However, the atypical regulation of ERK partner proteins, cell death kinetics, and cell line sensitivity spectra support a distinct mode of action. Our research shows that EI-52 induces a mislocalization of activated ERK, accompanied by an integrated stress response that triggers rapid apoptosis (Fig. 8). These findings align with previous research indicating that activation of ERK in inappropriate subcellular locations leads to cell death26,27.

Biological outcomes of ERK-MYD88 PPI vs ERK enzymatic inhibition.
ERK PPI inhibition has recently emerged as a promising alternative to direct kinase inhibition strategies. For example, selective ERK docking inhibition has been explored by Shapiro et al.28, who described ERK PPI inhibitors that mainly inhibit the interaction between ERK and RSK, leading to RSK inactivation. More recently, Kaoud and colleagues identified a small molecule that binds to the ED pocket of ERK2 DRS29. Treatment with this molecule inhibited ERK and RSK phosphorylation, resulting in a marked G1 growth arrest followed by late apoptosis. Herrero et al. developed an inhibitor of ERK dimerization, which binds outside the DRS and targets the cytoplasmic function of ERK without altering its nuclear function. This dimer inhibition compromises RSK activation and results in proliferation blockade and apoptosis specifically in cancer cells carrying RAS or BRAF mutations30. In contrast, EI-52, which docks in the CD pocket (Zone A) of the DRS, was selected based on inhibition of ERK-MYD88 PPI, without subsequent reduction of ERK or RSK phosphorylation. This difference in ERK partner disruption could explain the biological consequences of EI-52 treatment.
We have shown that EI-52 induces an ISR, a crucial cellular mechanism that helps cells cope with adverse conditions. The primary role of ISR is to enable cells to adapt, survive, and, when possible, recover from stressors31. However, if the cellular stress is severe, either in intensity or in duration, it will overwhelm the capacity of the adaptive response to resolve it and additional components become activated to execute cell death31. The tightly regulated nature of the ERK MAPK signaling pathway may explain why disruption of ERK complexes results in the activation of the integrated stress response, leading to cell death.
Moreover, cancer cells, which exhibit increased growth rates, also have heightened protein synthesis, resulting in a higher baseline level of the ISR in comparison to normal cells32,33. This observation could provide an explanation for the selective killing of cancer cells by EI-52, which induces a stronger ISR.
Unexpectedly, we found that ERK-MYD88 PPI inhibition induces NF-κB-dependent CXCL8 secretion, as well as ATP and HMGB1 release. These factors are mediators of an immunogenic death, which was previously known to be associated with chemotherapy, oncolytic viruses, antibiotics, cardiac glycosides, and nano pulse activation34,35,36,37,38. Interestingly, several studies have linked ICD to ISR39,40, raising the possibility that ISR activation following EI-52 treatment participates in ICD. Regardless of the precise mechanism underlying EI-52-induced ICD, our in vivo data showing that the T-cell compartment significantly contributes to the efficacy of EI-52, in addition to the beneficial combination with anti-PD1, suggests a role for ICD in the overall efficacy of EI-52.
In conclusion, our findings suggest that disrupting ERK interaction with MYD88 could be an attractive approach in cancer treatment.
Methods
This research complies with all relevant ethical regulations.
Ethics approvals
Mouse experiments were conducted in agreement with the local ethics committee, (Comité d’Evaluation Commun au Centre Léon Bérard, à l’Animalerie de transit de l’ENS, au PBES et au laboratoire P4; CECCAP), authorization numbers #5681, #9283. The limit for tumor volume is 2500 mm3, which was respected in all experiments.
Ex-vivo and Patient-derived tumoroid experiments were conducted according to the French regulations on the protection of persons. For the BRC of CLB (n°BB-0033-00050) biological material collection and retention activity is declared to the Ministry of Research (DC-2008-99 and AC-2019-3426). Samples were used in the context of patient diagnosis. Non used part of the samples might be used for research if patient is not opposed to it (information notice transmitted to each patient).
The BRC is compliant to GDPR requirements and the CNIL (French National Commission for Computing and Liberties) law. The BRC is certified according to AFNOR NFS96900 (N° 2009/35884.2) and ISO 9001 (Certification N° 2013/56348.2).
Cell lines and reagents
Human HCT116 colorectal cancer, HeLa cervical cancer, and HEK293T embryonic kidney cell line; murine colon carcinoma CT26 and Lewis lung carcinoma LLC1, were purchased from ATCC. ERK inhibitors (GDC-0994, LY3214996, VX-11e, SCH772984) were purchased from Selleckchem. Plasmids pCMV5-rat ERK2-WT and pCMV5-rat ERK2-D319N were a gift from Natalie Ahn (Addgene plasmid # 40812 and # 40820). PKR (L-003527), HRI (L-005007), PERK (L-004883), and RELA (L-003533) ON-TARGETplus SMARTpool human siRNAs were purchased from Dharmacon,. GCN2 siRNA was purchased from Santa Cruz (sc-45644) and non-targeting siRNA from Eurofin Genomics.
Antibodies used are listed in Supplementary Table 4.
Homogeneous time-resolved fluorescence (HTRF) assay
The HTRF assay was performed in white low volume 384-well microplates (Greiner). First, 18.75 nM His-ERK1 protein (Origene) and 50 µM of drugs were mixed and incubated in a total volume of 8 µL of optimized buffer 20 mM Tris-HCl pH 8.0, 0.05% Tween20, 0.1% BSA, 2.5% DMSO for 30 min at room temperature. Then, 4 µL/well of 125 nM biotinylated MYD88 peptide (Biotin-Ahx-PLAALNMRVRRRLSLFLNVR), 8 µL/wells of 6.66 nM anti-6His-Eu cryptate (Cisbio) 3.125 nM SA-XL Streptavidine-XL665 (Cisbio) in 20 mM Tris-HCl pH 8.0, 0.05% Tween20, 0.1% BSA were added into each well and incubated for 2 h at room temperature. After incubation, HTRF signals were measured using an Infinite F500 microplate reader (TECAN) at 620 nm and 665 nm emission. Signals were analyzed with an integration time of 500 µs, and the DeltaF and Z’ values were calculated.
Fluorescence spectroscopy and titration
For fluorescence spectroscopy, recombinant ERK was diluted into 20 mM Tris-HCl pH 8. Titration was performed by adding increasing concentrations of EI52 compound diluted in 70% PEG400/30% Propylene glycol in presence of 5 µM of His-ERK. The excitation wavelength was 295 nm and the emission was recorded between 310 nm – 380 nm in a spectrofluorimeter (SAFAS). The fluorescence of free Tryptophane was also recorded using a solution of 30 µM of NATA with or without the same concentrations of compounds. Ratio of F/F0 were calculated and the dissociation constant KD were determined using the equation Y = (Bmax * X) / (KD + X) where X is the concentration of the ligand and Y the specific interaction. The curves were drawn with GraphPad Prism software.
In silico docking
Several experimental structures of ERK co-crystallized with different peptides were analyzed with the molecular graphic system PyMol (Schrödinger), and per-residue interaction energies between the peptides and the proteins were computed with pyDockEneRes7 to evaluate hot residues in the peptides and proteins. The protonation states of protein titratable residues were predicted with the PCE server41. The ligand-binding pockets on ERK structures were predicted and further investigated using the machine learning-based pocket prediction tool P2Rank (Python standalone version 2.4)4. With such a tool, the top 2 or top 3 predicted binding pockets are generally true ligand binding sites. Docking of EI-52 or was then performed with Surflex-Dock (Surflex-scoring function)42, Vina (Vina scoring function)43,44,45 and Smina (Vinardo scoring function)46,47. The search region for the docking included Zones A and B and extended in each direction by about 5 Å. The poses obtained with the different docking tools were similar and we decided to compute the binding free energy for the poses generated using the Smina-Vinardo docking-scoring approach. The top 10 ERK-EI-52 docked poses and top 10 ERK-SP-26-docked poses were then processed using the GROMACS simulation software48 and the standalone Uni-GBSA python package49. The Molecular Mechanics/Generalized-Born-Surface Area (MM/GBSA) approach was applied and the binding free energy was estimated as ∆G = ∆Gprotein-ligand - ∆Gprotein - ∆Gligand. Optimization of the docked binding poses was here performed with energy minimization and not molecular dynamics as it has been repeatedly that energy minimization protein-ligand complexes yielded results as good as or better than those obtained after molecular dynamics49.
Bimolecular fluorescence complementation (BiFC)
The BiFC method was used as described50. Briefly, HEK293T cells were treated with DMSO or with increasing concentrations of EI-52 2 h prior to transfection. Cells were co-transfected for 24 h with PCDNA3-hERK1 and pCDNA3-hMyD88 tagged with N-and C-fragments of the VENUS protein, respectively. Fluorescence intensity was measured by flow cytometry on a BD Fortessa Horizon and analyzed using the FlowJo software.
Proximity ligation assay (PLA)
The PLA method was used as described51. Briefly, cells were treated with DMSO or EI-52 at the indicated concentrations, washed with PBS1X and fixed with 4% PFA.
MYD88/ERK interaction: cells were permeabilized with methanol for 2 min at -20 °C, then washed 3 times with PBS1X and blocked with blocking solution for 30 min at 37 °C. Cells were then incubated with primary antibodies against MYD88 (Invitrogen) and ERK (Cell Signaling technology), then with the appropriate DNA-linked secondary antibodies according to the manufacturer’s instructions. Transfected MYD88-Flag/ERK-Histidine interaction: cells were permeabilized with PBS-Triton 0.3%. Then washed 3 times with PBS1X and blocked with blocking solution for 30 min at 37 °C. Cells were then incubated with primary antibodies against Flag (Sigma) and Histidine (Clonetech), then with the appropriate DNA-linked secondary antibodies according to the manufacturer’s instructions. Fluorescence was quantified using the ImageJ software.
Cell recovery and cell death
105 HCT116 cells were seeded onto 96-well plates. The cells were then incubated with or without 8 μM of ERK kinase inhibitors or EI-52 in medium containing propidium iodide at 3.5 μg/mL. Cell confluence and the number of dead cells were determined using the IncuCyte™ Kinetic Live Cell Imaging System (Essen BioScience) at 2 h-intervals and up to 48 h.
Apoptosis detection
For apoptosis detection, 2 × 105 HCT116 cells were treated 24 h with DMSO or 8 μM EI-52, then stained with AnnexinV–fluorescein isothiocyanate (FITC) and propidium iodide (Apoptosis Detection Kit, Abcys). Fluorescence was acquired on a BD FACScalibur and analyzed using FlowJo software.
Eurofins Oncopanel Activity Data
Cells were grown in standardized media, seeded into 384-well plates and incubated in a humidified atmosphere of 5% CO2 at 37 °C. Compounds were added 3 days following cell seeding. At the same time, a time zero untreated cell plate was generated. EI52 was serially diluted in 2-fold steps from the highest test concentration (30 μM), and assayed over 10 concentrations with a maximum assay concentration of 0.1% DMSO. After a 2 day incubation period, cells were lysed with cell viability detection reagent CellTiter-Glo® (Promega). Bioluminescence was read by a PerkinElmer Envision® microplate reader. Cell viability was measured by the bioluminescence signal generated by the production of ATP in viable cells. The output is referred to as the relative cell count, where measured bioluminescence intensity was transformed to percent of control. Cellular response parameters were calculated using nonlinear regression to a sigmoidal single-site dose response model. IC50, defined as the test compound concentration at 50% of the maximum possible response.
Kinase assay
ERK1 kinase activity assay was performed in white 384-well microplates (Greiner) using Myelin Basic Protein as substrate (MBP, Sigma), active ERK1 protein (Millipore), and ADP-GloTM Kinase Assay (Promega). 15 ng of ERK1 were incubated at room temperature for 1 h in the dark with 1 µg of MBP in 40 mM Tris-HCl buffer pH 7.5, 20 mM MgCl2, 50 µM DTT, 0.1 mg/mL of BSA with various concentrations of test compounds or known ERK1 inhibitors K252a and FR180204 (Sigma). 5 µL of ADP-Glo® reagent was incubated for 40 min at room temperature, then with 10 µL of Kinase Detection Reagent for 30 min at room temperature. Luminescence was measured using an Infinite M200 microplate reader (TECAN). Signals were converted to % of ATP/ADP production and response curves were drawn using GraphPad Prism.
Transcriptomic analysis
Messenger RNA was extracted from HCT116, after 18 h treatment in triplicate with 6 µM of EI52 (CER) or DMSO using NucleoSpin RNA Kit (Macherey-Nagel) according to manufacturer instructions. Gene-expression profiling was first done using the Affymetrix GeneChip Human Genome U133 Plus 2.0 array (ThermoFischer Scientific).
Gene expression analysis was performed using Array Studio software (Omicsoft Corporation). Raw microarray data (CEL files) were processed using quantile normalization and robust multiarray average algorithm. Standard quality controls were performed (median average deviation score, principal component analysis) to identify outlier samples.
GSEA Analysis
Functional analyses were performed using Gene Set Enrichment Analysis (GSEA) software v2.0.452. GSEA is a robust computational method that determines whether an a priori defined set of genes shows statistically significant, concordant differences between two biologic states (e.g., high risk v low risk). GSEA aims to interpret large-scale expression data by identifying pathways and process. The main advantage of this method is its flexibility in creating molecular signature database of gene sets, including ones based on biologic pathways, chromosomal ___location, or expression profiles in previously generated microarray data sets. The input data for GSEA procedure were the following: (1) a complete table of genes ranked according to the log2 fold change between HCT116 treated 16 h with DMSO and EI-52, (2) a mapping file for identifying transcripts in U133 Plus 2.0 array platform, and (3) a catalog of 50 “hallmark” gene sets from the Molecular Signature Database (MSigDB)53 (MSigDB, version 5.1 release, www.broad.mit.edu/gsea/msigdb/msigdb_index.html). Default parameters were used. Inclusion gene set size was set between 15 and 500 and the phenotype was permutated 1,000 times. Gene sets that met the FDR 0.25 criterion were considered.
Western blot
Cells were lysed in Laemmli buffer (125 mM Tris-HCL (pH6,8), SDS 2%) and supplemented with a mixture of protease and phosphatase inhibitors. Lysates were sonicated, and then centrifuged for 15 min at 15,000 × g at 4 °C. Proteins were resolved on SDS- PAGE, transferred onto PVDF membranes by electroblotting, and nonspecific binding sites were blocked using Tris-buffered saline containing 5% (w/v) nonfat dry milk and 0.1% Tween. After incubation with primary antibodies against p-ERK, Erk, p-RSK, RSK, ATF4, p-EIF2α, CHOP, PKR, PERK, GCN2 (Cell Signaling Technology), DUSP5, (Abcam), HRI (Proteintech), histidine (Clontech), Flag (Sigma), tubulin (Sigma), or actin (Sigma) overnight at 4 °C, membranes were rinsed, incubated with horseradish peroxidase-conjugated secondary antibody and revealed by chemiluminescence using Bio-Rad Clarity Western ECL and BIO-Rad Chemidoc.
Cell cycle
Cells were plated and treated with DMSO, MEK inhibitor (U0126) at 10 µM, paclitaxel (Sigma) at 25 µM or EI-52 at 8 µM. Simultaneous staining for BrdU incorporation and DNA content was performed. Briefly, cells were pulsed with 1 µg/ml BrdU (Sigma) for 1 h prior to harvesting. Cells were then fixed in 70% ethanol for at least 30 min and the DNA was partially denatured in 3 N HCl, then neutralized with 0.1 M Na2B4OH. The cells were subsequently stained with FITC-coupled anti-BrdU (BD Biosciences), resuspended PBS containing 2.5% FCS and Propidium Iodide 2,5 μg/ml. Cells were acquired on a BD Fortessa Horizon flow cytometer equipped with a doublet discrimination module and analyzed using the FlowJo software.
Immunofluorescence
HeLa cells were serum starved for 24 h and treated as described in the legend. Then cells were fixed with 4% paraformaldehyde, and permeabilized with ice-cold methanol 5 min. The immunofluorescence staining with p-ERK and ERK antibodies (Cell Signaling Technology) was performed according to the protocol of the antibody supplier and fluorescence was observed by microscopy (Nikon).
Toxicity on chicken embryos
Toxicity evaluation was performed by INOVOTION (Grenoble). Briefly, fertilized White Leghorn eggs were incubated at 37,5 °C, 50% relative humidity for 9 days. At day 10, embryos were treated every two days for 10 days by dropping 100 μl of medium with DMSO, or 8 or 12 μM EI-52. The number of dead embryos and organ abnormalities in surviving embryos reflect toxicity.
Caspase 3/7 activity
104 cells were seeded onto 96-well plates. The cells were then incubated with or without 8 μM of EI-52 for 16 h. Caspase 3/7 activity was evaluated using Caspase-Glo 3/7 (Promega) according to the manufacturer’s protocol and using a Tecan Infinite M200 Pro.
Chemokine mRNA quantification
HCT116 cells were seeded onto 12-well plates. The next day, cells were treated with vehicle (DMSO) or 8 μM EI-52 for 16 h. For qPCR, total cellular RNA was extracted using the NucleoSpin RNA Kit (Macherey-Nagel). Then 0.5 µg of RNA were reverse transcribed using the High-Capacity cDNA reverse transcription Applied Biosystems kit (ThermoFischer Scientific) and quantified by real time PCR using specific primers for CXCL1-Fwd TCCTGCATCCCCCATAGTTA, CXCL1-Rev CTTCAGGAACAGCCACCAGT, CXCL2-Fwd CCCATGGTTAAGAAAATCATCG, CXCL2-Rev CTTCAGGAACAGCCACCAAT, CXCL3-Fwd TGAATGTAAGGTCCCCCGGA, CXCL3-Rev ACCCTGCAGGAAGTGTCAAT and CXCL8-Fwd AGACAGCAGAGCACACAAGC, CXCL8-Rev ATGGTTCCTTCCGGTGGT and the SYBR Green Master Mix (Applied Biosystems). qPCR was performed using the CFX connect real-time PCR system (Biorad). Expression of target genes was normalized against endogenous HPRT mRNA levels, used as an internal control, and assessed using the comparative 2-ΔΔCt method.
Chemokine, ATP and HMGB1 quantification
105 HCT116 cells were seeded onto 12-well plates. The next day, cells were treated with vehicle (DMSO), EI-52 8 μM, or MEK inhibitor (U0126) 10 μM for 24 h. ATP was quantified in the supernatant with Cell Titer-Glo (Promega) according the manufacturer’s instructions, using Tecan Infinite M200 Pro. ELISA was used to quantify secreted CXCL8 (Biolegend), CXCL1, CXCL2 (R&D Systems) and HMGB1 (IBL).
Patient-derived tumor organoids
Patient-derived lung tumor organoids were maintained in advanced DMEM containing HEPES, GlutaMax, Penicilline/Streptoimycine and primocione and B27, and supplemented with R-spondin-1, NOGGIN, A83-01 and Y-27632 from Miltenyi Biotec, SB202190 (Sigma), FGF7 and FGF10 (Thermo Fisher) and Nicotinamide (Sigma).
Patient-derived colon tumor organoids were maintained in advanced DMEM containing HEPES, GlutaMax, Penicilline/Streptoimycine primocine and B27, and supplemented with NOGGIN, A83-01 and Y-27632 from Miltenyi Biotec, SB202190 (Sigma), EGF (Miltenyi), Gastrin and PGE2 (Sigma).
One hundred tumoroids per well are seeded in 96-well plates (Greiner) in 100 µL of medium containing 5% Matrigel and incubated at 37 °C for 24 h. EI-52 or vehicle treatment was added using the HP D300 Digital Dispenser. After 48 h of treatment, cell death was evaluated by imaging caspase 3/7 activity (CellEvent™ Caspase-3/7 Detection Reagents, Invitrogen) and propidium iodide (Sigma) incorporation with OPERA Phoenix (Perkin Elmer) using an objective magnitude of 5 (non-confocal mode). The tumoroids described in this study can be obtained upon request to the 3D-Onco platform.
Organotypic culture
Patients signed a written informed consent agreeing to the use of tumor samples for research, according to the French regulations on the protection of persons (French Ethics Committee). Tumor tissue from surgical resections was obtained. Excess necrotic tissue was discarded and a 200 mm3 tumor fragment was included in an agarose gel. Automated slicing was performed using Microm HM650V Vibratome with slice thickness set at 250 μm. Adjacent slices were treated with DMSO or EI-52 8 µM for 24 hrs in DMEM culture medium supplemented with 10% Fetal Bovine Serum, penicillin/streptomycin/neomycin, and 1% glutamine at 37 °C in 5% CO2.
After 24 h, the culture supernatant was harvested and the tissue was fixed in 4% formalin, then imbedded in paraffin. Histological sections were performed and stained for hematoxylin-phloxin-saffron or cleaved-PARP (Cell Signaling Technology) on an automated Ventana Discovery XC (Roche) according to the manufacturer’s instructions. Cleaved-PARP was measured using a IHC score where 0 = no staining, 1 = 1–9%, 2 = 10–19%, and 3 ≥ 30% of cells are stained. The fresh human biological material used in the ex vivo experiments was used extemporaneously and cannot be stored to be subsequently distributed. It is therefore not available.
In vivo models
Animals were maintained in a specific pathogen-free animal facility (P-PAC, Lyon-France) and stored in sterilized filter-topped cages. Mice were handled in agreement with the institutional recommendations and procedures approved by the animal care committee (Comité d’Evaluation Commun au Centre Léon Bérard, à l’Animalerie de transit de l’ENS, au PBES et au laboratoire P4; CECCAP). The housing conditions of all animals were strictly following the ethical regulations. The room temperature ranged from 20 and 24 °C. The relative ambient humidity at the level of mouse cages was 55 ± 10%. Each cage was provided with food, water and two types of nesting material. A semi-natural light cycle of 12:12 was used. All the experiments complied strictly with the protocols approved by the ethics committee.
Syngeneic graft models
5 × 105 Lewis Lung Carcinoma (LLC) or CT26 colon carcinoma cells were implanted subcutaneously into the flanks of 8 week-old C57BL/6 or BALB/c (WT or nude) female mice, respectively (Charles River). When tumors reached 100 mm3, mice were treated by daily intraperitoneal injections of vehicle or EI-52 at the indicated dose. Tumor volume was measured twice a week with an electronic caliper.
Spontaneous model
Five-week-old K-rasLA2 female mice were treated intraperitoneally 5 times a week for 10 weeks with 25 mg/kg EI-52 or vehicle. Tumor load was evaluated at regular intervals on a Perkin Elmer Quantum FX microCT scan. Lung tumor load was quantified using the CALIPER software.
Combination therapy
CT26 colon carcinoma cells (5 × 105) were implanted subcutaneously into the flanks of 8 week-old BALB/c female mice (Charles River). When tumors reached 100 mm3, mice were treated by daily intraperitoneal injections of vehicle or EI-52 at 25 mg/kg and received 3 intraperitoneal injections of anti-PD1 (clone RPMI-14, BioXcell) (200 μg) or isotype control antibody (Rat IgG2a, κ clone 2A3, BioXcell) at days 0, 3, and 5. Tumor volume was measured twice a week with an electronic caliper.
Statistical and reproducibility
All the statistical analyses were performed on GraphPad Prism v.10 by using unpaired t-test, one- or two-way ANOVA, Wilcoxon, or Mann-Whitney. P-values < 0.05 were considered statistically significant. Only significant values are indicated and represented by (*) symbols. The exact p-values are indicated when provided by the analysis software (GraphPad Prism).
No statistical method was used to predetermine sample size. Experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment. No data were excluded from the analysis except one mouse from Fig. 7d that was excluded due to abnormal growth (ten-fold in two days).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided with this paper. The microarray data generated in this study have been deposited in the GEO database under accession code GSE153759. All remaining data can be found in the Article, Supplementary and source Data files. Source data are provided with this paper.
References
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Coste, I. et al. Dual function of MyD88 in RAS signaling and inflammation, leading to mouse and human cell transformation. J. Clin. Invest. 120, 3663–3667 (2010).
Kfoury, A. et al. MyD88 in DNA repair and cancer cell resistance to genotoxic drugs. JNCI J. Natl. Cancer Inst. 105, 937–946 (2013).
Krivák, R. & Hoksza, D. P2Rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure. J. Cheminform. 10, 39 (2018).
Liu, S., Sun, J.-P., Zhou, B. & Zhang, Z.-Y. Structural basis of docking interactions between ERK2 and MAP kinase phosphatase 3. Proc. Natl. Acad. Sci. USA 103, 5326–5331 (2006).
Gógl, G., Törő, I. & Reményi, A. Protein–peptide complex crystallization: a case study on the ERK2 mitogen-activated protein kinase. Acta Crystallogr. D Biol. Crystallogr. 69, 486–489 (2013).
Romero-Durana, M., Jiménez-García, B. & Fernández-Recio, J. pyDockEneRes: per-residue decomposition of protein-protein docking energy. Bioinforma. Oxf. Engl. 36, 2284–2285 (2020).
Zhou, T., Sun, L., Humphreys, J. & Goldsmith, E. J. Docking interactions induce exposure of activation loop in the MAP kinase ERK2. Structure 14, 1011–1019 (2006).
Garai, Á. et al. Specificity of linear motifs that bind to a common mitogen-activated protein kinase docking groove. Sci. Signal. 5, ra74 (2012).
Kase, H. et al. K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochem. Biophys. Res. Commun. 142, 436–440 (1987).
Ohori, M. et al. Identification of a selective ERK inhibitor and structural determination of the inhibitor-ERK2 complex. Biochem. Biophys. Res. Commun. 336, 357–363 (2005).
Robinson, F. L., Whitehurst, A. W., Raman, M. & Cobb, M. H. Identification of novel point mutations in ERK2 that selectively disrupt binding to MEK1. J. Biol. Chem. 277, 14844–14852 (2002).
Dimitri, C. A., Dowdle, W., MacKeigan, J. P., Blenis, J. & Murphy, L. O. Spatially separate docking sites on ERK2 regulate distinct signaling events in vivo. Curr. Biol. 15, 1319–1324 (2005).
Murphy, L. O. & Blenis, J. MAPK signal specificity: the right place at the right time. Trends Biochem. Sci. 31, 268–275 (2006).
Kidger, A. M. et al. Dual-specificity phosphatase 5 controls the localized inhibition, propagation, and transforming potential of ERK signaling. Proc. Natl. Acad. Sci. USA. 114, E317–E326 (2017).
Bastola, P., Neums, L., Schoenen, F. J. & Chien, J. VCP inhibitors induce endoplasmic reticulum stress, cause cell cycle arrest, trigger caspase-mediated cell death and synergistically kill ovarian cancer cells in combination with Salubrinal. Mol. Oncol. 10, 1559–1574 (2016).
Taniuchi, S., Miyake, M., Tsugawa, K., Oyadomari, M. & Oyadomari, S. Integrated stress response of vertebrates is regulated by four eIF2α kinases. Sci. Rep. 6, 32886 (2016).
Collin, G. et al. Intestinal epithelial cells adapt to chronic inflammation through partial genetic reprogramming. Cancers 15, 973 (2023).
Johnson, L. et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 410, 1111–1116 (2001).
Kue, C. S., Tan, K. Y., Lam, M. L. & Lee, H. B. Chick embryo chorioallantoic membrane (CAM): an alternative predictive model in acute toxicological studies for anti-cancer drugs. Exp. Anim. 64, 129–138 (2015).
Kepp, O. et al. Consensus guidelines for the detection of immunogenic cell death. OncoImmunology 3, e955691 (2014).
Sukkurwala, A. Q. et al. Immunogenic calreticulin exposure occurs through a phylogenetically conserved stress pathway involving the chemokine CXCL8. Cell Death Differ 21, 59–68 (2014).
Lavoie, H., Gagnon, J. & Therrien, M. ERK signalling: a master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 21, 607–632 (2020).
Yang, L., Zheng, L., Chng, W. J. & Ding, J. L. Comprehensive analysis of ERK1/2 Substrates for potential combination immunotherapies. Trends Pharmacol. Sci. 40, 897–910 (2019).
Sharrocks, A. D., Yang, S.-H. & Galanis, A. Docking domains and substrate-specificity determination for MAP kinases. Trends Biochem. Sci. 25, 448–453 (2000).
Bartholomeusz, C. et al. PEA-15 induces autophagy in human ovarian cancer cells and is associated with prolonged overall survival. Cancer Res. 68, 9302–9310 (2008).
Cagnol, S. & Chambard, J.-C. ERK and cell death: mechanisms of ERK-induced cell death - apoptosis, autophagy and senescence: ERK and cell death. FEBS J 277, 2–21 (2010).
Hancock, C. N. et al. Identification of novel extracellular signal-regulated kinase docking ___domain. Inhibitors. J. Med. Chem. 48, 4586–4595 (2005).
Kaoud, T. S. et al. Modulating multi-functional ERK complexes by covalent targeting of a recruitment site in vivo. Nat. Commun. 10, 5232 (2019).
Herrero, A. et al. Small molecule inhibition of ERK dimerization prevents tumorigenesis by RAS-ERK pathway oncogenes. Cancer Cell 28, 170–182 (2015).
Pakos-Zebrucka, K. et al. The integrated stress response. EMBO Rep. 17, 1374–1395 (2016).
McConkey, D. J. The integrated stress response and proteotoxicity in cancer therapy. Biochem. Biophys. Res. Commun. 482, 450–453 (2017).
Tameire, F. et al. ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat. Cell Biol. 21, 889–899 (2019).
Menger, L. et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci. Transl. Med. 4, 143ra99 (2012).
Nuccitelli, R. et al. Nano-pulse stimulation is a physical modality that can trigger immunogenic tumor cell death. J. Immunother. Cancer 5, 32 (2017).
Tesniere, A. et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 29, 482–491 (2010).
Schiavoni, G. et al. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res. 71, 768–778 (2011).
Zhou, H. et al. The oncolytic peptide LTX-315 triggers immunogenic cell death. Cell Death Dis. 7, e2134 (2016).
Püschel, F. et al. Starvation and antimetabolic therapy promote cytokine release and recruitment of immune cells. Proc. Natl. Acad. Sci. USA 117, 9932–9941 (2020).
Zhu, Q. C. et al. Induction of the proinflammatory chemokine interleukin-8 is regulated by integrated stress response and AP-1 family proteins activated during coronavirus infection. Int. J. Mol. Sci. 22, 5646 (2021).
Miteva, M. A., Tufféry, P. & Villoutreix, B. O. PCE: web tools to compute protein continuum electrostatics. Nucleic Acids Res. 33, W372–W375 (2005).
Spitzer, R. & Jain, A. N. Surflex-Dock: Docking benchmarks and real-world application. J. Comput. Aided Mol. Des. 26, 687–699 (2012).
Lagarde, N. et al. Online structure-based screening of purchasable approved drugs and natural compounds: retrospective examples of drug repositioning on cancer targets. Oncotarget 9, 32346–32361 (2018).
Labbé, C. M. et al. MTiOpenScreen: a web server for structure-based virtual screening. Nucleic Acids Res. 43, W448–W454 (2015).
Forli, S. et al. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 11, 905–919 (2016).
Koes, D. R., Baumgartner, M. P. & Camacho, C. J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 53, 1893–1904 (2013).
Quiroga, R. & Villarreal, M. A. Vinardo: A scoring function based on AutoDock vina improves scoring, docking, and virtual screening. PLoS One 11, e0155183 (2016).
Pronk, S. et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinforma. Oxf. Engl. 29, 845–854 (2013).
Yang, M. et al. Uni-GBSA: an open-source and web-based automatic workflow to perform MM/GB(PB)SA calculations for virtual screening. Brief. Bioinform. 24, bbad218 (2023).
Kerppola, T. K. Visualization of molecular interactions using bimolecular fluorescence complementation analysis: characteristics of protein fragment complementation. Chem. Soc. Rev. 38, 2876–2886 (2009).
Söderberg, O. et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 (2006).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 (2005).
Liberzon, A. et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425 (2015).
Acknowledgements
We thank the Research Pathology, P-PAC, P-PAC-Imaging, EX-VIVO, and Organoïd 3D-ONCO plateforms, Université de Lyon, Université Claude Bernard Lyon 1, INSERM 1052, CNRS 5286, Centre Léon Bérard, Centre de recherche en Cancérologie de Lyon (CRCL), Lyon, 69373, France, and the Synergy Lyon Cancer Foundation. We also thank Lucas Pons for his assistance in the BiFc experiment. This work was supported by funding from La Ligue Nationale Contre le Cancer EL2014-2 (TR), Institut National du Cancer PLBIO (TR, 2020-119), Fondation ARC pour la recherche sur le cancer (IC, PJA20171206416), the Integrated Cancer Research Site LYriCAN (TR, INCa-DGOS-Inserm_12563), ANR (TR, ANR -11-EQPX-0035), and Pulsalys (TR, D02576).
Author information
Authors and Affiliations
Contributions
M.B., L.Mag., L.Mar., T.H.P., N.S., S.D., R.J., R.F., N.H.S., T.F., N.H., C.C., N.A., and V.C. performed experiments and analyzed data. B.V. performed docking studies. F.V. and P.S. performed and analyzed transcriptomic analysis. F.V., S.G., S.M., I.T., T.R., and I.C. analyzed data. F.V., S.G., T.R., and I.C. wrote the manuscript. S.L., S.M., S.A., F.V., S.G., T.R., and I.C. edited the manuscript. T.R. and I.C. conceived the project and managed all studies. F.V. and S.G. have contributed equally to this work.
Corresponding authors
Ethics declarations
Competing interests
I.C., S.G., and T.R. are co-founders of TheraPPI Bioscience, a spin-off of their host academic institutions. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Melanie Cobb, who co-reviewed with Ana Martin-Vega and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Virard, F., Giraud, S., Bonnet, M. et al. Targeting ERK-MYD88 interaction leads to ERK dysregulation and immunogenic cancer cell death. Nat Commun 15, 7037 (2024). https://doi.org/10.1038/s41467-024-51275-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51275-z
