
Abstract
Tandem repeats (TRs) are genomic regions that tandemly change in repeat number, which are often multiallelic. Their characteristics and contributions to gene expression and quantitative traits in rice are largely unknown. Here, we survey rice TR variations based on 231 genome assemblies and the rice pan-genome graph. We identify 227,391 multiallelic TR loci, including 54,416 TR variations that are absent from the Nipponbare reference genome. Only 1/3 TR variations show strong linkage with nearby bi-allelic variants (SNPs, Indels and PAVs). Using 193 panicle and 202 leaf transcriptomic data, we reveal 485 and 511 TRs act as QTLs independently of other bi-allelic variations to nearby gene expression, respectively. Using plant height and grain width as examples, we identify and validate TRs contributions to rice agronomic trait variations. These findings would enhance our understanding of the functions of multiallelic variants and facilitate rice molecular breeding.
Similar content being viewed by others


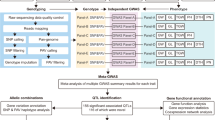
Introduction
Alterations in gene expression levels can generate diverse plant phenotypes. Genome sequence variations that influence gene expression are important drivers of differences in organismal traits1. Precisely modulating plant traits are essential for breeding new and desirable traits into crops. For example, Ideal Plant Architecture 1(IPA1) was a typical pleiotropic gene in rice (Oryza sativa L.) that increases grain number per panicle but reduces tiller number2. Ideal IPA1 expression levels could optimize the combination of tiller number and panicle size to achieve the highest yield potential3. However, very few biallelic variations can be exploited to precisely regulate endogenous gene expression in plants to produce ideal traits. Genome editing techniques could be used to generate plants with quantitative trait variations along a continuum4,5. However, due to a lack of data for regions that could be mutated to quantitatively tune gene expression, researchers have to create lots of gene editing lines to identify lines with expected rice traits. Growing evidence has verified that bi-allelic variations such as single nucleotide polymorphisms (SNPs), small insertions and deletions (Indels) and presence/absence variations (PAVs) only partially explain heritability; multiallelic variants are a hidden source of complex trait heritability6.
Tandem repeats (TRs) are stretches of DNA in which the nucleotide sequences are repeated several times in tandem. TRs are classified as short tandem repeats (STRs), also known as microsatellites, or variable number tandem repeats (VNTRs), also known as minisatellites, based on the length of the tandemly repeat units (1–6 bp and ≥7 bp, respectively)7. TR variations are one of the most abundant variation types in animal and plant genomes. They are highly unstable in length and are typically multiallelic8. Growing evidence has demonstrated that TR variants can mediate gene expression in species such as humans, pigs, Caenorhabditis elegans, sunflowers, and Arabidopsis thaliana9,10,11,12,13,14,15. At least 50 human disorders associated with TR polymorphisms have been described to date8. Compared with bi-allelic variants, multiallelic TR variations have a unique ability to drive a range of phenotypic variations16, which cannot be easily explained by nearby bi-allelic variants. Although TR variations have previously been exploited in rice primarily as genetic markers for quantitative trait locus (QTL) mapping17, researchers have also found that TR variations have the potential to fine-tune rice gene expression and phenotypes. For example, a TR variation of the CCATTC sequence in the 5′ untranslated region (UTR) of OsSPL13 was demonstrated that the allele of one copy leads to increased transcription of this gene and results in longer rice grain length and higher yield, when comparing to the allele of two copies18. By map-based cloning, Zhang et al.3 found rice lines carrying alleles with three tandemly repeat copies (3137 bp repeat) upstream of IPA1 showed higher expression of IPA1 by creating an open chromatin structure which attenuates the epigenetic repression, and lead to wide stem and high panicle primary branch number, compared to rice lines carrying alleles with one repeat copy3. In FRIZZY PANICLE (FZP) gene, rice lines containing two copies of 18 bp TRs at ~5.3 kb upstream repressed its expression comparing to rice lines containing only one copy19. Another research showed the near isogenic lines containing a 4 bp TR deletion at ~2.7 kb upstream of FZP gene decrease its expression level by affecting the binding activities of auxin response factors to its promoter, when compared to the near isogenic lines without the TR deletion20. Both TR variations affect grain yield19,20. Rice accessions with more than three GCG sequence repeats in the first exon of COLD11 have higher chilling tolerance21. However, due to technical limitations, TR variations are often overlooked in genomic studies. Although gene copy number variations have previously been characterized in rice22,23, systematic genome-wide TR variation identification and functional contribution analyses have not been conducted.
Due to their repetitive nature and tendency to contain high GC content, it is challenging to accurately identify genome-wide TR polymorphisms24. The classic method of TR genotyping such as HipSTR25, lobSTR26, popSTR27, GangSTR28, ExpansionHunter29, adVNTR30, is to identify TRs in the reference genome, then map short reads generated from an accession of interest back to the reference14. Tools developed for this method have a strong reference bias because only TRs present in the reference genome can be genotyped. Besides that, mapping short reads to repetitive regions and inferring expanded repeat lengths is challenging due to reads that map to multiple loci are typically removed during data processing31,32. These factors make it impossible to precisely determine the repeat number. However, long-read sequencing technologies and the corresponding high-quality genome assemblies avoid the drawbacks of the traditional TR genotyping methods, providing a unique opportunity to systematically investigate TR polymorphisms at the population-scale33.
In the present study, we annotate TR loci in 231 rice genome assemblies, which include the Nipponbare genome and 230 assemblies based on ONT long reads. We then integrate the TR annotations into a pan-genome graph to identify TR variation loci. From these data, a pan-TR dataset is constructed, including 227,391 TR variation loci with multiple alleles and 54,416 loci that are missing from the reference genome. Using transcriptomic data generated from the panicle tissue of 193 accessions and the leaf tissue of 202 accessions, we conduct a genome-wide analysis of TR variations that affected expression levels of adjacent genes. We also evaluate the effects of TR variations on gene expression while controlling for the effects of nearby bi-allelic variants. Finally, we conduct a colocalization analysis to identify eTRs that had the potential to regulate rice phenotypes. These findings enhance our understanding of the functions of multiallelic variants and provide candidate targets for genome editing to fine-tune gene expression and optimize quantitative traits in rice.
Results
The pan-TR dataset
We previously collected and assembled the genomes of a set of 230 rice accessions that are highly representative of the global genetic and phenotypic diversity of rice germplasm34 (Supplementary Data 1). For each of these accessions and the O. sativa subsp. japonica cv. ‘Nipponbare’ reference genome (Nipponbare IRGSP 1.0)35, we annotated known TR loci with RepeatMasker and performed de novo TR identification with two programs: Tandem Repeats Finder36 and ULTRA37. There was an average of 379,176 tandem repeat loci per accession, ranging from 346,269–416,093 and comprising an average of 22.9 Mbp in each genome (range = 20.6–24.8 Mbp) (Supplementary Data 2). To check the assembly quality of the TR loci in each accession, we evaluated TRs identified in the assemblies of the 93-11 accession and the corresponding large-insert bacterial artificial chromosome (BAC) clones38. With a threshold of 100% length and sequence concordance between our genome assembly and the corresponding BACs, the validation rate of the 231,898 putative STR loci was 89.8%. The genome assemblies were therefore determined to be of high quality in the regions classified as TRs.
We have constructed a pan-genome graph of the 230 accessions and the Nipponbare reference genome which was used to represent multiple genomes while preserving the coordinates of the linear genome34. After integrating the TR annotations with the pan-genome graph, we identified 531,575 polymorphic TR loci (Fig. 1a), including 227,391 loci with at least three alleles (referred to as multiTRs). Because we aimed to evaluate the role of multiallelic variants in regulating gene expression and phenotypes, we focused primarily on the multiTR loci in further analyses. To assess multiTR quality, we manually evaluated two previously-characterized TR variations around OsSPL13 and COLD11. We found that both TR variations were present in our TRs call set and novel alleles existed in our accession panel for both loci (Fig. 1b–e). We next randomly selected 218 TR variation loci ranging in size from 2 to 1564 bp and manually evaluated them through multiple sequence alignments of the corresponding genome assemblies. 90.4% of the TR variation loci could be validated (Supplementary Data 3). We also randomly selected eight TR variations for manual validation with PCR followed by gel electrophoresis or Sanger sequencing (Supplementary Fig. 1).

a Schematic of the pan-TR polymorphism dataset. In a previous study, we assembled the genomes of 230 rice accessions with broad genetic diversity (including 202 O.sativa accessions and 28 O.rufipogon accessions) to construct a pan-genome graph34. In the present study, we conducted de novo whole-genome tandem repeat annotation for each accession and the Nipponbare genome. After integrating the TR annotations into the pan-genome graph to get TR variation loci, we obtained the pan-TR polymorphism dataset, which included TR loci absent from the reference genome. Known TR variations that are causal for rice phenotypes were validated in the pan-TR dataset. Alleles for TRs around OsSPL13 (b, c) and COLD11 (d, e) and their distribution among rice subpopulations.
Characteristics of the TR dataset
The multiTR dataset included 145,279 STRs and 82,112 VNTRs (Fig. 2a). Previously, researchers identified TR loci in the reference genome, then mapped resequencing data to the reference genome in plants14,39. Identification of TR polymorphisms from the pan-genome graph reduced the reference bias and allowed us to find TR variations that were absent from the Nipponbare reference genome. In the dataset generated for this study, 10.0% (14,544/145,279) of the STRs and 48.6% (39,872/82,112) of the VNTRs were not present as TR arrays in the Nipponbare reference genome (Fig. 2a). For the TRs that were present in the Nipponbare genome, the major alleles of 28.5% (37,316/130,735) of STRs and 21.0% (8886/42,240) of VNTRs differed from the alleles in the Nipponbare genome (Fig. 2b). The motif (or repeat unit) lengths of TR variants varied from 2 to 2581 bp (Fig. 2c) and the allele numbers varied from 3 to 105, the lower bound of which was due to we mainly focusing on TR loci with at least three alleles. Of the multiTR variations, 31.7% had more than five alleles (Fig. 2d). 15.9% of STR loci and 8.7% of VNTR loci here had a major allele frequency of less than 0.5 in the present dataset, while the ratio for STR loci was only 4% in C. elegans40, which may be due to the comparatively high genetic diversity included in the present study (Fig. 2e).

a Distribution of each TR type. The inner pie chart indicates the ratio of short TRs (STRs) (red) and variable number TRs (VNTRs) (blue) in the pan-TR dataset. The outer pie chart indicates the ratio of TRs that were present in the Nipponbare reference genome (dark green) and TRs absent from the reference genome (light green). b Statistics summarizing TR copy number differences between the major allele and the reference allele. Red and blue dots indicate STRs and VNTRs, respectively. c Distribution of the repeat motif length at each TR locus. d Distribution of allele numbers at each TR locus. e Distribution of the frequency of the major alleles at each TR locus. The dashed line indicates a major allele frequency of 0.5. f The distribution of genetic variants’ distance to the nearest transcription start site (TSS). Each color indicates a genomic variant. The overlap between genetic variants indicates similar distribution between variants. g Distribution of genomic variations along each chromosome. p-values indicate differences in distribution between TRs and other bi-allelic variants (Wilcoxon rank sum test). h Distribution of linkage disequilibrium (LD) values between TRs and bi-allelic variants within 100 kb. LD was calculated as the absolute value of a pairwise Pearson’s correlation test (|R|). For each TR, the maximum |R| value with adjacent variants on either side is recorded. The dashed line indicates |R| = 0.30 and |R| = 0.70. Source data are provided as a Source Data file.
Of the multiTRs, a total of 13.31% (30,274/227,391) were located in the coding region of 17,376 genes (in total), compassing almost 1/3 of the genes present in the Nipponbare reference genome (Supplementary Fig. 2a). GO enrichment analysis showed these genes were associated with reproductive structure development and DNA binding process (Supplementary Fig. 2b). We also found that among the TR variations located in gene coding sequence, the alleles of 8593 loci may cause frameshift mutation (Supplementary Fig. 2c). The allele frequency of 96.42% (8285/8593) of such alleles were <5% in the Pan-TR dataset (Supplementary Fig. 2d). We also identified a total of 21,681 TR variation loci with alleles that may cause amino acid insertion or deletion variations, but only 3443 of them were located in the Pfam functional ___domain (Supplementary Fig. 2c).
Most TRs were not sufficiently represented by nearby bi-allelic variants
To explore whether TR distribution suggested a regulatory role in gene expression, we examined how the TR variations were distributed compared to annotated genes in the rice reference genome. In general, the TR sites tended to cluster upstream of TSSs (Fig. 2f), similar to their distribution in A. thaliana14. However, we also found that bi-allelic variants showed a statistically similar distribution pattern as TR variations (Wilcoxon rank sum test statistics p = 0.780, 0.200, and 0.140 for PAVs, SNPs, and Indels, respectively) (Fig. 2f). If the TR variations were in strong linkage disequilibrium (LD) with these bi-allelic variants, it would be unlikely that the analysis would reveal new TR variants responsible for differences in gene expression due to the high resolution of SNPs, PAVs, and Indels across the rice genome. Thus, to establish whether the TR variations were in strong LD with these bi-allelic variants, we first compared the distribution of multiTRs and bi-allelic variants (namely SNPs, Indels, and PAVs) along each chromosome. Eleven of the chromosomes (all except Chromosome 12) showed significant differences between the distribution of multiTRs and at least one type of bi-allelic variants (Fig. 2g) (Wilcoxon rank sum test statistics). This indicated a lack of strong LD between the TR variations and nearby bi-allelic variants (classified as those within 100 kb). A small proportion of TR variations were in strong LD (r ≥ 0.7) with each type of bi-allelic variant: 31.3% with SNPs, 15.0% with Indels, and 26.4% with PAVs. More than a third of TR variations were weakly correlated (Pearson’s r < 0.3, or not statistically significant) with adjacent SNPs, Indels, or PAVs (38.3%, 58.8%, and 40.6%, respectively) (Fig. 2h). These results indicated that the gene expression effects driven by variations in repeat length were not likely to be fully captured by bi-allelic variants.
Profiling of expression level variations in young leaves and panicles
To compare the effects of TR variations and those of bi-allelic variants on gene expressions, we performed a genome-wide analysis to identify associations between these variants and expression levels of nearby genes. The expression data were generated from leaves at the young seedling stage34 (202 accessions) and from young panicles (10–20 mm) at the early developmental stages41 (193 accessions).
For TR variations, we performed a linear regression between the repeat number of the TR and normalized expression of the gene, controlling for population structure. For SNPs, PAVs and Indels, the associations were performed in the same way using genotype dosages (0,1,2). Using this approach, we identified 9343 genes for which expression levels were significantly associated with genetic variants (eGenes) in the leaf and 12,823 eGenes in young panicles. This included 6232 eGenes that overlapped between the two tissues (Fig. 3a). As expected, functional enrichment analysis indicated that the leaf-specific eGenes were enriched in the biological process of photosynthesis (EnrichmentScore = 1.66, p = 0.004), whereas eGenes specific to the young panicle were enriched in flower development (EnrichmentScore = 1.18, p = 0.017) (Supplementary Data 4 and 5). Because the young panicle is directly involved in flower and grain development, eGenes specific to the young panicle were expected to be more informative in identifying elite alleles that contributed to rice grain yield and quality. DHT1 (LOC_Os04g54440) mediates the splicing of pre-mRNA for the strigolactone receptor D14; mutants for this gene have small panicles and grains, dwarfism, and more tillers compared to wild-type plants42. We found that DHT1 expression in the young panicle was associated with the number of TC repeats in its promoter and 5’UTR region (Fig. 3b, c). Elevated GW6a (LOC_Os06g44100) expression enhances grain weight and yield43; GW6a expression levels were positively correlated with the TR variants in the promoter region (Supplementary Fig. 3).

a Statistical comparison of genes for which expression levels were significantly associated with genetic variants (eGenes) in the panicle and young leaf tissues. b Manhattan plot of a panicle-specific eGene DHT1(LOC_Os04g54440). DHT1 was expressed in both leaf and panicle tissues. Genetic variants were significantly associated with its panicle expression variations, other than leaf expression variation. c Pearson correlation analysis between TR repeat numbers and normalized panicle expression of the gene DHT1(LOC_Os04g54440). The error bands indicate 95% confidence intervals. The p-values were calculated by two-sided t test. d, e Statistical comparison of eGenes identified using different genomic variants as markers in the panicle (d) and the young leaf tissues (e). f Example of eGenes identified only with TR markers in the panicle. g Pearson correlation analysis between the TR repeat number and LOC_Os01g02910 expression in the panicle. The error bands indicate 95% confidence intervals. The p-values were calculated by two-sided t test. Source data are provided as a Source Data file.
Among all variants, we identified 10,810, 10,221, 8409, and 5789 eGenes associated with TR variations, SNPs, PAVs, and Indels, respectively, in the panicle (Fig. 3d). In the leaves, there were 7685, 7249, 6224, and 4027 eGenes associated with TR variations, SNPs, PAVs, and Indels, respectively (Fig. 3e). Of these eGenes, 1392 in the panicle and 1049 in the leaf were only associated with nearby TR polymorphisms rather than bi-allelic variants. For example, TA-repeat variants in the first intron of LOC_Os01g02910 were negatively associated with expression levels of this gene in the panicle (Fig. 3f, g). LOC_Os01g02910 gene encodes a glycosyltransferase, and its homologous gene, GSA1 (LOC_Os03g55040), regulates grain size and abiotic stress tolerance in rice44. Expression levels of LOC_Os04g52630 in the seedling leaves were associated with GA-repeat variants in the promoter region (Supplementary Fig. 4). This gene encodes a leucine-rich repeat-containing protein kinase and is associated with drought tolerance45.
A large proportion of eGenes were associated with both TR variations and the bi-allelic variants (Fig. 3d, e). To fine-map the potential causal variants of the candidate 6636 and 9418 genes associated with both TR variations and bi-allelic variants in leaf and panicle, we inferred credible sets of variants using Bayesian fine-mapping with susieR46. 4964 leaf genes and 7369 panicle genes had 95% credible sets, and 1867 leaf genes and 1642 panicle genes contained TR variations in their credible sets. To determine whether the leading TR variations (TR variations that most significantly associated with expression for each gene, eTRs) explained variations in gene expression better than the leading bi-allelic variants (bi-allelic variants that most significantly associated with gene expression, eBi-allelic variants), we compared regression models of gene expression as a function of bi-allelic variants (namely SNPs, Indels, or PAVs) with models of gene expression as a function of both TR variations and bi-allelic variants. In the panicle, 570 TR-gene models explained variations in gene expression better than the models including only the eBi-allelic variants (ANOVA q value < 0.05) (Supplementary Fig. 5a). In the leaf, this number decreased slightly to 550 TR-gene pairs (Fig. 4a).

a Comparison of gene expression models with or without eTR variant. R2 indicated expression variations explained by the model. X-axis and Y-axis indicate R2 of models including only the eBi-allelic variant and both eBi-allelic variant and eTR, respectively. Red dots indicate genes for which models including eTRs were significantly better than models including only eBi-allelic variants (Benjamini–Hochberg test, q value < 0.05); blue dots indicate those without significantly differences. b Original (unconditioned) eTR effect sizes (β) compared to conditioned eTR β. Red points indicate eTRs with consistent effect directions between conditioned and unconditioned analysis; the rest points indicate those with discordant effect directions. Not significant β are represented as 0. c Manhattan plot for OsPRR1. Cx represents eTR repeat number. Pie chart shows eTR and eSNP distribution. d Pearson correlation analysis between eTR repeat number and OsPRR1 leaf expression. Red and green regression lines indicate analyses including all accessions and only accessions with the major SNP type, respectively. e Plant height among accessions with different eTR repeat numbers. f Pearson correlation analysis between the eTR repeat number and plant height. Red and green regression lines indicate analyses including all accessions and only accessions with the major SNP type, respectively. g Schematic diagram indicating that mutated site of osprr1. h Morphologies of the osprr1 mutant and the wild type (Xiushui134). i Plant height of the osprr1 mutants (n = 36) and the wild type (n = 38) plants after heading stage. j Schematic diagram of the recombinant vectors containing OsPRR1 promoter for firefly luciferase complementation assay. REN Renilla luciferase, LUC firefly luciferase, pOsPRR1C3(NH242) the vector containing the OsPRR1 promoter region with 3 TR copies, pOsPRR1C4(NH027) the vector containing 4 TR copies. k Relative LUC/REN activity in tobacco protoplast transformed with pOsPRR1C3 and pOsPRR1C4 vector (n = 3). In d and f, the error bands indicate 95% confidence intervals (two-tailed t test). In i and k, data presented as mean ± SD, p-values were generated using two-tailed t test. Source data are provided as a Source Data file.
We also considered the possibility that the observed TR associations may have been indirect correlations driven by LD between the TR variations and other uninvestigated but causal bi-allelic variants. To assess whether TR variations acted as QTLs independent of other local genetic variation, for each gene that was putatively associated with TR variants, we performed conditional analyses by removing the effect of the strongest bi-allelic variants associated with it. Using the accessions with the common versions of the leading bi-allelic variants, we tested the TR-gene pairs. Only TR-gene pairs that had at least 3 TR alleles in the accessions with the common bi-allelic variants were tested. In the panicle and the leaf, 485 and 511, respectively, of the fine-mapped TR–gene pairs showed the same directional effects after controlling for the lead bi-allelic variants (Fig. 4b, Supplementary Fig. 5b).
For example, in leaf tissues, we identified both TR variations and SNPs were significantly associated with OsPRR1 (LOC_Os02g40510) expression (Fig. 4c). Both the leading SNP (C/A at position 24569295 on Chr2) and the leading TR variation ((CGTCC)n from positions 24569319–24569333 on Chr2) were in the 5′ UTR. The copy number of the leading TR variation was significantly positively associated with OsPRR1 expression (Fig. 4d). After removing the effects of the leading SNP, the positive association of the TR variation with gene expression was still significant. OsPRR1 is a member of the pseudo-response regulator (PRR) gene family. Other members of the same gene family, such as OsPRR37 and OsPRR59, are strong contributors to the regulation of photoperiodic flowering, plant height, and grain productivity in rice47,48. Consistent with those prior findings, we here found that accessions with more copies of the leading TR variation were shorter in plant height (Fig. 4e). The negative associations were still significant even after controlling for the leading SNPs (Fig. 4f). To verify the role of OsPRR1 in regulating plant height, we generated the CRISPR knock-out line osprr1 containing a frame-shift mutation caused by 1 bp deletion (Fig. 4g; Supplementary Fig. 6). The homozygous lines were subsequently employed for examining the plant height phenotype. The plant height of osprr1 plants were significantly higher than the wild plants, as expected from the association results (Fig. 4h, i). We also conducted luciferase reporter assays to validate the effects of TRs located in OsPPR1 promoter region on its transcriptional activation. Compared to the one of accessions (such as NH242) with 3 copies of the repeat motif, the one (such as NH027) of accessions with 4 copies in the OsPRR1 promoter fragment showed significantly higher relative expression activity (Fig. 4j, k). We also got a homozygous editing line with a 2 bp deletion in the TR region, making one copy of the CGTCC repeats were destructed (Supplementary Fig. 7a). The TR editing plants showed reduced gene expression of OsPRR1 in young leaf tissue (Supplementary Fig. 7b) and higher plant height than the wild type plants (Supplementary Fig. 7c, d).
To understand the functional mechanisms by which eTRs may regulate gene expression, we analyzed eTR distribution among regions with histone markers and accessible chromatin. Although in the whole genome background, TR variations were not enriched or slightly enriched in the regions with histone markers and chromatin accessibility, we found eTRs showed higher enrichment in these regions compared to the TR variation background (Supplementary Fig. 8). Thus, epigenetic modifications may have been involved in TR-mediated gene expression regulation.
eTRs are potential regulator of phenotypes
We next wondered whether the identified eTRs directly affected rice phenotypes. We conducted a genome-wide association study (GWAS) to identify TR variations associated with grain width. SNPs, Indels, and PAVs significantly associated with grain width were also identified as a control. Using all of these markers, we identified the previously-characterized signal GW5 on Chr5, which is a major gene that contributes to grain size diversity in rice49 (Supplementary Fig. 9). In addition to the GW5 signal, we found a peak on Chr6 using the TR variants; the peak was not statistically significant in the bi-allelic variant dataset (Fig. 5a). The region within 500 kb around the peak contained 122 genes that were expressed in the panicles. The expression of five of these genes were significantly associated with grain width (Fig. 5a, Supplementary Fig. 10). Three of the five genes had eQTL signals in panicles based on TR variants (Fig. 5b). To test whether these eTRs were causal, we used the R package ‘coloc’ to quantify the posterior probability of causality for each variant. The causal variants for both the grain width phenotype and expression of the three genes with significant eQTL signals were fine-mapped (Fig. 5c, d). Only LOC_Os06g03850 expression shared the same candidate causal TR variant with the grain width phenotype. The ‘coloc’ package was also used to compare association statistics at each TR for grain width and LOC_Os06g03850 expression. As expected, the posterior probability of grain width and LOC_Os06g03850 expression in the panicle were colocalized, reaching the maximum value (1) for the TR variant (GT)n at position 1552149 on Chr6 (Fig. 5e), which prompted us to focus further on LOC_Os06g03850, named TRGW6, and the TR variation. This TR variant was not identified as a TR in the Nipponbare reference genome because there was only one copy of the repeat motif (Fig. 5f). However, we identified seven TR alleles for this locus in the pan-TR dataset. The TR variant was located in the promoter region of TRGW6, which encodes a putative impaired sucrose induction protein. A homolog in Arabidopsis (AT4G27750) is involved in sugar sensing and carbohydrate synthesis, and mutants for the gene have restricted seed setting50. In rice, grain width generally differs between indica and japonica accessions, to the extent that this characteristic can be used to distinguish between the subpopulations. Here, positive associations between TR variations and grain width were significant in both indica and japonica accessions (Fig. 5g). The associations between the TR variants and gene expression levels were also independent of the subpopulations (Fig. 5h).

a Manhattan plot showing associations between grain width and TR variations, PAVs, SNPs and Indels. Dashed lines of each color indicate the genome-wide threshold value for the corresponding variant type. Around the 1 Mbp region of the leading peak, 122 genes were expressed in the panicle tissue (Median of FPKM > 0 and Maximum of FPKM ≥ 1). Expression levels of the five indicated genes were significantly associated with grain width. b Manhattan plot showing associations between TRs and expression levels of the five genes in the panicle associated with grain width. The dashed line indicates the genome-wide threshold value. Posterior probability of causality for grain width (c) and expression levels of the three genes with significant eTRs (d). “#” indicates the value of the posterior probability. e Posterior probability of a TR being the causal variant for both grain width and LOC_Os06g03850 expression. The dashed line across c–e indicates the position of the same TR variant. f The candidate causal TR was in the promoter region of LOC_Os06g03850 and 7 alleles existed in the present dataset. g Associations between TR repeat numbers and grain width in the indica and japonica subpopulation of Oryza sativa. h Associations between TR repeat numbers and LOC_Os06g03850 expression in the panicle tissue among members of the indica and japonica subpopulation (green and red lines, respectively). In g and h, the error bands indicate 95% confidence intervals, p-values were calculated by two-sided t test. Source data are provided as a Source Data file.
To validate the gene functions and the effects of TR on TRGW6 expression, we conducted transgenics-based function validation experiment. We generated the CRISPR knock-out line trgw6 containing a frame-shift mutation caused by 31 bp deletion (Fig. 6a, Supplementary Fig. 11). The homozygous lines were subsequently employed for examining the grain width. The grain width of trgw6 were significantly lower than the wild plants (Fig. 6b). We also generated TR copy editing lines using CRISPR-Cas12a system. The mutant lines with a copy of TR deletion showed lower expression of TRGW6 and narrower grain width than the wild plants (Fig. 6c–f). The mutant lines with a copy of TR insertion showed higher expression of TRGW6 and wider grains than the wild plants (Fig. 6g–j).

a Schematic diagram indicating that the target and mutated site of TRGW6 by CRISPR/Cas9 technology. b The grain width phenotype of trgw6 and its wild type (ZH11). Bar = 10 mm. The average grain width of all seeds in a single plant represents the grain width value of the plant (n = 10). c The TR sequence edit diagram of TRGW6 by CRISPR–Cas12a promoter editing (CAPE) system in NH142 background. TR position was shown in (Fig. 5f). The background accession NH142 contains 9 TR copies in TRGW6 promoter region, and 1 copy was removed in the editing lines (NH142-1copy). d Expression level of TRGW6 in plants of NH142 and NH142-1copy. Rice ACTIN was used for the internal reference. e, f The grain width phenotype of NH142 and NH142-1copy. The average grain width of all seeds in a single plant represents the grain width value of the plant. Three plants were measured from each of the two materials (n = 3). g The TR sequence edit diagram of TRGW6 by CAPE in NH072 background. The background accession NH072 contains 9 TR copies in TRGW6 promoter region and 1 TR copy was added in the editing lines (NH072+1copy). h Expression level of TRGW6 in NH072 and NH072+1copy (n = 3). Rice ACTIN was used for the internal reference. i, j The grain width phenotype of NH072 (n = 5) and NH072+1copy (n = 3). The average grain width of all seeds in a single plant represents the grain width value of the plant. In b, d, e, h and i, data presented as mean ± SD. The p-values were calculated by two-sided t test. Source data are provided as a Source Data file.
Discussion
Plant breeding efforts to optimize agronomic traits require minor but significant changes in quantitative traits. Multiallelic variants have the potential to cause slight phenotypic changes in both animals and plants21,51. Tandem repeat variants were polymorphic and often multiallelic14. Their contributions to rice phenotypes have previously been overlooked in population-scale studies due to the difficulty of analyzing these complex regions39. De novo TR annotation based on high-quality genome assemblies is a reliable way to overcome such technical difficulties31. In a previous study, our lab assembled the genomes of 230 Asian rice accessions based on ONT long reads and constructed a pan-genome graph from those data34. In the present study, we conducted de novo TR annotation in each genome, identifying TR variation loci based on genome alignment and the rice pan-genome graph. Using 395 transcriptomic datasets from panicle and young leaf tissues, we identified the contributions of TR variations to variations in rice gene expression. In comparing those contributions to the effects of bi-allelic variants such as SNPs, PAVs, and Indels, we quantified the independent contribution of TR variations to gene expression. We also identified TR variations that contributed to key rice phenotypes by analyzing the colocalization of GWAS and eQTL signals.
The traditional methods of identifying TR variations involve detecting or genotyping TR variants with short or long sequencing reads based on comparison to a single reference genome assembly27. These methods limit the detection of TR variations; for example, only those that vary in copy number compared to the reference genome and those that are shorter than the sequencing read length can be identified31. Full genome assemblies have accurate consensus sequences and Mbp-scale contiguity, and therefore great advantages in the comprehensive detection of TR variants31. Although tools for the construction of TR pan-genome graphs have been published, these have previously been limited to the VNTR regions present in the reference genome31. Here, we conducted de novo TR annotations in 231 genomes (the Nipponbare reference genome and 230 rice assemblies). We integrated these TR annotations into the pan-genome graph of the 231 accessions to identify all variants at each TR locus. This yielded 227,391 multi-allelic TR loci, including 145,279 STRs and 82,112 VNTRs. The TR motif length varied from 2 to 2581 bp. Using the pan-genome graph, we could reduce the reference bias, leading to the identification of 54,416 TR variation loci that were missing from the reference genome. Using this TR dataset, we established the genomic distribution of TR variations and determined that they existed in low LD with bi-allelic variants, indicating that it may be possible to identify causal TR variants that are distinct from the bi-allelic variants.
In contrast to genomic resources for humans, there are rare population-level transcriptional data available for rice52,53, in some cases at the tissue-specific level. Plant growth can be divided into the vegetative and reproductive stages. We previously collected leaf tissue from 202 accessions during the vegetative growth stage34, and young panicle tissue from 193 accessions. In those tissues, we identified 2441 genes for which expression levels were only associated with TR variants. Among the genes for which expression levels were associated with both bi-allelic and TR variants, for 485 panicle and 511 leaf genes TRs had additional contributions to gene expression beyond the bi-allelic variants’ contributions. Using the grain width phenotype as an example, we identified TR variants regulated rice phenotypes and gene expressions. Genome editing techniques have been developed to introduce precise DNA modifications into plants; precise editing of the eTRs identified here could allow researchers to fine-tune key rice phenotypes, thus optimizing yield.
Several limitations of this study should be noted. Although we used multiple methods to identify TRs, extra-long VNTRs could not be fully captured here; considering the computing resources, the maximum repeat motif length that we could identify was only of <4 Kbp. We also used a linear regression model to identify variants contributing to variations in gene expression. In human research, a couple of disease instances (e.g. ALS) showed sigmoidal relationship between expression and TRs, meaning some TRs that effect gene expressions and phenotypes may be missed in the present modeling approach54,55. A large sample size is required to identify TR variations. Thus, in the coming years, the release of additional genomes and transcriptomes will allow for further identification of rare TR variants in particular. TR variations are sequence insertion and deletion variations. Whether the mechanisms of insertion and deletion variations that caused by TR variations and those not caused by TRs on effecting gene expressions and phenotypes were different will be a fundamental and interesting question to identify the independent roles of TR variations in the future. Although we here demonstrated several examples of multi-allelic TR-mediated quantitative regulation of gene expression and phenotypes, more experimental evidence will be needed to test their effects in rice breeding.
Despite these technical limitations, our findings clearly show that multiallelic TR variations broadly contribute to variations in rice gene expression and have a unique ability to quantitatively drive phenotypic variation. Our future work will focus on utilizing these TR variations to fine-tune key agronomic traits in rice to optimize yield and quality. Such studies will not only contribute answers to basic research questions regarding the genetic mechanisms of phenotype regulation but will help to ensure food security with this critical crop.
Methods
Samples
A total of 231 accessions were used in this study, including Nipponbare, 202 Asian cultivated rice (O.sativa), and 28 Asian wild rice (O.rufipogon). The genomes of the 202 Asian cultivated rice and 28 Asian wild rice were assembled using long-read data produced in a previous study34, and downloaded from the Genome Warehouse (GWH) database (https://bigd.big.ac.cn/gwh/) under accession PRJCA004295. The Nipponbare IRGSP 1.0 genome35 was downloaded from http://rice.uga.edu/pub/data/Eukaryotic_Projects/o_sativa/annotation_dbs/pseudomolecules/version_7.0/.
TR identification
Tandem repeats present in the Nipponbare reference genome and each of the different assemblies were identified using a combination of three distinct software. Specifically, each assembly was de novo annotated using the Tandem Repeats Finder (TRF version 4.09.1)36 with parameters ‘2 7 7 80 10 50 2000 -f -d -h’, as well as the ULTRA software (version 0.99.17)37 with parameters ‘-mi 2 -md 2 -p 4001 -mu 2 -ws 90000 -os 10000 -n 4 -f’. TRs were also initially annotated in a RepeatMasker (version open-4.0.7; http://www.repeatmasker.org) run using the RepBase library (Edition-20170127, parameter: -species rice). For each accession, we used the command BEDTools56 merge (parameter: -d 1 -c 4 -o distinct) to merge the TR annotations identified with each from three software. If there were differences in the annotated TR repeat motifs for the same position, all potential repeat motifs were temporarily kept and subsequently corrected in the construction of the pan-TR dataset (see below).
Pan-TR dataset construction
To identify the TR variants segregating across the population, we used the pan-genome graph from our previous work34. To integrate the TR annotations identified in each assembly with the pan-genome graph, the bubbles, overlapping with the TR annotations on each assembly were extracted using the command BEDTools intersect56. The pan-genome ___location and allele information (contig name, contig start, and contig end through the bubble) of these bubbles were extracted afterwards. As most bubbles spanned several kilobases, multiple TRs identified on each assembly may belong to the same bubble. To distinguish between these TRs, we constructed a pairwise whole genome alignment between each two assemblies using the minimap2 (version 2.17-r941) software57 (parameters: -t 2 -x asm5 -L -c --cs=long). After this, we used the liftover function available in the paftools.js tool from minimap2 to identify the accurate collinear position of each TR in the other assemblies. This allowed us to obtain the initial TR loci.
To correct the TR repeat motif and genotyping for each TR locus, we first collected all the possible motifs according to the initial TR annotation. Because the original pattern of repetition for VNTRs was more likely to be obscured when compared to STRs, we employed two distinct strategies to genotype STRs and VNTRs. Specifically, we obtained the genomic sequence of each STR in the different assemblies, including 100 bp flanking regions, using SAMtools (version 1.9)58. Next, we measured the length of the longest continuous repeat sequence for each possible repeat motif. To identify the loose STRs, we treated it as a part of the tandem repeats if the sequence between two target motifs was shorter than 1.1 times the target motif length. Finally, we compared the length of the repeat arrays of all possible motifs for each STR locus, selected the longest one as the final motif, and genotyped the motif repeats across all accessions.
For VNTRs, we obtained the sequence of the TR locus on each assembly, including the 500 bp flanking sequence, using SAMtools (version 1.9). For each possible motif, we repeated the motif to create a pseudo sequence longer than all the sequences from all assemblies in the current locus. We used the BLASTn tool to find regions of similarity with the pseudo sequence in the target sequence from each assembly (parameter ‘-dust no -task blastn-short -word_size 7’)59, And solely kept those with ≥90% identity for genotyping the corresponding accession. The motif that was present in most accessions were selected, as the final VNTR motif.
TR validation
We used three different strategies to validate the TR dataset. We began by downloading the BAC insert sequence of accession 93-11(https://www.ncbi.nlm.nih.gov/nuccore/AAAA00000000.2) and identified TR loci in the BAC sequence as described above. We the employed minimap257 to map the BAC sequence against the 93-11(NH231) assembly, and used the mapping coordinates to identify all overlapping TR loci. We directly compared the tandem repeat sequences from the BAC sequences to the 93-11 assembly. In order for a sequence to be considered validated, we required 100% length concordance and sequence identity between the BACs and the genome assembly used in our study.
After this, we randomly selected 218 TR variation loci and validated their genotypes. Specifically, for each of these loci, we obtained the TR sequences, including 500 bp flanking sequences on each assembly. Next, we used the clustalo software (version 1.2.4)60 to conduct multiple sequence alignment, and the Geneious Prime software (version 2021.0.3; Biomatters, New Zealand) to visualize the alignment and manually checked the TR genotypes.
Finally, we randomly selected a total of 8 TR variation loci to perform PCR and Sanger sequencing validation. To achieve this, total DNA was extracted using the CTAB method. PCR was performed using 2 × Hieff PCR Master Mix (With Dye) according to the manufacturer’s instructions. All PCR programs comprised a 95 °C/5 min denaturation step, followed by 35 cycles of 95 °C/30 s, 55 °C/100 s, and 72 °C/30 s, and a final elongation step of 72 °C/7 min. For agarose gel electrophoresis analysis, a total of 8 μL of each PCR reaction mixture was used and placed on a 1% agarose gel containing Tris-borate-EDTA buffer (45 mM Tris, 45 mM borate, 1 mM EDTA, pH 8.0) at 110 V for 15 min. The PCR products were sent to the Sangon Biotech (Shanghai) for Sanger sequencing. The primer sequences used in this experiment and the genomic ___location of TRs are listed in Supplementary Tables 1 and 2 as well as Supplementary Data 6.
Bi-allelic variant calling
We used sentieon (version 202112.02) bwa to map the filtered clean short-reads to the Nipponbare reference genome using the parameters ‘bwa mem -M -R -t 16 -K 10000000’61. The resulting sam files were transformed into bam files by running the command ‘sentieon util sort --bam_compression 1 -r --sam2bam -i’. After this, sentieon driver was used to tag and remove the repeat sequences with parameters ‘--algo LocusCollector --algo Dedup --rmdup’. We used the sentieon driver --algo Haplotyper to identify both SNPs and Indels.
The PAV dataset was obtained from the publish data in our previous work34. Briefly, PAVs were called by mapping the Nanopore reads to the Nipponbare genome using minimap2 and NGMLR (version 0.2.7)62 and then called using Sniffles (version 1.0.11)62 with parameters ‘-l 50 -genotype’. The PAVs were then merged using SURVIVOR (version 1.0.7) with parameters ‘1000 2 1 -1 -1 50’63.
Transcriptome sequencing
Panicle tissues were collected for 193 accessions (Supplementary Data 1) in a field experiment in Shenzhen, which took place in 2021. Panicles (10–20 mm) for each accession were collected at the heading stage. Total panicle RNAs were extracted using TRIzol kits (15596-018). After measuring RNA quality by performing agarose gel electrophoresis, Nanodrop, Qubit 2.0, and Agilent 2100 bioanalyzer, we constrcuted RNA libraries with a size of 300 bp per insert using the TruSeq RNA Library Preparation Kit (Version 2, Illumina, USA). RNA was sequenced using the Illumina high-throughput sequencing platform NovaSeq 6000. Finally, a total of 1.33 Tb RNA-seq of clean RNA reads were obtained (Supplementary Data 2). Transcriptomic sequencing for young leaves of one-month-old seedlings of 202 accessions was conducted in a previous study34.
eQTL analysis
Clean RNA-seq reads were mapped to the Nipponbare genome with TopHat2 (version 2.0.12)64. Based on the alignments, raw read counts were derived for each gene and normalized to FPKM using Cufflinks with default parameters65. For eQTL analysis, leaf expression data from 202 O.sativa accessions and panicle expression data from 193 O.sativa accessions were used. Genes with a mean FPKM value larger than 0.1 were used in the downstream analysis. 26,853 genes in panicle tissues and 23,736 genes in leaf tissues met this condition. Expression values were adjusted separately for each tissue to control for population structure and expression technical variations as covariates, which was similar to previous studies9,66. Briefly, we used the top 5 principal components as the covariates for population structure, and the top 20 hidden and confounding factors in the expression data were inferred using the probabilistic estimation of expression residuals (PEER) method67.
We performed linear regression with adjusted expression values for the TR variations, SNPs, Indels, and PAVs identified within 100 kb of each gene. We used the TR repeat numbers as TR genotypes and the variant dosages (0, 1, or 2) for SNPs, Indels, and PAVs genotypes. Adjusted expression values were Z-scaled to obtain a mean value of 0 and a variance of 1, before performing regression to ensure the effect sizes ranged between −1 and 1. Linear regression was performed using the OLS function available from the Python statsmodels.api module (https://www.statsmodels.org). The model returns estimated regression coefficients computed using ordinary least squares and two-sided p-values for each regression coefficient.
We used a similar strategy to previous research to identify the significant variant-gene pairs9,66. Briefly, for each gene, we selected the variant with the strongest Bonferroni-corrected p-value for each gene, and used the adjusted p-values to conduct FDR correction using the Benjamini–Hochberg method with α = 0.05 (the fdrcorrection function in the statmodels.stats.multitest module).
Fine-mapping analysis
The susieR package46 was used to identify all potential causal variants for each gene among those located within 100 kbp surrounding region of the gene. 95% credible sets (CS) for all variants containing a potential causal variant within a locus, were generated.
Comparing with the lead bi-allelic variant
We used a model comparison to determine whether eTRs could explain additional variation in gene expression beyond that explained by the leading bi-allelic variant for each gene. We used ANOVA to test whether the joint model performed significantly better than the bi-allelic-only model.
Phenotype collection
We collected plant height and grain width phenotypes for 202 Os accessions at the maturity stage. The measurements were retrieved from field work experiments conducted in the city of Mianyang, Sichuan Province, in 2020. Each phenotype was collected from a total of 6 plants for each accession.
Correlation analysis of TRs and genomic features
The histone markers and chromatin accessibility of the Nipponbare reference genome were downloaded from the RiceENCODE database (http://glab.hzau.edu.cn/RiceENCODE/). We used the two-sided Fisher exact test available in R to evaluate whether TRs were enriched in these genomic features compared to the whole genome background and whether eTRs were enriched in these genomic features compared to the TR dataset background.
TR characteristic analysis
The physical distribution of TR variations across chromosomes was identified by analyzing the ratio of TRs in 200 kb sliding windows (using 100 kb steps) compared to the pan-TR dataset. The spatial distribution of SNPs, PAVs, and Indels was identified in a similar manner. For each chromosome, Wilcoxon rank sum test statistics were conducted between the distribution of TRs and each bi-allelic variant.
Linkage disequilibrium between TRs and nearly bi-allelic variants (100 kb) was identified by calculating Pearson correlations for the genotype. For each TR, we recorded the maximum |R| within adjacent variants on either side.
Transient dual-luciferase (dual-LUC) assay
The OsPRR1 promoter sequence with different TR copies was cloned into pGreenII-0800-LUC, and subsequently transformed into tobacco protoplasts. The primer sequences are listed in Supplementary Table 1. The Renilla luciferase (REN) gene directed by the 35S promoter in the pGreenII 0800-LUC vector was used as an internal control. Firefly LUC and REN activities were measured using the Dual-Luciferase reporter assay kit (Beyotime) and a GloMax 20/20 luminometer (Promega). LUC activity was normalized to REN activity, which allowed us to calculate LUC/REN ratios. The reported data represents the averages of at least three independent replicates.
Access to transgenic materials
The knock-out mutant plants of OsPRR1 and TRGW6 were generated using the CRISPR/Cas9 technology68. The binary vector pVK005-1 (ViewSolid Biotech, VK005-1) were transfected into Xiushui134 and ZH11 using Agrobacterium infection, respectively. NH168 was chosen for editing the TR regions at the promoter of OsPRR1 using CRISPR/Cas9 technology. 51 T0 plants were obtained, and only one homozygous line carried reduced TR copies due to a 2 bp deletion. And the TR regions of TRGW6 were edited using a CRISPR–Cas12a promoter editing (CAPE) system4 with the vector pGEL589. NH142 and NH072 were chosen as background materials. We obtained > 60 T0 plants from each material. From the sequencing results, we selected 3 homozygous lines carrying the TR deletion and 4 homozygous lines carrying the TR insertion. All selected T0 lines were planted in Lingshui City, Hainan Province to obtain T1 plants and measured the plant height and grain size traits. The single-guide RNAs (sgRNAs) and sequencing primer used were listed in Supplementary Table 1.
Real-time PCR
To investigate whether TR copy numbers affect the expression level of OsPRR1 and TRGW6 gene, we extracted total RNA from leaves using the TRIzol reagent (Invitrogen). And reverse-transcribed it into the fist-strand cDNA with a Kit (YEASEN, 11141ES60). Real-time PCR was performed in an i-Cycler Bio-Rad machine, with each reaction containing 7.5 µL of SYBR green MIX (YEASEN, 11201ES08), 3 µL cDNA that was diluted 10-folds, and 0.3 µL 10 mmol−1 of primers in a final volume of 15 µL. The PCR amplification program was set at 95 °C for 2 min, followed by 40 cycles of 95 °C for 15 s, 55 °C for 15 s, and 72 °C for 15 s. The ACTIN gene was used as internal reference. The primers used for qPCR are listed in Supplementary Table 1.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The rice leaf transcriptome data and the panicle transcriptome generated in this study have been deposited in the NCBI Sequence Read Archive database under accession PRJNA692672 and PRJNA682327, respectively. Source data are provided with this paper.
Code availability
All data were analyzed with standard programs and packages, as detailed above. The code is available on zenodo (https://doi.org/10.5281/zenodo.11227547) and Supplementary Code 1.
References
Albert, F. W. & Leonid, K. The role of regulatory variation in complex traits and disease. Nat. Rev. Genet. 16, 197–212 (2015).
Song, X. G. et al. IPA1 functions as a downstream transcription factor repressed by D53 in strigolactone signaling in rice. Cell Res. 27, 1128–1141 (2017).
Zhang, L. et al. A natural tandem array alleviates epigenetic repression of IPA1 and leads to superior yielding rice. Nat. Commun. 8, 14789 (2017).
Zhou, J. P. et al. An efficient CRISPR-Cas12a promoter editing system for crop improvement. Nat. Plants 9, 588–604 (2023).
Xue, C. X. et al. Tuning plant phenotypes by precise, graded downregulation of gene expression. Nat. Biotechnol. 41, 1758–1764 (2023).
Wainschtein, P. et al. Assessing the contribution of rare variants to complex trait heritability from whole-genome sequence data. Nat. Genet. 54, 263–273 (2022).
Tautz, D. Notes on the definition and nomenclature of tandemly repetitive DNA sequences. Exs 67, 21–28 (1993).
Depienne, C. & Mandel, J. L. 30 years of repeat expansion disorders: What have we learned and what are the remaining challenges? Am. J. Hum. Genet. 108, 764–785 (2021).
Fotsing, S. F. et al. The impact of short tandem repeat variation on gene expression. Nat. Genet. 51, 1652–1659 (2019).
Bakhtiari, M. et al. Variable number tandem repeats mediate the expression of proximal genes. Nat. Commun. 12, 2075 (2021).
Wu, Z. Z. et al. Mapping short tandem repeats for liver gene expression traits helps prioritize potential causal variants for complex traits in pigs. J. Anim. Sci. Biotechnol. 13, 8 (2022).
Zhang, G. T. & Andersen, E. C. Interplay Between Polymorphic Short Tandem Repeats and Gene Expression Variation in Caenorhabditis elegans. Mol. Biol. Evol. 40, msad067 (2023).
Ranathunge, C. et al. Transcribed microsatellite allele lengths are often correlated with gene expression in natural sunflower populations. Mol. Ecol. 29, 1704–1716 (2020).
Reinar, W. B., Lalun VO, Reitan, T., Jakobsen, K. S. & Butenko, M. A. Length variation in short tandem repeats affects gene expression in natural populations of Arabidopsis thaliana. Plant Cell 33, 2221–2234 (2021).
Shi, Y. et al. Characterization of genome-wide STR variation in 6487 human genomes. Nat. Commun. 14, 2092 (2023).
Mukamel, R. E. et al. Protein-coding repeat polymorphisms strongly shape diverse human phenotypes. Science 373, 1499–1505 (2021).
Kaur, S., Panesar, P. S., Bera, M. B. & Kaur, V. Simple Sequence Repeat Markers in Genetic Divergence and Marker-Assisted Selection of Rice Cultivars: A Review. Crit. Rev. Food Sci. Nutr. 55, 41–49 (2015).
Si, L. Z. et al. OsSPL13 controls grain size in cultivated rice. Nat. Genet. 48, 447–456 (2016).
Bai, X. et al. Duplication of an upstream silencer of FZP increases grain yield in rice. Nat. Plants 3, 885–893 (2017).
Huang, Y. Y. et al. Variation in the regulatory region of FZP causes increases in secondary inflorescence branching and grain yield in rice domestication. Plant J. 96, 716–733 (2018).
Li, Z. et al. Natural variation of codon repeats in COLD11 endows rice with chilling resilience. Sci. Adv. 9, eabq5506 (2023).
Zhao, F. et al. A genome-wide survey of copy number variations reveals an asymmetric evolution of duplicated genes in rice. BMC Biol. 18, 73 (2020).
Qin, P. et al. Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell 184, 3542–3558 e3516 (2021).
Rajan-Babu IS, Dolzhenko, E., Eberle, M. A. & Friedman, J. M. Sequence composition changes in short tandem repeats: heterogeneity, detection, mechanisms and clinical implications. Nat. Rev. Genet. 25, 476–499 (2024).
Willems, T. et al. Genome-wide profiling of heritable and de novo STR variations. Nat. Methods 14, 590–592 (2017).
Gymrek, M., Golan, D., Rosset, S. & Erlich, Y. lobSTR: A short tandem repeat profiler for personal genomes. Genome Res. 22, 1154–1162 (2012).
Kristmundsdóttir, S., Sigurpálsdóttir, B. D., Kehr, B. & Halldórsson, B. V. popSTR: population-scale detection of STR variants. Bioinformatics 33, 4041–4048 (2017).
Mousavi, N., Shleizer-Burko, S., Yanicky, R. & Gymrek, M. Profiling the genome-wide landscape of tandem repeat expansions. Nucleic Acids Res. 47, e90 (2019).
Dolzhenko, E. et al. ExpansionHunter: a sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics 35, 4754–4756 (2019).
Bakhtiari, M., Shleizer-Burko, S., Gymrek, M., Bansal, V. & Bafna, V. Targeted genotyping of variable number tandem repeats with adVNTR. Genome Res. 28, 1709–1719 (2018).
Lu, T. Y., Chaisson, M. J. P. & Human Genome Struct Variation C. Profiling variable-number tandem repeat variation across populations using repeat-pangenome graphs. Nat. Commun. 12, 4250 (2021).
Chintalaphani, S. R., Pineda, S. S., Deveson, I. W. & Kumar, K. R. An update on the neurological short tandem repeat expansion disorders and the emergence of long-read sequencing diagnostics. Acta Neuropathol. Commun. 9, 2–20 (2021).
De Coster, W., Weissensteiner, M. H. & Sedlazeck, F. J. Towards population-scale long-read sequencing. Nat. Rev. Genet. 22, 572–587 (2021).
Shang, L. et al. A super pan-genomic landscape of rice. Cell Res. 32, 878–896 (2022).
Kawahara, Y. et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6, 2–10 (2013).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Olson, D. & Wheeler, T. ULTRA: A model based tool to detect tandem repeats. ACM BCB 2018, 37–46 (2018).
Yu, J. et al. The genomes of Oryza sativa: A history of duplications. Plos Biol. 3, 266–281 (2005).
Mei, H. et al. Population-scale polymorphic short tandem repeat provides an alternative strategy for allele mining in cotton. Front. Plant Sci. 13, 916830 (2022).
Zhang, G. T., Wang, Y. & Andersen, E. C. Natural variation in C. elegans short tandem repeats. Genome Res. 32, 1852–1861 (2022).
Zhang, H. et al. Population-level exploration of alternative splicing and its unique role in controlling agronomic traits of rice. Plant Cell, koae181, https://doi.org/10.1093/plcell/koae181 (2024).
Liu, T. Z. et al. Dwarf and High Tillering1 represses rice tillering through mediating the splicing of D14 pre-mRNA. Plant Cell 34, 3301–3318 (2022).
Song, X. J. et al. Rare allele of a previously unidentified histone H4 acetyltransferase enhances grain weight, yield, and plant biomass in rice. Proc. Natl Acad. Sci. USA 112, 76–81 (2015).
Dong, N. Q. et al. UDP-glucosyltransferase regulates grain size and abiotic stress tolerance associated with metabolic flux redirection in rice. Nat. Commun. 11, 2629 (2020).
Rabello, A. R. et al. Identification of drought-responsive genes in roots of upland rice (Oryza sativa L). BMC Genomics 9, 485 (2008).
Wang, G., Sarkar, A., Carbonetto, P. & Stephens, M. A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Soc. Ser. B Stat. Methodol. 82, 1273–1300 (2020).
Wang, Y. P. et al. Clock component OsPRR59 delays heading date by repressing transcription of Ehd3 in rice. Crop J. 10, 1570–1579 (2022).
Yan, W. H. et al. Natural variation in Ghd7.1 plays an important role in grain yield and adaptation in rice. Cell Res. 23, 969–971 (2013).
Duan, P. G. et al. Natural Variation in the Promoter of GSE5 Contributes to Grain Size Diversity in Rice. Mol. Plant 10, 685–694 (2017).
Rook, F. et al. Impaired sucrose induction1 encodes a conserved plant-specific protein that couples carbohydrate availability to gene expression and plant growth. Plant J. 46, 1045–1058 (2006).
Malik, I., Kelley, C. P., Wang, E. T. & Todd, P. K. Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat. Rev. Mol. Cell Biol. 22, 589–607 (2021).
Ming, L. et al. Transcriptome-wide association analyses reveal the impact of regulatory variants on rice panicle architecture and causal gene regulatory networks. Nat. Commun. 14, 7501 (2023).
Liu, C. et al. eQTLs play critical roles in regulating gene expression and identifying key regulators in rice. Plant Biotechnol. J. 19, 2357 (2022).
Elden, A. C. et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–U1077 (2010).
Lee, T. et al. Ataxin-2 intermediate-length polyglutamine expansions in European ALS patients. Hum. Mol. Genet. 20, 1697–1700 (2011).
Quinlan, A. R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinforma. 47, 11.12.11–34 (2014).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Kendig, K. I. et al. Sentieon DNASeq Variant Calling Workflow Demonstrates Strong Computational Performance and Accuracy. Front. Genet. 10, 736 (2019).
Sedlazeck, F. J. et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 15, 461 (2018).
Jeffares, D. C. et al. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun. 8, 14061 (2017).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Gymrek, M. et al. Abundant contribution of short tandem repeats to gene expression variation in humans. Nat. Genet. 48, 22–29 (2016).
Stegle, O., Parts, L., Piipari, M., Winn, J. & Durbin, R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat. Protoc. 7, 500–507 (2012).
Ma, X. L. et al. A Robust CRISPR/Cas9 System for Convenient, High-Efficiency Multiplex Genome Editing in Monocot and Dicot Plants. Mol. Plant 8, 1274–1284 (2015).
Acknowledgements
This research was supported by the National Natural Science Foundation of China (32188102, 32372148), Innovation Program of Chinese Academy of Agricultural Sciences (CAAS-CSIAF-202303). Innovation Program of Chinese Academy of Agricultural Sciences, the Youth innovation of Chinese Academy of Agricultural Sciences (Y2023QC36) and Guangdong Basic and Applied Basic Research Foundation (2023A1515110429) and the Youth Program of Guangdong Basic and Applied Research (2021A1515111123).
Author information
Authors and Affiliations
Contributions
L.S. and Q.Q. conceived and directed the project. H.H. performed all TR annotation and downstream analyses and wrote the manuscript. Y.Leng, X.C. and Y.Zhu designed and coordinated field experiments, and draw the figures. X.Li, Q.Y., Bin Zhang, W.H. helped accomplish pan-TR datasets construction. H.W., X.Liu, Q.X., M.G., and H.Z. collected and propagated the accessions and investigated phenotypes. L.Y., Y.Lv., X.W., C.S. processed bi-allelic variants calling and eQTL analysis. Z.Z., W.C., Bintao Zhang, T.W., and X.Y. collected panicle tissues and extracted RNA for transcriptome sequencing. H.Q., Q.Z., X.D., C.L., and Y.C. assisted in the TR characteristic analysis. Y.W., X.Z, G.X. and Y.Zhou revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xuehui Huang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
He, H., Leng, Y., Cao, X. et al. The pan-tandem repeat map highlights multiallelic variants underlying gene expression and agronomic traits in rice. Nat Commun 15, 7291 (2024). https://doi.org/10.1038/s41467-024-51854-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51854-0
