
Abstract
Increased fatty acid synthesis benefits glioblastoma malignancy. However, the coordinated regulation of cytosolic acetyl-CoA production, the exclusive substrate for fatty acid synthesis, remains unclear. Here, we show that proto-oncogene tyrosine kinase c-SRC is activated in glioblastoma and remodels cytosolic acetyl-CoA production for fatty acid synthesis. Firstly, acetate is an important substrate for fatty acid synthesis in glioblastoma. c-SRC phosphorylates acetyl-CoA synthetase ACSS2 at Tyr530 and Tyr562 to stimulate the conversion of acetate to acetyl-CoA in cytosol. Secondly, c-SRC inhibits citrate-derived acetyl-CoA synthesis by phosphorylating ATP-citrate lyase ACLY at Tyr682. ACLY phosphorylation shunts citrate to IDH1-catalyzed NADPH production to provide reducing equivalent for fatty acid synthesis. The c-SRC-unresponsive double-mutation of ACSS2 and ACLY significantly reduces fatty acid synthesis and hampers glioblastoma progression. In conclusion, this remodeling fulfills the dual needs of glioblastoma cells for both acetyl-CoA and NADPH in fatty acid synthesis and provides evidence for glioma treatment by c-SRC inhibition.
Similar content being viewed by others


Introduction
Glioma exhibits high aggressiveness and recurrence rate, even in response to improved therapies like temozolomide (TMZ) combined with radiation therapy (RT)1,2. Consequently, it is considered as one of the most intractable tumors, with devising novel therapeutic approaches remaining an arduous task3. Since the discovery of the renowned Warburg effect, the phenomenon that tumor cells prefer glucose for ATP production and biological macromolecule synthesis even under well-oxygenated conditions4,5, a growing body of evidence has indicated metabolic reprogramming as a distinct hallmark of tumors6,7,8. Beyond the activation of aerobic glycolysis, another metabolic trait of tumors is enhanced lipid metabolism, particularly de novo fatty acid synthesis9,10. This activation of fatty acid synthesis (FAS) is crucial for rapidly proliferating tumor cells to build substantial bio-membrane. It has been revealed that tumor cells can promote de novo fatty acid synthesis by elevating the expression of a series of lipogenic enzymes, including ATP-citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), and fatty acid synthase (FASN)11,12,13. Unraveling the mechanism underlying this promotion of de novo fatty acid synthesis in glioma cells might provide insights for the formulation of new therapeutic strategies against glioma.
De novo fatty acid synthesis is well-known process that takes place in the cytosol, with cytosolic acetyl-CoA being the exclusive substrate for this anabolic event. For a considerable period, cytosolic acetyl-CoA was thought to be solely produced via the ACLY-catalyzed classic reaction, exploring citrate as a substrate in mammals. Recent studies, however, have illuminated a new avenue. These studies show that acyl-CoA synthetase short-chain family member 2 (ACSS2) can facilitate fatty acid biosynthesis by converting acetate to acetyl-CoA in hepatic cell carcinoma and glioma14,15,16,17. This discovery establishes an alternative route for cytosolic acetyl-CoA synthesis, revealing an additional function of acetate in supporting tumor progression by replenishing the acetyl-CoA pool for fatty acid synthesis. Acetate, found in blood plasma, generally originates from the breakdown of dietary fibers in the intestine, oxidation of alcohol in the liver, and dietary source such as vinegar, with concentrations ranging from approximately 50–200 μM18,19. Cells make use of plasma acetate through monocarboxylate transporters (MCTs) and subsequently convert it to acetyl-CoA by ACSS220,21,22. In 2018, Liu et al. uncovered that intracellular acetate is produced from glucose by two mechanisms: the ROS-mediated oxidation of glucose-derived pyruvate and the dehydrogenation of pyruvate by PDH23. According to this study, it can be inferred that ROS oxidation of pyruvate to acetate is a potential method for ROS elimination. Given that increased aerobic glycolysis and relatively high ROS levels are characteristic of many tumor cells, it’s plausible that tumor cells can generate substantial acetate through ROS-mediated pyruvate oxidation. This acetate can then be converted to acetyl-CoA by cytosolic-localized ACSS2 for fatty acid synthesis. We observed significant acetate production from glucose in glioblastoma cell lines. Additionally, Maher et al. reported that in clinical glioma samples, glucose oxidation in the tricarboxylic acid (TCA) cycle only contributes to less than 50% of the acetyl-CoA pool24. This observation suggests that more than half of the acetyl-CoA in glioma originates from alternative sources, such as acetate. However, the detailed mechanism underlying ACSS2 activation for the utilization of endogenous and exogenous acetate to synthesize acetyl-CoA for fatty acid synthesis in glioma cells remains unclear. Furthermore, the coordinated regulation of ACSS2 and ACLY to meet the substantial acetyl-CoA demands of glioma cells is still largely unexplored.
The non-receptor tyrosine kinase c-SRC is encoded by SRC, which was the first proto-oncogene discovered in animal cells and implicated in a wide range of human cancers25,26. In normal conditions, the kinase activity of c-SRC is inhibited by the intramolecular interaction between its C-terminal and N-terminal c-SRC homology 2 (SH2) domains. This intramolecular interaction is disrupted upon stimulation by mitogenic growth factors such as EGF and PDGF, resulting in c-SRC activation27,28. Additionally, c-SRC can be constitutively activated due to mutations or deletions in the C-terminal ___domain29. Activated c-SRC plays a role in promoting tumorigenesis and tumor progression by influencing various cellular processes, including cell adhesion, metastasis, angiogenesis, and metabolic reprogramming. Numerous studies demonstrate that c-SRC is abnormally activated in various tumors, including colon cancer, bladder cancer, pancreatic cancer, and glioma29. Previous research reported that c-SRC phosphorylates and activates glycolytic enzymes (HK1/2 and PFKFB3) and G6PD, thereby facilitating tumorigenesis30,31,32. Subsequently, we performed a screening for c-SRC-interacting proteins using affinity purification and liquid chromatograph-mass spectrometry (LC–MS) and identified ACSS2 and ACLY as two c-SRC-interacting proteins. Since these two proteins serve to produce cytosolic acetyl-CoA, we were prompted to investigate whether c-SRC promotes glioblastoma progression by regulating them to enhance fatty acid synthesis.
In this work, we observed that c-SRC is widely activated in glioma and stimulates the production of acetate-derived acetyl-CoA by phosphorylating ACSS2 at Tyr530 and Tyr562. Surprisingly, c-SRC also phosphorylates ACLY at Tyr682 but inhibits its enzymatic activity. This inhibition diverts citrate to the production of NADPH catalyzed by IDH1 for fatty acid synthesis. Consequently, c-SRC simultaneously satisfies the glioblastoma cell’s need for both acetyl-CoA and NADPH in de novo fatty acid synthesis by coordinately regulating ACSS2 and ACLY. Our research uncovers the detailed mechanism by which c-SRC contributes to glioblastoma progression and suggests the potential development of specific c-SRC inhibitors as a therapeutic strategy for glioma therapy.
Results
c-SRC promotes glioblastoma progression
To investigate the contribution of c-SRC to glioblastoma progression, we assessed the activation status of c-SRC in 33 clinical glioma samples by detecting the level of c-SRC phosphorylation at Tyr419 using the p-c-SRC antibody. As shown in Fig. 1a, c-SRC was found to be activated in glioma samples particularly in high-grade glioma (WHO Grade III and IV) (Fig. 1b). This suggests a potential correlation between c-SRC activation and glioma malignancy. Subsequently, we isolated primary glioma cells (designated glioma cells A–F) from clinically excised glioma samples and found c-SRC activation in 4 out of 6 primary glioma cells (Supplementary Fig. 1a). To ascertain the impact of c-SRC on glioma cell proliferation, glioma cells C and D were treated with c-SRC inhibitors (PP2 and Saracatinib) and their growth curves were determined. Notably, the proliferation of c-SRC-activated glioma cells C and D was dramatically suppressed (Supplementary Fig. 1b–d). Similarly, the proliferation of commercially available glioma cell lines CHG-5 and U87, both characterized by active c-SRC, was markedly inhibited by PP2 and Saracatinib but not U118 with inactive c-SRC (Supplementary Fig. 1e–i). In line with these findings, c-SRC knockdown retarded the proliferation of U87 and CHG-5 cells (Fig. 1c–e). These results underscore the contribution of c-SRC to glioblastoma cell proliferation. To further validate this observation, we reconstituted c-SRC expression in triple knockout mouse embryonic fibroblasts (SYF-KO MEFs) lacking c-SRC, YES, and FYN. As expected, the reconstitution of either c-SRC wild-type (c-SRC-WT) or constitutively activated c-SRC (Y529F, c-SRC-CA), but not kinase-dead c-SRC (K297R, c-SRC-KD), restored their proliferative ability (Supplementary Fig. 1j). Furthermore, we performed colony formation assay and orthotopic xenograft assay to assess the oncogenic potential of c-SRC in glioma. c-SRC knockdown significantly reduced the colony formation capacity of U87 cells (Supplementary Fig. 1k, l) and impaired the formation of mouse orthotopic tumor (MOT) (Fig. 1f, g and Supplementary Fig. 1m, n). Additionally, c-SRC knockdown improved the overall health status (referred to mice’s body weight) and survival of mice subjected to U87-induced MOT formation (Supplementary Fig. 1o–q). In summary, c-SRC plays a pivotal role in promoting glioblastoma progression.

a Western blot analysis of clinical glioma samples to determine c-SRC expression and activation (p-c-SRC). IDH1 R132 mutation occurs in two cases of low-grade gliomas (#23 and #32). IDH1 mutations were not found in high-grade gliomas. The protein marker kilodalton (kDa) in Western Blot is abbreviated as Mr (K). The same applies to the WB figure below. The samples derive from the same experiment but different gels for p-c-SRC and c-SRC. N: normal tissue; WHO glioma grade: I–IV, n = 35. b The correlation between c-SRC activation and glioma malignancy (WHO glioma grade: I–IV) was analyzed based on optical density integrity (ODI) with ImageJ in clinical tissue (patient 3–35, n = 33 clinical samples). c–e, c-SRC in U87 and CHG-5 was knocked down by two independent short hairpin RNA (shSRC #1 and shSRC #2; shCtrl: pLL3.7-based scramble shRNA as control) (e). 1 × 104 U87 and 1 × 104 CHG-5 cells were seeded in 6-well plates, and the cell numbers were quantified at the indicated time. Growth curves illustrating cell proliferation of U87 (c) and CHG-5 (d) are depicted, based on cell numbers (n = 3 independent experiments). f, g MRI images of brain sections were captured 30 days after intracranial infusion of 1 × 105 U87 cells (f). Tumor volume was calculated based on the sphere radius of brain tumors (g) (n = 6 independent mice). Statistical data in (b, c, d, and g) are presented as the mean ± SD. p-values were calculated based on unpaired and two-sided Student’s t-test.
As aforementioned, we identified ACSS2 and ACLY as c-SRC-interacting proteins through an earlier screening by virtue of affinity purification of c-SRC followed by LC–MS analysis. Both ACSS2 and ACLY are involved in the production of cytosolic acetyl-CoA, which serves as the direct substrate for fatty acid synthesis13,33. Given the pivotal role of fatty acid synthesis in glioblastoma malignancy, we were prompted to direct our focus towards investigating whether c-SRC promotes glioblastoma malignancy by co-regulating ACSS2 and ACLY to facilitate acetyl-CoA production for fatty acid synthesis.
c-SRC stimulates de novo fatty acid synthesis by increasing the utilization of glucose-derived acetate
A previous study has reported that acetate is produced from glucose through the oxidation of pyruvate23. In our study, we cultured glioblastoma cells and assessed acetate concentration in cell-cultured medium using nuclear magnetic resonance (NMR) (Fig. 2a). As shown in Fig. 2b and Supplementary Fig. 2a, acetate is abundantly secreted into the extracellular medium. Additionally, we cultured cells with media (without glucose, pyruvate, and glutamine) supplemented with different carbon sources, finding that glucose, pyruvate, and lactate contribute to acetate secretion (Fig. 2c). Lactate contributes to acetate secretion, which is attributed to the conversion of lactate to pyruvate by lactate dehydrogenase. Based on our experimental results and the study by Liu et al. 23, we propose that glucose-derived acetate can be secreted into the extracellular matrix and recycled for reuse. Since acetate serves as a critical substrate for fatty acid synthesis and plays a vital role in glioblastoma malignancy15, our initial exploration focused on whether c-SRC promotes glioblastoma malignancy by increasing the utilization of glucose-derived acetate. Notably, c-SRC knockdown results in a sharp increase in the secretion of [U-13C]-acetate from [U-13C]-glucose (Fig. 2d) and a decrease in consumption of exogenous [2-13C]-acetate (Fig. 2e), indicating that c-SRC stimulates acetate utilization. To eliminate interference from endogenous glucose-derived acetate, [2-13C]-acetate was used for cell culture to directly analyze the impact of c-SRC on acetate utilization and recycling (Fig. 2f). As shown in Fig. 2g, h and Supplementary Fig. 2b, the level of acetyl-CoA (M + 1) derived from [2-13C]-acetate was significantly decreased when c-SRC is knocked down in primary cell and glioblastoma cell lines. Additionally, the levels of malonyl-CoA (M + 1) were also reduced in U87 and CHG-5 cells with c-SRC knockdown (Fig. 2i and Supplementary Fig. 2c). In alignment with these findings, 13C incorporation in myristic acid (C14:0), palmitic acid (C16:0) and stearic acid (C18:0) were remarkedly reduced by c-SRC knockdown in both CHG-5 and U87 cells after treatment with [2-13C]-acetate for 3 days, as determined by gas chromatography-mass spectrometry (GC–MS) (Fig. 2j, Supplementary Fig. 2d). The total levels of fatty acids were also substantially decreased by c-SRC knockdown (Supplementary Fig. 2e). Considering the cytoplasmic localization of ACSS2 and mitochondrial localization of ACSS1 and ACSS334,35, we embarked on investigating whether acetate-derived acetyl-CoA contributes to TCA cycle. Interestingly, the intensity of 13C-labeled TCA cycle intermediates (M + 1) is much less than that of 13C-labeled acetyl-CoA in U87 cells upon the treatment with [2-13C]-acetate (Supplementary Fig. 2f). Collectively, these findings underscore that the predominant fate of acetate-derived acetyl-CoA is to feed de novo fatty acid synthesis but not TCA cycle. In conclusion, c-SRC stimulates de novo fatty acid synthesis by increasing the utilization of glucose-derived acetate (Fig. 2k).

a The NMR 1H spectrum of unlabeled acetate ([U-12C]-acetate) and 13C-labeled acetate ([U-13C]-acetate). b Media were collected after 12 h of cell culture with complete medium and subjected to NMR analysis. The concentration of acetate (13C-unlabeled) in medium (n = 3 independent experiments) was quantified according to an acetate standard curve. c U87 cells were cultured with DMEM (no glucose, no glutamine, no sodium pyruvate, no phenol red, cat. A1443001) supplemented with glucose (5 mM), pyruvate (10 mM) and lactate (10 mM) individually for 24 h, followed by the NMR analysis of acetate in the cell-cultured medium (n = 3 independent experiments). Fresh medium as control (ND: not detected). d c-SRC in CHG-5 and U87 was knocked down. The cells were cultured with 10 mM [U-13C]-glucose for 24 h, followed by [U-13C]-acetate analysis in the medium using NMR (n = 3 independent experiments). e c-SRC in U87 was knocked down by shSRC #1. Cells were incubated with 1 mM [U-13C]-acetate for 24 h. The medium was collected and analyzed by NMR to calculate the acetate consumption after c-SRC knockdown (n = 3 independent experiments). f Schematic representation of the anabolic fate of [2-13C]-acetate (red dot represents 13C). g–i Primary glioma cells and U87 cells, both are knocked down for c-SRC, were cultured in a complete medium containing 5 mM [2-13C]-acetate for 24 h. The acetyl-CoA (M + 1) derived from [2-13C]-acetate in primary glioma cells (g) and U87 cells (h) were analyzed, along with the determination of the malonyl-CoA (M + 1) in U87 cells (i) (n = 3 independent experiments). j CHG-5 cells with c-SRC knockdown were cultured in a complete medium containing 5 mM [2-13C]-acetate for 3 days. Afterward, the indicated free fatty acids that derived from [2-13C]-acetate were extracted and analyzed using GC–MS (n = 3 independent experiments). k A diagram illustrating acetate production and c-SRC-mediated utilization and recycling (depicted by our team using Microsoft PowerPoint software). Statistical data are presented as the mean ± SD (n = 3). p-values were calculated based on unpaired and two-sided Student’s t-test.
c-SRC phosphorylates ACSS2 at Tyr530 and Tyr562
To investigate whether c-SRC promotes acetate utilization in fatty acid synthesis by regulating ACSS2, we conducted reciprocal co-immunoprecipitation (co-IP) assays with overexpressed proteins to examine the interaction between c-SRC and ACSS2. Our findings indicated a robust interaction between these two proteins (Fig. 3a, b, and Supplementary Fig. 3a, b). This interaction was further substantiated through the co-IP assay with endogenous proteins and the GST-pulldown assay with bacterially purified proteins (Fig. 3c, d). Domain mapping assays indicated that ACSS2 binds to c-SRC SH2, but not the kinase ___domain (SH1 ___domain) (Supplementary Fig. 3c). These results underscore the interaction between c-SRC and ACSS2.

a, b U87 cells were transfected with ACSS2 and c-SRC as indicated. After 24 h of transfection, cells were lysed and subjected to anti-Flag M2 beads for reciprocal immunoprecipitation (IP) assays, followed by Western blot to analyze the interaction between ACSS2 and c-SRC. c Endogenous IP between ACSS2 and c-SRC was performed in U87 cells, with ACSS2 knockdown U87 cells serving as a control. d GST, GST-ACSS2 and His-c-SRC were expressed and purified from BL21 E.coli competent cells. GST-pulldown assay of GST-ACSS2 and His-c-SRC was performed with GST as control. e U87 cells were transfected with HA-ACSS2 and Flag-c-SRC, followed by immunoprecipitation with anti-HA beads. The tyrosine phosphorylation of ACSS2 was detected with or without (w/wo) calf-intestinal alkaline phosphatase (CIAP) treatment. f Different forms of HA-c-SRC (WT, CA, and KD) were individually co-transfected with Flag-ACSS2 into U87 cells. The tyrosine phosphorylation levels of ACSS2 were determined after immunoprecipitation with anti-Flag M2 beads. WT: wild-type; CA: constitutive activated (Y529F); KD: kinase-dead (K297R) g, h Representative mass spectra of ACSS2 peptides, showing phosphorylation residues at Tyr530 (g) and Tyr562 (h). i Flag-ACSS2-WT or Flag-ACSS2-DMut (double mutation, Y530F + Y562F) were co-transfected with HA-c-SRC in U87 cells. Phosphorylation levels of Tyr530, Tyr562, and total tyrosine (p-Tyr) of ACSS2 were detected with corresponding antibodies after immunoprecipitation with anti-Flag M2 beads. j U87 cells transfected with c-SRC-WT and c-SRC-KD were analyzed for the phosphorylation levels of ACSS2-Tyr530 and ACSS2-Tyr562 with specific antibodies. The samples derive from the same experiment but different gels for p-Tyr530, p-Tyr562, ACSS2, and Flag. KD: kinase dead. k GST-ACSS2 and His-c-SRC were purified from BL21 E.coli competent cells. In vitro kinase assay was performed to determine ACSS2 phosphorylation by c-SRC. The samples derive from the same experiment but different gels for p-Tyr530, p-Tyr562, and p-Tyr. l Phosphorylation levels of ACSS2-Tyr530, ACSS2-Tyr562, and c-SRC in U87 cells were analyzed after treatment with c-SRC inhibitor Saracatinib (10 μM) for 24 h. The samples derive from the same experiment but different gels for p-Tyr530, p-Tyr562, ACSS2, and p-c-SRC. m c-SRC is further knocked down in ACSS2 knockdown U87 cells, followed by phosphorylation analysis of p-Tyr530 and p-Tyr562 on ACSS2. The samples derive from the same experiment but different gels for p-Tyr530, p-Tyr562, ACSS2, and c-SRC. n, o The alignments of the residues (Tyr530 and Tyr562 of ACSS2 in homo sapiens) across different species were analyzed by Snapgene software (6.0.2). Experiments in this figure were performed three times, except those in (a, b, e, g, h, and i). Most of these experiments were also repeated in 293 T cells (Supplementary Fig. 3).
Given that c-SRC is a tyrosine kinase that often promotes oncogenic processes through the phosphorylation of its target proteins, we conducted a series of experiments to ascertain whether c-SRC regulates ACSS2 via phosphorylation. Overexpressed ACSS2 could be robustly phosphorylated by c-SRC, as indicated by a positive signal detected with a pan-tyrosine phosphorylation antibody (p-Tyr) (Fig. 3e, Supplementary Fig. 3d). This phosphorylation was effectively abolished by either treatment of cell lysate with calf-intestinal alkaline phosphatase (CIAP) (Fig. 3e, Supplementary Fig. 3d) or treatment of cell with c-SRC inhibitors (Supplementary Fig. 3e). Notably, c-SRC-CA (c-SRC constitutively activated form) showed an enhanced ability to phosphorylate ACSS2, whereas the kinase-dead mutant c-SRC-KD lost this ability when compared to c-SRC-WT (Fig. 3f). Based on these findings, we propose that ACSS2 serves as a substrate for c-SRC kinase. Next, we conducted affinity purification of ACSS2 followed by LC–MS analysis to identify the tyrosine sites on ACSS2 phosphorylated by c-SRC. This analysis identified Tyr530 and Tyr562 as two c-SRC phosphorylation sites on ACSS2 (Fig. 3g, h). In comparison to ACSS2-WT, single mutant ACSS2-Y530F or ACSS2-Y562F (tyrosine-to-phenylalanine mutant) exhibited reduced phosphorylation levels when co-expressed with c-SRC. In contrast, the double mutant ACSS2-Y530F/Y562F (ACSS2-DMut) nearly completely abolished such phosphorylation, indicating that both Tyr530 and Tyr562 are c-SRC phosphorylation sites (Supplementary Fig. 3f). For practical determination of ACSS2 phosphorylation status under endogenous conditions, we commercially synthesized peptides containing phosphorylated Tyr530 or Tyr562, immunized rabbits and raised antibodies capable of specifically recognizing phosphorylation at these sites (designated as p-Tyr530 and p-Tyr562, respectively). When co-expressed with c-SRC, these two antibodies detected strong phosphorylation signals on ACSS2-WT but not ACSS2-DMut (Fig. 3i, Supplementary Fig. 3g). Moreover, c-SRC, but not c-SRC KD, induced robust phosphorylation of ACSS2 at Tyr530 and Tyr562 (Fig. 3j), and these two sites of phosphorylation by c-SRC were further validated in vitro kinase assay (Fig. 3k). Conversely, inhibition or knockdown of endogenous c-SRC in glioblastoma cells dramatically decrease the phosphorylation level of ACSS2 (Fig. 3l, m). Furthermore, two other members of the SRC family, BLK and HCK, were also found to phosphorylate ACSS2 at Tyr530 and Tyr562 residues (Supplementary Fig. 3h, i). And these two phosphorylation residues of ACSS2 are highly conserved among mammals (Fig. 3n, o). These results highlight the specificity of these two antibodies in recognizing phosphorylation at the corresponding sites induced by c-SRC and underscore the significance of c-SRC’s regulation on ACSS2.
ACSS2 is activated to facilitate fatty acid synthesis by c-SRC-mediated phosphorylation
Having clarified the phosphorylation of ACSS2 by c-SRC, we proceeded to explore the regulatory role of this phosphorylation in de novo fatty acid synthesis using acetate as a precursor. ACSS2 knockdown in U87 cells was cultured with [2-13C]-acetate and quantified for [2-13C]-acetate-derived acetyl-CoA (M + 1), malonyl-CoA (M + 1) and fatty acids (myristic acid, palmitic acid, and stearic acid, M + 1). The results indicated that ACSS2 knockdown remarkably abrogated the synthesis of [2-13C]-acetate-derived acetyl-CoA, malonyl-CoA, and fatty acids, affirming that ACSS2 is the pivotal enzyme driving acetate-based de novo fatty acid synthesis (Supplementary Fig. 4a, b). In further experiments, we observed that co-expression of c-SRC-WT, but not c-SRC-KD, dramatically activated the function of ACSS2 in promoting the production of acetyl-CoA, malonyl-CoA and fatty acids (Fig. 4a–d). Additionally, c-SRC exhibited the capability to substantially enhance the function of ACSS2-WT, albeit to a lesser degree for c-SRC-Y530F and c-SRC-Y562F mutants (Fig. 4e, Supplementary Fig. 4c). Intriguingly, c-SRC’s stimulatory effect on ACSS2 was entirely absent for the ACSS2-DMut, despite the latter maintaining its basal activity (Fig. 4e, f, Supplementary Fig. 4c and Supplementary Fig. 4d). These findings suggest that the necessity of both Tyr530 and Tyr562 phosphorylation by c-SRC to promote ACSS2’s function in fatty acid synthesis.

a, b Plasmids expressing ACSS2, c-SRC-WT and c-SRC-KD were transfected in U87 cells as indicated. After 6 h of transfection, cells were shifted to a complete medium containing 5 mM [2-13C]-acetate for 24 h of cell culture. The levels of acetate-derived acetyl-CoA (M + 1) (a) and malonyl-CoA (M + 1) (b) were analyzed by LC–MS (n = 3 independent experiments). KD: kinase dead. c, d U87 cells were transfected with ACSS2, c-SRC-WT, and c-SRC-KD as indicated (d) and incubated with 5 mM [2-13C]-acetate for 3 days. The levels of acetate-derived fatty acids (C16:0 and C18:0, M + 1) were analyzed by GC–MS (n = 3 independent experiments) (c). The samples derive from the same experiment but different gels for Flag (d). e U87 cells were transfected with c-SRC and different mutants of ACSS2 as indicated. After 24 h of transfection, cells were cultured in a fresh complete medium containing 5 mM [2-13C]-acetate for another 24 h. [2-13C]-acetate-derived acetyl-CoA (M + 1) was detected by LC–MS (n = 3 independent experiments). f U87 cells were transfected with ACSS2-WT, ACSS2-Dmut and c-SRC and incubated with 5 mM [2-13C]-acetate for 3 days. C16:0 (M + 1) and C18:0 (M + 1) were analyzed by GC–MS (n = 3 independent experiments). g ACSS2 knockdown of CHG-5 cells were constructed and re-expressed for ACSS2-WT (re-ACSS2-WT) or its double mutation (re-ACSS2-DMut). The same samples derived from same experiment but different gels for p-Tyr530, p-Tyr562, and ACSS2. h, i CHG-5 cells after reconstitution with ACSS2-WT or ACSS2-DMut were incubated with medium containing 5 mM [2-13C]-acetate for 24 h. The acetyl-CoA (M + 1) (h) and malonyl-CoA (i) were analyzed by LC–MS (n = 3 independent experiments). j CHG-5 cells as indicated in (g) were incubated with a complete medium containing 5 mM [2-13C]-acetate for 3 days. Then [2-13C]-acetate-derived fatty acids (C14:0, C16:0, and C18:0) (M + 1) were determined by GC–MS (n = 3 independent experiments). k U87 cells were transfected with ACSS2. After 24 h of transfection, cells were cultured in a fresh medium containing 5 mM [2-13C]-acetate for another 24 h. The levels of acetate-derived acetyl-CoA (M + 1) (left) and malonyl-CoA (M + 1) (right) for de novo fatty acid synthesis were then analyzed by LC–MS (n = 3 independent experiments). Statistical data are presented as the mean ± SD (n = 3). p-values were calculated using unpaired and two-sided Student’s t-test.
Re-expression of ACSS2-DMut in ACSS2 knockdown cells, as expected, failed to fully reinstate ACSS2’s capacity to drive acetate-derived acetyl-CoA production for the synthesis of malonyl-CoA and fatty acids (Fig. 4h–k, Supplementary Fig. 4e and Supplementary Fig. 4f). This finding reinforces the necessity of c-SRC in enhancing ACSS2’s function via phosphorylation at Tyr530 and Tyr562. To confirm that the unresponsiveness of ACSS2-DMut to c-SRC is due to its inability to be phosphorylated by c-SRC, rather than any structural disruption caused by the double mutation per se, we subjected ACSS2-DMut and ACSS2-WT to trypsin digestion in vitro and analyzed the digested fragments via SDS-PAGE. The digestion patterns were identical for both proteins (Supplementary Fig. 4g). Protein structure of ACSS2-WT and ACSS2-Dmut were predicted and analyzed by AlphaFold, implying that no significant structural deviation in ACSS2-DMut when compared to ACSS2-WT (Supplementary Fig. 4h–j). These results provide evidence to exclude the possibility of enzyme structure and resultant activity being influenced by double mutation per se, indicating that ACSS2-DMut can be used as a c-SRC unresponsive mutant that retains basal activity for further investigations. Importantly, the overexpression of ACSS2 dramatically increased the level of [2-13C]-acetate-derived acetyl-CoA (M + 1) and malonyl-CoA (M + 1) but not the metabolites in TCA cycle (Fig. 4k, Supplementary Fig. 4k). These results indicate that ACSS2 primarily functions to divert acetate to cytosolic acetyl-CoA for fatty acid synthesis, rather than mitochondrial acetyl-CoA for TCA cycle. In summary, both Tyr530 and Tyr562 phosphorylation on ACSS2 are imperative for c-SRC’s stimulation of acetate-based de novo fatty acid synthesis.
c-SRC phosphorylates ACLY at Tyr682
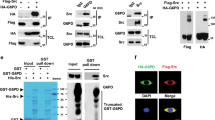
Cytosolic acetyl-CoA is known to be produced through two distinct pathways: ACSS2-catalyzed generation from acetate and ACLY-mediated lysis of citrate (Fig. 5a). After establishing the role of c-SRC in stimulating ACSS2-catalyzed acetyl-CoA production, our focus shifted towards understanding whether c-SRC is also implicated in the regulation of ACLY, as ACLY was identified as a potential c-SRC-interacting protein in our previous investigation. In order to ascertain the interaction between ACLY and c-SRC, ACLY and the other two enzymes involved in de novo fatty acid synthesis, ACC1 and FASN, were individually co-expressed with c-SRC, followed by IP assay. The result reveals a specific interaction between ACLY and c-SRC (Fig. 5b), further substantiated by IP assays and GST-pulldown experiment (Fig. 5c–f, and Supplementary Fig. 5a, b). Furthermore, our findings suggest that both the kinase activity ___domain (SH1 ___domain) and SH2 ___domain were involved in c-SRC’s interaction with ACLY, as evidenced by the disruption of this interaction caused by c-SRC inhibitors or its kinase-dead mutation (Supplementary Fig. 5c–e). Domain mapping assays indicate that ACLY binds to c-SRC through its C-terminal citryl-CoA lyase (CCL) ___domain (Supplementary Fig. 5f). We then investigated whether c-SRC can phosphorylate ACLY. Our results demonstrate that c-SRC effectively stimulated tyrosine phosphorylation of ACLY in vitro, and this phosphorylation was abolished by CIAP treatment or c-SRC inhibitors (PP2 and Saracatinib) (Fig. 5g–i and Supplementary Fig. 5g, h). Furthermore, in comparison with c-SRC-WT, c-SRC-CA reveals enhanced phosphorylation of ACLY, while c-SRC-KD fails to phosphorylate ACLY (Supplementary Fig. 5i). Interestingly, three other members of SRC family, namely YES, BLK and HCK, also exhibit the ability to phosphorylate ACLY (Supplementary Fig. 5j). Then, we identified Try682 and Try854 as two potential sites on ACLY that could be phosphorylated by c-SRC. We constructed these two mutants of ACLY, Y682F, and Y854F, and found that the Y682F mutant, rather than Y854F, significantly diminished the c-SRC-mediated phosphorylation of ACLY (Fig. 5j, k). This result indicates that c-SRC phosphorylates Tyr682 of ACLY, and this phosphorylation site is highly conserved among mammals (Fig. 5l). Our trypsin digestion assay suggests that the Y682F mutation may not affect the structure of ACLY (Supplementary Fig. 5k), which is also validated by AlphaFold analysis (Supplementary Fig. 5l). In conclusion, c-SRC phosphorylates ACLY at the Tyr682 residue.

a A diagram illustrating the role of key enzymes (ACSS2 and ACLY) in cytosolic acetyl-CoA production during de novo fatty acid synthesis (depicted by our team using Microsoft PowerPoint software). b Enzymes involved in de novo fatty acid synthesis, including ACLY, ACC1, and FASN, were co-expressed with Flag-c-SRC in HEK-293T cells, followed by IP assay to investigate whether c-SRC interacts with ACLY, ACC1 and FASN. c HA-ACLY and Flag-c-SRC were co-transfected into U87 cells as indicated. Cells were lysed, and the lysates were subjected to anti-Flag M2 beads to enrich Flag-tagged c-SRC. The total lysate and immunoprecipitates were analyzed by Western blot. d Reciprocal immunoprecipitation was performed with anti-Flag M2 beads after transfection with HA-c-SRC and Flag-ACLY in U87 cells. e Endogenous IP assay was performed in U87 cells using an ACLY primary antibody (with rabbit IgG as negative control). f Recombinant proteins (GST, GST-ACLY, and His-c-SRC) were purified from BL21 E.coli competent cells. GST-pulldown assay was performed to determine the interaction between ACLY and c-SRC in vitro. g Recombinant GST-ACLY and His-c-SRC were purified from BL21 E.coli competent cells, followed by in vitro kinase assay to determine the direct phosphorylation of ACLY by c-SRC. h U87 cells were transfected with HA-c-SRC and Flag-ACLY and subjected to anti-Flag M2 beads for immunoprecipitation. Tyrosine phosphorylation of ACLY was detected after the precipitates were treated with or without CIAP. i HA-c-SRC and Flag-ACLY were transfected into U87 cells. After 12 h of transfection, cells were incubated with c-SRC inhibitor Saracatinib (10 μM) for another 8 h. Then IP assay was performed and tyrosine phosphorylation of ACLY was detected by WB. j Representative mass spectrum of the specific ACLY peptide with phosphorylated Tyr682 residual (red). k Flag-tagged ACLY mutants (Y854F or Y682F) were co-transfected with HA-c-SRC in HEK-293T cells. After 24 h of transfection, ACLY was immunoprecipitated and its tyrosine phosphorylation was analyzed by WB. l Alignment analysis of the residues (Tyr682, ACLY) across different species by SnapGene software (6.0.2). Experiments in (e, f, g, and k) were performed three times. Experiments in (c, d, h, and i) were repeated in 293 T cells (Supplementary Fig. 5).
c-SRC phosphorylation of ACLY inhibits its activity and facilitates NADPH production for fatty acid synthesis
ACLY-catalyzed acetyl-CoA production is a canonical resource for de novo fatty acid synthesis, so we were intrigued by the possibility that c-SRC stimulates fatty acid synthesis by activating ACLY activity through phosphorylation. Surprisingly, enzymatic activity assay revealed an unexpected suppression of ACLY activity by c-SRC-WT and c-SRC-CA (Fig. 6a). Conversely, c-SRC knockdown or inhibition dramatically increased ACLY activity (Fig. 6b, Supplementary Fig. 6a). However, ACLY activity was not suppressed by c-SRC when the Y682F mutation was introduced, indicating that c-SRC inhibits ACLY by phosphorylation at Tyr682 (Fig. 6c). In line with this, endogenous ACLY activity was markedly suppressed by the reconstituted expression of c-SRC-WT and c-SRC-CA in SYF-KO MEFs, but not by c-SRC-KD (Supplementary Fig. 6b). These observations collectively suggest that c-SRC-mediated phosphorylation negatively regulates the enzymatic activity of ACLY. To delve into the inhibitory mechanism, ACLY is immunoprecipitated and eluted by Flag-peptide, followed by subjection to ACLY activity assay with various concentrations of citrate to determine the maximum velocities of ACLY (Vmax) and analyze the affinity to substrate (Km value). Our findings indicated that c-SRC inhibits ACLY by decreasing the Vmax (Fig. 6d, e). The inhibitory effect of c-SRC on ACLY activity might seem counterintuitive, given ACLY’s crucial role in catalyzing acetyl-CoA production for fatty acid synthesis. This contradiction prompted us to further explore the biological significance of ACLY inhibition by c-SRC. A possible explanation lies in the metabolic fate of elevated cytosolic citrate following ACLY inhibition. It has been well established that cytosolic citrate can be successively converted to isocitrate and α-ketoglutarate (α-KG) by aconitase 1 (ACO1) and isocitrate dehydrogenase 1 (IDH1). Importantly, IDH1-catalyzed the conversion of isocitrate to α-KG, which is accompanied by the production of NADPH. NADPH is the reducing equivalent as a critical factor required for fatty acid synthesis. To examine whether c-SRC shunts citrate to IDH1-catalyzed reactions for NADPH generation by inhibiting ACLY and consequently facilitating fatty acid synthesis, we analyzed NADPH levels and found a decreased in NADPH level (Fig. 6f, g), an increase in NADP+ level (Supplementary Fig. 6c) and a sharp decrease in the NADPH/ NADP+ ratio (Supplementary Fig. 6d) when c-SRC is knocked down in cells. Similarly, rescuing the expression of ACLY-Y682F in ACLY knockdown cells (Supplementary Fig. 6e) resulted in diminished NADPH levels (Fig. 6h) and a decreased NADPH/ NADP+ ratio (Supplementary Fig. 6f). Even though the abundance of acetyl-CoA and malonyl-CoA increased after cells were reconstituted with ACLY-Y682F (Fig. 6i), glucose-derived fatty acids (myristic acid, palmitic acid and stearic acid) were still significantly reduced (Fig. 6j), which can be attributed to the reduction of NADPH. Notably, the rescue of ACLY-Y682F did not affect the levels of intermediates in the TCA cycle (Fig. 6k). These results indicate that c-SRC phosphorylation of ACLY inhibits its activity and shunts citrate to NADPH production for fatty acid synthesis.

a Flag-ACLY was co-transfected with HA-tagged c-SRC-WT, c-SRC-CA and c-SRC-KD in U87 cells. After 24 h of transfection, cells were lysed and subjected to malate dehydrogenase (MDH)-coupled ACLY activity assay (n = 3 independent experiments). b U87 cells with c-SRC knockdown were analyzed for ACLY activity (n = 3 independent experiments). c ACLY-WT or ACLY-Y682F were co-transfected with HA-c-SRC in U87 cells, followed by ACLY activity analysis (n = 3 independent experiments). d ACLY activity assay with various concentrations of citrate was performed in U87 cells to analyze velocities of ACLY after c-SRC-mediated phosphorylation (n = 3 independent experiments). e The double-reciprocal plot of ACLY after phosphorylation by c-SRC is calculated based on the result in (d) (n = 3 independent experiments). f Primary glioma cells with c-SRC knocked down by two independent shRNAs were analyzed for NADPH levels using LC–MS (n = 3 independent experiments). g NADPH level was determined by LC–MS after c-SRC knockdown in U87 and CHG-5 cells (n = 3 independent experiments). h ACLY knockdown U87 and CHG-5 cells were reconstituted with ACLY-WT (re-ACLY-WT) and ACLY-Y682F (re-ACLY-Y682F). Cells were then analyzed for NADPH level by LC–MS (n = 3 independent experiments). i The levels of acetyl-CoA and malonyl-CoA in re-ACLY-WT and re-ACLY-Y682F U87 cells were analyzed by LC–MS (n = 3 independent experiments). j U87 cells reconstituted with ACLY-WT (re-ACLY-WT) or ACLY-Y682F (re-ACLY-Y682F) were incubated in fresh medium containing 20 mM [U-13C]-glucose for 3 days. The [U-13C]-glucose-derived fatty acids were quantified by GC–MS (n = 3 independent experiments). k The abundance of metabolites in the TCA cycle was determined after reconstitution with ACLY-WT or ACLY-Y682F in U87 cells (n = 3 independent experiments). Statistical data are presented as the mean ± SD (n = 3). p-values were calculated using unpaired and two-sided Student’s t-test.
c-SRC stimulates glioblastoma progression by regulating both ACSS2 and ACLY and optimizing acetyl-CoA and NADPH production for fatty acid synthesis
Bringing together all the aforementioned evidence, we propose that c-SRC fine-tunes the production of cytosolic acetyl-CoA and NADPH to facilitate fatty acid synthesis. To validate this concept, we conducted a double knockdown of ACLY and ACSS2 (shACSS2 + shACLY), which are subsequently reconstituted with the expression of ACSS2-WT and ACLY-WT (re-ACSS2WT + ACLYWT, abbreviated as re-WT + WT) or c-SRC unresponsive mutants of ACSS2 and ACLY (re-ACSS2Y530F/Y562F + ACLYY682F, abbreviated as re-Mut+Mut) (Fig. 7A). As we expected, the levels of both acetyl-CoA and NADPH were dramatically decreased in re-Mut+Mut cells (Fig. 7b, c). Correspondingly, the synthesis of fatty acids derived from [U-13C]-glucose and proliferative capacity were notably diminished in re-Mut+Mut cells (Fig. 7d, e). These findings provide confirmation that c-SRC plays a key role in stimulating fatty acid synthesis by co-regulating both ACSS2 and ACLY to optimize acetyl-CoA and NADPH production. Subsequently, we performed a mouse orthotopic tumor formation assay by infusing re-WT + WT or re-Mut+Mut U87 cells in mice brains. MRI imaging revealed that intracranial tumors generated by re-Mut+Mut cells were notably smaller than those generated by re-WT + WT cells (Fig. 7f). This result suggests that c-SRC stimulates glioblastoma progression by the phosphorylation of both ACSS2 and ACLY. To confirm the contribution of ACSS2 and ACLY to the oncogenic effects of c-SRC, we compare the proliferation of glioblastoma cells with the knockdown of ACSS2 and ACLY in constitutively activated c-SRC U87 cells. The double knockdown of ACSS2 and ACLY almost completely abolishes the oncogenic effects of c-SRC (Supplementary Fig. 7a). This result indicates that ACSS2 and ACLY are two key regulators in c-SRC-promotion of tumorigenesis. To ascertain the clinical relevance of this regulation, we analyzed the correlation between c-SRC activation (indicated by its phosphorylation level) and the tyrosine phosphorylation levels of ACLY and ACSS2 in clinical glioma samples. Tyrosine phosphorylation of ACLY and ACSS2 was found to be enhanced in high-grade glioma and as significantly correlated with c-SRC activity (Fig. 7g, h). This correlation of ACSS2 phosphorylation with c-SRC activity was further validated using our custom-made antibodies specifically recognizing the phosphorylation of ACSS2 at Tyr562 and Tyr530 (Fig. 7i, j). Importantly, c-SRC did not exhibit a close correlation with ACLY and ACSS2 according to database (Supplementary Fig. 7b, c). In conclusion, c-SRC stimulates glioblastoma progression by phosphorylating both ACSS2 and ACLY, thus optimizing acetyl-CoA and NADPH production for maximal fatty acid synthesis.

a U87 and CHG-5 cells were subjected to double knockdown of both ACSS2 and ACLY (shACSS2+shACLY, D-KD). These D-KD cells were then reconstituted with ACSS2-WT + ACLY-WT (abbreviated as re-WT + WT) or ACSS2-Y530F/Y562F + ACLY-Y682F (abbreviated as re-Mut+Mut). The samples derive from the same experiment but different gels for ACLY and ACSS2. b CHG-5 and U87 with re-WT + WT and re-Mut+Mut as shown in (a) were incubated with 10 mM [U-13C]-glucose for 24 h and determined for [U-13C]-glucose-derived acetyl-CoA (M + 2) by LC–MS (n = 3 independent experiments). c NADPH level in re-WT + WT U87 cells and re-Mut+Mut U87 cells were determined by LC–MS (n = 3 independent experiments). d U87 cells as shown in (a) were cultured in fresh medium containing 20 mM [U-13C]-glucose for 3 days, followed by analysis of [U-13C]-glucose-derived fatty acids with GC–MS (n = 3 independent experiments). e U87 cells were constructed as described in (a). 1 × 104 cells were seeded to analyze for proliferation by growth curve (n = 3 independent experiments). f Intracranial xenografts were established by injecting 1 × 105 of the U87 cells with re-WT + WT and re-Mut+Mut into nude mice’s brains. MRI images of the xenografts were acquired 30 days post-injection (left). The volume of the tumors in (e) was quantified by the sphere radius (right, n = 6 independent mice). g, h ACSS2 and ACLY in clinical samples were immunoprecipitated, followed by detection of their tyrosine phosphorylation level with p-Tyr antibody (N: normal tissue; WHO glioma grade: I–IV; n = 12 independent samples). The samples derive from the same experiment but different gels for c-SRC and p-c-SRC, another for ACLY, p-Tyr (IP-ACLY), and another for ACSS2, p-Tyr (IP-ACSS2) were processed in parallel. c-SRC activity was indicated by determination of its phosphorylation with p-c-SRC antibody (g). The correlation between c-SRC activity and tyrosine phosphorylation of ACSS2 or ACLY was analyzed according to relative optical density integrity (ODI) normalized to the corresponding total protein level (h). i, j Phosphorylation of endogenous ACSS2 in clinical glioma tissues was analyzed using specific phosphorylation antibodies (p-Tyr530 and p-Tyr562) (i). The correlation between c-SRC activity and phosphorylation of ACSS2-Tyr530 or ACSS2-Tyr562 was analyzed as in (j). N: normal tissue; WHO glioma grade: I–IV; n = 24 glioma samples. The samples derive from the same experiment but different gels for p-Tyr530, p-Tyr562, ACSS2, p-c-SRC. Data (b–f) are mean ± SD. p-values were calculated based on unpaired and two-sided Student’s t-test. The correlation analysis in (h, j) was performed by two-tailed Pearson r test.
Discussion
Glioma is one of the most intractable tumors for its high invasion and resistance to clinical treatment1,2. Metabolic remodeling is a distinct hallmark of cancer, thus identifying biomarkers or protein targets associated with this reprogramming could offer new avenues for cancer therapy. There’s accumulating evidence suggesting that increased fatty acid synthesis benefits the glioma malignancy36,37. Clarification of the pivotal regulatory mechanism underlying fatty acid synthesis may be helpful for glioma treatment. Cytosolic acetyl-CoA, the unique substrate for fatty acid synthesis, has been considered for a long time being exclusively produced via ACLY-based reaction with citrate from the TCA cycle. However, the studies in 2014 revealed that ACSS2 can convert acetate to acetyl-CoA in the cytosol14,15. Nonetheless, how ACSS2 is activated and how ACLY and ACSS2 are coordinately regulated in the fatty acid synthesis of glioma cells remain largely unknown.
Our study uncovers that c-SRC is activated in glioblastoma and remodels the production of cytosolic acetyl-CoA for fatty acid synthesis by activating ACSS2 and inhibiting ACLY (Extended Fig. 8). This metabolic remodeling has profound biological significances and highlights the significance of acetate as a precursor for cytosolic acetyl-CoA and fatty acids to support glioblastoma growth. Acetate could be acquired by glioblastoma cells through two ways: (1) endogenous conversion from the oxidation of pyruvate by ROS and keto acid dehydrogenases; (2) uptake from blood plasma. Our preliminary data shows that large amounts of acetate were produced from glucose in U87 and CHG-5 glioblastoma cells (Fig. 2b, c and Supplementary Fig. 2a). Since oxidation of pyruvate by hydrogen peroxide to acetate is an important manner to eliminate ROS, we propose that ROS elimination is coupled to acetyl-CoA production by acetate utilization, facilitated by c-SRC-mediated activation of ACSS2. Acetate concentrations in blood plasma have been reported to range from 50 to 200 μM, and cells may potentially uptake acetate through monocarboxylate transporters (MCTs)18,20. However, the regulation of MCTs responsible for acetate transportation, which is crucial for fulfilling the acetate demands of glioma cells, is still not well understood. Acetate utilization was reported to be increased when glucose, growth factors, and oxygen were deprived, suggesting that acetate is an alternative carbon source to produce acetyl-CoA for fatty acid synthesis under distinct nutritional stresses16,17,38. According to our investigation, c-SRC is the potential master to fine-tune the acetate utilization in response to various cellular stresses. Moreover, the inhibition of ACLY by c-SRC and resultant blockage of citrate-derived acetyl-CoA production serves to shunt citrate to IDH1-catalyzed dehydrogenation for NADPH production. NADPH, the essential and exclusive reducing equivalent for fatty acid synthesis, is in high demand by rapidly proliferating glioma cells. Therefore, its abundant supply becomes necessary and is achieved through multiple pathways. For example, our previous study indicated that c-SRC can stimulate glucose-6-phosphate dehydrogenase to increase NADPH production32. Likewise, in this context, c-SRC also acts to enhance NADPH production to support optimal fatty acid synthesis by dampening ACLY activity, which shunts citrate to the IDH1-catalyzed dehydrogenation pathway. However, it remains unknown whether IDH1 is also influenced by c-SRC in this metabolic remodeling, and this aspect awaits investigation in future studies. It’s noteworthy that the IDH1 Arg132 mutation occurs in both low-grade and high-grade gliomas, with mutated IDH1 showing a propensity to convert α-KG to 2-HG39,40,41,42,43. In our study, we observed wild-type IDH1 in high-grade glioma tissues, U87 cells, and CHG-5 cells. Therefore, we speculate that wild-type IDH1 in high-grade gliomas (III and IV) is responsible for the conversion of isocitrate to α-ketoglutarate, thereby generating NADPH. In summary, c-SRC remodels fatty acid synthesis by effectively coordinating the regulation of both ACSS2 and ACLY, ensuring the simultaneous provision of acetyl-CoA and NADPH to meet the specific requirements of glioblastoma cells. Therefore, our study presents a paradigm for the “on-off mechanism” in metabolic regulation, shedding light on our understanding of c-SRC’s role in modulating metabolic flexibility in gliomas.

Glioma cells exploit glucose and acetate, which permeate the blood-brain barrier, to fulfill their metabolic requirements. Specifically, these cells utilize glucose to generate acetate, releasing a substantial amount of acetate into the extracellular matrix. In glioma, c-SRC is extensively activated and phosphorylates ACSS2 to stimulate the synthesis of cytoplasmic acetyl-CoA from glucose or extracellular matrix-derived acetate. Concurrently, c-SRC-mediated phosphorylation of ACLY inhibits its activity, diverting citrate towards IDH1-catalyze NADPH production, thereby providing reducing equivalents for fatty acid synthesis. Consequently, c-SRC simultaneously satisfies the glioma cell’s need for both acetyl-CoA and NADPH in de novo fatty acid synthesis by coordinately regulating ACSS2 and ACLY, remodeling fatty acid synthesis and fostering the glioblastoma progression. Figure 8 created with BioRender.com released under a Creative Commons Attribution-NonCommerical-NoDerivs 4.0 International license.
The abnormal activation of c-SRC is prevalent in a wide array of human cancers, and its oncogenic effect makes it an attractive target for cancer treatment29. The potential advantages of inhibiting c-SRC have been demonstrated in clinical cancer treatments44. Several c-SRC inhibitors, such as dasatinib, saracatinib, and bosutinib, have been developed and subjected to clinical trials45,46,47. However, most of these clinical trials have shown that the efficacy of single-agent c-SRC inhibitors is moderate, yet they are generally well-tolerated in solid tumors48,49,50. This limited effect of c-SRC inhibitors could be partially attributed to the multiple members of the SRC family with different sensitivity to the same one. Recently, TAT-Cx43266-283, a peptide introduced as an effective agent for inhibiting c-SRC according to the gap junction protein connexin43 (CX43), exerts remarkable anti-tumor effects on glioma stem cell both in vitro and in vivo51,52,53. These studies, combined with our findings, unequivocally underscore the critical role of c-SRC as a driving factor in glioblastoma progression, implying that inhibiting c-SRC could potentially emerge as a promising therapeutic strategy for glioblastoma treatment. Moreover, the recognition of the key role played by ACSS2 in the regulation of fatty acid synthesis highlights its potential as a pivotal target in the development of novel anti-tumor drugs.
Methods
Cell culture
U118, U87, HEK-293T, and SYF-KO MEF were purchased from the American Type Culture Collection (ATCC). CHG-5 was purchased from GIWEIBio. SHG-44 was obtained from Procell. Each of the cell lines was cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum and maintained in a humidified incubator containing 5% CO2 at 37 °C. All cell lines were authenticated by STR analysis and routinely tested to be free of mycoplasma.
Antibodies and reagents
Rabbit anti-SRC (Cat. 2123, 1:1,000 for WB), rabbit anti-phospho-SRC (Tyr416) (Cat. 6943 S, 1:1,000 for WB), mouse anti-Phospho-Tyrosine (Cat. 9411 S, 1:1,000 for WB) mouse anti-β-actin (Cat. A1978, 1:5,000 for WB) and mouse anti-FLAG (Cat. F1804, 1:2,000 for WB) were purchased from Cell Signaling Technology. Rabbit anti-ACLY (Cat. 15421-1-AP,1:1,000 for WB), rabbit anti-ACSS2 (Cat.16087-1-AP, 1:1,000 for WB), rabbit anti-HA (Cat. 51064-2-AP, 1:2,000 for WB), and rabbit anti-FLAG (Cat. 20543-1-AP,1: 1,000 for WB) were purchased from Proteintech. Mouse monoclonal anti-SRC (Cat. sc-8056, 1:500 for WB) and mouse anti-HA (F-7) (Cat. sc-7392, 1:1,000 for WB) were purchased from Santa Cruz. Goat anti-mouse IgG (H + L) secondary antibody (Cat. 31430, 1:10,000 for WB) and goat anti-rabbit IgG (H + L) secondary antibody (Cat. 31460, 1:10,000 for WB) were purchased from Invitrogen. p-Tyr530-ACSS2 and p-Tyr562-ACSS2 were generated by immunizing rabbits with synthetic peptides (CGIMRTVY[pTyr]GNHE, CDGY[pTyr]WITGRI).
DMEM (no glucose, no glutamine, no sodium pyruvate, no phenol red, cat. A1443001), DMEM (Cat. 12800082), and DMEM/F12 (Cat# A4192001) were purchased from Gibco. TurboFect transfection agent (Cat. R05312), protein A/G PLUS agarose (Cat. 20423), GSH agarose (Cat. G2879), Ni-NTA agarose (Cat. 25215), and puromycin (Cat. A11138-03) were purchased from Thermo Fisher Scientific. Collagenase I (Cat# SCR103) was purchased from Sigma-Aldrich. PP2 (Cat. HY-13805) was purchased from MedChemExpress. Saracatinib (Cat. 379231-04-6) was purchased from Selleck Chemicals. [U-13C]-glucose (Cat. 110187-42-3) and [2-13C]-acetate (Cas# 13291-89-9) were purchased from Cambridge Isotope Laboratories.
Plasmids construction and mutagenesis
Full-length complementary DNAs (cDNA) encoding ACLY, ACSS2, c-SRC, ACC1, and FASN were amplified using mRNA purified from HEK-293T cells. For the purpose of conducting immunoprecipitation assays through transient expression, the cDNAs were integrated into the pCDNA3.3 vector and transfected into the HEK-293T cells by polyethylenimine (PEI) transfection or U87 cells by lipofectamine. To establish stable cell lines via lentivirus packaging, the cDNAs were inserted into the pBOBI vector. Multiple forms of c-SRC including c-SRC-CA, c-SRC-KD, and deletion mutants were generated and widely used in previous work.
All mutants of ACLY and ACSS2 were generated by a mutated-site primer-mediated PCR mutagenesis, employing a high-fidelity DNA polymerase (PrimeSTAR, Takara). The primers used for mutagenesis as follows: ACSS2-Y530F forward: 5′-AGTCTtTGGGAACCACGAAC-3′, ACSS2-Y530F reverse: 5′-TCCCAaAGACTGTGCGCATG-3′; ACSS2-Y562F forward: 5′-ATTtCTGGATCACTGGCAGG-3′, ACSS2-Y562F reverse: 5′-ATCCAGaAATAGCCATCCTGGTC-3′; ACLY-Y682F forward: 5′-TCTtTGAGGGCGTGGCCATTGGTGG-3′, ACLY-Y682F reverse: 5′-TCAaAGACGCCATCCGTGGTCCGAG-3′.
For shRNA-mediated knockdown, the plasmids including shACSS1, shACSS2, shACSS3, shACLY, and shSRC were generated by inserting 19-21 specific oligonucleotides into pLL3.7 and pLKO.1. The target sequence used for knockdown as follows: shACSS1: 5′-CCAGTTAAATGTCTCTGTCAA-3′; shACSS2: 5′-CAGGATTGATGACATGCTCAA-3′; shACSS3: 5′-GCCGTTGATCGTCATATTGAA-3′; shACLY: 5′-GGCATGTCCAACGAGCTCAA-3′; shSRC #1: 5′-GCTCCAGATTGTCAACAAC-3′; shSRC #2: 5′-GGACCTTCCTCGTGCGAGA-3′.
For reconstitution plasmids of ACSS2 (re-ACSS2) and ACLY (re-ACLY), synonym mutagenesis by primer-mediated PCR was performed with the corresponding primer as follows: ACSS2 forward: 5′-CAGaATcGAcGAtATGCTCAATGTATCTGGACACCTGCTG-3′, ACSS2 reverse: 5′-CATaTCgTCgATtCTGCCAGTGATCCAGTAATAGCCATCC-3′; ACLY forward: 5′-GGtATGTCtAAtGAaCTCAACAATATCATCTCTCGGACCACGGAT-3′, ACLY reverse: 5′-GtTCaTTaGACATaCCTCCGGAACGTGAGACATAGGCCACGCTGC-3′. The synonymous mutant bases in all primers above are shown in lower-case. The above primers used for plasmids construction were synthesized by Sangon Biotech.
Lentivirus package, infection, and transfection
Lentivirus package was performed in HEK-293T by lipofectamine transfection. In brief, the target plasmid and lentivirus elements (PMDL: VSVG: REV = 5:3:2) were transfected by using Turbofect lipofectamine (Cat. #R0532, Thermo Scientific). After 48 h of virus packaging, the supernatant was purified by centrifugation (3000 × g, 5 min), stored at −80 °C, and prepared for subsequent infection. Prior to infection, cells were cultured at 50% confluency, and the medium was replaced with fresh DMEM containing polybrene (10 μg/mL). Subsequently, the cells were infected with the prepared lentivirus and incubated for 24 h before proceeding to further analysis. Polyethyleneimine (PEI, Polysciences) is widely used for HEK-293T transfection. For this purpose, HEK-293T cells were cultured to around 70% confluency (35 mm dish) before transfection. A specific amount of plasmid expressing ACSS2, ACLY or c-SRC were added to 180 μL HBS solution (150 mM NaCl, 20 mM Hepes, pH 7.37–7.38). Subsequently, 20 μL PEI (40,000 MW, 10 μM in HBS) was added. The transfection mixture was then added to HEK-293T cells 30 min later. For U87 and CHG-5 transfection, lipofectamine (Turbofect, Thermo Scientific) was used. In detail, the plasmid was added to fresh DMEM without serum, followed by the addition of lipofectamine as the manufacturer’s recommendations. After 6 h of transfection, cells were changed with a fresh medium. Protein expression assays and immunoprecipitation assays were carried out 24 h after transfection.
Immunoprecipitation and Western blot
Cells were lysed and harvested in cell lysis buffer (1% Triton-X 100, 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 2.5 mM sodium pyrophosphate, 1 mM DTT, 1 mM β-glycerophosphate, 1 mM sodium orthovanadate, 1 μg/mL leupeptin, 1 mM phenyl-methyl-sulfonyl-fluoride). Subsequently, the cell lysates were subjected to sonication for 12 s (sonic for 2 s, stop for 1 s) to completely release the cell component, followed by centrifugation at 20,000 × g for 15 min. For immunoprecipitation, the collected supernatant was subjected to anti-Flag M2 beads (Sigma) for IP-Flag or A/G beads with HA antibody (Proteintech, cat. 51064-2-AP) for IP-HA. In the case of endogenous IP, the total cell lysate was pre-cleaned with 10 μL A/G beads (GE, NY, USA) for 1 h and then subjected to 10 μL A/G beads with 1 μg primary antibody or IgG (as control). Then the mixture was rotated at 4 °C for 3 h and the immunoprecipitates were washed 3 times with fresh lysis buffer. The SDS loading buffer was added to the immunoprecipitates, followed by boiling for 10 min to completely denature the protein.
For Western blot, proteins were separated by SDS-PAGE and transferred to polyvinylidene fluoride membranes (PVDF, Roche). After blocking with 5%(w/v) milk or bovine serum albumin (BSA, for phosphorylation signal analysis) dissolved in TBST (20 mM Tris-base, 135 mM sodium chloride, and 0.1% Tween 20), the membranes with specific proteins were incubated with corresponding primary antibodies diluted in TBST supplemented with 5% BSA (Sigma). The HRP-conjugated secondary antibodies (Thermo Fisher) were used to visualize the specific protein based on enhanced chemiluminescence system. X-ray films were scanned by HP LaserJet MFP M227 to present the blots in figures. ImageJ 1.53k was used to quantify the greyscale (ODI) of blots.
Glioma sample processing for Western blot
Glioma samples were collected and processed for Western blot with the approval of the Medical Ethics Committee of the First Affiliated Hospital of Xiamen University (XMYY-2022KY076). No sex and gender-based analysis have been performed in this study. In addition, the glioma patients include both males and females. All our procedures have been authorized by signed informed consent from the patients, and the donation of glioma tissue is completely voluntary and without compensation. The excised glioma tissues were rinsed with ice-cold PBS three times, followed by homogenization in lysis buffer (1% Triton-X 100, 20 mM Tris-HCl (pH-7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 2.5 mM sodium pyrophosphate, 1 mM DTT, 1 mM β-glycerophosphate, 1 mM sodium orthovanadate, 1 μg/mL leupeptin, 1 mM phenyl-methyl-sulfonyl-fluoride). Subsequently, the lysates were subjected to sonication for 1 min on ice (sonic for 2 s, stop for 1 s) to completely release the cell component, followed by centrifugation at 20,000 × g for 15 min. The supernatant was collected to determine the protein concentration by R250 (OD595). Same protein mass was used to prepare the sample for Western blot by adding SDS loading buffer. To further accurately analyze the activation status of c-SRC in clinical glioma tissues, we initially detected the expression of β-actin (internal reference). Subsequently, we adjusted the sample loading quantity to ensure consistency in β-actin based on grayscale scanning analysis. Then, same volume of samples was added to analyze the expression of c-SRC and its phosphorylation level.
Generation of mice orthotopic tumor (MOT) of glioma
All experimental procedures involving mice were conducted in accordance with the guidelines and approvals of The Laboratory Animal Center of Xiamen University (XMULAC20180041). The mice orthotopic tumor generation was carried out following established protocols54. Cells were infected with lentivirus for 48 h, digested with trypsin, and collected in ice-cold PBS. An incision longitudinally in the middle of the scalp was made, followed by drilling a hole with a diameter of 4 μm in the parietal bone. The hole was specifically located 1 mm posterior to the bregma and 1 mm to the right of the sagittal suture. Subsequently, a single-cell suspension (1 × 105 U87 cells, 2 × 105 primary glioma cells) in a volume of 5 μL of PBS was intracranially infused into the brain using a 10 μL Hamilton syringe, at a depth of 2.5 mm. Each cell line was injected into a cohort of 6-week-old female BALB/c nude mice. All orthotopic tumor transplantations were completed within 1 h of the surgical procedure, with mice kept warm on a heating pad and provided special care during the post-operative recovery period. Mice were housed under specific pathogen-free conditions with free access to water and a standard diet comprising 65% carbohydrate, 11% fat, and 24% protein. The light cycle was set from 8:00 to 20:00, maintaining a temperature of 21–24 °C and humidity between 40–70%. After the intracranial injection of glioma cells in mice, we monitored their eating and movement twice daily to ensure their well-being. If a mouse’s weight fell below 13 g, we humanely euthanized it to terminate the experiment for that mouse. For MRI analysis of mice orthotopic tumor, the images were collected by ParaVision 6.0.1 with Bruke 9.4 T MicroMRI. And the size of tumors in each brain did not exceed the maximum limit set (1 cm, diameter of tumor) by the Animal Ethics Committee of Xiamen University.
Isolation and culturing of primary glioma cells
All experimental processes were conducted with the support and approved of the Medical Ethics Committee of the First Affiliated Hospital of Xiamen University (XMYY-2022KY076). No sex and gender-based analysis have been performed in isolation and culturing of primary glioma cells. All our procedures have been authorized by signed informed consent from the patients, and the donation of glioma tissue is completely voluntary and without compensation. In this study, primary glioma cells were isolated from individual patient glioma tissues according to previous study55. In brief, glioma tissues were carefully isolated and immediately processed for the establishment of primary glioma cells within 2 h after surgical resection. The tumor tissues were minced using a scalpel in a solution of PBS and then digested with 0.1% collagenase I at 37 °C for 30 min. The enzymatic digestion was blocked by adding fetal bovine serum (FBS). The minced and digested tissue was passed through 100 µm cell strainers to achieve a single-cell suspension. Following three rinses with PBS, the cells were plated in DMEM/F12 medium supplemented with 10% FBS, 2 mM L-glutamine, 10 ng/mL EGF/FGF-2, and 1% penicillin-streptomycin. The plated cells were cultured in a humidified incubator at 37 °C with 5% CO2 for 72 h. Unattached cells were removed by changing the media, while the attached cells were continued to be cultured for subsequent passages and analysis.
Protein purification and GST-pulldown assay
Plasmids encoding GST, GST-ACLY, GST-ACSS2, and His-c-SRC were transformed into BL21 E.coli competent cells. The E.coli cells were grown in shaking LB medium to OD600 value between 0.7 and 0.8. Next, isopropyl-β-D-thiogalactoside (IPTG, 500 μM) was added to stimulate protein expression for 18 h. Proteins were purified with glutathione sepharose beads or Ni-NTA beads. GST-pulldown assays were performed by incubating His-c-SRC with GST, GST-ACLY, and GST-ACSS2 (GST as control) in GST-pulldown buffer (50 mM Tris-HCl (pH 7.6), 120 mM NaCl, 5 mM MgCl2, 0.5% Nonidet P-40, 1 mM EDTA) for 3 h at 4 °C, followed by pulldown with glutathione sepharose beads. The precipitates were washed three times with above GST-pulldown buffer. Then, input components and the precipitates were added with SDS loading buffer and boiled for 10 min for Western blot analysis.
LC–MS-based identification of tyrosine phosphorylation
ACSS2 and ACLY were co-transfected with c-SRC individually in U87 cells. ACSS2 and ACLY were immunoprecipitated with anti-Flag M2 beads after 24 h transfection, followed by SDS-PAGE separation. ACSS2 and ACLY were excised from Coomassie-stained gels and subjected to trypsin digestion. Digested peptides were extracted using 0.15% formic acid, and 67% acetonitrile and dried for analysis with AB SCIEX Triple TOF 5600 system. The protein mass spectrometry data was collected by Analyst TF 1.7 and analyzed by ProteinPilot 4.0. These analytical procedures were conducted by the Analysis and Testing Center at the School of Life Sciences (Xiamen University).
ACLY activity
The enzymatic activity of ACLY was performed according to previous studies56,57. Cells were lysed and harvested in cell lysis buffer. The total cell lysate or eluted ACLY were added to 200 μL ACLY reaction buffer (200 mM Tris-HCl (pH 8.0), 20 mM sodium citrate, 10 mM MgCl2, 10 mM DTT, 0.25 mM NADH, 10 mM ATP, 0.2 mM CoA and malate dehydrogenase (MDH) 5 U/mL). To determine the initial velocities and Km value of ACLY, various concentration of citrate was added to ACLY reaction buffer (Fig. 6d, e). ACLY activity was spectrophotometrically determined based on MDH-coupled reaction from ACLY-catalyzed oxaloacetate and NADH (absorbance at OD340) to malate and NAD+. The slope of decrease in OD340 which was measured every 30 s for 30 min reflects the reduction of NADH. Relative ACLY activity was calculated according to the slope of the resulting curve.
Colony formation assay
Cell number (as described in figure legends) was calculated and seeded in 6-well plates (n = 3), followed by cell culture for specific days. After crystal violet staining, the colony number was calculated for analysis.
In vitro kinase assay
In vitro kinase assay was performed based on a previously described protocol58. In brief, recombinant proteins, including His-c-SRC, GST-ACSS2 and GST-ACLY, were purified in BL21 E.coli competent cells and incubated in a kinase buffer (25 mM Hepes (pH 7.4), 50 mM NaCl, 5 mM MgCl2, 1 mM DTT, 0.5 mg/mL BSA, 1 mM Na3VO4, 200 μM ATP) for 30 min at 28 °C. SDS sample buffer was added to stop the reaction, followed by WB to determine the phosphorylation of ACSS2 or ACLY.
AlphaFold analysis of protein structure
To ascertain whether the mutations ACSS2-Y530F/562 F and ACLY-Y682F induce alterations in the protein structure, we predicted the protein structures of both wild-type ACSS2 (ACLY) and the ACSS2-Y530F/562 F (ACLY-Y682F) mutants by AlphaFold analysis (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb#scrollTo=UGUBLzB3C6WN). We typed in the protein sequences and five protein structures were predicted. And the first structure of each protein was chosen for structural analysis. The influence of tyrosine-to-phenylalanine mutations at positions 530 and 562 in ACSS2 was analyzed by PYmoL 2.3.4. Similarly, the impact of the tyrosine-to-phenylalanine mutation at position 562 in ACLY was analyzed using the same methodology.
Analysis of acetate in medium by NMR
Cells were cultured as described in figure legends. The cell-cultured medium was collected and centrifuged at 12,000 × g for 5 min. 400 μL supernatant was transferred to 5 mm NMR tubes. An internal tube containing 200 μL D2O (used for field-frequency lock) with 1 mM sodium 3-(trimethylsilyl) propionate-2,2,3,3-d4 (TSP) was used to provide the chemical shift reference (δ 0.00) for quantification of metabolites. One dimensional (1D) CPMG spectra were acquired using the pulse sequence [RD-90°-(τ-180°-τ)n-ACQ] with water suppression for culture medium on a Bruker Avance III 850 MHz spectrometer (Bruker BioSpin, Germany) equipped with a TCI cryoprobe at 25 °C provided by College of Chemistry and Chemical Engineering (Xiamen University) or a Bruker Avance III 600 MHz spectrometer (Bruker BioSpin, Germany) provided by Core Facility of Biomedical Sciences (Xiamen University). NMR data was collected by TopSpin (version 3.0). The identified metabolites were confirmed by a combination of 2D NMR data and the Human Metabolome Data Base (HMDB).
Quantification of metabolites based on LC–MS
The sample preparation of liquid chromatography-mass spectrometry (LC–MS) for metabolic analysis was performed with some modifications based on previous study59. To analysis the 13C-labeled metabolites derived from [2-13C]-acetate, the cells were seeded at 60–70% confluency and the medium was changed with complete DMEM containing 5 mM [2-13C]-acetate for another 24 h. The cells were washed with pre-cold PBS 3 times and quenched by ice-cold 80% methanol solution. The mixture was vortexed for 20 s to totally release the cell metabolites and centrifuged at 20,000 × g for 30 min at 4 °C. The collected supernatant was dried by vacuum centrifugal concentrator at 4 °C. The dried cell extracts were dissolved in 50% acetonitrile solution and prepared for subsequent LC–MS analysis.
For metabolites analysis, the liquid chromatography with SCIEX ExionLC AD was prepared and all chromatographic separations were performed with a Millipore ZIC-pHILIC column (5 μm, 2.1 × 100 mm internal dimensions, PN: 1.50462.0001). The column was maintained at 40 °C and the injection volume of all samples was 2 μL. The mobile phase consisted of 15 mM ammonium acetate and 3 mL/L Ammonium Hydroxide (>28%) in LC–MS grade water (mobile phase A) and LC–MS grade 90% (v/v) acetonitrile–HPLC water (mobile phase B) run at a flow rate of 0.2 mL/min. The analysts were separated with the following gradient program: 95% B held for 2 min, increased to 45% B in 13 min, held for 3 min, and the post time was set 4 min.
The gradient was: 95% B for 2 min, then to 45% B within 13 min (linear gradient), then maintained for 3 min, then to 95% B directly, and maintained for 4 min. The flow rate was 0.2 mL/min. The QTRAP mass spectrometer using an Turbo V ion source. The ion source was run in negative mode with a spray voltage of −4500 V, Gas1 40 psi and Gas2 50 psi, and Curtain gas 35 psi, Metabolites were measured using the multiple reactions monitoring mode (MRM). The relative abundance of metabolites was analyzed by MultiQuant Software (AB SCIEX).
For metabolites quantification, the levels of metabolites rely on the integrated area, which is subsequently normalized to protein mass.
Metabolite extraction and GC–MS analysis
Gas chromatography-mass spectrometry (GC–MS) is widely used in isotope-labeled fatty acid analysis and performed according to previous study60. Generally, cells were seeded in 6-well plates at 10–20% confluency. To analysis the 13C-labeled fatty acids derived from [2-13C]-acetate, the cells was changed with 4 mL complete medium containing 5 mM [2-13C]-acetate and maintained in a humidified incubator for 3 days. The cells were rinsed with pre-cold PBS, quenched by 800 μL ice-cold 80% methanol solution with 10 μg/mL Myristic-d27 acid and scraped to 1.5 mL EP tubes. 500 μL volume of chloroform were added and the mixture were vortexed for 20 s and kept on ice for 30 min to extract the free fatty acids. Next, the mixture was centrifuged at 20,000 × g for 30 min at 4 °C. The non-polar fraction dissolved in chloroform was transferred to a new tube and evaporated under nitrogen airflow. Fatty acid methyl ester was generated by dissolving and reacting dried chloroform fraction in 100 μL 1% H2SO4 methanol solution for 30 min at 70 °C. The quantification of metabolites relies on the integrated area, which is subsequently normalized to protein mass.
The detailed analysis of GC–MS was performed using an Agilent 7000D equipped with a capillary column under electron impact ionization at 70 eV. 1 μL of the sample was injected in column box under the splitless mode, using helium as the carrier gas. The GC temperature was held at 70 °C for 2 min after injection and increased to 250 °C at the rate of 5 °C/min. The GC–MS data was collected by MassHunter GC/MS acquisition for fatty acid analysis.
Statistics & reproducibility
No statistical method was used to predetermine the sample size. Results are derived from independent experiments and are represented as dots in the quantification histograms. Most experiments were not randomized; however, randomization was employed in some key LC–MS experiments (Figs. 4h, i, 6h, i, k, 7b, c, and Supplementary Fig. 6f). This randomization provides a more objective reflection of the effects of ACSS2 and ACLY on fatty acid synthesis. p-values were calculated based on unpaired and two-tailed Student’s t-test using GraphPad Prism Software or Microsoft Excel. p < 0.05 was considered statistically significant. To analyze the correlation of c-SRC phosphorylation with ACSS2/ACLY phosphorylation, as well as c-SRC activation in high-grade and low-grade glioma, we used two-tailed Pearson’s r test. The p-values are shown in the figures. Most immunoblots are representative of at least three independent experiments, except for the experiments which have been repeated in two cell lines.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Protein phosphorylation raw data have been deposited to the ProteomeXchange Consortium (http://www.proteomexchange.org/) through the PRIDE partner repository with the dataset identifier PXD054314. Metabolic LC–MS raw data, NMR raw data, GC–MS raw data, and the structural prediction of ACSS2-WT/DMut and ACLY-WT/Y682F by AlphaFold are deposited in Figshare repository (https://figshare.com/s/804ab10e1d0af9989f72). Source data are provided as a Source Data file. Uncropped scans of all blots generated in this study are provided in the Source Data. The correlation analysis between c-SRC and ACSS2/ACLY are available in the Chinese Glioma Genome Atlas (CGGA) database (http://cgga.org.cn/). Source data are provided with this paper.
References
Wen, P. Y. & Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 359, 1850–1850 (2008).
Stupp, R. et al. Radiotherapy plus concomitant and adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 352, 987–996 (2005).
Gilbert, M. R. et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 370, 699–708 (2014).
Warburg, O., Wind, F. & Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 8, 519–530 (1927).
Warburg, O. On the origin of cancer cells. Science 123, 309–314 (1956).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Kroemer, G. & Pouyssegur, J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell 13, 472–482 (2008).
DeBerardinis, R. J., Lum, J. J., Hatzivassiliou, G. & Thompson, C. B. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7, 11–20 (2008).
Koundouros, N. & Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 122, 4–22 (2019).
Efeyan, A., Comb, W. C. & Sabatini, D. M. Nutrient-sensing mechanisms and pathways. Nature 517, 302–310 (2015).
Zaidi, N., Swinnen, J. V. & Smans, K. ATP-citrate lyase: a key player in cancer metabolism. Cancer Res. 72, 3709–3714 (2012).
Wellen, K. E. et al. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080 (2009).
Currie, E., Schulze, A., Zechner, R., Walther, T. C. & Farese, R. V. Jr Cellular fatty acid metabolism and cancer. Cell Metab. 18, 153–161 (2013).
Comerford, S. A. et al. Acetate dependence of tumors. Cell 159, 1591–1602 (2014).
Mashimo, T. et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 159, 1603–1614 (2014).
Schug, Z. T. et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 27, 57–71 (2015).
Gao, X. et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat. Commun. 7, 11960 (2016).
Schug, Z. T., Vande Voorde, J. & Gottlieb, E. The metabolic fate of acetate in cancer. Nat. Rev. Cancer 16, 708–717 (2016).
Bloemen, J. G. et al. Short chain fatty acids exchange across the gut and liver in humans measured at surgery. Clin. Nutr. 28, 657–661 (2009).
Jeon, J. Y. et al. Regulation of acetate utilization by monocarboxylate transporter 1 (MCT1) in hepatocellular carcinoma (HCC). Oncol. Res. 26, 71–81 (2018).
Li, M. et al. Monocarboxylate transporter 1 promotes proliferation and invasion of renal cancer cells by mediating acetate transport. Cell Biol. Int. 45, 1278–1287 (2021).
Sivaprakasam, S., Bhutia, Y. D., Yang, S. & Ganapathy, V. Short-chain fatty acid transporters: role in colonic homeostasis. Compr. Physiol. 8, 299–314 (2017).
Liu, X. et al. Acetate production from glucose and coupling to mitochondrial metabolism in mammals. Cell 175, 502–513.e513 (2018).
Maher, E. A. et al. Metabolism of [U-13C]glucose in human brain tumors in vivo. NMR Biomed. 25, 1234–1244 (2012).
Martin, G. S. The hunting of the Src. Nat. Rev. Mol. Cell Biol. 2, 467–475 (2001).
Tice, D. A., Biscardi, J. S., Nickles, A. L. & Parsons, S. J. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc. Natl Acad. Sci. USA 96, 1415–1420 (1999).
Brown, M. T. & Cooper, J. A. Regulation, substrates and functions of src. BBA Rev. Cancer 1287, 121–149 (1996).
Mori, S. et al. Identification of two juxtamembrane autophosphorylation sites in the PDGFβ-receptor; involvement in the interaction with Src family tyrosine kinases. EMBO J. 12, 2257–2264 (1993).
Yeatman, T. J. A renaissance for SRC. Nat. Rev. Cancer 4, 470–480 (2004).
Zhang, J. et al. c-Src phosphorylation and activation of hexokinase promotes tumorigenesis and metastasis. Nat. Commun. 8, 13732 (2017).
Ma, H. et al. c-Src promotes tumorigenesis and tumor progression by activating PFKFB3. Cell Rep. 30, 4235–4249.e4236 (2020).
Ma, H. et al. c-Src facilitates tumorigenesis by phosphorylating and activating G6PD. Oncogene 40, 2567–2580 (2021).
Menendez, J. A. & Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 7, 763–777 (2007).
Schwer, B., Bunkenborg, J., Verdin, R. O., Andersen, J. S. & Verdin, E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc. Natl Acad. Sci. USA 103, 10224–10229 (2006).
Yoshimura, Y., Araki, A., Maruta, H., Takahashi, Y. & Yamashita, H. Molecular cloning of rat acss3 and characterization of mammalian propionyl-CoA synthetase in the liver mitochondrial matrix. J. Biochem. 161, 279–289 (2017).
Eyme, K. M. et al. Targeting de novo lipid synthesis induces lipotoxicity and impairs DNA damage repair in glioblastoma mouse models. Sci. Transl. Med. 15, eabq6288 (2023).
Miska, J. & Chandel, N. S. Targeting fatty acid metabolism in glioblastoma. J. Clin. Invest. https://doi.org/10.1172/JCI163448 (2023).
Bulusu, V. et al. Acetate recapturing by nuclear acetyl-CoA synthetase 2 prevents loss of histone acetylation during oxygen and serum limitation. Cell Rep. 18, 647–658 (2017).
Yan, H. et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773 (2009).
Tommasini-Ghelfi, S. et al. Cancer-associated mutation and beyond: the emerging biology of isocitrate dehydrogenases in human disease. Sci. Adv. 5, eaaw4543 (2019).
Dang, L. et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744 (2009).
Reuss, D. E. Updates on the WHO diagnosis of IDH-mutant glioma. J. Neurooncol. 162, 461–469 (2023).
Berger, T. R., Wen, P. Y., Lang-Orsini, M. & Chukwueke, U. N. World Health Organization 2021 classification of central nervous system tumors and implications for therapy for adult-type gliomas: a review. JAMA Oncol. 8, 1493–1501, (2022).
Zhang, S. & Yu, D. Targeting Src family kinases in anti-cancer therapies: turning promise into triumph. Trends Pharmacol. Sci. 33, 122–128 (2012).
Montero, J. C., Seoane, S., Ocana, A. & Pandiella, A. Inhibition of SRC family kinases and receptor tyrosine kinases by dasatinib: possible combinations in solid tumors. Clin. Cancer Res. 17, 5546–5552 (2011).
Mayer, E. L. & Krop, I. E. Advances in targeting SRC in the treatment of breast cancer and other solid malignancies. Clin. Cancer Res. 16, 3526–3532 (2010).
Wheeler, D. L., Iida, M. & Dunn, E. F. The role of Src in solid tumors. Oncologist 14, 667–678 (2009).
Daud, A. I. et al. Phase I study of bosutinib, a src/abl tyrosine kinase inhibitor, administered to patients with advanced solid tumors. Clin. Cancer Res. 18, 1092–1100 (2012).
Baselga, J. et al. Phase I safety, pharmacokinetics, and inhibition of SRC activity study of saracatinib in patients with solid tumors. Clin. Cancer Res. 16, 4876–4883 (2010).
Belli, S. et al. c-Src and EGFR inhibition in molecular cancer therapy: what else can we improve? Cancers 12, (2020).
Jaraiz-Rodriguez, M. et al. Connexin43 peptide, TAT-Cx43266-283, selectively targets glioma cells, impairs malignant growth, and enhances survival in mouse models in vivo. Neuro Oncol. 22, 493–504 (2020).
Talaveron, R., Matarredona, E. R., Herrera, A., Medina, J. M. & Tabernero, A. Connexin43 region 266-283, via Src inhibition, reduces neural progenitor cell proliferation promoted by EGF and FGF-2 and increases astrocytic differentiation. Int. J. Mol. Sci. https://doi.org/10.3390/ijms21228852 (2020).
Jaraiz-Rodriguez, M. et al. A short region of connexin43 reduces human glioma stem cell migration, invasion, and survival through Src, PTEN, and FAK. Stem Cell Rep. 9, 451–463 (2017).
Jiang, N. et al. Fatty acid oxidation fuels glioblastoma radioresistance with CD47-mediated immune evasion. Nat. Commun. 13, 1511 (2022).
Kim, S. S., Pirollo, K. F. & Chang, E. H. Isolation and culturing of glioma cancer stem cells. Curr. Protoc. Cell Biol. https://doi.org/10.1002/0471143030.cb2310s67 (2015).
Srere, P. A. The citrate cleavage enzyme. I. Distribution and purification. J. Biol. Chem. 234, 2544–2547 (1959).
Bauer, D. E., Hatzivassiliou, G., Zhao, F., Andreadis, C. & Thompson, C. B. ATP citrate lyase is an important component of cell growth and transformation. Oncogene 24, 6314–6322 (2005).
Lin, S. Y. et al. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science 336, 477–481 (2012).
Chaneton, B. et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 491, 458–462 (2012).
Metallo, C. M. et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384 (2011).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (U21A20373 (Q.L.), 32070749 (B.J.), 82072777 (Z.W.), 91857102 (Q.L.)), Fujian Provincial Clinical Research Center for Brain Diseases (2021FJSLCYX01 (Z.W.)), Xiamen Clinical Research Center for Neurological Diseases (2021XMSLCYX01 (Z.W.)), China Postdoctoral Science Foundation (2022M712672 (Wentao Z.)) and Project of Xiamen Cell Therapy Research Center (Grant No.3502Z20214001 (Z.W.)). We appreciate the help from the Analysis and Testing Center of Xiamen University.
Author information
Authors and Affiliations
Contributions
Z.W., Q.L., and Wentao Z. conceived this project and designed the experiments. Wentao Z., C.O., L.Z., J.W., and J.Z. contributed to the performance of the research. C.O., J.Z., J.W., Y.Z., C.H., Q.X., B.J., and F.L. contributed to the analysis and interpretation of data and critical review of the manuscript. C.Z. and M.Z. performed LC–MS and GC–MS respectively. C.X. identified the phosphorylation sites of ACSS2 and ACLY by LC–MS. X.H. designed the peptides and raised the specific phosphorylation antibodies of ACSS2. B.Z., Wenpeng Z., J.H., S.C., and X.L. were responsible for the collection and excise of clinical patient’s glioma samples. B.Z. generated clinical primary glioma cells. J.W. and D.L. analyzed acetate by NMR. Wentao Z. wrote the original draft. Q.L. Z.W. and Wentao Z. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, W., Ouyang, C., Zhang, L. et al. The proto-oncogene tyrosine kinase c-SRC facilitates glioblastoma progression by remodeling fatty acid synthesis. Nat Commun 15, 7455 (2024). https://doi.org/10.1038/s41467-024-51444-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51444-0
