
Abstract
Due to low success rates and long cycles of traditional drug development, the clinical tendency is to apply omics techniques to reveal patient-level disease characteristics and individualized responses to treatment. However, the heterogeneous form of data and uneven distribution of targets make drug discovery and precision medicine a non-trivial task. This study takes pyroptosis therapy for triple-negative breast cancer (TNBC) as a paradigm and uses data mining of a large TNBC cohort and drug databases to establish a biofactor-regulated neural network for rapidly screening and optimizing compound pyroptosis drug pairs. Subsequently, biomimetic nanococrystals are prepared using the preferred combination of mitoxantrone and gambogic acid for rational drug delivery. The unique mechanism of obtained nanococrystals regulating pyroptosis genes through ribosomal stress and triggering pyroptosis cascade immune effects are revealed in TNBC models. In this work, a target omics-based intelligent compound drug discovery framework explores an innovative drug development paradigm, which repurposes existing drugs and enables precise treatment of refractory diseases.
Similar content being viewed by others
Introduction
Drug development, an important research area for chemical scientists and pharmaceutical companies, has been plagued by pain points such as low success rates, high costs, and long development cycles1. Traditional drug development often fails due to inaccurate targets, poor safety, unsatisfactory efficacy, and the inability to identify suitable patient populations2. In light of advancing technologies and the transformative impact of data science on drug development, there is an urgent need to change the status quo with innovative technologies and models to open the era of intelligent drug development.
Upon reviewing nearly two decades of observations, it has been discovered that single-target therapeutics often fail to produce satisfactory outcomes due to the intricate nature of disease systems. Rather than focusing solely on modulating a predetermined single target, most first-in-class medications approved by the U.S. Food and Drug Administration (FDA) have concentrated on influencing disease phenotypes or biomarkers to provide therapeutic benefits3. The key to achieving drug innovation and personalized treatment lies in utilizing phenotypic screening to proactively discover original medications. Target omics-based smart medicine involves utilizing genomic, proteomic, metabolomic, or other omics data to provide personalized healthcare4. Omics technologies enable the analysis of large-scale biological data, identifying specific molecular targets linked to disease susceptibility and predicting drug response based on an individual’s unique genetic makeup, thereby facilitating diagnosis, prognosis, and treatment selection5,6. For example, genome sequencing can locate mutated genes in cancer cells that drive tumor growth, enabling the selection of targeted therapies accordingly. Metabolomic analysis can determine changes in the levels of specific metabolites in blood or urine during disease progression7,8. Given the vast amount of complex clinical data, the explosion of biological knowledge, and the creation of various databases, it is crucial to identify high-risk biomarkers associated with diseases as target omics and find the appropriate medications to effectively intervene with these targets, thus fostering precision medicine and aiding in drug discovery.
Theories based on modulating disease phenotypes or biomarkers have led to the development of molecularly targeted combination therapies in the clinic9. Although potential drug combinations can traditionally be tested based on biological or clinical expertise through methods such as high-throughput screening, the limited number of experimental filters and the heterogeneity of the physical environment still hinder the discovery and optimization of compound drugs10,11. More systematic predictive methods are needed to efficiently navigate and explore this vast space, ultimately reducing development costs and time. In recent years, artificial intelligence (AI) has shown promising applications in compound validation, target identification, drug discovery, dosage design, and drug repositioning, significantly contributing to the drug development process12,13,14,15,16. Comprehensive benchmarking of several computational models for drug screening was performed in two DREAM Challenges originated from the public. With sufficient drug information and augmented training data, computational predictions can attain high accuracy for sorting chosen drugs17. Deep neural networks have recently outperformed other less complex models and shown state-of-the-art performance in predicting drug combination synergy18. However, accurate modeling of drug combination effects remains a challenge due to the scarcity of high-quality training data, and the “black box” character of machine learning models presents difficulties in providing transparent and interpretable biological insights for human understanding. Therefore, developing integrated and robust models that can generalize, learn from a large amount of available data, and interpretably identify biosignatures to facilitate exploration of the broad combinatorial drug space is of great importance for compound drug development.
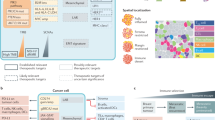
In this work, we propose a framework for intelligent compound drug discovery based on target omics and evaluate the advancement of this technology platform, which integrates bioinformatics analysis, artificial intelligence, and experimental validation for the discovery and optimization of compound drugs (Fig. 1a). We specifically focus on the paradigm of pyroptosis therapy for triple-negative breast cancer (TNBC), a subtype with highly malignancy and poor prognosis, lacking effective therapeutic targets and treatments19. Pyroptosis has recently been recognized in recent years as a powerful pathway for cancer eradication, an inflammatory death modality mediated by the gasdermin (GSDM) protein family in tumor cells that initiates a robust anti-tumor immune response20,21. We perform a bioinformatic analysis of the typical pyroptosis genes to generate target omics associated with TNBC and then identify corresponding potential pyroptosis-inducing drugs based on a large TNBC cohort and drug databases. Subsequently, a biofactor-regulated neural network AI model, BFReg-NN, is established to screen and optimize pyroptosis compound drugs rapidly, followed by preliminary experimental validation (Fig. 1b). A representative preferred drug pair, mitoxantrone (MIT) and gambogic acid (GA) is formulated into a bionic nanococrystal (MG@PM) to promise the rational drug delivery with optimal synergistic ratios. The potent pyroptosis-inducing and anti-tumor immunological effects of MG@PM are confirmed in the TNBC model (Fig. 1c). We also carefully identify the presence of pyroptosis cascade effect in TNBC metastatic models after MG@PM treatment. In addition, the unique mechanism by which compound drugs disrupt tumor cellular redox homeostasis to stimulate ribosomal stress, thereby effectively modulating specific pyroptosis genes, has been carefully explored. Overall, this study provides an innovative methodological strategy for compound drug development in TNBC or other refractory diseases.

a The schematic illustration of the target omics-based intelligent compound drug discovery framework. b The workflow for discovering and optimizing anti-TNBC compound pyroptosis drugs based on big data and artificial intelligence. c Construction of MG@PM and the proposed mechanism of MG@PM for promoting systemic anti-tumor immune response by evoking tumor cell pyroptosis-induced cascade effect.
Results
Identification of pyroptosis genes and drugs across TNBC cohorts
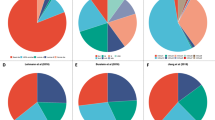
To assess the pyroptosis characterization and probe potential opportunities for precise targeting of pyroptosis in TNBC, we analyzed transcriptomic data from 360 patients with TNBC at Fudan University Shanghai Cancer Center (FUSCC) using bioinformatics techniques (Fig. 2a)22. We summarized 45 specific pyroptosis-related genes by KEGG pathway enrichment and previous literature23,24,25. Figure 2b illustrates the pathways discovered through Gene Ontology (GO) functional enrichment analysis of these genes. Notably, the pyroptosis and NOD-like receptor signaling pathways exhibit high enrichment levels, indicating a positive correlation with pyroptosis. We found that these pyroptosis-related genes showed significant differential expression in the transcriptome profiles of 360 TNBC tissues versus 88 normal breast tissues, and most of them showed marked upregulation (Fig. 2c). Depicting the transcriptome features of 45 genes in a relational map presents that they had extensive interactions in TNBC, suggesting that pyroptosis could be an option for TNBC treatment (Fig. 2d). By applying the Cox proportional hazards model and Akaike information criterion (AIC), we identified a set of nine genes (CASP1, BCL2, CTSD, CASP5, TRADD, NFKB2, CASP9, TNF, and GSDMD) as target omics of TNBC pyroptosis that significantly predicted recurrence-free survival (RFS) of TNBC patients. Our pyroptosis-based survival model for TNBC RFS showed a concordance index (c-index) of 0.7. The expression profiles of these nine key pyroptosis genes are shown in Supplementary Fig. 1. The fact that the pyroptosis genes significantly affected patient RFS compared to other factors, such as age and therapy (radiotherapy/chemotherapy), also further suggested that pyroptosis phenotype modulation strategies could be more important in treating TNBC (Supplementary Fig. 2). Furthermore, the hazard ratio of the nine identified genes indicating that CASP9 increased the risk of TNBC RFS, whereas CASP5 significantly reduced it (Fig. 2e). We predicted the Kaplan-Meier curves and immune scores for TNBC RFS based on the high and low risk of key pyroptosis genes, emphasizing their significant influence on the survival and immune profile of TNBC patients (Fig. 2f and Supplementary Fig. 3)26. These results suggest that pyroptosis therapy can be an effective treatment for TNBC. Next, we utilized various drug databases and filtering methods to discover drug candidates associated with the nine key pyroptosis regulators. The Comparative Toxicogenomics Database (CTD) dataset was used to identify drugs associated with at least one of these regulators and to calculate the mean z-score of therapeutic drug activity (DTP NCI-60) based on RNA expression of pyroptosis regulators, deriving 133 potential pyroptosis-related drugs from 20348 drugs (Supplementary Fig. 4). We then used the Swiss Target Prediction tool to obtain the targets of these drugs and confirmed their association with pyroptosis regulators using the distance in the protein-to-protein interaction (PPI) network. Thus, we narrowed our search to 35 drugs, which demonstrated a significant correlation with the nine key pyroptosis regulators (Fig. 2g).

a The workflow for screening of signature genes and corresponding drugs. b Gene Ontology (GO) enrichment analysis of 45 typical pyroptosis-associated genes. c Heat map of differentially expressed pyroptosis regulator genes in TNBC and normal breast tissues from the FUSCCTNBC cohort. d Diagrams of the correlations between the expression levels of pyroptosis regulators. The scatter plot represented the correlation between CASP1 and NLRP3, GSDMD and CASP1, TRADD, and NFKB2 (NLRP3 vs. CASP1: P < 0.0001; CASP1 vs. GSDMD: P < 0.0001; NFKB2 vs. TRADD: P < 0.0001). Data are analyzed with Spearman correlation. e The distribution diagram (forest plots) of hazard ratios across typical pyroptosis genes. f The Kaplan-Meier overall survival curve of two clusters distinguished by pyroptosis strata (risk=high vs. risk = low: P < 0.0001). Data are analyzed with Log-rank test. g Correlation between IC50 values of 35 predicted pyroptosis inducers and the expression levels of typical pyroptosis genes. * P < 0.05, ** P < 0.01, and *** P < 0.001. Data are represented as mean ± SD and analyzed with two-way ANOVA followed by multiple comparisons test. Source data are provided as a Source Data file.
Screening compound medications and predicting effects through machine learning
Based on the set of potential pyroptosis drugs obtained, we proposed to use a biological factor-regulated neural network model (BFReg-NN) to learn and obtain excellent compound medications27. Specifically, BFReg-NN used the existing knowledge (e.g., protein-protein interaction network) to build a “white-box” architecture of biological factors, and learn the factor functions in cells by graph neural networks, ultimately giving insight into the mechanisms of the whole system (Fig. 3a, b). BFReg-NN is able to simulate interactions between biological factors (e.g., genes, proteins, or drug targets) in the cell transparently and accurately. In many tasks (e.g., TNBC subtype classification), BFReg-NN outperformed the traditional methods of logistic regression, neural network, and random forest in terms of accuracy (Fig. 3c). Herein, BFReg-NN was instantiated by restricting biological factors as drug targets and pyroptosis genes and learning how targets influence pyroptosis-related genes and ultimately affect TNBC RFS (Fig. 3a). Since our goal was to predict the effect of compound drugs through pyroptosis signature genes, we constructed the architecture of BFReg-NN from four layers: (1) the layer for the relationship among two drug targets, (2) links from drug targets to genes, (3) the layer for the relationship among pyroptosis genes and (4) survival layer. A more detailed diagram is shown in Supplementary Fig. 5. We trained BFReg-NN for a survival task by TNBC RFS data. Thus, we could constrain drug targets to influence TNBC RFS through pyroptosis signature genes (Fig. 3b). More details on the AI procedures can be found in Methods.

a The workflow for screening effective compound drugs affecting pyroptosis signature genes by artificial intelligence. b Schematic diagram of the architecture of BFReg-NN. As an example, drug targets for MIT and GA were shown on the left, and the nine identified pyroptosis signature genes were on the right. c Accuracy analysis of the BFReg-NN model comparing logistic regression, neural network, and random forest in the task of TNBC subtype classification (n = 100 technical replicates). Central line indicates median, box indicates interquartile range, whiskers show 1.5x the IQR. Data are analyzed with one-way ANOVA followed by multiple comparisons test. d Histogram of the ranking of BFReg-NN predicting the correlation between each drug combination and TNBC RFS. The calculation was repeated ten times. e Phase contrast microscopy images of MDA-MB-231 cells treated with twelve sets of compound drugs randomly selected from the top 10% of the ranking. Scale bar = 5 μm. The experiments were repeated three times independently. f Visualization images of important targets of MIT and GA to regulate pyroptosis signature genes using Integrated Gradient prediction. The warm-toned colors represented the targets of GA, while the cool-toned colors indicated the targets of MIT. The width of the lines represented the importance score. g Diagram of drug and protein combination. i) GA and TXN, ii) GA and MCL1, iii) MIT and BTK, iv) MIT and TOP2A. Source data are provided as a Source Data file.
We applied BFReg-NN to screen all possible drug combinations and calculated their c-index scores to indicate the correlation between drug targets and TNBC RFS. The ranking results are shown in Fig. 3d, where we considered the compound drugs ranked in the top 10% as the most promising combinations for treating TNBC by affecting the signature pyroptosis genes. To examine the effectiveness of the compound drugs by the BFReg-NN screening, we performed the intervention treatment on MDA-MB-231, a human-derived TNBC cell line. These results yielded surprising findings as all twelve randomly selected drug pairs, within the top 10% of drug combinations, caused varying degrees of balloon-like changes in MDA-MB-231, a typical feature of pyroptosis (Fig. 3e, Supplementary Fig. 6). Collectively, our approach to compound drug discovery that integrates big data bioinformatics analysis and neural network artificial intelligence is reliable and quite promising.
In order to verify the pyroptosis effect and anti-tumor efficacy of these preferred pyroptosis compound drugs, we conducted more intensive experimental exploration for the representative drug pair of mitoxantrone (MIT) and gambogic acid (GA) (Supplementary Fig. 7). Recent studies have demonstrated that both MIT and GA have multiple tumor-killing mechanisms. MIT is a first-line chemotherapeutic for breast cancer, and its in vivo toxicity may be attributed to its potential pyroptosis-inducing ability24,28. GA, a natural product with strong anticancer activity, could significantly enhance the sensitivity of tumor cells to chemotherapeutics29,30. The combination of MIT and GA has a c-index value of 0.90 (ranked in the top 8%), and both have relatively low IC50 values (Supplementary Fig. 8). The architecture was visualized by BFReg-NN to explore how the targets of MIT and GA interact together and to predict important targets using Integrated Gradient31. As shown in Fig. 3f, we found that GA targets MCL1 and TXN, as well as MIT targets BTK and TOP2A, had high importance scores, indicating that they will strongly impact the pyroptosis signature genes. Figure 3g showed the three-dimensional structures of GA and MIT and the non-covalent bonding sites of TXN, MCL1, BTK, and TOP2A, respectively.
Combination of MIT and GA synergistically induces pyroptosis
We combined MIT with GA as compound drugs for the effective induction of pyroptosis and investigated whether they synergistically affected malignant cancer cells. Through analyzing the cell viability of 4T1 cells incubated with different mass ratios of drug combinations for 24 h, we found that MIT and GA produced different synergistic effects (Fig. 4a, b). The most decisive synergistic impact was observed when the mass ratio of MIT to GA was 1:1.5 (Supplementary Table. 1), with a combination index (CI) value of 0.599, which was much lower than 1. More importantly, at this ratio, the IC50 of MIT and GA decreased to 0.30 μg/mL and 0.45 μg/mL, respectively, much lower than that of free MIT and GA. Low dose but high efficacy would greatly reduce drug side effects and satisfy the prerequisite for chemotherapeutics-induced pyroptosis24. Observation by phase-contrast microscopy revealed a massive ballooning of cells at the mass ratio of MIT to GA 1:1.5, suggesting a strong pyroptosis effect induced by the combination of both chemotherapeutics (Fig. 4c). Pyroptosis occurs when the N-terminal fragment of the GSDM family is activated to cleave to form pores in the plasma membrane, causing the inward flow of aqueous fluid and cell swelling, and eventually leading to cell membrane rupture20. Considering that the degrees of pyroptosis varied with different drug ratios, we used the pyroptosis index (percentage of pyroptosis cells to all cells in the photos at the indicated time point) to screen the optimal drug ratio for inducing pyroptosis32. As shown in Fig. 4d, the maximum value of the pyroptosis index was 61.7% at the mass ratio of MIT to GA 1:1.5. With this ratio, the drug combination could induce the maximal pyroptosis, which was consistent with the cell viability results.

a Cytotoxicity assay of 4T1 cells treated with a combination of MIT and GA at variable mass ratios and b the corresponding combination index (CI) was plotted with fraction affected (Fa) (n = 3 independent experiments). c Phase contrast microscopy images of 4T1 cells treated with a combination of MIT and GA at variable mass ratios. Scale bar = 5 μm. d Pyroptosis index of 4T1 cells after different treatments (n = 3 independent experiments). The experiments were repeated three times independently. Data are analyzed with one-way ANOVA followed by multiple comparisons test. e TEM images of MG, MG@PM. Scale bar = 100 nm. f The particle size and photographs of MG@PM. g Zeta potential of MG@PM (n = 3 independent experiments). Data are represented as mean ± SD and analyzed with one-way ANOVA followed by multiple comparisons test (PM vs. MG: P < 0.001; PM vs. MG@PM: P = 0.1337). h The particle size variation of MG or MG@PM in PBS during seven days (n = 3 independent experiments). i UV absorption spectra of MIT, GA, and MG. j Western blot analysis of platelet signature proteins including CD47, CD41, and P-selectin on MG@PM. k Quantitative analysis of the band intensity in image J (n = 3 independent experiments). Data are represented as mean ± SD and analyzed with unpaired student’s t tests (PM vs. MG@PM in CD47: P = 0.033; PM vs. MG@PM in CD41: P = 0.7546; PM vs. MG@PM in P-selectin: P = 0.1184). l Pharmacokinetic properties of ICG-labeled MG (IMG) and MG@PM (IMG@PM) in vivo. Inset was the blood retention of IMG and IMG@PM 24 h post-injection (n = 3 mice). Data are represented as mean ± SD and analyzed with unpaired student’s t tests. m Fluorescence imaging of 4T1 tumor-bearing mice at preset time points after administration (left) and ex vivo imaging of major organs at 24 h after administration (right). n The semi-quantification results of the fluorescence intensity of major organs (n = 3 mice). Data were represented as mean ± SD. Data are analyzed with unpaired student’s t tests (IMG vs. IMG@PM in tumor: P < 0.001). Source data are provided as a Source Data file.
Synthesis and characterization of nanococrystals
Despite the great potential of MIT in combination with GA to achieve promising cell pyroptosis effects, the poor solubility of both drugs limits their clinical application. Furthermore, different pharmacokinetics and tissue distribution characteristics of both free drugs make it impossible to guarantee an optimal synergistic ratio at the tumor site after intravenous injection of both free drugs33. Therefore, to guarantee the rational tumor drug delivery with optimal synergistic ratios, we formed nanococrystals (MG) of MIT and GA by carrier-free self-assembly and coated them with platelet membranes by extrusion to achieve stable bionic nanocrystals (Fig. 1c). The prepared nanococrystals were spherical and had a good monodispersity. A 10-nm layer of cell membrane coating was observed on the surface of MG@PM compared with MG, demonstrating the successful coating of platelet membranes (Fig. 4e). Dynamic light scattering (DLS) indicated that the particle size of MG and MG@PM were separately 125.6 nm and 155.3 nm, while the corresponding zeta potentials were −5.8 mV and −22.1 mV, respectively (Fig. 4f, g). The surface of MG@PM was successfully coated with negatively charged platelet membranes, which would avoid plasma protein adsorption and maintain nanococrystal stability in the circulation. The particle size of MG and MG@PM in PBS was measured for seven consecutive days, and it was found that the particle size of MG increased to 1175.2 nm while that of MG@PM only increased to 315.0 nm, indicating that MG@PM has superior stability under physiological conditions due to the prevention of nanococrystal aggregation by the platelet membrane coating (Fig. 4h). In addition, MG@PM kept stable in H2O, PBS, DMEM, and FBS, and the presence of the Tyndall effect under laser irradiation also confirmed the dimensional stability of MG@PM (Supplementary Fig. 9a). The characteristic absorption peaks of MIT (610 nm) and GA (368 nm) were visible after the formation of MG nanococrystals, indicating that both drugs were successfully loaded in the nanococrystals (Fig. 4i and Supplementary Fig. 9b). The standard curves of MIT and GA were established based on the absorbance values (Supplementary Fig. 9c, d), and the specific synergistic ratio (MIT/GA = 1:1.5) was achieved by the current method. The drug loading for MIT and GA in nanococrystals was 40.0% and 60.0%, respectively, and the encapsulation rate for MIT and GA was 24.7% and 57.3%, respectively. Additionally, the results of release profile showed that MIT and GA were rapidly and almost synchronously released from MG@PM in PBS with a pH of 5.5 and reached the maximum of 73.6% and 66.5% at 8 h, respectively (Supplementary Fig. 10). In contrast, the release of MIT and GA was much lower in PBS with a pH of 7.4, with both being less than 20% at 8 h, suggesting that MG@PM can maintain stability under normal physiological conditions, minimize drug leakage, and keep the drugs at the appropriate ratios upon reaching the tumor foci to ensure optimal therapeutic efficacy.
Since nanococrystals camouflaged by platelet membranes are an emerging bionic nano platform, we verified whether MG@PM had physicochemical properties similar to platelets, such as tumor targeting, low immunogenicity, and high compatibility. Analysis of the protein composition in MG@PM using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) revealed protein expression similar to those of platelet membranes (Supplementary Fig. 9e), suggesting effective migration of platelet membranes on the surface of MG@PM. Specific protein markers of platelets such as CD4734,35,36, CD41, and P-selectin were not significantly differentially expressed in PM and MG@PM (Fig. 4j, k). Based on the excellent in vitro physicochemical characteristics of bionic nanococrystals, we further examined their in vivo behavioral properties in 4T1 tumor-bearing mice. For this purpose, we constructed ICG-labeled MG (IMG) and MG@PM (IMG@PM) (Supplementary Fig. 9f). As shown in Fig. 4l, IMG@PM exhibited a longer blood circulation time than IMG, which was attributed to the stabilizing effect of platelet membranes on nanococrystals and the immune evasion ability of the platelet membrane coating. Due to the simultaneous tumor-targeting ability of platelet membranes, the accumulation of IMG@PM at the tumor site was significantly enhanced at 24 h after intravenous administration, with a 6.5-fold higher fluorescence intensity than IMG (Fig. 4m, n). In order to more accurately detect the behavior of the drug in the body, we used LC-MS/MS to reassess the changes in blood concentrations at different time points after systemic administration, as well as organ-specific distribution. The standard curves and the drug concentration-time curves for MIT and GA were plotted as shown in Supplementary Fig. 11a–d. We analyzed the main pharmacokinetic parameters including area under the curve (AUC), blood circulation half-life (t1/2), clearance (CL), and mean residence time (MRT) (Supplementary Table. 4). The AUC for MIT and GA in the MG@PM group were 3.28 and 1.82-fold higher than those in the MG group, respectively. In addition, MIT and GA in MG@PM had longer t1/2 than those in MG (P < 0. 01). Elimination of MIT and GA in the MG@PM group was also slower than those in the MG group (P < 0.001 for CL), and MRT of MIT and GA in the MG@PM was observed to be 1.92-fold and 1.54-fold longer than in those of the MG group, respectively. Interestingly, MIT has a higher CL and a lower MRT than GA in the MG group, indicating that MG is not stable in the blood and at least part of MG disassembles during circulation. In contrast, MIT had CL and MRT consistent with GA in the MG@PM group, indicating that MG@PM is stable in the bloodstream, maintaining a consistent mass ratio of MIT and GA throughout circulation. Together, these data demonstrate that platelet modification improves the in vivo behavioral profile of MG and that the long-lasting blood circulation of bionic nanocrystals will ensure superior targeting of tumors. The results of drug distribution in various organ tissues showed that the accumulation of MIT and GA in tumor tissues was increased about 3-fold with the help of the targeting ability of platelet membranes, and the accumulation ratio of MIT and GA was as expected (Supplementary Fig. 11e, f).
In vitro cytotoxic and pyroptosis effects of nanococrystals
After the successful construction of bionic nanococrystals, we evaluated whether the platelet membrane coating could facilitate the internalization of the MG payload. As shown in Fig. 5a, IMG@PM was found to be more uptake by 4T1 cells than IMG at 4 h, which could be due to the interaction of P-selectin on platelet membranes and CD44 receptors on tumor cells that promotes the cellular uptake of MG37. Given the excellent cellular uptake of MG@PM, we further investigated the cytotoxicity of nanococrystals on 4T1 cells. As shown in Fig. 5b, compared to the weak cytotoxicity in the MIT and GA groups, cell viability was significantly diminished in the MG and MG@PM groups in a concentration-dependent manner. At minute concentrations (0.25 μg/mL of MIT, 0.375 μg/mL of GA), the cell viability of 4T1 cells after MG treatment decreased to 39.7%. Due to the platelet membrane coating, MG@PM had higher stability, cellular uptake, and lower cell viability (32.3%) compared with MG. Meanwhile, the strongest killing effect on 4T1 cells was also found in the MG@PM group by live/dead cell staining experiments (Supplementary Fig. 12a), consistent with the above results.

a Uptake of ICG-labeled MG (IMG) and MG@PM (IMG@PM) in 4T1 cells at 1 h and 4 h, respectively. Scale bar = 50 μm. b Viability of 4T1 cells after different treatments (n = 5 independent experiments). MIT vs. MG@PM: P < 0.001; GA vs. MG@PM: P < 0.001. Data are analyzed with two-way ANOVA followed by multiple comparisons test. c Phase contrast microscopy observation of 4T1 cell morphology under different treatments. White arrows pointed to pyroptosis cells. Scale bar = 15 μm. d LDH release from 4T1 cells after different treatments (n = 5 independent experiments). MIT vs. MG@PM: P < 0.001; MIT vs. MG: P < 0.001; GA vs. MG@PM: P < 0.001; GA vs. MG: P < 0.001. Data are analyzed with one-way ANOVA followed by multiple comparisons test. e Intracellular ATP content in 4T1 cells after different treatments (n = 5 independent experiments). MIT vs. MG@PM: P < 0.001; MIT vs. MG: P < 0.001; GA vs. MG@PM: P < 0.001; GA vs. MG: P < 0.001. Data are analyzed with one-way ANOVA followed by multiple comparisons test. f Flow cytometry assay of the ROS production in 4T1 cells after different treatments. g Intracellular GSH level in 4T1 cells after different treatments (n = 5 independent experiments). MIT vs. MG@PM: P < 0.001; MIT vs. MG: P < 0.001; GA vs. MG@PM: P = 0.031; GA vs. MG: P = 0.145. Data are analyzed with one-way ANOVA followed by multiple comparisons test. h Heat map of the differential expression of pyroptosis-related proteins in the MG@PM group compared with the control group. i Quantitative protein analysis of predicted target genes for GA and MIT (n = 3 independent experiments. Control vs. MG@PM in Txn: P < 0.0001; Control vs. MG@PM in Mcl1: P = 0.0059; Control vs. MG@PM in Top2a: P = 0.0157; Control vs. MG@PM in Erbb2: P = 0.0425). Data are analyzed with unpaired student’s t tests. j Bubble plot of the KEGG pathway analysis for differentially expressed proteins enriched in the MG@PM group compared to the control group. The colors of the nodes reflected the P-values of the designated pathways, and the sizes of the nodes indicated the number of differentially expressed proteins enriched in the pathways. Data are analyzed with two-way ANOVA followed by multiple comparisons test. k Analysis of the protein functional interaction network of the MG@PM group using the Search Tool for the Retrieval of Interacting Genes / Proteins (STRING) algorithm. Data were represented as mean ± SD. Source data are provided as a Source Data file.
Since a combination of MIT and GA with a specific mass ratio (MIT/GA = 1:1.5) had prominent pyroptosis-inducing properties, we further validated whether MG@PM could efficiently activate pyroptosis. 4T1 cells treated with MG and MG@PM swelled and generated typical spherical bubbles, while no significant changes were observed in cells treated with the same amount of free MIT or GA (Fig. 5cand Supplementary Movie. 1). In addition, flow cytometry assay showed that MG@PM significantly increased the number of PI-positive cells, indicating that pyroptosis leads to the loss of cell membrane integrity (Supplementary Fig. 12b). When pyroptosis occurs, a large amount of intracellular lactate dehydrogenase (LDH), ATP, and other contents leak into the supernatant. We examined the LDH release and intracellular ATP content in cells after different treatments. It was found that the release of LDH from the MG and MG@PM groups was much higher than that from the MIT and GA groups, while intracellular ATP concentration in the MG and MG@PM groups decreased significantly (Fig. 5d, e). We applied Western blot experiments to investigate the effects of MIT and GA on pyroptosis-associated proteins. Firstly, through the detection of the expression levels of several GSDM-family proteins, we found that both MIT and GA could enhance the amount of GSDME-N-terminal but could not increase the GSDMD and GSDMB-N-terminal. Meanwhile, the expressions of GSDME-FL and Pro-Caspase-3 in the MG and MG@PM groups were decreased compared with that in the free drug groups, whereas the amount of GSDME-N and Cleaved-Caspase-3 were significantly upregulated (Supplementary Figs. 13, 14). By immunofluorescence staining we found that the amount of GSMDE-N increased in the cells after MG and MG@PM treatment (Supplementary Fig. 15). Quantitative analysis showed that the number of GSDME-N positive cells increased to 49.7% and 59.5% in MG and MG@PM group, respectively (Supplementary Fig. 16).
To verify that MG@PM-triggered pyroptosis was dependent on the Caspase-3/GSDME pathway, we knocked down the expression of Caspase-3 in 4T1 cells using siRNA technology (Supplementary Fig. 17a). We found that Caspase-3 knockdown sharply inhibited the production of GSDME-N fragments in MG@PM-treated 4T1 cells (Supplementary Fig. 17b), and eliminated the morphological features of cell pyroptosis (Supplementary Fig. 17c). In addition, we continued to use siRNA technology to knock down the expression of GSDME in 4T1 cells (Supplementary Fig. 18a). The results showed that after GSDME knockdown, the pyroptosis swelling feature disappeared, and instead, the apoptosis morphology feature appeared in MG@PM-treated 4T1 cells. In addition, the level of LDH release was significantly reduced in MG@PM-treated 4T1 cells (Supplementary Fig. 18b, c). The above results demonstrated that MG@PM-triggered pyroptosis was dependent on the Caspase-3/GSDME pathway. Additionally, we also scrutinized the dose-dependent effects of MG@PM. The results showed that both medium (0.625 μg/mL) and high doses (1.25 μg/mL) of MG@PM caused balloon-like alterations of cells and release of LDH, whereas the low-dose (0.25 μg/mL) group was not significantly altered compared with the control group (Supplementary Fig. 19). The cleavage of Caspase-3 and GSDME was initiated in 4T1 cells after treatment of both medium and high doses of MG@PM, and the highest amount was observed in the high-dose group. The above results indicate that MG@PM can significantly induce cell pyroptosis at a 1.25 μg/mL concentration.
A disruption of the intracellular redox environmental balance often accompanies the onset of pyroptosis38. We detected the production of ROS in 4T1 cells after different treatments using a fluorescent probe (2,7-dichlorofluorescein diacetate, DCFH-DA). As shown in Fig. 5f, the mean fluorescence intensity of the MG and MG@PM groups was much higher than that of the MIT and GA groups. Subsequently, the detection of intracellular GSH content revealed a significant decrease of 43% in the GA group and 60% in the MG@PM group compared with the control group, indicating that MG@PM treatment has a stronger GSH depletion capacity (Fig. 5g). We inferred that the MG@PM might induce pyroptosis by disrupting intracellular redox balance.
Mechanism of nanococrystals-mediated pyroptosis
Based on the prominent pyroptosis induction of 4T1 cells by MG@PM treatment, we conducted tandem mass-tagging (TMT)-based proteomics to uncover the related mechanisms. Principal component analysis (PCA) was performed on 6114 plausible proteins detected in the control and MG@PM groups, accounting for 68.45% of the variables (Supplementary Fig. 20). Clustered heat map results showed substantial differences in primary protein sources between the control and MG@PM groups (Supplementary Fig. 21). Compared with the control group, 558 proteins were differentially expressed in the MG@PM group, of which 275 proteins were upregulated and 283 were down-regulated (Supplementary Fig. 22). GO enrichment analysis revealed that differential proteins mainly affected cytoplasm, cell death, mitochondria-related operations, and protein transport (Supplementary Fig. 23). To investigate the mechanism in-depth, we focused on the expression of proteins involved in pyroptosis. As shown in Fig. 5h, after MG@PM treatment, Bax (Bcl2-associated X protein), Bcl2l13 (Bcl2-like 13), Bcl2l15 (Bcl2-like 15), and other proteins involved in apoptosis regulatory pathways did not show significant activation but decreased instead (Supplementary Fig. 24a)39,40. In contrast, the expressions of inflammatory signaling molecules such as Hmgb1 (High mobility group protein B1) and Il1α (Interleukin-1 alpha) were enhanced. In addition, the changes in cellular redox homeostasis-related differential proteins were shown in Supplementary Fig. 24b. Notably, there were significant differences in typical pyroptosis-associated proteins, such as Tradd, Ctsd, Nfkb2, Tnf, Bcl2, and Gsdmd. Next, we focused on the proteins of the target genes of MIT and GA (Supplementary Tables 2 and 3) and found that significant changes in the expressions of Txn, Mcl1, Top2a, and Erbb2 were detected in the 4T1 samples after MG@PM treatment (Fig. 5i). Txn (Thioredoxin), a potential target of GA, is actively involved in cellular redox response and regulates mitochondrial ROS content. Reduced expression of Txn deteriorates oxidative stress and affects several pyroptosis genes, such as Nlrp3, Casp1, Gsdmd, Casp3, and related pathways41,42. Mcl1 (Induced myeloid leukemia cell differentiation protein Mcl-1), as another target of GA, is a member of the Bcl-2 family, which could induce Casp3/Gsdme pathway-dependent pyroptosis upon inactivation43. Top2a (DNA topoisomerase 2-alpha) and Erbb2 (Receptor tyrosine-protein kinase erbB-2), a pair of often-jointly mutated proto-oncogenes, are both major targets of MIT. This pair of targets can alter the sensitivity of tumor cells to chemotherapeutic agents after cell cycle disruption while perturbing Casp1 triggers pyroptosis44,45. This identification of such drug targets was generally consistent with the predicted results of BFReg-NN (Fig. 3f). Missing results for other targets might be due to non-negligible differences between cellular and tissue samples. To further clarify whether MG@PM affected these target proteins, we detected the activity levels of relevant target proteins in 4T1 cells after treatment with different formulations by using enzyme immunoassay kits. GA could deplete 45.3% of Mcl1 and 25.8% of Txn in 4T1 cells, and MIT could deplete 39.3% of Top2a and 22.7% of Erbb2 in 4T1 cells, respectively, compared with the Control group (Supplementary Fig. 25). Both MG and MG@PM groups had significant effects on these four target protein activities.
The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis showed that MG@PM treatment mainly affected pathways involving T cell receptor signaling pathway, TNF signaling pathway, ribosome, oxidative phosphorylation, inflammatory mediator regulation of TRP channels compared to untreated cells (Fig. 5j). We finally visualized the up-down regulation and interconnections of proteins by identifying the differential protein interaction network of MG@PM, thus helping to reveal the hidden therapeutic targets (Fig. 5k). According to the protein interaction network results, ribosome-related proteins, especially mitochondrial ribosomal protein (MRP) deficiency played a key role in activating cancer cell pyroptosis by MG@PM treatment. Since mitochondrial ribosome biogenesis is essential for cellular respiration, defects in MRPs induce mitochondrial dysfunction, alter oxidative phosphorylation (OXPHOS) activity and NAD+/NADH ratio, as well as increase ROS production46,47. Several studies demonstrated that the silencing of Mrpl41, Mrpl43, and Mrps5, etc., which were involved in cell cycle regulation, led to specific downregulation of the pro-apoptotic factor Bax, while ribosomal stress triggered by perturbed ribosome biosynthesis could initiate cell pyroptosis48,49,50. Taken together with these analyses, we suggested that MG@PM could induce extensive ROS toxicity by disrupting redox homeostasis, making MRPs defective, and disrupting the mitochondrial respiratory chain, while MG@PM eventually induced the tumor cell pyroptosis by triggering intracytoplasmic ribotoxic stress. To verify the changes in intracellular homeostasis, we monitored the intracellular levels of NAD+ and NADH and found that the NAD+/NADH ratio was significantly elevated in the MG and MG@PM groups compared to the Control group (Supplementary Fig. 26). This suggested that the cells are in an oxidative environment and it has been reported that an elevated NAD+/NADH ratio could contribute to the inhibition of rRNA synthesis51,52. In JC-1 staining experiments, a large number of red-to-green shifts were detected in the MG and MG@PM groups, indicating the occurrence of mitochondrial membrane potential damage (Supplementary Fig. 27). Thus, we clarified the effects of MG@PM on ribosomal and mitochondrial stress by observing changes in energy metabolism and mitochondrial function. In conclusion, we validated the pathological processes related to tumor cell pyroptosis by proteomics analysis and tapped the main targets of nanococrystals and their role in regulating ribosomal stress to trigger pyroptosis.
In vivo antitumor effects of nanococrystals
We evaluated the therapeutic effect of nanocrystals MG@PM as promising pyroptosis activators on the orthotopic 4T1 model. The regimens shown in Fig. 6a were followed to treat the tumor-bearing mice, and tumors were removed for ex vivo photography on day 15 (Fig. 6c, d). Compared with PBS, GA and MIT had a weak inhibitory effect on tumor growth. The reason might be attributed to the fact that MIT and GA, both small-molecule chemotherapeutics, decayed rapidly in circulation and accumulated little in tumors53. Moreover, due to the systemic toxicity of MIT in vivo, mice in the MIT group showed significant weight loss after treatment (Fig. 6b), and one died on day 9. Hypertrophic cardiomyocytes in heart tissues, increased cytoplasmic eosinophilic granules in lung tissues, and increased glomerular volume and cell count in kidney tissues were observed in the MIT group, which indicated that MIT could cause varying degrees of damage to multiple organs (Supplementary Fig. 29). In contrast, MG and MG@PM treatments displayed stronger antitumor effects than GA or MIT treatment with significantly smaller tumor volume and stronger tumor necrosis and apoptosis (Fig. 6e). The striking therapeutic effect was observed in the MG@PM group, where the tumor volume and tumor weight were approximately 7 times smaller than those of the PBS group at the end of this experiment (Fig. 6c, Supplementary Fig. 28). However, the MG group had a relatively weaker therapeutic effect due to the lack of stability and targeting in vivo. Notably, there was no significant weight loss in the mice of the nanococrystal groups, and no distinct pathological changes were observed in the organ sections compared to the PBS group. The superior biosafety makes MG@PM a promising treatment for tumors. We performed Western blot experiments on tumor samples from mice to identify important pyroptosis target proteins. As shown in Supplementary Fig. 30, the amount of Cleaved-Caspase-3 and GSDME-N were significantly increased in the MG@PM group. In contrast, neither the MIT, GA, nor MG groups were able to effectively induce tumor pyroptosis due to low drug accumulation and synergistic effects. At the same time, we were surprised to find significant inhibition of hepato-pulmonary metastasis of tumor cells in the MG@PM group (Supplementary Fig. 29). We speculated that MG@PM elicited a potent antitumor immune response and eliminated tumor metastasis.

a Schematic diagram of the therapeutic regimen of murine 4T1 tumor models. b Changes in the body weight of tumor-bearing mice during the therapeutic period (n = 5 mice). Data are analyzed with two-way ANOVA followed by multiple comparisons test. c Tumor volume change curves after different treatments (n = 5 mice). d Images of the excised orthotopic tumors in various groups on day 15. The red dashed rectangle indicated mouse death. e Pathological H&E staining and TUNEL staining of tumor slices on day 15. Scale bar = 100 μm. f The expression levels of inflammatory factors INF-γ (MG@PM: P < 0.001) and g TNF-α (MG@PM: P < 0.001) in the serum of mice treated with different formulations on days 6, 7, and 8 (n = 3 mice). Data are analyzed with two-way ANOVA followed by multiple comparisons test. h Flow cytometry analysis of the proportion of mature DC cells in tumor-draining lymph nodes (TDLN) (n = 4 mice). Data are analyzed with one-way ANOVA followed by multiple comparisons test. i Flow cytometry analysis of the ratio of CD4+ T cells in tumors (n = 4 mice). MIT vs. MG: P < 0.001; GA vs. MG: P = 0.0038. Data are analyzed with one-way ANOVA followed by multiple comparisons test. j Flow cytometry analysis of the ratio of CD8+ T cells in tumors (n = 4 mice). MIT vs. MG: P < 0.001; GA vs. MG: P = 0.0015. Data are analyzed with one-way ANOVA followed by multiple comparisons test. k Flow cytometry analysis of the proportion of Treg cells in tumors (n = 4 mice). MIT vs. MG: P < 0.001; GA vs. MG: P = 0.0016. Data were represented as mean ± SD and analyzed with one-way ANOVA followed by multiple comparisons test. Source data are provided as a Source Data file.
Nanococrystals-mediated pyroptosis for in vivo immune system activation
As tumor cell pyroptosis releases large amounts of contents, it could promote the exposure of tumor-associated antigens and alleviate the immunosuppressive microenvironment23. The content of high mobility group box 1 (HMGB1), a representative molecule of damage-associated molecular patterns (DAMPs) in tumor tissues54, was found to be significantly increased in the MG@PM group compared with other groups (Supplementary Fig. 31). To investigate the ability of MG@PM as an innovative pyroptosis inducer to stimulate the host immune response, we then examined cytokines, including INF-γ and TNF-α, in the serum of mice on day 6, 7, and 8 after different treatments. Compared with the other groups, the secretion of INF-γ and TNF-α in the MG@PM group on day 6 after treatment was significantly higher (Fig. 6f, g) and peaked on day 7. Accordingly, we investigated the immune environment of Balb/C tumor-bearing mice after seven days of treatment. DCs in lymph nodes and the spleen were isolated and collected for single-cell suspension preparation and antibody staining. Flow cytometry assay showed that the subclusters of mature DC cells (CD11c+CD80+CD86+) in lymph nodes of the MG@PM group significantly increased (21.7%) by 6.06 folds compared with that of the control group (3.58%) (Supplementary Fig. 32a, Fig. 6h). Similar results were also reflected in the DC subpopulation of splenic lymphocytes (Supplementary Figs. 32b, 33). These results confirmed the activation of DCs and their positive involvement in the pyroptosis-induced immune process. To further evaluate the alterations of the tumor immune microenvironment, we observed the changes in immune cell clusters in tumor tissues (Fig. 6i, j, Supplementary Fig. 32c, d). It was revealed that the proportion of both helper CD4+ T cells and killer CD8+ T cells was significantly increased in the MG@PM group compared with other groups, and the cell population was up to 24.9% and 18.4%, respectively, suggesting a potent immune response within the tumor tissue. Otherwise, regulatory T cells (Treg), a subset of T cells that suppress the antitumor immune processes, reduced 2-3 folds in the MG@PM group relative to the control group, demonstrating the alleviation of the tumor immunosuppressive microenvironment (Fig. 6k, Supplementary Fig. 32e). In conclusion, MG@PM-mediated tumor pyroptosis releases tumor-associated antigens that trigger efficient immune activation in vivo. Furthermore, we found that MG@PM induced a substantial elevation of GZMB content in tumor tissues (Supplementary Fig. 34), consistent with cohort analysis data (Supplementary Fig. 35). It has been reported that GZMB released by killer lymphocytes can induce tumor cell pyroptosis by cleaving GSDME at the same site as Caspase-353,54. Thus, we reasonably have to propose the hypothesis that GZMB might have contributed to the pyroptosis effect triggered by MG@PM.
In vivo anti-metastasis efficacy and the cascade pyroptosis effect
The high metastasis of breast cancer has been a significant challenge for clinical treatment55. Based on the potent immune response of the pyroptosis inducer, we investigated the prospect of MG@PM versus clinical first-line therapy, anti-PD-1/Abraxane, in murine lung metastasis models56,57. Following the model construction and the treatment schedule shown in Fig. 7a, the expansion and growth of tumor metastases were monitored by bioluminescence imaging. The results showed that 5 days after injection of 4T1-Luc cells, there were obvious tumor bioluminescence signals in the lungs of the PBS group, which gradually increased over time (Fig. 7b). Compared with the PBS group, the anti-PD-1 and anti-PD-1/Abraxane groups started to present bioluminescence signals on days 19 and 24, respectively, which slowly increased with time, indicating that anti-PD-1 and anti-PD-1/Abraxane can restrain tumor metastasis to a certain extent. Surprisingly, mice in the MG@PM group showed almost no apparent bioluminescence signals at the end of the bioluminescence observation (days 29), indicating that tumor metastasis was notably suppressed by MG@PM treatment. The number of metastatic nodules in the lungs was recorded by Bouin’s staining and photographed after the final bioluminescence observation (Fig. 7c, d). The number of metastatic nodules in the MG@PM group was deficient and significantly less than that in the anti-PD-1/Abraxane group. H&E staining of pathological sections showed pulmonary tissue in the PBS group was primarily dominated by metastases, while almost no metastases were seen in the MG@PM group (Fig. 7c). Encouragingly, all mice in the PBS and anti-PD-1 groups died within 60 days, while the survival rate in the MG@PM group was 83.4%, higher than that in the anti-PD-1/Abraxane group (50%), indicating the effectiveness of MG@PM treatment against lung metastases (Fig. 7e). We found that although MIT, GA, and MG had a slight therapeutic effect on orthotopic tumors, they did not significantly inhibit tumor metastasis and did not show prolonged survival (Supplementary Figs. 36, 37). The therapeutic effect in orthotopic tumors suggested that MG@PM significantly induced tumor tissue pyroptosis and provoked potent immunotherapeutic effects, which were ineffective in the MIT, GA, and MG groups. In orthotopic tumor tissues of the lung metastasis model, the clinical therapeutic agent Anti-PD-1/Abraxane caused a slight up-regulation of Cleaved-Caspase-3, which may be caused by apoptosis (Supplementary Fig. 38). While only MG@PM caused a significant up-regulation of GSDME-N, suggesting that a significant pyroptosis effect occurred in the orthotopic tumor tissues of the MG@PM group.

a Schematic illustration of dosing regimens in 4T1-luc metastatic tumor models. b In vivo bioluminescence images of mice in different groups. c Representative lung photographs and H&E staining images of lung slices in different groups. Scale bar = 500 μm. d Number of lung metastasis nodules in different groups (n = 5 mice). Anti-PD-1/Abraxane vs. MG@PM: P < 0.001. Data are analyzed with one-way ANOVA followed by multiple comparisons test. e Survival curves of mice in different groups in 60 days (n = 6 mice). PBS vs. MG@PM: P < 0.001; Anti-PD-1/Abraxane vs. MG@PM: P = 0.180. f Immunohistochemistry analysis of GSMDE at the lung site after the indicated treatments. The dashed line referred to the metastatic tumor area. Scale bar = 100 μm. g Fluorescence images of GSDME (red) and GZMB (green) distribution in metastatic lung tissues after the indicated treatments. The nuclei were stained with DAPI (blue). Scale bar = 50 μm. Data were represented as mean ± SD. Source data are provided as a Source Data file.
Considering our previously hypothesized adjuvant role of GZMB on MG@PM-induced pyroptosis, we examined the immune microenvironment in metastatic tumor tissues of the lungs and noticed a distinct population of CD8+ T cells in MG@PM group (Supplementary Fig. 39)58,59. The expression of GSDME in the metastatic lung region was significantly higher in the MG@PM group (Fig. 7f). In the present study we applied the rechallenge model, where lung tumor elimination is dependent on the systemic immune response induced by orthotopic tumor pyroptosis, and the presence of GSDME in the lungs suggests that MG@PM produced a durable and effective stimulatory response. In addition, immunofluorescence staining of lung tissue sections revealed enhanced expression and overlapping of GSDME and GZMB in the MG@PM group (Fig. 7g). The above results suggested that MG@PM induces an efficient pyroptosis within the orthotopic tumor, leading to appreciable local exposure of tumor-associated antigens, which ignites a broad tumor immune response like a match, and a large number of CD8+ T cells are recruited and release GZMB, which further shears the GSDME protein of the tumor cells, thus amplifying the pyroptosis effect. We defined this process as the MG@PM-triggered cascade pyroptosis loop. This cascade pyroptosis loop also provides a theoretical basis for tumor elimination by ultra-low doses of MG@PM.
Discussion
While recent studies have defined certain targets that may affect pyroptosis and developed corresponding targeted agents, they all failed due to inefficient induction, poor safety, and lack of a holistic view of tumor pyroptosis characteristics60,61. Here, we systematically identified target omics of TNBC pyroptosis based on the genetic profile characteristics of TNBC patients and established a collection of potential pyroptosis inducers according to these genes. Screening for disease-specific risk genes or molecular biomarkers based on a patient’s unique molecular profile is consistent with the concept of precision medicine3. Machine learning provides powerful support for rapidly optimizing medication combinations from a pool of drugs62. In the context of biomedical artificial intelligence, the importance of interpretability is increasingly recognized, and BFReg-NN is biologically relevant to accurately characterize the mapping between drug targets and signature genes and efficacy while retaining the high accuracy of the “black box” model. On the basis of rapid access to the compound drug synergy scores, can also help researchers to explore combinations with similar predictive mechanisms. In the subsequent experiments, omics explored the regulatory mechanisms of the key targets of the preferred drugs MIT and GA, closing the loop from prediction to theoretical validation. Due to non-negligible differences between cell samples and tumor tissue samples, the detailed mechanism by which MG@PM triggers pyroptosis of tumor cells requires further clarification.
Based on the physicochemical properties of the preferred drug pair MIT and GA, we customized the carrier-free bionic nanococrystal with the optimal synergistic ratio. While significantly increasing the drug loading, we attenuate the limited efficacy and systemic toxicity of small-molecule drugs due to their different pharmacokinetic properties in vivo63,64. Based on AI target prediction and proteomics results, we hypothesized that MIT increases the sensitivity of 4T1 to GA by inducing cell cycle disruption. Meanwhile, GA disrupts cellular redox homeostasis, regulates ROS content, and ultimately causes Casp3/Gsdme-dependent pyroptosis. In addition, when the ratio of MIT to GA is 1:1.5, nanocrystals with the most suitable particle size and stability can be prepared, which may have optimal cell membrane penetration. All these potential mechanisms could affect their synergistic effect as well as the effectiveness of inducing pyroptosis. Both in vitro and in vivo experiments have demonstrated the superior antitumor efficacy of MG@PM even at small doses, particularly in metastatic tumors, where the MG@PM activated cascade pyroptosis loop has shown the potential to outperform the clinical treatment regimen Anti-PD-1/Abraxane. This drug formulation strategy is reasonable and concise for the in vivo application of compound drugs and facilitates clinical translation. We will also continue to explore the efficacy and mechanism of action of other predicted potent compound drugs on the TNBC model.
This compound drug discovery technology platform we have established, which integrated bioinformatics analysis, artificial intelligence, dosage form development, and experimental validation, has the potential to drive breakthrough innovations and enhance efficiency in the drug development process. The target omics-based intelligent drug discovery framework is not only applied to the genome but will provide medical treatments more compatible with disease requirements in the emerging assay technologies, prospectively selecting personalized treatment options by modulating phenotypes or biomarkers. Meanwhile, AI technology empowers rapid drug development, providing interpretable biological meaning alongside efficient precision. Having attempted pyroptosis therapy for TNBC, our target omics-based intelligent drug discovery framework holds promise for addressing a wider range of other refractory diseases in the future. This proposed methodology will profoundly impact human health by providing a paradigm for intelligent drug discovery concepts while enhancing close interactions between multiple disciplines.
Methods
Ethical statement
All the animal experiments were performed in accordance with the guidelines evaluated and approved by Institutional Animal Care and Use Committee (IACUC), Fudan University School of Pharmacy (Shanghai, China).
Materials
All chemical agents were of analytical grade. Mitoxantrone (MIT) was purchased from Meilunbio, Co., Ltd (Dalian, China). Gambogic acid (GA), Indocyanine green (ICG), and Hoechst 33258 were obtained from Aladdin Reagent Co. Ltd. (Shanghai, China). Cell counting kit-8 (CCK-8), Calcein-AM/PI, Annexin-V-FITC/PI, JC-1, and DAPI were from KeyGen Biotech (Nanjing, China). DCFH-DA was acquired from Sigma-Aldrich (St. Louis, MO, USA). Abraxane (nanoparticle albumin-bound paclitaxel) was purchased from Celgene Co. Ltd. (New Jersey, USA). Anti-CD47 antibody (ab108415), anti-CD41 antibody (ab134131), and anti-CD62P (ab255822) were purchased from Abcam (Cambridge, UK). Anti-PD-1 antibody was purchased from Bio X Cell Biotechnology (New Hampshire, USA). Dulbecco’s modified eagle medium (DMEM), certified fetal bovine serum (FBS), phosphate Buffered Saline (PBS), penicillin-streptomycin stock solutions, and trypsin-EDTA (0.25%) were obtained from Invitrogen Co., (Carlsbad, CA, USA). All the other chemical solvents and agents were acquired from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China).
Cell culture
The tumor cell line MDA-MB-231, 4T1, and 4T1-Luc were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM with the addition of 10% FBS (v/v), 100 mg/mL of streptomycin, and 100 U/mL of penicillin. Cells were incubated at 37°C in a humidified environment with 5% CO2. Cells were tested monthly and found to be negative for mycoplasma contamination.
Experimental animals
Female Balb/C mice of 7-week-old (20 ± 1 g) and female Sprague-Dawley (SD) rats of 10-week-old (200 ± 10 g) were both obtained from SLAC Animal Ltd. (Shanghai, China) and raised under standard housing conditions in the Department of Experimental Animals, Fudan University (Shanghai, China). Female mice were chosen because the majority of breast cancers is seen in female patients. According to the guidelines of the ethics committee, the maximal tumor size permitted was 1500 mm3. Mice were euthanized when the tumor burden exceeded this threshold. Due to the blood volume requirements, female SD rats were used for the LC-MS/MS analysis.
Exploration of the drug candidates
The gene expression of 360 TNBC patients and 88 normal breast tissues was pre-processed by Transcripts Per Million (TPM) normalization. Given 45 pyroptosis genes, we used the R package “survival” to analyze the survival of TNBC patients according to the gene expression of these genes. Then we selected the key nine pyroptosis genes as target omics associated with TNBC with the highest Akaike Information Criterion (AIC), indicating the best trade-off between the survival’s goodness of fit and the model’s simplicity65. According to the nine key pyroptosis genes, we identified 3804 drug candidates by chemical–gene interactions in the Comparative Toxicogenomics Database (http://ctdbase.org/downloads/). To filter these drug candidates, we obtained the RNA composite expression of the pyroptosis regulators and the corresponding compound activity data (DTP NCI-60) from the CellMiner database (https://discover.nci.nih.gov/cellminer/home.do). We conducted a correlation analysis between the average z-score of compound activity and the RNA composite expression of the pyroptosis regulators. Then we identified 133 drug candidates significantly related to at least one pyroptosis gene (P < 0.05). To further narrow down the list of drug candidates, we analyzed the targets of these compounds using the SwissTargetPrediction database (http://www.swisstargetprediction.ch/) and PPI networks obtained from the STRING database (https://string-db.org/)66. We qualified 35 drug candidates with at least one target associated with pyroptosis genes whose distance was smaller than or equal to 2 of the PPI value.
Introduction of BFReg-NN
BFReg-NN could define the neural network architecture by existing biological knowledge. For example, we divided biological factors into distinct levels, L = {Gene, Protein, Pathway,…}. The modulatory relationships of internal factors were presented as a matrix set A = {AGene, AProtein, APathway,…,AL}. AGene could be a genetic relationship determined by the Gene Regulatory Network (GRN), while AProtein was defined by PPI. Since APathway was a hypergraph, every edge could link over two nodes, called hyperedges. The factors in a hyperedge propagated the information to directly affect others. Values in Al were binary, representing the presence of relations. We also identified the binary mapping matrixes M = {M1, M2, …, ML−1} from level l to its upper-level l+1 as the interaction between levels. We programmed M1 as a mapping from genes to proteins and M2 to be a straightforward mapping across levels of proteins and pathways. Both A and M determined the structure of the neural network, which included neurons and connections between neurons.
BFReg-NN used gene expression data \(x\) as input. We encoded each gene individually using the embedded layer, where \({H}_{i}^{{{\mathrm{0,0}}}}={emb}({x}_{i})\). At the intra-level \(l\), we aimed to help each biological factor interact with others by \({A}_{l}\). Thus, we used graph neural networks and message-passing mechanisms to update the embedding \({H}^{l}\), where \({\widehat{H}}_{i}^{l}={{{\rm{update}}}}({\sum}_{j\in {A}_{l}(i)}{{{\rm{meassage}}}}({H}_{i}^{l},{H}_{j}^{l}),{H}_{i}^{l})\). The message function was to produce messages from factor j to factor i, where \({A}_{l}(i)\) decided which factors were neighbors for factor i. The updating function was to renew the embedding of factor i with the acquired message and the prior hidden embedding. The embedding was learned level by level. As \({M}_{l}\) was inter-layer relationship between l and l + 1, we utilized the shielded deep neural network at the next layer to update the original expression \({H}_{i}^{l+{{\mathrm{1,0}}}}\), where \({H}^{l+1}={{{\rm{activation}}}}(({M}_{l}\odot {W}^{l}){\widehat{H}}^{l,}+{b}^{l})\). The element-wise multiplication \({M}_{l}\odot {W}^{l}\) ensured that absent relations were not applied to update. \({W}^{l}\) and \({b}^{l}\) were parameters that can be learned in deep neural networks.
We further enhanced BFReg-NN by adding extra edges in A. The available biological knowledge reflected the hidden relationships between factors detected by biotechnology. Nevertheless, owing to technical limitations, certain knowledge remained challenging to be detected. Hence, we set up the interaction in two ways. One was generalized conditioning sustained by available knowledge A. The other was a partial interaction concealed at the non-existent edge of A, predicting fresh biological knowledge. We used \({A}_{l}\) to restrain the learnable matrix \({A^{\prime} }_{l}\) rather than the binary matrix \({A}_{l}\) employed in the basic model to find fresh knowledge. For non-existent edges, we reweighed it by a small value \(\alpha=0.005\) due to it being less convincing. As a result, the edge intensities depending on both types of knowledge were revised as follows:
The parameter \({\beta }_{i,j}^{l}\) was learned by a Multilayer Perceptron based on the embeddings \({H}_{i}^{l}\) and \({H}_{j}^{l}\). Thus, we learned the factor embeddings while using these embeddings and transitions to deduce the strength of implicit interactions among factors. Finally, we used some downstream tasks to train BFReg-NN and obtain the weights in BFReg-NN. For example, we could train it to classify the cell types based on gene expression if we know the labels of cells. By analyzing the weights, we obtained the important genes and how these genes led to the cell type.
We compared our proposed BFReg-NN model with various types of methods on three different tasks. These methods include: (1) traditional methods (MLP, LSTM, Random Forest, XGBoost, Transformer), (2) two GNN methods (GCN and GAT), and (3) two classical medical models (DCell and P-NET)67,68. For both the GNN and medical models, we provided gene expression data and graph topology as inputs. The graph topology included gene regulatory networks, protein-protein interaction networks, and Gene Ontology (GO) term hierarchy, etc. Therefore, we ensured a fair evaluation of its performance. The detailed experimental procedure can be found at this link (https://arxiv.org/pdf/2304.04982.pdf).
Utilization of BFReg-NN to predict the drug effects
Recall the four layers are (1) relations among drug targets, (2) links between drug targets and pyroptosis genes, (3) relations among pyroptosis genes (4) the survival layer. In the first and third layers, we use GNN-based equations to obtain the overall situation among drug targets or pyroptosis genes. In the second layer, we use a DNN-based equation to transform the overall situation reflected from drug targets into the individual situation of each pyroptosis gene. The final layer is a CoxPH model to transform the overall situation reflected from the pyroptosis gene into patient survival time. Here we limited the neural network architecture of BFReg-NN by drug targets and pyroptosis genes. We defined \({A}_{1}\) as the relationship between drug targets, \({A}_{2}\) as the relationship between pyroptosis genes, and \({M}_{1}\) as the association between drug targets and pyroptosis genes. All the relationships were extracted by PPI networks (https://string-db.org/). We set the combined score of an edge as larger than 0.6 in \({A}_{1}\), \({A}_{2}\) and \({M}_{1}\). To enrich the association in \({M}_{1}\), factors were also connected if they are 2-hop neighbors. The downstream task was a survival task to predict recurrence-free survival (RFS) of TNBC patients. For all possible drug combinations, we trained the corresponding BFReg-NNs by different architectures decided by different input drug targets. If the drug targets can accurately predict the survival of TNBC patients, it indicates that these two drugs have highly relevant targets for TNBC patient survival. The c-index was used as an indicator to evaluate the fitness of the model69. It quantifies the level of concordance between predicted and observed survival times, with 1 indicating perfect concordance, and 0.5 suggesting no better than random chance. The c-index of the final selected drug pairs is larger than 0.9, which indicates the high association of drug pairs and the survival of TNBC patients. We also implemented Integrated Gradient to analyze BFReg-NN after it was well-trained. We obtained the important score of each drug target through different pyroptosis genes to influence TNBC RFS.
Preparation and characterization of nanococrystals
Preparation of platelet membrane-derived vesicles
Platelets were separated from the entire blood of female Balb/C mice of 7-week-old (20 ± 1 g) by centrifugation to separate erythrocytes and leukocytes and suspended in PBS with protease inhibitor tablets35. The purified platelet-rich plasma was subsequently placed in ice-cold PBS containing EDTA and prostaglandin E1 (PGE1, Sigma Aldrich, USA) to prevent platelet activation and then centrifuged at 800 × g for 20 min at room temperature to collect platelet precipitates. Platelet membranes were obtained via a repeat freeze-thaw cycle. Briefly, platelet suspensions were frozen in liquid nitrogen, thawed at 37°C, and centrifuged at 4000 × g for 5 min. After washing three times, it was resuspended in water and set aside.
Preparation of MG and MG@PM
MG nanococrystals were prepared by a one-step self-assembly method. The dimethyl sulfoxide (DMSO) solution containing 5 mg/mL of MIT and the ethanol solution containing 3 mg/mL of GA were mixed in equal volumes, and 100 µL of the mixture was added dropwise to 2 mL of ddH2O. Then stirred at room temperature for 5 h, and MG gradually appeared. After centrifugation (15,000 × g), the precipitate was collected and repeatedly rinsed in water three times to remove DMSO and unassembled drug. After the last centrifugation, MG was resuspended with 1 mL of sterile PBS and stored at 4°C for subsequent use. To prepare MG@PM, 100 μL of MG suspension (3.5 mg/mL) was mixed with 150 µL of platelet membrane suspension (1 mg/mL of membrane proteins) and then successively squeezed through 1 µm, 400 nm, and 200 nm polycarbonate porous membranes (Avanti Polar Lipids Inc, GE Healthcare, USA). ICG-labeled MG or MG@PM were obtained by the same procedure as MG and MG@PM, except that 1 mg/mL of ICG was added to the ethanol solution during the self-assembly process of MG.
Nanococrystal characterization
The morphology of MG and MG@PM was characterized by using transmission electron microscopy (TEM) (TEM-1400 Plus electron microscope, Leica, Germany). The diameter, polydispersity index, and zeta potential of nanococrystals were measured by a dynamic light scattering detector (Zetasizer, Malvern, UK). To examine the stability of MG and MG@PM in PBS, the Z-average diameter was measured for seven consecutive days, while the steady state of MG@PM in different media was observed and photographed. The loading capability (LC) and encapsulation efficiency (EE) of MIT and GA in MG@PM were measured by the UV-vis spectrophotometer (PerkinElmer Lambda 750). The characteristic absorption peaks of MIT and GA were 610 nm and 370 nm, respectively.
EE (%) = (Amount of drugs in nanococrystals)/(Total amount of drugs input) × 100%
LC (%) = (Amount of drugs in nanococrystals)/(Nanococrystals weight) × 100%
We performed drug release of MG@PM with a dialysis method. Briefly, 2 mL of MG@PM suspension (containing 1 mg of MIT and 1.5 mg of GA) was put in dialysis bags (ThermoFisher, MWCO = 3 kDa) and incubated within 50 mL of release medium (0.01 M PBS, pH = 5.5 or 7.4) at 37 °C at 100 rpm for 24 h. The release profiles of MIT and GA from MG@PM was tested.
Membrane protein characterization
The membrane proteins of MG@PM nanococrystals were characterized by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). MG@PM nanococrystals were lysed with the RIPA lysate, and protein concentrations were quantified, with platelet membranes and MG serving as controls. The presence of specific protein markers was identified by Western blot analysis. In brief, the resulting gels were shifted onto polyvinylidene difluoride (PVDF) membranes and hatched with antibodies (anti-CD47, anti-CD41, and anti-P-selectin) overnight at 4 °C. The membranes were then incubated with horseradish peroxidase-labeled goat anti-rabbit lgG H&L (HRP) antibody (G-21234, InvitrogenTM, USA) at room temperature for 1 h. GAPDH antibody (MA5-15738, InvitrogenTM, USA) was used as a control. The resulting bands were tested with an ECL developer (Beyotime, China) and quantified using ImageJ software.
Cellular uptake
To determine the cellular uptake of MG and MG@PM, 4T1 cells were seeded into confocal dishes (2×104 cells per dish) and cultured for 24 hours. Then ICG-labeled MG and MG@PM (50 ng/mL of ICG) were added to cells and incubated for 1 h and 4 h. To acquire fluorescence signals, cells were fixed with 4% paraformaldehyde before nuclei staining with Hoechst 33258 for 8 min. Photos were subsequently taken with a confocal laser scanning microscope (CLSM) (Leica, Germany).
Cytotoxicity assay
A total of 5 × 103 4T1 cells per well were seeded overnight in 96-well plates, which were subsequently subjected to various treatments for 24 h. Afterward, the CCK-8 reagent was applied to the cell and hatched for 1.5 h. Then the absorbance at 490 nm of each well was determined using the microplate reader (Multiskan MK3, Thermos, USA).
Besides, the live/dead cell staining experiment was carried out using CLSM. Briefly, 4T1 cells were seeded into confocal dishes (2 × 104 cells/dish) and incubated for 24 h. Then cells were cocultured with various formulations for 24 h, staining with Calcein-AM and PI according to the protocol, followed by imaging with the CLSM.
Synergistic effect evaluation
4T1 cells were treated with compound drugs of MIT and GA at different mass ratios, and cell viability was examined as described above. The combination index (CI) of various combinations was examined using the Chou-Talalay method, and the results of the CompuSyn software were classified as synergistic (CI < 1), additive (CI = 1), and antagonistic (CI > 1)70. Fraction affected (Fa) between 0.2 and 0.8 was deemed to be valid.
Flow cytometry
4T1 cells with different treatments (Control, MIT, GA, MG, MG@PM) were harvested, stained with Annexin V-FITC/PI, and subjected to flow cytometry for quantitative analysis of cell pyroptosis (CytoFLEX S, Beckman, USA). For quantitative analysis of ROS production in tumor cells, 4T1 cells were treated with different formulations for 6 h and stained with DCFH-DA (10 µM) for 30 min, after which they were washed with PBS several times and collected for flow cytometry.
4T1 cells with different treatments (Control, MIT, GA, MG, MG@PM) were fixed, permeabilized, and blocked. The samples were hatched with anti-GSDME-N antibody overnight at 4 °C, then stained with anti-FITC-IgG (H + L) antibody at room temperature. After that, the cells were collected and washed with PBS several times and analyzed with flow cytometry.
Pyroptosis assay
To observe cell morphological changes, 4T1 cells were seeded into 6-well plates, after which they were cultured for 24 h in different preparations respectively. The cell morphology was visualized under a phase contrast microscope (Olympus, Japan). Cellular LDH and ATP levels were tested using a firefly luciferase-based ATP assay kit (Beyotime, China) and LDH cytotoxicity assay kit (Beyotime, China) according to both manufacturers’ instructions. The luminescent value and absorbance were measured using a microplate reader (Multiskan MK3, Thermos, USA).
Western blotting analysis
The cell or tissue samples were lysed with the RIPA lysate, and protein concentrations were quantified. The presence of specific protein markers was identified by Western blot analysis. The resulting gels were shifted onto PVDF membranes and hatched with antibodies overnight at 4 °C. Primary antibody against GSDMB (ab215729, 1:1000), GSDMD (ab209845, 1:1000), and GSDME (ab215191, 1:1000) was obtained from Abcam. Caspase-3 (#9662, 1:1000) and GAPDH antibody (#2118, 1:1000) were purchased from Cell Signaling Technology. The resulting bands were incubated with goat anti-rabbit lgG H&L (HRP) antibody and tested with an ECL developer (Beyotime, China) and quantified using ImageJ software.
siRNA-mediated knockdown
4T1 cells were seeded into 6-well plates (1 × 105/well) for siRNA-mediated knockdown. After 24 h in culture, 3 μL of siCasp3 or siGsdme (RiboBio, Guangzhou, China) was transfected with Lipo3000 according to the manufacturer’s instructions. After 72 h, transfected 4T1 cells were treated with PBS, MIT, GA, MG, or MG@PM for subsequent experiments.
Consumption of intracellular GSH
Cells after different treatments were collected and subjected to two freeze-thaw circles to release intracellular content, and the supernatant was removed after centrifugation and processed according to the kit instructions. Intracellular GSH concentrations were measured by the GSH assay kit (Keygen Biotech, China).
TMT-based proteomics analysis
Proteomics features of 4T1 cells treated with MG@PM were analyzed by liquid chromatography-mass spectrometry (RIGOL L-3000, RIGOL TECHNOLOGIES, Beijing, China)35. The 4T1 cells without any treatment were taken as control. Protein samples were electrophoresed by SDS-PAGE to examine the concordance of each group. Protein samples (100 µg) were reduced, alkylated, and digested overnight with trypsin at 37°C. Then, each sample solution was tagged with TMT labeling reagent. After mixing and labeling, the samples were dissolved with 100 µL of mobile phase A (water containing 10 mM ammonium formate), centrifuged at 14000 × g for 20 min, and the supernatant was extracted and graded using a high-performance liquid chromatography phase. The solubilized powder was dissolved in 10 µL of liquid A, centrifuged at 14000 × g for 20 min at 4°C, and 1 µg of the supernatant was injected into the sampler for liquid mass detection using an Orbitrap Fusion Lumos mass spectrometer. The NSI source was operated in positive mode. Other ionization source parameters are listed as follows, spray voltage: 2000 V, capillary temperature: 320 °C. The scan ranges of full scan mode were set at m/z 407–1500. The raw data of mass spectrometry detection was analyzed using the Mus musculus UniProt database71. The parameters for peptide identification using Proteome Discoverer 2.4 software (Thermo Fisher Scientific) were set as follows: Carbamidomethyl(C) as static modification; The was M Oxidation (15.995 Da), TMT-6plex (K, N-terminal), Acetyl (Protein N-terminal) as dynamic modification;Precursor ion mass tolerance was ± 15 ppm; Fragment ion mass tolerance was ± 0.02 Da; Max missed cleavages was 2. Peptide- and protein-level false discovery rates (FDRs) were filtered to 1%. Statistical analysis of the identification and quantization results was done using Perseus 1.6.7.0 software. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE72 partner repository with the dataset identifier PXD053939.
Protein activity assay
4T1 cells were seeded in 12-well plates at a density of 1 × 105 cells per well and cultured overnight. Afterward, cells were treated with different formulations for 12 h. After that, cells were collected, lysed in an ice bath, and spun down. The concentration of Txn, Mcl1, Top2a, and Erbb2 in the supernatant was analyzed by using the ELISA assay kits (Jianglai Biology, China) according to the manufacturer’s instructions. Then optical density (OD) value at 450 nm of each well was determined using the microplate reader.
NADH and NAD+ measurement
4T1 cells were cultured in 12-well plates (5 × 104 cells per well). After being treated with PBS, MIT, GA, MG, or MG@PM for 12 h, the cells were washed with PBS three times. NADH and NAD+ levels were assessed using the NAD+/NADH assay kit (WST-8) (Beyotime, China) according to the manufacturer’s instructions. Then optical density (OD) value at 450 nm of each sample was determined using the microplate reader.
Mitochondrial Membrane Potential Monitor
4T1 cells were seeded into 12-well plates (1 × 105/well) for 24 h at 37 °C and incubated with different formulations. Finally, cells were dyed with JC-1 dyestuff for 30 min and the fluorescence signal was obtained by flow cytometry. The change of the JC-1 indicator from red to green can facilely detect the decrease of cell membrane potential.
Pharmacokinetics and biodistribution study
To confirm that the platelet membrane modification could endow the nanococrystal with prolonged blood circulation, six female Balb/C mice of 7-week-old (20 ± 1 g) were intravenously injected with IMG and IMG@PM (1 mg/kg of ICG) separately. At the indicated time points (0.017, 0.25, 0.5, 1, 2, 4, 8, 12, and 24 hours after injection), blood samples (50 μL) were taken from the fundic venous plexus and placed into heparin-preprepared polyethylene tubes. Plasma was collected by centrifugation and then subjected to fluorescence analysis of ICG on a microplate reader (Ex = 785 nm, Em = 810 nm). The major pharmacokinetic parameters were analyzed with Drug and Statistics (DAS) software.
The orthotopic breast tumor model was established by inoculating 3 × 106 4T1 cells into one mammary fat pad of female Balb/C mice of 7-week-old (20 ± 1 g). To identify the tumor-targeting efficiency of nanococrystals, IMG and IMG@PM (1 mg/kg of ICG) were intravenously injected in tumor-bearing mice, respectively. Fluorescence imaging of mice was performed with the IVIS Spectrum imaging system (PerkinElmer, USA) at pre-designed time points (1, 2, 4, 8, 12, and 24 h) post-injection. After 24 h, tumors and major organs (heart, liver, spleen, lung, and kidneys) were isolated from the mice and subjected to ex vivo fluorescence imaging using the IVIS Spectrum imaging system.
LC-MS/MS analysis
Six SD rats were randomly divided into 2 groups (n = 3 rats) and i.v. injected with MG and MG@PM (3 mg/kg of MIT, 4.5 mg/kg of GA), respectively. Blood samples (200 μL) were collected at 0.017, 0.25, 0.5, 1, 2, 4, 8, 12, and 24 h after injection and then centrifuged at 5000 × g for 10 min immediately. The supernatant plasma samples were stored at −20 °C until analysis. To detect the concentrations of MIT and GA in plasma, 150 μL of methanol was added to 50 μL of plasma samples in order to precipitate proteins. Then, the samples were vortexed for 1 min and centrifuged at 14,000 × g for 10 min. The supernatant was subsequently subjected to liquid chromatography–tandem mass spectrometry (LC-MS/MS, SCIEX Triple Quad™ 5500, USA) for analysis. Chromatographic separation was carried out on an Acquity UPLC column (2.1 × 100 mm, 1.8 µm). The mobile phase A was ultrapure water containing 2 mM FA, and the mobile phase B was ACN. Quantification of the ions was achieved by the multiple reaction monitoring (MRM) mode, in positive mode for mitoxantrone (monitoring the transition of the m/z 445 precursor ion to the m/z 88) and gambogic acid (monitoring the transition of the m/z 629.4 precursor ion to the m/z 545). The major pharmacokinetic parameters were analyzed with Drug and Statistics (DAS) software.
Drug distribution in various organ tissues was examined in the orthotopic breast tumor model. Tumors and major organs (heart, liver, spleen, lungs, and kidneys) were isolated from the Balb/C mice after tail vein injection of MG and MG@PM (3 mg/kg of MIT, 4.5 mg/kg of GA) for 24 h. The tissues were homogenized by a homogenizer in PBS and precipitated with methanol. After centrifugation at 12,000 × g for 10 min, the supernatant was taken and then the levels of MIT and GA in the respective tissues were detected by LC-MS/MS following the above method.
Anti-tumor efficacy
On day 10 after tumor implantation, murine orthotopic tumor models were assigned randomly to 5 groups, and then PBS, MIT, GA, MG, and MG@PM (3 mg/kg of MIT, 4.5 mg/kg of GA) were injected intravenously once two days for 3 times. Tumor volume was measured with calipers every two days during the period. After 15 days, tumors were isolated for photographing and weighing. The stripped tumors were soaked in 4% paraformaldehyde for 48 h, and paraffin sections were prepared to study the destruction of tumor cells induced by different treatments. Moreover, the sections were evaluated through terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) and hematoxylin-eosin staining (H&E) and observed by an Inverted fluorescence Microscope (Nikon, Japan).
In vivo immunostimulation experiment
For in vivo immunostimulation studies, the lymph nodes, spleens, and tumor tissues of murine orthotopic tumor-bearing models were collected on day 7, cut into pieces, and incubated in DMEM containing collagenase type I (Sigma, USA), collagenase type IV (Biosharp, China), hyaluronidase (Sigma, USA), and of DNase (Biosharp, Germany) at 37°C for 40 min. Then the sample was passed through 200 mesh nylon strainers to prepare single-cell suspensions and analyzed by flow cytometry. For the analysis of T cells, the single-cell suspensions were stained with anti-CD3-PerCP, anti-CD4-FITC, and anti-CD8-APC antibodies. For the analysis of regulatory T cells (Treg), the single-cell suspensions were stained with anti-CD25-APC, anti-CD4-FITC, and anti-Foxp3-PE antibodies. To assess the maturity of DCs, the single-cell suspensions were dyed with anti-CD11c-FITC, anti-CD86-APC, and anti-CD80-PE antibodies and analyzed by flow cytometry. In addition, sera from different groups of mice were collected on days 6, 7, and 8, respectively. The secreted cytokines in the serum, including TNF-α, IFN-γ, and GZMB were determined by ELISA kits (Absin, Shanghai).
In vivo antimetastatic studies
Ten days after the orthotopic breast tumor model was established, mice received different treatments on days 0, 2, 4, 6, and 8: 1) PBS, 2) Anti-PD-1 (200 µg/kg), 3) Abraxane (20 mg/kg) + Anti-PD-1 (200 µg/kg), 4) MG@PM (3 mg/kg of MIT, 4.5 mg/kg of GA). 1 × 106 4T1-Luc cells were injected intravenously on day 9 to construct a lung metastasis model. Bioluminescence imaging of mice was recorded by the IVIS Spectrum imaging system every five days. Each mouse received an intraperitoneal injection of D-luciferin sodium solution (150 mg/kg) 20 min before bioluminescence imaging. Mice were executed on day 29. Pulmonary tissues were captured, colored with Bouin’s solution (Servicebio, China), and imaged to calculate lung metastases. Lung tissue sections were also stained with H&E and observed with Olympus Slide View VS200 (Tokyo, Japan). As for survival analysis, the lifespan of the murine tumor-bearing model adopted with the same administration schedule was recorded during the treatment period.
Statistics and reproducibility
No statistical method was used to predetermine sample sizes. For in vitro studies, treatment groups were randomly assigned and were not changed when treatment was given on the culture day indicated. These experiments were completed in replicates and independent experiments. For animal studies, mice were randomly assigned to treatment groups after tumor inoculation. The starting tumor burden in the treatment and control groups was similar before treatment. The investigators were blinded to allocation during experiments and outcome assessment. Overall survival was estimated by Kaplan–Meier methods and compared with log-rank tests. Cox proportional hazard models were used for multivariate survival analysis. Multivariate logistic regression models were used to assess binary outcomes of response to treatment. Pearson’s correlation coefficient was used to assess linear correlations between variables. Two-group was compared using unpaired student’s t-test, and multiple-group analysis was performed using one-way ANOVA followed by multiple comparisons test. In vitro experiments were carried out at least triplicate independent experiments as indicated. All in vivo studies included a minimum of five mice per group for drug therapy studies. All statistical analyses were performed using GraphPad Prism 9 software.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Gene expressions of these genes on TNBC and normal breast tissues were obtained from Fudan University Shanghai Cancer Center TNBC datasets (FUSCCTNBC, http://www.biosino.org/; OEP000155 node; GEO: GSE118527; SRA: SRP157974; figshare: https://doi.org/10.6084/m9.figshare.19783498.v5). Gene Ontology (GO) was performed using the Metascape database (http://metascape.org/). A protein-protein interaction (PPI) network was constructed based on relationship data obtained from the STRING database (https://cn.string-db.org/). The proteomics data have been deposited to the ProteomeXchange with the dataset identifier PXD053939. Source data are provided with this paper. All remaining data can be found in the Article, Supplementary and Source Data files. Source data are provided with this paper.
Code availability
The work informed BFReg-NN developments, which are available from GitHub (https://github.com/BoshuOuyang/BFregNN-Cox-for-pyroptosis-in-TNBC/).
References
Nass, S. J. et al. Accelerating anticancer drug development — opportunities and trade-offs. Nat. Rev. Clin. Oncol. 15, 777–786 (2018).
Ocaña, A., García-Alonso, S., Amir, E. & Pandiella, A. Refining early antitumoral drug development. Trends Pharmacol. Sci. 39, 922–925 (2018).
Vincent, F. et al. Phenotypic drug discovery: recent successes, lessons learned and new directions. Nat. Rev. Drug Discov. 21, 899–914 (2022).
Gandomi, A. & Haider, M. Beyond the hype: big data concepts, methods, and analytics. Int. J. Inf. Manag. 35, 137–144 (2015).
Edgar, R., Domrachev, M. & Lash, A. E. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 (2002).
Parkinson, H. et al. ArrayExpress-a public database of microarray experiments and gene expression profiles. Nucleic Acids Res 35, D747–D750 (2007).
Moffat, J. G., Vincent, F., Lee, J. A., Eder, J. & Prunotto, M. Opportunities and challenges in phenotypic drug discovery: an industry perspective. Nat. Rev. Drug Discov. 16, 531–543 (2017).
Yang, F. et al. Ferroptosis heterogeneity in triple-negative breast cancer reveals an innovative immunotherapy combination strategy. Cell Metab. 35, 84–100.e108 (2023).
Al-Lazikani, B., Banerji, U. & Workman, P. Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol. 30, 679–692 (2012).
Chen, Y., Pal, S. & Hu, Q. Cell-based relay delivery strategy in biomedical applications. Adv. Drug Deliv. Rev. 198, 114871 (2023).
Holbeck, S. L. et al. The National Cancer Institute ALMANAC: a comprehensive screening resource for the detection of anticancer drug pairs with enhanced therapeutic activity. Cancer Res 77, 3564–3576 (2017).
Lavecchia, A. Machine-learning approaches in drug discovery: methods and applications. Drug Discov. Today 20, 318–331 (2015).
Guvenc Paltun, B., Mamitsuka, H. & Kaski, S. Improving drug response prediction by integrating multiple data sources: matrix factorization, kernel and network-based approaches. Brief. Bioinform 22, 346–359 (2021).
Reker, D. et al. Computationally guided high-throughput design of self-assembling drug nanoparticles. Nat. Nanotechnol. 16, 725–733 (2021).
Sadybekov, A. A. et al. Synthon-based ligand discovery in virtual libraries of over 11 billion compounds. Nature 601, 452–459 (2022).
Lutz, I. D. et al. Top-down design of protein architectures with reinforcement learning. Science 380, 266–273 (2023).
Menden, M. P. et al. Community assessment to advance computational prediction of cancer drug combinations in a pharmacogenomic screen. Nat. Commun. 10, 2674 (2019).
Preuer, K. et al. DeepSynergy: predicting anti-cancer drug synergy with Deep Learning. Bioinformatics 34, 1538–1546 (2018).
Waks, A. G. & Winer, E. P. Breast cancer treatment: a review. JAMA 321, 288–300 (2019).
Broz, P., Pelegrin, P. & Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 20, 143–157 (2020).
Chen, Y. et al. Small-molecule ferritin degrader as a pyroptosis inducer. J. Am. Chem. Soc. 145, 9815–9824 (2023).
Jiang, Y. Z. et al. Genomic and transcriptomic landscape of triple-negative breast cancers: subtypes and treatment strategies. Cancer Cell 35, 428–440.e425 (2019).
Zhang, Z., Zhang, Y. & Lieberman, J. Lighting a fire: can we harness pyroptosis to ignite antitumor immunity? Cancer Immunol. Res. 9, 2–7 (2021).
Wang, Y. et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103 (2017).
Ouyang, B. et al. Albumin-based formononetin nanomedicines for lung injury and fibrosis therapy via blocking macrophage pyroptosis. Mater. Today Bio 20, 100643 (2023).
Yoshihara, K. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612 (2013).
Xinnan, D., Caihua, S., Jie, Z., Xiaoxiao, L. & Dongsheng, L. Biological Factor Regulatory Neural Network. arXiv e-prints. https://doi.org/10.48550/arXiv.42304.04982 (2023).
Paciucci, P. A. Sklarin, N.T. Mitoxantrone and hepatic toxicity. Ann. Intern Med. 105, 805–806 (1986).
Xia, G. et al. Gambogic acid sensitizes gemcitabine efficacy in pancreatic cancer by reducing the expression of ribonucleotide reductase subunit-M2 (RRM2). J. Exp. Clin. Cancer Res. 36, 107 (2017).
Su, X. et al. Localized disruption of redox homeostasis boosting ferroptosis of tumor by hydrogel delivery system. Mater. Today Bio 12, 100154 (2021).
Sundararajan, M., Taly, A. & Yan, Q. Axiomatic attribution for deep networks. International conference on machine learning. PMLR. 70, 3319–3328 (2017).
Xiao, Y. et al. Microenvironment-Responsive Prodrug-Induced Pyroptosis Boosts Cancer Immunotherapy. Adv. Sci. 8, e2101840 (2021).
Guo, J., Yu, Z., Das, M. & Huang, L. Nano codelivery of oxaliplatin and folinic acid achieves synergistic chemo-immunotherapy with 5-fluorouracil for colorectal cancer and liver metastasis. ACS Nano. 14, 5075–5089 (2020).
Zhuang, J. et al. Targeted gene silencing in vivo by platelet membrane-coated metal-organic framework nanoparticles. Sci. Adv. 6, eaaz6108 (2020).
Jiang, Q. et al. Platelet membrane-camouflaged magnetic nanoparticles for ferroptosis-enhanced cancer immunotherapy. Small 16, e2001704 (2020).
Hu, Q. et al. Engineered nanoplatelets for enhanced treatment of multiple myeloma and thrombus. Adv. Mater. 28, 9573–9580 (2016).
Liu, Y. et al. Erythrocyte-platelet hybrid membranes coating polypyrrol nanoparticles for enhanced delivery and photothermal therapy. J. Mater. Chem. B. 6, 7033–7041 (2018).
Bertheloot, D., Latz, E. & Franklin, B. S. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol. Immunol. 18, 1106–1121 (2021).
Kataoka, T. et al. Bcl-rambo, a novel Bcl-2 homologue that induces apoptosis via its unique C-terminal extension. J. Biol. Chem. 276, 19548–19554 (2001).
Meng, F. et al. BCL2L13: physiological and pathological meanings. Cell Mol. Life Sci. 78, 2419–2428 (2021).
Jia, Y. et al. Metformin protects against intestinal ischemia-reperfusion injury and cell pyroptosis via TXNIP-NLRP3-GSDMD pathway. Redox Biol. 32, 101534 (2020).
Chen, H., Peng, Y., Wang, L. & Wang, X. Sevoflurane attenuates cognitive dysfunction and NLRP3-dependent caspase-1/11-GSDMD pathway-mediated pyroptosis in the hippocampus via upregulation of SIRT1 in a sepsis model. Arch. Physiol. Biochem 128, 1413–1420 (2022).
Orzalli, M. H. et al. Virus-mediated inactivation of anti-apoptotic Bcl-2 family members promotes Gasdermin-E-dependent pyroptosis in barrier epithelial cells. Immunity 54, 1447–1462 e1445 (2021).
Nielsen, K. V. et al. Aberrations of ERBB2 and TOP2A genes in breast cancer. Mol. Oncol. 4, 161–168 (2010).
Cruz-Lopez, O. et al. Design, synthesis, HER2 inhibition and anticancer evaluation of new substituted 1,5-dihydro-4,1-benzoxazepines. J. Enzym. Inhib. Med Chem. 36, 1553–1563 (2021).
Lee, H. Y., Nga, H. T., Tian, J. & Yi, H. S. Mitochondrial metabolic signatures in hepatocellular carcinoma. Cells 10, 1901 (2021).
Yang, J., Chen, Z., Liu, N. & Chen, Y. Ribosomal protein L10 in mitochondria serves as a regulator for ROS level in pancreatic cancer cells. Redox Biol. 19, 158–165 (2018).
Pecoraro, A., Pagano, M., Russo, G. & Russo, A. Ribosome biogenesis and cancer: overview on ribosomal proteins. Int J. Mol. Sci. 22, 5496 (2021).
Robinson, K. S. et al. FL. ZAKα-driven ribotoxic stress response activates the human NLRP1 inflammasome. Science 377, 328–335 (2022).
Kang, J. et al. Ribosomal proteins and human diseases: molecular mechanisms and targeted therapy. Signal Transduct. Target Ther. 6, 323 (2021).
Wu, J. et al. PHA-4/FoxA senses nucleolar stress to regulate lipid accumulation in Caenorhabditis elegans. Nat. Commun. 9, 1195 (2018).
Murayama, A. et al. Epigenetic control of rDNA loci in response to intracellular energy status. Cell 133, 627–639 (2008).
Chen, Y. et al. Proteolysis-targeting chimera (PROTAC) delivery system: advancing protein degraders towards clinical translation. Chem. Soc. Rev. 51, 5330–5350 (2022).
Chen, R., Kang, R. & Tang, D. The mechanism of HMGB1 secretion and release. Exp. Mol. Med. 54, 91–102 (2022).
Wan, L., Pantel, K. & Kang, Y. Tumor metastasis: moving new biological insights into the clinic. Nat. Med. 19, 1450–1464 (2013).
Gradishar, W. J. et al. Breast cancer, Version 3.2020, NCCN clinical practice guidelines in oncology. J. Natl Compr. Canc Netw. 18, 452–478 (2020).
Schmid, P. et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 21, 44–59 (2020).
Zhang, Z. et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579, 415–420 (2020).
Liu, Y. et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci. Immunol. 5, eaax7969 (2020).
Yang, F., Bettadapura, S. N., Smeltzer, M. S., Zhu, H. & Wang, S. Pyroptosis and pyroptosis-inducing cancer drugs. Acta Pharmacologica Sin. 43, 2462–2473 (2022).
Su, X. et al. A Carbonic Anhydrase IX (CAIX)-Anchored Rhenium(I) Photosensitizer Evokes Pyroptosis for Enhanced Anti-Tumor Immunity. Angew. Chem. Int. Ed. Engl. 61, e202115800 (2021).
Xie, C. et al. Amelioration of Alzheimer’s disease pathology by mitophagy inducers identified via machine learning and a cross-species workflow. Nat. Biomed. Eng. 6, 76–93 (2022).
Da Silva, C. G., Rueda, F., Lowik, C. W., Ossendorp, F. & Cruz, L. J. Combinatorial prospects of nano-targeted chemoimmunotherapy. Biomaterials 83, 308–320 (2016).
Chen, Y. et al. Tailored chemodynamic nanomedicine improves pancreatic cancer treatment via controllable damaging neoplastic cells and reprogramming tumor microenvironment. Nano Lett. 20, 6780–6790 (2020).
Tang, B. et al. Integrative analysis of the molecular mechanisms, immunological features and immunotherapy response of ferroptosis regulators across 33 cancer types. Int J. Biol. Sci. 18, 180–198 (2022).
Daina, A., Michielin, O. & Zoete, V. SwissTargetPrediction: updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res 47, W357–W364 (2019).
Ma, J. et al. Using deep learning to model the hierarchical structure and function of a cell. Nat. Methods 15, 290–298 (2018).
Elmarakeby, H. A. et al. Biologically informed deep neural network for prostate cancer discovery. Nature 598, 348–352 (2021).
Steck, H., et al. In advances in neural information processing systems 20. 21st Annual Conference on Neural Information Processing Systems (2008).
Chou, T. C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharm. Rev. 58, 621–681 (2006).
Gessulat, S. et al. Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods 16, 509–518 (2019).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: A Hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, 543–552 (2022).
Acknowledgements
The authors deeply thank Meigui Zhu and the teamwork (Bionovogene Co., Ltd., Suzhou, China) for proteomics characterization. The authors also thank Shiyanjia Lab (www.shiyanjia.com) for the TEM analysis. This work was supported by the National Science Foundation of China (82172073, 51933002, 8217070298), the National Key R&D Program of China (2022YFF0708700), the Guangdong Basic and Applied Basic Research Foundation (2021A1515220011), the Program of the Shanghai Academic Research Leader (20XD1400400). This work was partially funded by Microsoft Research Asia.
Author information
Authors and Affiliations
Contributions
B.O. and S.S. designed the experiments. B.O. and C.S. performed the experiments and collected the data. X.D., Q.C., W.Y., T.L., and L.S. provided equipment and technical support. X.S., Y.C., Y.H., S.W., X.Q., R.X., and R.H. assisted in the experimental validation. All authors provided adequate support in writing the manuscript. J.Z. and S.P. gave guidance in data analysis. Z.P., D.L., and J.W. guided in the conceptualization and methodological part of this research. All authors gave their approval to the final manuscript version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jinyao Liu, Xiaochen Bo and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ouyang, B., Shan, C., Shen, S. et al. AI-powered omics-based drug pair discovery for pyroptosis therapy targeting triple-negative breast cancer. Nat Commun 15, 7560 (2024). https://doi.org/10.1038/s41467-024-51980-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51980-9
This article is cited by
-
Cancer chemoprevention: signaling pathways and strategic approaches
Signal Transduction and Targeted Therapy (2025)
-
RETRACTED ARTICLE: Genetic inference and single cell expression analysis of potential targets in heart failure and breast cancer
Journal of Cancer Research and Clinical Oncology (2024)
