
Abstract
Replacement of expensive and rare platinum with metal–nitrogen–carbon catalysts for oxygen reduction reactions in proton exchange membrane fuel cells is hindered by their inferior activity. Herein, we report a highly active iron-nitrogen-carbon catalyst by optimizing the carbon structure and coordination environments of Fe-N4 sites. A critical high-temperature treatment with ammonium chloride and ammonium bromide not only enhances the intrinsic activity and density of Fe-N4 sites, but also introduces numerous defects, trace Br ions and creates mesopores in the carbon framework. Notably, surface Br ions significantly improve the interaction between the ionomer and catalyst particles, promoting ionomer infiltration and optimizing the O2 transport and charge transfer at triple-phase boundary. This catalyst delivers a high peak power density of 1.86 W cm−2 and 54 mA cm−2 at 0.9 ViR-free in a H2-O2 fuel cells at 80 °C. Our findings highlight the critical role of interface microenvironment regulation.
Similar content being viewed by others

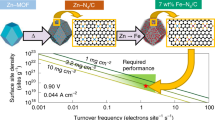

Introduction
Proton exchange membrane fuel cells (PEMFCs), as zero-emission power generation systems, are highly desirable for a variety of applications due to their environmental friendliness1,2,3. Achieving mass production and commercialization of PEMFCs is heavily contingent on their cost competitiveness4,5,6,7,8. Currently, the utilization of Pt-based catalysts remains inevitable, accounting for 41% of the total stack costs. Approximately 80% of the total Pt loading at is required for the PEMFCs cathode due to the sluggish kinetics of the oxygen reduction reaction (ORR)9,10,11,12. The development of PGM-free ORR catalysts to replace Pt has been pursued for over a decade13. The US Department of Energy (DOE) has set ambitious target for PGM-free catalysts; for instance, they have targeted a current density of 44 mA cm−2 at absolute 150 kPa H2–O2 and 0.9 ViR-free (where iR-free indicates that the internal resistance is compensated for) by 202514.
The M–NC catalysts, where M is a transition metal such as Fe and Co, have demonstrated encouraging activity levels15,16,17,18,19,20,21. The nitrogen-doped carbon matrix (NC) is able to stabilize metal ions, such as Fe, through the formation of Fe–Nx coordination with its nitrogen ligands22,23,24. This coordination is strikingly similar to the metal-N4 core found in metal porphyrins and phthalocyanines. Significant advancements have been achieved recently, especially with Fe–NC, to improve the acidic ORR activity of M–NC materials25,26,27,28,29,30,31. However, their activity and stability levels, remain well below the necessary performance standards required to replace PGM catalysts at the cathode, underscoring the significance of further improving activity. Numerous synthesis techniques have been used in an attempt to fine-tune the Fe–NC structures in order to increase the ORR activity. The primary objective is to enhance their intrinsic activity and optimize the amount of atomically distributed iron as Fe–Nx28. Only patchy success has been seen thus far, with the majority of documented Fe–NC reaching atomically scattered iron concentrations of 0.5 to 2.0 wt%32. Higher Fe content causes partial or total Fe clustering during pyrolysis, resulting in the formation of metallic nanoparticles, iron carbide, iron nitride, and other compounds33. Studies have shown that single-atom Fe loading may be increased by up to 7.0 wt%28,34,35. When these Fe–NC catalysts are tuned, they exhibit half-wave potentials similar to those of Pt measured at the rotating disk electrode (RDE)27. More significantly, their fuel cell current densities, which stand at 41.3 mA cm−2 at 0.9 ViR-free, have reached previously unheard-of heights28. Though significant advancements in Fe–NC catalysts have been made, the DOE 2025 target for PGM-free catalysts has not yet been met. For this purpose, it is necessary to further improve the intrinsic activity and active site density of Fe–NC catalysts. In addition, the reaction environment under the operating conditions of PEMFCs also needs to be regulated by special strategies.
Here, we developed a Fe–NC electrocatalyst with trace Br ion distribution, which exhibited excellent fuel cell performance. NH4X (X = Cl, Br) salts were added to the pyrolysis process, and decomposed to produce the gases such as NH3, HCl, and HBr. These gases not only etched the carbon substrate to form more carbon defects and mesopores, but also successfully distributed trace Br ions evenly on the surface of the Fe–NC catalyst. Based on the subsequent characterization results, the intrinsic activity and site density of the prepared Fe–NCBrCl catalyst were improved. Interestingly, the trace Br ions distributed on the surface promoted the uniform distribution of the ionomers on the surface of the catalyst particles, and even penetrated into the interior of the particles. Benefitting from the smooth mass transfer and faster interfacial electron transfer in the catalytic layer of Fe–NCBrCl cathode, the constructed Fe–NCBrCl catalyst reached a high peak power density of 1.86 W cm−2. And the current density of 54 mA cm−2 at 0.9 ViR-free was attained following the DOE test protocol, exceeding the DOE 2025 target (44 mA cm−2 at 0.9 ViR-free).
Results
Catalyst synthesis and structures
Supplementary Fig. 1 describes the two-stage strategy for the synthesis of highly active Fe–N–C catalysts. Initially, a ZIF-8@Phen composite precursor was thermally activated to form NC carriers and the micropores in the carriers accounted for more than 90% of NC carriers (Supplementary Figs. 2 and 3, Table 1). The Fe–NC catalyst was then treated at 900 °C in an Ar atmosphere with ammonium chloride and ammonium bromide salt (NH4Cl and NH4Br, named BrCl), and the final catalyst was named as Fe–NCBrCl (Supplementary Fig. 4). Mesopores and abundant carbon defects emerged during the BrCl treatment due to the etching effect of HCl, HBr, and NH3, which are the decomposition products of NH4Cl and NH4Br (Supplementary Figs. 5–7, Supplementary Movie 1). Figure 1a demonstrated the well-dispersed atomic Fe sites (bright dots) and some holes within the Fe–NCBrCl catalyst via an aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image. The broad distribution of Fe, N and Br was further demonstrated by the energy-dispersive X-ray spectroscopy (EDS) elemental mapping and XRD (Fig. 1b, c and Supplementary Fig. 8). Figure 1d, e indicated that BrCl treatment can change the N structure and cause the production of pyridine N based on X-ray absorption near edge spectroscopy (XANES) obtained for the N K-edge and X-ray photoelectron spectroscopy (XPS) N 1s spectrum. The formation of additional edge sites is facilitated by the etching effect of NH3, which is generated from the decomposition of NH4X, thereby promoting the incorporation of pyridinic N (Supplementary Fig. 9)36. In addition, as demonstrated by the hysteresis curve, BrCl treatment inhibited the creation of superparamagnetic iron species (Fe derivative clusters) and increased the number of Fe–Nx sites (Fig. 1f, Supplementary Figs. 10 and 11). Fe K-edge energies in X-ray spectra of Fe–NC and Fe–NCBrCl were compared to ferrous and ferric standards including FePc and Fe2O3. The results revealed that Fe was mostly in the Fe3+ oxidation state in Fe–NC catalysts, whereas Fe in Fe–NCBrCl had a lower oxidation state (Fig. 1g). A first coordination shell consisting of four nitrogen atoms can faithfully duplicate the Fourier transform of the extended X-ray absorption fine structure (EXAFS) of Fe–NC and Fe–NCBrCl (Fig. 1h, i, Supplementary Table 2). The Fe–N bond length in Fe–NCBr and Fe–NCBrCl treated with NH4Br was expanded relative to that in Fe–NC. NH4Cl and NH4Br played distinct roles in the synthesis of catalysts and reaction mechanisms. NH4Cl was especially effective in fostering the development of mesopores and carbon defects, which were vital for improving mass transport in fuel cells and providing doping sites for Br. In contrast, NH4Br, while less efficient at creating mesopores and carbon defects than NH4Cl, benefits from its ability to decompose into bromine more readily, as HBr yielded Br2 with relative ease (Supplementary Note 1, Figs. 12–14, and Tables 3, 4 for details). When a Br atom was doped adjacent to the Fe–N4 site, its larger atomic radius induced spatial distortion in the Fe–N4 plane, resulting in the elongation of the Fe–N bond (Supplementary Fig. 15 and Table 5). Furthermore, high-sensitivity low-energy ion scattering spectroscopy (HS-LEIS) results revealed that 92% of the Br ions were confined to the surface region (less than 1 nm) of the Fe–NCBrCl particles, with a surface bromine content of 1.51 wt% and a bulk bromine content of 0.12 wt% (Supplementary Figs. 16–18 and Table 6).

a Aberration-corrected atomic resolution HAADF-STEM micrograph of Fe–NCBrCl. b, c HAADF-STEM images and corresponding EDS mapping. d XAS spectra collected at N K-edge. e The deconvoluted high-resolution N 1s XPS. f The hysteresis curves. g Normalized Fe K-edge XANES spectra. h Fourier transform of k3-weighted EXAFS spectra. i FT-EXAFS fitting curve, gray ball represents C atom, blue ball represents N atom, red ball represents Fe atom, yellow ball represents Br atom.
ORR activity, SD and TOF in RDE
ORR activity of different Fe–NCs was assessed in a 0.1 M H2SO4 electrolyte by rotating disk electrode (RDE, Supplementary Fig. 19). ORR voltammetry exhibits a strong ORR performance illustrated in Fig. 2a in the order of Fe–NCBrCl > Fe–NCBr ≈ Fe–NCCl > Fe–NC. With a half-wave potential (E1/2) of 0.838 VRHE and a mass activity of 4.302 A g−1 at 0.85 VRHE, the best performing Fe–NCBrCl catalyst demonstrated high activity. These values are significantly higher than that of the Fe–NC (Fig. 2b). An in-situ nitrite stripping technique was used to assess how catalyst activation affected site activity and site density (SD)37. The SD values of Fe–NC was 5.74 × 1019 sites g−1 (0.05 sites nm−2), lower than that of the Fe–NCBrCl (6.31 × 1019 sites g−1, 0.06 sites nm−2), which reflects the importance of NH4Cl and NH4Br during pyrolysis (Fig. 2c, Supplementary Figs. 20, 21, Table 7). Furthermore, the SD values of Fe–NCBrCl showed a remarkable degree of agreement with the edge iron site count (6 ± 2 × 1019 sites g−1) calculated for this catalyst using a ___domain size of La = 7.3 ± 2.3 nm acquired from Anthony et al.28. The TOFs on nitrite poisoning were calculated by dividing the reduction in ORR kinetic current by SD. Fe–NCBrCl at 0.80 and 0.85 VRHE had computed TOF values of 4.18 and 0.90 e site−1 s−1, respectively, which were higher than Fe–NC (Fig. 2d). This suggests that doping Br atoms increased the TOF of the electrochemically accessible Fe sites38. The results from density functional theory (DFT) calculations substantiate that Fe–N4 sites doped with halogen atoms, such as Cl and Br, exhibit an enhanced ability to activate O2 molecules. Nonetheless, the formation of Cl doping is hindered during pyrolysis due to the recalcitrance of HCl. Consequently, Br2, which arises from the decomposition of HBr, is more readily incorporated into Fe–N–C materials36 (Supplementary Note 2, Figs. 22–26, Table 8).

a RDE ORR curves measured in O2-saturated 0.1 M H2SO4 at 10 mV s−1 with a rotation speed of 900 rpm. The catalyst loading was 0.6 mg cm−2. b The kinetic ORR activities of Fe–NCs. c Comparison of SD values determined by nitrite stripping method for Fe–NC and Fe–NCBrCl catalysts. d Comparison of TOF at 0.80 and 0.85 VRHE for Fe–NC and Fe–NCBrCl catalysts. Error bars represent the standard deviation for three separate measurements.
Performance characterization in a fuel cell
Fe–NC and Fe–NCBrCl served as the cathode catalysts in single-cell PEMFCs tests. Fe–NCBrCl exhibited superior performance under 250 kPaabs H2-O2 conditions, reaching a peak power density (Pmax) of 1.86 W cm−2 at 0.45 V. This value is approximately 0.6 W cm−2 higher than that of Fe–NC (Fig. 3a), which exceeded the commercial Pt/C prepared under the same conditions (0.2 mgPt cm−2, 150 kPa absolute O2 pressure). Fe–NCBrCl cathode has achieved the highest values reported for PGM-free catalysts in recent years (Supplementary Fig. 27 and Table 9)29,32,39,40,41,42,43. At 0.8 and 0.7 ViR-free, the current densities of Fe–NCBrCl were 0.781 and 3.111 A cm−2, respectively (Fig. 3b, c), surpassing the value achieved by Fe–NC (0.147 and 0.826 A cm−2 at 0.8 and 0.7 ViR-free). Figure 3d showed that the Tafel slope of Fe–NCBrCl was 84 mV dec−1, which was lower than that of Fe–NC (97 mV dec−1), reflecting the enhanced kinetic activity.

a H2-O2 PEMFCs polarization curves. Cathode, ~3.5 mgcat cm−2 for Fe–NC and 0.2 mgPt cm−2 for Pt/C; anode, 0.4 mgPt cm−2 Pt/C; GORE-SELECT® membrane (15 μm thickness); 0.3 L H2 min−1 and 0.4 L O2 min−1 feed, 100% relative humidity (RH), 250 kPa absolute partial pressure H2 and O2, 80 °C, electrode area 1.21 cm2. The cell voltage and power density are not iR corrected. b The polarization curves without (solid line) and with (dotted line) iR-correction and HFR under 250 kPaabs H2-O2 PEMFCs. c Current densities at 0.8 ViR-free and 0.7 ViR-free. d Tafel plots derived from the ORR polarization curves displayed in (c).
The superior activity of Fe–NCBrCl was further proven in the evaluation of PGM-free catalysts based on the DOE test protocol. As seen in Fig. 4a, Fe–NCBrCl demonstrated a Pmax of 1.29 W cm−2 at 0.45 V. Repeated tests demonstrated excellent repeatability of the high performance (Supplementary Fig. 28). Fe–NC, on the other hand, had a lower Pmax of 0.97 W cm−2. And for Fe–NCBrCl, the current density at 0.9 ViR-free was 54 mA cm−2, exceeding the DOE 2025 target of 44 mA cm−2 at 0.9 ViR-free (Fig. 4b, Supplementary Fig. 29, Table 10). In the H2–air operation (Fig. 4c), Fe–NCBrCl exhibited a Pmax of 0.88 W cm−2, which was considerably higher than most of the previously reported PGM-free catalysts (Supplementary Table 11). In addition, a measured current density of 259 mA cm−2 and 300 mA cm−2 was reached at 0.8 and 0.78 V (or 300 mA cm−2 at 0.8 ViR-free), only 41 mA cm−2 (or 0.02 V) lower than the DOE 2025 target (300 mA cm−2 at 0.8 V) (Fig. 4d). In order to better evaluate the practical application performance of Fe–NCBrCl, a three-piece PEMFCs stack with a MEA area of 25 cm2 was assembled (Supplementary Figs. 30 and 31). To mitigate mass transport limitations at high current densities, which were critical in practical situations involving lower O2 stoichiometry operation, the Fe–NCBrCl loading in the catalyst layer was set at 2.0 mg cm−2. The polarization curves of stack were acquired in an O2 atmosphere without background pressure. At 1.8 and 1.4 V (without iR-correction), the PEMFCs stack demonstrated a power of 25.3 and 36.2 W, respectively (Supplementary Fig. 32). It meant that the Fe–NCBrCl catalyst showed promise for practical applications, suggesting the feasibility of widespread application.

a H2–O2 polarization curves, acquired from OCV to 0.70 V in 25 mV steps and 0.70 V to 0.25 V in 50 mV steps, with a hold time of 45 s per point. 0.3 L H2 min−1 and 0.4 L O2 min−1 feed, 100% relative humidity (RH), 150 kPa absolute partial pressure H2 and O2, 80 °C. b H2–O2 polarization curve with iR-correction. c H2–air polarization curves. d Tafel plots derived from the ORR polarization curves displayed in (c).
Mechanistic insights into the PEMFCs activity enhancement
Within the cathode catalyst layer, the spatial distribution of Fe sites and ionomer was verified by time-of-flight secondary ion mass spectrometry (ToF-SIMS). Two-dimensional images of spatial arrangement of the components on the catalytic layer surfaces corresponding to Fe–NC and Fe–NCBrCl were shown in Fig. 5a, b. S− ions (green region) indicated sulfonic groups on ionomers, whereas Fe+ ions (red region) indicated Fe sites on Fe–NCs catalysts. It was evident that the thicker ionomer coating on the Fe–NC surface resulted in a sparse dispersion of Fe+ (Fig. 5a). On the surface of the Fe–NCBrCl catalytic layer, trace amounts Br− ions can be detected (Supplementary Fig. 33). The Br− ions in Fe–NCBrCl repelled the sulfonic groups of the ionomer, causing a more even dispersion of the ionomer throughout the surface of the catalytic layer (Fig. 5b). The distribution of ionomers in the pores was further investigated by examining the depth distribution curves of Fe+ and SO3− ions. The ionomer polymer was typically 2–5 nm in size, too tiny to penetrate the core of the particles and could only be coated on their surface44,45. The pores on the surface of Fe–NC particles were mostly micropores, accounting for about 92.8% (Supplementary Table 1). Within the Fe–NC particles, the SO3− distribution intensity was essentially nonexistent (Fig. 5c). Nevertheless, Fe–NCBrCl particles formed mesopores as a result of NH4Cl and NH4Br breaking down, and the surface Br− ions on the particles aid in the uniform dispersion of ionomers. Consequently, mesopores allowed a little quantity of ionomers to enter the particles. As a result, the SO3− intensity distribution of Fe–NCBrCl particles was also found inside of it (Fig. 5c). Upon analyzing the quantitative correlation between the surface Br content and the Pmax, a parabolic relationship was observed. The Pmax attained its apex at an equimolar ratio of NH4Br to NH4Cl (1:1) (Supplementary Note 3, Figs. 34–36, Table 12).

a, b The secondary ion two-dimensional imaging from ToF-SIMS, a Fe–NC, b Fe–NCBrCl. The green is S−, the red is Fe+. c Integral SO3− distribution in cathode catalyst layer shows intensity gradients from both Fe–NC (blue) and Fe–NCBrCl (red) catalysts. d Relationship between Rtotal and absolute gas pressure obtained by the limiting current method. Insets were Rnp and Rp. e An Arrhenius plot of i0 obtained from the Nyquist plots shown in Supplementary Fig. 37. f Nyquist plots for PEMFCs of indicated cathode catalysts at the current density of 1.0 A cm–2, the inset shows the equivalent circuit model, Rct and Rmt of Fe–NC and Fe–NCBrCl obtained by EIS fitting. Error bars represent the standard deviation of the fitting results.
By using the limiting current method46,47, the oxygen transport behavior of membrane electrodes was typically studied (Supplementary Fig. 37). Traditionally, Rtotal was the total of the pressure-dependent transfer impedance Rp and the pressure-independent transfer impedance Rnp48. The oxygen molecules diffusing through the mesopores in the gas diffusion layer (GDL) and the cathode catalytic layer (CCL) were the primary source of the resistance of oxygen diffusion, or Rp. On the other hand, oxygen was transported in the micropores of the catalyst aggregate and the ionomer layer, which caused the resistance to oxygen diffusion, or Rnp49,50,51. Rnp somewhat increased as a result of the ionomer seeping into the interior of Fe–NCBrCl particles (Fig. 5d). Lower Rp will arise from improved oxygen transport in the catalytic layer due to the mesoporous Fe–NCBrCl. It was established that the oxygen transport limitation in CCL was predominantly caused by the diffusion resistance of O2 in the mesopores of the thick cathode catalytic layer, when comparing Rp and Rnp. The effect of cell temperature on the charge transfer of Fe–NC and Fe–NCBrCl was also examined. As the temperature rose, the high-frequency limit shifted on the real axis in a negative direction (Supplementary Fig. 38). It indicated that the ohmic resistance, that is, the resistance of the membrane, decreased as the temperature of the cell increased. The charge-transfer mechanism was responsible for the depressed ellipse that was seen in all charts in the high-frequency range. The charge transfer resistance Rct was estimated by fitting these impedance measurements to the equivalent circuit (Supplementary Table 13). By replacing the constant phase angle element (CPE) with the double layer capacitance of the Randles equivalent circuit, this equivalent circuit was created. A porous electrode causes a depressed semicircle, which is frequently described by the CPE. The exchange current i0 was calculated from Rct according to the Eq. (1)52:
The temperature dependency of i0 might be assessed. In Supplementary Fig. 39, the Arrhenius plot of i0 was displayed52. In addition, good linearity was noted. For Fe–NCBrCl, the apparent activation energy (Ea) of charge transfer was determined to be 8.56 kJ mol−1 based on the angle of the plot (Fig. 5e). Compared to the value of 10.56 kJ mol−1 for Fe–NC, this value showed a faster charge transfer for Fe–NCBrCl, which was compatible with the lower Tafel slope and lower resistivity (Supplementary Fig. 40). Electrochemical impedance spectroscopy Nyquist plots at 1.0 A cm−2 and a Randles model simulation in Fig. 5f further supported the quicker mass and charge transfer. As a result of a richer triple-phase boundary (TPB), faster interfacial charge transfer, better combination of ionomers and Fe active sites, and a more porous catalytic layer, fuel cells assembled with Fe–NCBrCl demonstrated an extremely high levels of operational performance.
Discussion
In summary, we demonstrated a highly active Fe–NC catalyst by adjusting the structure of the carbon carrier and the chemical environment of the active center, representing a critical step towards replacing Pt and enabling large-scale applications of PEMFCs. We explored how the intrinsic activity and site density of Fe–N4 sites in the catalyst can be dramatically enhanced by regulating the catalyst’s local carbon structure. The pyrolysis treatment with NH4Cl and NH4Br was essential in creating a high density of carbon defects, mesopores, and trace Br doping on the catalyst surface, which significantly boosted ORR activity of Fe–NC catalyst. A broader dispersion of the ionomer across the surface of catalytic layer was achieved by the Br− ions on the surface of Fe–NCBrCl, which repelled the sulfonic groups of the ionomer. This resulted in a richer TPB, faster O2 transport, proton transport, and interface charge transfer channels. Thus, the performance of the synthesized Fe–NCBrCl catalyst outperformed the DOE 2025 target, achieving a high peak power density (1.86 and 0.88 W cm−2 in O2 and air environment, respectively) and 54 mA cm−2 at 0.9 ViR-free. Furthermore, employing H2–air, a measured current density of 259 mA cm−2 (300 mA cm−2 at 0.8 ViR-free) was attained at 0.8 V, which is just 41 mA cm−2 less than the DOE 2025 target (300 mA cm−2 at 0.8 V). In light of this, the achievement of promising activity of a single PGM-free catalyst described here which is equivalent to that of Pt catalysts showed tremendous promise for Pt catalyst replacement in PEMFCs and, eventually, for overcoming the cost barrier of existing PEMFCs.
Methods
Materials
Zinc nitrate hexahydrate (Zn (NO3)2·6H2O, ≥99.99%), 2-methylimidazole (98%), 1,10-phenanthroline monohydrate (Phen, ≥99.99%), ferrous chloride tetrahydrate (FeCl2·4H2O, 98%), ammonium chloride (NH4Cl, 99.5%) ammonium bromide (NH4Br, 99.0%) were sourced from Aladdin. Commercial 20 wt% Pt/C (Johnson Matthey) served as a comparison benchmark. 5 wt% Nafion ionomer was purchased from DuPont. GORE-SELECT® membrane (15 µm thickness) was purchased from Sinero Co. Methanol (CH3OH, 99.5%), Isopropanol (C3H8O, 99.5%) were purchased from Chemical Reagent Co., Ltd. Ultrapure water (Millipore, 18.25 MΩ cm) was used throughout all experiments.
Catalyst synthesis
The NC substrate was synthesized through the pyrolysis of a ZIF-8@Phen composite precursor, which included 1,10-phenanthroline monohydrate and ZIF-8 nanocrystals29. To prepare the ZIF-8@Phen composite precursors, Zn(NO3)2·6H2O (10 mmol, 2.975 g) and 2-mIm (80 mmol, 6.568 g) were each dissolved separately in 100 mL of methanol using ultrasound for 5 min. The Zn(NO3)2 solution was then rapidly poured into the 2-mIm solution, and the mixture was stirred vigorously for 16 h at room temperature. The resulting white ZIF-8 precipitate was collected by centrifugation, washed several times with methanol, and dried overnight under vacuum at 60 °C. Subsequently, 1.0 g of the obtained ZIF-8 and 0.3 g of 1,10-phenanthroline were dispersed in a 2:1 ethanol and deionized water solution. This mixture was magnetically stirred for 12 h at room temperature and then evaporated in an oil bath at 80 °C. The dry powders were thoroughly ground and pyrolyzed under an Ar flow at 1000 °C (5 °C·min⁻¹) for 1 h, followed by natural cooling to room temperature. The resulting black products were designated as NC.
Next, the NC substrate (100 mg) was ground together with NH4Cl (150 mg), NH4Br (150 mg), and FeCl2·4H2O (6 mg), followed by a heat treatment at 900 °C (10 °C·min⁻¹) under an Ar flow for 1 h to obtain the Fe–NC catalyst, referred to as Fe–NCBrCl. During this process, NH4Cl and NH4Br decomposed to produce NH3, HCl, and HBr gases at high temperatures, generating significant internal stress and etching the carbon. This created numerous mesopores and carbon defects, similar to previously reported Fe–N–C catalysts treated with NH3 gas. In addition, HCl likely dissociated further at high temperatures to form H2 and Cl2 gases, where the Cl2 gas reacted with residual Fe aggregates in the catalysts, facilitating the formation of atomically dispersed Fe sites53. For comparison, Fe–NC catalysts were also synthesized without adding NH4Cl or NH4Br, while Fe–NCCl catalysts were produced with only NH4Cl, and Fe–NCBr catalysts with only NH4Br. ICP-MS analysis revealed Fe contents of 2.25 wt% for Fe–NC and 2.50 wt% for Fe–NCBrCl.
Physical characterization
Scanning electron microscopy (SEM) images were acquired using a Hitachi S4800 at an acceleration voltage of 15 kV. Transmission electron microscopy (TEM) and high-resolution TEM (HR-TEM) analyses were conducted on a TECNAI F20 operating at 200 kV. Scanning transmission electron microscopy (STEM) and elemental mapping were performed with an FEI Talos F200s at 200 kV. High-angle annular dark-field STEM (HAADF-STEM) was carried out on an FEI Themis Z at 200 kV, equipped with a cold field-emission gun and an aberration corrector. For these observations, samples were prepared by depositing ethanol-dispersed samples onto a copper microgrid. Atomic-resolution images were captured with an FEI Titan Themis 60–300 at 200 kV. Nitrogen physisorption measurements were conducted using a Micromeritics Tristar 3020 Surface Area Analyzer after drying the samples under vacuum at 200 °C for 3 h. X-ray powder diffraction (XRD) patterns were obtained using a Rigaku SmartLab-SE Powder X-ray diffractometer with Cu-Kα radiation (λ = 1.5406 Å), scanned at a rate of 10° min⁻¹ over a 2θ range of 10–90°. X-ray photoelectron spectroscopy (XPS) was performed on a PHI Quantum-2000 using Al Kα radiation (1486.6 eV), with binding energies referenced to C 1s at 284.8 eV. The iron concentration in the samples was determined using inductively coupled plasma mass spectrometry (ICP-MS) on a Perkin Elmer NexION 300. Fe K-edge X-ray absorption spectra (XAS) were collected in fluorescence mode at beamline 1W1B of the Beijing Synchrotron Radiation Facility (BSRF). N K-edge XAS spectra were acquired at the TPS 44 A station of the National Synchrotron Radiation Research Center (NSRRC) in Hsinchu, Taiwan.
Electrochemical measurements
At room temperature, all electrochemical curves were conducted using a three-electrode setup with a Pine on a CHI 760E electrochemical workstation. The reference and counter electrodes were a saturated calomel electrode (KCl-saturated) and a graphite rod, respectively. To prepare a uniform catalyst ink, 6 mg of catalyst was sonicated for 30 min in 1 mL of a mixture containing 600 μL isopropanol, 380 μL ultrapure water, and 20 μL of 5 wt% Nafion solution. For the commercial 20 wt% Pt/C sample, 1 mg of catalyst was dispersed in 1 mL of a 0.05 wt% Nafion solution. A specific volume of the catalyst ink was applied to the polished glassy carbon rotating ring-disk electrode (RRDE, diameter 5.61 mm, area 0.2475 cm2) to achieve the desired catalyst loading. For all ORR experiments, a glassy carbon RRDE coated with PGM-free catalyst ink was used as the working electrode, with a loading of 0.6 mg cm−2 at 900 r.p.m. in 0.1 M H2SO4 electrolyte and the measurements are conducted at 30 °C. A commercial 20 wt% Pt/C catalyst served as a reference, tested in 0.1 M HClO4 with a charge of 15 µgPt cm−2. The reference electrode was calibrated to a reversible hydrogen electrode (RHE) in the same electrolyte before each test. ORR polarization curves for PGM-free catalysts were obtained using linear sweep voltammetry (LSV) from 0.1 to 1.1 V (vs. RHE) at 900 r.p.m. with a scan rate of 10 mV s−1. The resistance is automatically compensated by 80%, and the resistance in the impedance spectrum mode is measured by the pine system. The electrolyte was measured by pH instruments to ensure a constant test environment (pH = 0.7 for 0.1 M H2SO4). All potentials were converted to RHE potentials using the equation:
Quantification of site density and turnover frequency by nitrite stripping
The SD was determined using the nitrite reduction method by Kucernak37. Briefly, nitrite interacts with the Fe metal center to form stable poisoned adducts, which can be fully stripped within the potential range of 0.35 to −0.35 V (vs. RHE). The excess coulometric charge (Qstrip) from the stripping peak is proportional to the SD:
where nstrip (=5) is the number of transferred electrons per stripped one nitrite. NA is Avogadro constant (6.02 × 1023 sites mol−1). F is Faraday’s constant (96485 C mol−1). Δjm is the disparity in mass activity of the catalyst at 0.8 V or 0.85 V.
Fuel cell MEA tests
Membrane electrode assembly (MEA): To prepare the cathode ink, Fe–NC catalysts were ultrasonically blended with 0.2 mL of deionized water, 0.8 mL of isopropanol, and a 5 wt% Nafion solution in an ice bath for 1 h29. The ink was applied to a gas diffusion layer (GDL) made from PTFE-treated Toray 060 carbon paper, achieving a catalyst loading of 3.5 mg cm⁻². The cathode catalyst layer contained about 50 wt% Nafion. The anode catalyst consisted of 40 wt% Pt/C, with a platinum loading of 0.4 mgPt cm⁻². The MEA assembly involved hot-pressing the fabricated cathode with an anode, a 15-µm-thick GORE-SELECT® membrane, and a gasket at 130 °C and a pressure of 3 MPa for 90 s. The MEA had an active area of 1.1 × 1.1 cm².
MEA test: A Model 850e fuel cell test system (Scribner Associates, Inc.) was utilized to obtain polarization curves at 80 °C. Initially, the cell was brought to 80 °C without any flow, followed by passing humidified N₂ at 0.3 L min⁻¹ over both the cathode and anode for 1 h to ensure membrane and ionomer hydration. The cathode received air/oxygen at flow rates of 1.0/0.4 L min⁻¹, while the anode was supplied with H₂ at a flow rate of 0.3 L min⁻¹. During the fuel cell tests, reactant gases were subjected to absolute pressures of either 150 kPa or 250 kPa. Throughout the polarization curve measurements, the dew points of the cathode and anode gases, as well as the cell temperature, were kept constant at 80 °C. MEA performance measurements under H2–O2 and H2–air conditions adhered to protocols recommended by the US ElectroCat Consortium27. Specifically, a 45-s holding time at each voltage was implemented to attain a steady-state current density. Electrochemical impedance spectroscopy was performed at a steady current of 1.0 A cm−2, spanning a frequency range from 10 kHz to 0.1 Hz with an AC amplitude constituting 10% of the DC current.
PEMFCs stack test: A PEMFCs stack is made up of three MEAs (each active area is 25 cm2, cathode catalyst loading is 2.0 mg cm−2) connected in a series. O2 flowing at 1.0 L min−1 and H2 flowing at 0.8 L min−1 were supplied to the cathode and anode, respectively. The cathode and anode gas dew points and the cell temperature were maintained at 70 °C and the absolute pressures were 50 kPa for reactant gases (background pressure is 0 kPa) during the measurement of the polarization curves.
Computational details
All spin-polarized calculations were performed with the VASP54. The projector augmented wave method and the PBE functional of generalized gradient approximation (GGA) with were applied55,56. The Van der Waals interaction was included in this study57. The kinetic cut-off energy for the planewave basis was 400 eV. The vacuum layer between the periodic images of Fe–N4–C sheets was 20 Å. For the geometry optimization, the Brillouin zone was sampled with a 4 × 4 × 1 k-point mesh of the Monkhorst–Pack scheme, and all atoms in the supercell are allowed to relax. The energy convergence criterion is 10−5 eV, and the final forces on all atoms are less than 0.01 eV Å−1. The GGA + U method is applied to account for the strongly localized d orbitals of the Fe58. The Hubbard U values for the Fe atom of the Fe–N4 structure are chosen to be 3.3, attempting to have a brief insight into the intermediate adsorption at the Fe atom during O2 reduction. The first carbon shell of the Fe–N4 center was doped with the chlorine or bromine atom, respectively, forming the relevant Fe–N4–Cl and Fe–N4–Br structures.
Data availability
The data that support the findings of this study are available within the article and its Supplementary Information files. All other relevant data supporting the findings of this study are available from the corresponding authors upon request. Source data file has been deposited in Figshare, https://doi.org/10.6084/m9.figshare.25701651.
References
Kodama, K. et al. Challenges in applying highly active Pt-based nanostructured catalysts for oxygen reduction reactions to fuel cell vehicles. Nat. Nanotechnol. 16, 140–147 (2021).
Sui, S. et al. A comprehensive review of Pt electrocatalysts for the oxygen reduction reaction: Nanostructure, activity, mechanism and carbon support in PEM fuel cells. J. Mater. Chem. A 5, 1808–1825 (2017).
Kulkarni, A. et al. Understanding catalytic activity trends in the oxygen reduction reaction. Chem. Rev. 118, 2302–2312 (2018).
Yang, C. et al. Sulfur-anchoring synthesis of platinum intermetallic nanoparticle catalysts for fuel cells. Science 374, 459–464 (2021).
Chong, L. et al. Ultralow-loading platinum-cobalt fuel cell catalysts derived from imidazolate frameworks. Science 362, 1276–1281 (2018).
Chen, F. et al. Blocking the sulfonate group in Nafion to unlock platinum’s activity in membrane electrode assemblies. Nat. Catal. 6, 392–401 (2023).
Zhan, C. et al. Zinc intercalated lattice expansion of ultrafine platinum–nickel oxygen reduction catalyst for PEMFC. Adv. Funct. Mater. 33, 2212442 (2022).
Huang, L. et al. Progress of Pt-based catalysts in proton-exchange membrane fuel cells: a review. J. Electrochem. 28, 2108061 (2022).
Zhao, Z. et al. Graphene-nanopocket-encaged PtCo nanocatalysts for highly durable fuel cell operation under demanding ultralow-Pt-loading conditions. Nat. Nanotechnol. 17, 968–975 (2022).
Xiao, F. et al. Atomically dispersed Pt and Fe sites and Pt–Fe nanoparticles for durable proton exchange membrane fuel cells. Nat. Catal. 5, 503–512 (2022).
Zhao, Z. et al. Pt-based nanocrystal for electrocatalytic oxygen reduction. Adv. Mater. 31, 1808115 (2019).
Zhang, T. et al. Adjusting the alloying degree of Pt3Zn to improve acid oxygen reduction activity and stability. J. Electrochem 28, 2106091 (2022).
Jaouen, F. et al. Heat-treated Fe/N/C catalysts for O2 electroreduction: are active sites hosted in micropores? J. Phys. Chem. B 110, 5553–5558 (2006).
Thompson, S. T. et al. ElectroCat: DOE’s approach to PGM-free catalyst and electrode R&D. Solid State Ion. 319, 68–76 (2018).
Cheng, X. et al. Nano-geometric deformation synergistic Co nanoparticles & Co-N4 composite site for proton exchange membrane fuel cells. Energy Environ. Sci. 14, 5958–5967 (2021).
Yin, S. et al. Self-template synthesis of atomically dispersed Fe/N-codoped nanocarbon as efficient bifunctional alkaline oxygen electrocatalyst. ACS Appl. Energy Mater. 3, 625–634 (2020).
Lin, X. et al. Large-scale production of holey carbon nanosheets implanted with atomically dispersed Fe sites for boosting oxygen reduction electrocatalysis. Nano Res. 15, 1926–1933 (2021).
Qu, X. et al. In-situ growth of carbon nanotubes for improving the performance of Co-N/C catalysts in proton exchange membrane fuel cell. Chem. Eng. J. 461, 142054 (2023).
Yin, S. et al. Construction of highly active metal-containing nanoparticles and FeCo-N4 composite sites for the acidic oxygen reduction reaction. Angew. Chem. Int Ed. 59, 21976–21979 (2020).
Li, C. et al. Fe nanoparticles encapsulated in N-doped porous carbon for efficient oxygen reduction in alkaline media. J. Electrochem. 29, 2210241 (2023).
Li, Y. et al. Recent advances in exploring highly active & durable PGM-free oxygen reduction catalysts. J. Electrochem. 29, 2215002 (2023).
Zitolo, A. et al. Identification of catalytic sites for oxygen reduction in iron- and nitrogen-doped graphene materials. Nat. Mater. 14, 937 (2015).
Strickland, K. et al. Highly active oxygen reduction non-platinum group metal electrocatalyst without direct metal–nitrogen coordination. Nat. Commun. 6, 7343 (2015).
He, Y. et al. Atomically dispersed metal–nitrogen–carbon catalysts for fuel cells: advances in catalyst design, electrode performance, and durability improvement. Chem. Soc. Rev. 49, 3484–3524 (2020).
Zhang, H. et al. High-performance fuel cell cathodes exclusively containing atomically dispersed iron active sites. Energy Environ. Sci. 12, 2548–2558 (2019).
Jiao, L. et al. Chemical vapour deposition of Fe–N–C oxygen reduction catalysts with full utilization of dense Fe–N4 sites. Nat. Mater. 20, 1385–1391 (2021).
Liu, S. et al. Atomically dispersed iron sites with a nitrogen–carbon coating as highly active and durable oxygen reduction catalysts for fuel cells. Nat. Energy 7, 652–663 (2022).
Mehmood, A. et al. High loading of single atomic iron sites in Fe–NC oxygen reduction catalysts for proton exchange membrane fuel cells. Nat. Catal. 5, 311–323 (2022).
Yin, S. et al. Seizing gaseous Fe2+ to densify O2-accessible Fe-N4 sites for high-performance proton exchange membrane fuel cells. Energy Environ. Sci. 15, 3033–3040 (2022).
Cheng, X. et al. Instantaneous free radical scavenging by CeO2 nanoparticles adjacent to the Fe−N4 active sites for durable fuel cells. Angew. Chem. Int Ed. 62, e202306166 (2023).
Cheng, X. et al. Revealing the optimal configuration for synergy effect of metal nanoparticles and MN4 sites for oxygen reduction reaction. Nano Energy 100, 107440 (2022).
Wan, X. et al. Fe–N–C electrocatalyst with dense active sites and efficient mass transport for high-performance proton exchange membrane fuel cells. Nat. Catal. 2, 259–268 (2019).
Li, J. et al. Evolution pathway from iron compounds to Fe1(II)–N4 sites through gas-phase iron during pyrolysis. J. Am. Chem. Soc. 142, 1417–1423 (2019).
Jin, Z. et al. Understanding the inter-site distance effect in single-atom catalysts for oxygen electroreduction. Nat. Catal. 4, 615–622 (2021).
Hai, X. et al. Scalable two-step annealing method for preparing ultra-high-density single-atom catalyst libraries. Nat. Nanotechnol. 17, 174–181 (2021).
Li, Q. et al. Highly efficient Fe-N-C oxygen reduction electrocatalyst engineered by sintering atmosphere. J. Power Sources 449, 227497 (2020).
Malko, D. et al. In situ electrochemical quantification of active sites in Fe–N/C non-precious metal catalysts. Nat. Commun. 7, 13285 (2016).
Wang, L. et al. Phosphorus and halogen co-doped graphene materials and their electrochemistry. Chem. Eur. J. 22, 15444–15450 (2016).
Proietti, E. et al. Iron-based cathode catalyst with enhanced power density in polymer electrolyte membrane fuel cells. Nat. Commun. 2, 416 (2011).
Wang, Y. et al. S-Doping of an Fe/N/C ORR catalyst for polymer electrolyte membrane fuel cells with high power density. Angew. Chem. Int Ed. 127, 10045–10048 (2015).
Fu, X. et al. In situ polymer graphenization ingrained with nanoporosity in a nitrogenous electrocatalyst boosting the performance of polymer-electrolyte-membrane fuel cells. Adv. Mater. 29, 1604456 (2017).
Liu, Q. et al. The solid-phase synthesis of an Fe-N-C electrocatalyst for high-power proton-exchange membrane fuel cells. Angew. Chem. Int Ed. 57, 1204–1208 (2018).
Li, Y. et al. A general carboxylate‐assisted approach to boost the ORR performance of ZIF-derived Fe/N/C catalysts for proton exchange membrane fuel cells. Adv. Funct. Mater. 31, 2009645 (2021).
Men, S. et al. X-ray photoelectron spectroscopy of pyridinium-based ionic liquids: comparison to imidazolium- and pyrrolidinium-based analogues. ChemPhysChem 16, 2211–2218 (2015).
Rauf, M. et al. Insight into the different ORR catalytic activity of Fe/N/C between acidic and alkaline media: Protonation of pyridinic nitrogen. Electrochem. Commun. 73, 71–74 (2016).
Daniel, R. et al. Measurement of oxygen transport resistance in PEM fuel cells by limiting current methods. J. Electrochem. Soc. 156, B991–B1003 (2009).
Ohma, A. et al. Analysis of proton exchange membrane fuel cell catalyst layers for reduction of platinum loading at Nissan. Electrochim. Acta 56, 10832–10841 (2011).
Huang, J. et al. Review of characterization and modeling of polymer electrolyte fuel cell catalyst layer: The blessing and curse of ionomer. Front Energy 11, 334–364 (2017).
Dang, D. et al. High-performance, ultralow platinum membrane electrode assembly fabricated by in situ deposition of a Pt shell layer on carbon-supported Pd nanoparticles in the catalyst layer using a facile pulse electrodeposition approach. ACS Catal. 5, 4318–4324 (2015).
Ono, Y. et al. Influence of equivalent weight of ionomer on local oxygen transport resistance in cathode catalyst layers. J. Electrochem Soc. 160, F779–F787 (2013).
Kongkanand, A. et al. The priority and challenge of high-power performance of low-platinum proton-exchange membrane fuel cells. J. Phys. Chem. Lett. 7, 1127–1137 (2016).
Takahashi, M. et al. Reaction behavior of LiFePO4 as a cathode material for rechargeable lithium batteries. Solid State Ion. 148, 283–289 (2002).
Varnell, J. A. et al. Identification of carbon-encapsulated iron nanoparticles as active species in non-precious metal oxygen reduction catalysts. Nat. Commun. 7, 12582 (2016).
Kresse, G. et al. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Kresse, G. et al. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1999 (1758).
Perdew, J. et al. Generalized gradient approximation made simple. Phys. Rev. Lett. 78, 1396 (1997).
Grimme, S. et al. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Dudarev, S. L. et al. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study. Phys. Rev. B 57, 1505 (1998).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant no. 22288102, 22172134, U1932201, U2032202), the Science and Technology Planning Project of Fujian Province (2022H0002), the National Natural Science Foundation of China (grant no. 22202082). This research was thankful to the Beijing Synchrotron Radiation Facility (1W1B, BSRF), the National Synchrotron Radiation Research Center (NSRRC) in Hsinchu, Taiwan, R.O.C. and Shanghai Synchrotron Radiation Facility for help with characterizations. This research also employed the resources of Beijing Synchrotron Radiation Laboratory 4B7B and 4b9b beamline stations (under Contract No. 2021-BEPC-PT-005771, 2021-BEPC-PT-005765). We are very grateful for Dr. G.-L. M. help in the in-situ heating TEM experiment. S.Y. is thankful for the support from Xiamen University Nanqiang Cultivation Program for Outstanding Doctoral Students.
Author information
Authors and Affiliations
Contributions
The project was conceptualized by S.Y., R.H., Y.J., and S.S. R.H., Y.J., and S.S. supervised this project. S.Y. and L.C. synthesized the catalysts, conducted the electrochemical tests, and the related data processing. S.Y. performed materials characterization with the help of X.C., H.Z., and Y.H. H.H. performed the Fe K-edge XAS characterization. X.K. and Y.L. performed the N K-edge XANES. Y.J. and S.Y. performed the theoretical calculations and mechanistic analysis. R.H. and S.S. provided suggestions on the work. S.Y. and R.H. wrote the manuscript; and L.C., J.Y., R.H., Y.J., and S.S. helped with the revision of the manuscript. We appreciate the Beijing Synchrotron Radiation Facility (1W1B, BSRF) and the National Synchrotron Radiation Research Center (NSRRC) in Hsinchu, Taiwan, R.O.C. for help with XAS characterizations. All authors discussed the results.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jian Xie, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yin, S., Chen, L., Yang, J. et al. A Fe-NC electrocatalyst boosted by trace bromide ions with high performance in proton exchange membrane fuel cells. Nat Commun 15, 7489 (2024). https://doi.org/10.1038/s41467-024-51858-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51858-w
This article is cited by
-
From Single-atom to Bi-atom and Ordered Multi-atom: Not Just a Number Changing for Electrocatalysis
Chemical Research in Chinese Universities (2025)
