
Abstract
Influenza viruses cause substantial morbidity and mortality every year despite seasonal vaccination. mRNA-based vaccines have the potential to elicit more protective immune responses, but for maximal breadth and durability, it is desirable to deliver both the viral hemagglutinin and neuraminidase glycoproteins. Delivering multiple antigens individually, however, complicates manufacturing and increases cost, thus it would be beneficial to express both proteins from a single mRNA. Here, we develop an mRNA genetic configuration that allows the simultaneous expression of unmodified, full-length NA and HA proteins from a single open reading frame. We apply this approach to glycoproteins from contemporary influenza A and B viruses and, after vaccination, observe high levels of functional antibodies and protection from disease in female mouse and male ferret challenge models. This approach may further efforts to utilize mRNA technology to improve seasonal vaccine efficacy by efficiently delivering multiple viral antigens simultaneously and in their native state.
Similar content being viewed by others


Introduction
Seasonal influenza virus infection causes significant disease and economic burden globally every year. In the 2022–23 season, it was estimated there were over 30 million cases in the United States alone, and worldwide there can be between 250,000 and 500,000 deaths from influenza illness in a given season1,2. Seasonal influenza viruses also contribute to a significant economic burden of an estimated $25 billion USD in a typical flu season3. One of the main tools in battling seasonal influenza burden are vaccines. These vaccines typically induce a highly focused response to antigenic domains of the viral hemagglutinin (HA) and to a lesser extent, the viral neuraminidase (NA)4,5,6. The HA-directed antibodies elicited typically block host receptor engagement and thus, mediate virus neutralization7,8,9. The strong effect of these antibodies, however, places a significant selective pressure on antigenic sites in the head ___domain of HA, leading to rapid viral antigenic drift10,11,12. These changes necessitate seasonal influenza vaccine reformulation every year to target three or four virus strains that are expected to be circulating, typically an H1N1 and an H3N2 Influenza A virus (IAV) and a Victoria-lineage and a Yamagata-lineage Influenza B virus (IBV).
While subunit, recombinant protein, and live-attenuated FDA-approved influenza vaccines exist, the most widely administered seasonal influenza vaccines consist of influenza viral particles that are grown to scale in eggs then inactivated with detergents13. This process ‘splits’ the viral particles and subsequently exposes the immune system to multiple antigenic viral proteins, such as NA and HA, upon vaccination. Split inactivated vaccines are normalized only on HA content, leaving the amount and integrity of the other major glycoprotein, NA, uncertain. Indeed, it has been shown that NA-specific human antibodies poorly recognize NA within split inactivated vaccines and the NA-directed vaccine responses themselves can be highly variable14,15. Additionally, relatively long manufacturing pipelines necessitate 6–8 months between strain selection and vaccine administration, allowing time for mutations to arise in circulating strains that change the antigenicity relative to vaccine strains16. Egg-adapted mutations acquired by vaccine strains during production can also significantly alter antigenicity. These issues can lead to extreme examples where the vaccines end up being ineffective against certain subtypes of influenza viruses, shown recently in the 2014–15, 2016–17 and 2017–18 influenza seasons13,16,17,18,19,20,21. Even under more normal conditions, seasonal influenza vaccines still elicit only ~20–60% vaccine efficacy22. mRNA-based vaccines may represent a practical path to improved influenza vaccines. These vaccines consist of mRNA transcripts that encode for a particular antigen, encapsulated in lipid nanoparticles (LNPs). They can mediate extremely high vaccine efficacy rates (>90% for COVID-19) through induction of both robust humoral and cellular immunity23,24,25,26,27,28,29,30. Since they do not produce infectious viruses, these vaccines are considered safe to administer in immunocompromised and elderly individuals as well as children31,32,33. To make mRNA-LNPs, researchers only require the sequence of the intended antigen and then corresponding oligonucleotides can be synthesized without heavy reliance on external production factors such as egg availability. This relatively simple production process allows for more rapid updates in the event of viral mutations or changes in circulating strain prevalence34. The question of what influenza virus antigen(s) mRNA vaccine formulations should include to maximize vaccine-mediated protection, however, remains an open area of debate.
While the inclusion of an HA protein as an antigen is a forgone conclusion as vaccination with HA is essential for generating neutralizing antibodies, the inclusion of neuraminidase in the vaccine may also be beneficial as NA-directed responses have been shown to boost the breadth of protectivity and limit disease14,15,35. NA-directed antibodies have been shown to be important controllers of infection that limit disease in animal models5,36,37. Additionally, NA is a more conserved viral protein that accumulates slower antigenic drift than HA, therefore broadly reactive NA antibodies have been shown to confer cross-reaction protection both within and across NA subtypes38,39,40,41,42. Thus, it may be that the simultaneous delivery of multiple viral glycoproteins (as per natural infection), could allow for maximal vaccine-induced protection. Indeed, vaccination with non-HA or multiple influenza antigens via mRNA-LNPs is currently being investigated by numerous groups and companies43,44,45,46,47,48,49,50,51. For mRNA approaches delivering multiple antigens, viral proteins are often encoded in separate mRNA-LNPs, which are then mixed prior to vaccination. This method of vaccine production and delivery, while at least partially effective in eliciting antibody responses, is more complex, costly, and cannot control which transfected cells express which glycoprotein and fails to control the relative expression of viral proteins in cells that express both. This lack of coordinated co-expression of the glycoproteins ultimately fails to reconstitute the biological landscape of an infected cell and limits meaningful biological interactions between NA and HA52,53,54,55. While expressing multiple antigens from a single mRNA could potentially solve many of the problems of co-delivery of multiple antigens, a technical solution for our goal of creating such an mRNA vaccine, and one that would also more closely recapitulate an infected cell, remained unclear.
In this work, we develop an mRNA vaccine platform that delivers NA and HA as a single open reading frame (ORF) to enable expression of multiple unmodified antigens from a single mRNA, using a unique glycoprotein organization with an artificial furin cleavage site and 2A ribosome-skipping sequences. We find that this design is able to induce robust immune responses to both antigens and at levels consistent with vaccination of mRNA-LNPs encoding either viral protein alone. Furthermore, we show that this technology can be used to deliver the NA and HA of four different influenza viruses as a quadrivalent mRNA vaccine that delivers eight distinct antigens while only using four mRNA encapsulated in individual LNPs. In a single dose, this quadrivalent mRNA vaccine is effective and in some cases superior to a current inactivated industry standard vaccine comparator in immunogenicity for all vaccine strains in both mouse and ferret models. We find that our vaccine induces antibodies targeting not only the conserved stalk region of vaccine strain HAs, but also to antigenically diverse group 1 and 2 HAs, suggesting broader immunity against diverse influenza virus strains. Together, these data present the strategy suggested here as an effective and efficient way to improve current seasonal as well as broadly protective influenza vaccine strategies.
Results
NA and HA can be efficiently expressed from a single ORF
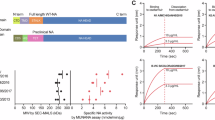
Our goal was to express completely native HA and NA glycoproteins from a single mRNA. While viral 2A sites have been widely utilized to facilitate expression of multiple individual proteins from the same transcript, they leave residual amino acids on the termini of the separated proteins56,57. It is known that some furin cleavage sequences (such as RKRR) preceeding a porcine teschovirus 2A (PTV-2A) site allow the furin protease to remove most of the residual PTV-2A amino acids, and carboxypeptidases can then eliminate the residual furin cleavage motif58. We previously demonstrated that we could adapt this approach to influenza viral glycoproteins in the context of an infectious virus by encoding NA followed by HA59. We therefore hypothesized that a similar approach could be used to generate unmodified NA and HA proteins in the context of an mRNA vaccine (Fig. 1a). To test this hypothesis, we first designed DNA plasmids that encoded for either the expression of NA, HA, or both proteins through the NA-F2A-HA construct using viral sequences from the contemporary H3N2 virus, A/Tasmania/503/2020, which was a component of the 2021–22 seasonal influenza vaccine. After validating that we had the proper tools to detect the A/Tasmania/503/2020 viral proteins (Supplementary Fig. 1A–D), we used cell-based ELISAs and microscopy to demonstrate that NA and HA were expressed from cells transfected with the NA-F2A-HA plasmid to similar levels as those transfected with plasmids encoding either antigen alone or as a mixture (Fig. 1b–e). Similarly, confocal microscopy revealed proper expression and trafficking of each of these proteins to the surface of cells transfected with our NA-F2A-HA plasmid (Fig. 1f). We also wanted to confirm that the NA and HA expressed from our NA-F2A-HA plasmid were functional. NA expressed from our NA-F2A-HA construct was found to mediate sialic acid removal in a lectin-based activity assay to the same degree as NA individually expressed from a plasmid or in combination with an HA-expressing plasmid (Fig. 1g). Similarily, HA expressed from the NA-F2A-HA plasmid was found to mediate similar membrane fusion activity as HA individually expressed from a plasmid or in combination with an NA-expressing plasmid under low pH conditions (Fig. 1h). These results suggested that indeed, the NA-F2A-HA design could be applied to vaccine design to express two functional, native viral proteins from a single ORF.

a Diagram of NA-F2A-HA expression vector. Open reading frame, ORF; furin protease, F; carboxypeptidase, CP; signal peptide peptidase, SPP. Created in BioRender. Leonard, R. (2024) BioRender.com/d14z470. b Cell-based ELISAs against 293T cells transfected with the indicated plasmids and stained with dilutions of an N2 polyclonal antibody. c Area under the curve-analysis (AUC) of 4 independent experiments represented in b. Arb. Units=Arbitrary Units. d Cell-based ELISAs against 293T cells transfected with the indicated plasmids and stained with an H3 HA-specific monoclonal antibody, 9H10. e AUC analysis of 4 independent experiments represented in d. f Confocal microscopy of cells transfected with the indicated plasmids, stained with Hoescht 33342 (blue), N2 polyclonal antibody (red), and H3 monoclonal antibody, 9H10 (green). g NA activity detected by ELLA assay using 293Ts transfected with plasmids expressing the indicated genes. h Confocal microscopy of HA fusion assay in 293Ts 24 h post transfection, stained with Hoescht 33342 (blue) and the fluorescent lectin, WGA (green). Panels b and d are representative of four independent experiments, and panels c, e, and g include means from all four independent experiments. For panels f and h, images were taken 24 h post transfection and are representative of two independent experiments. All scale bars = 30 μm. For all panels including statistics, significance was determined using Kruskal–Wallis test followed by pairwise Wilcoxon rank-sum tests with a Benjamini–Hochberg FDR correction. FDR-corrected p-values are reported above sample groups. Data are shown as means ± standard error of mean (SEM). Source data are provided as a Source Data file.
NA-F2A-HA mRNA-LNP is immunogenic in mice
We next wanted to investigate the immune response that an mRNA-LNP with this design would elicit compared to vaccination with mRNA-LNPs encoding either glycoprotein alone. Therefore, we generated mRNA-LNPs that encoded for either the NA, HA, or the NA-F2A-HA proteins. The LNP carriers themselves have been shown to act as strong adjuvants, therefore no additional adjuvants were included60. These mRNA-LNPs were delivered to mice, as well as control groups that included an mRNA-LNP encoding luciferase, a group where the individual HA and NA mRNA-LNPs were mixed prior to vaccination, as well as the 2021–22 split inactivated Flulaval vaccine and a matched bovine serum albumin (BSA) control (Fig. 2a). For the mRNA vaccines, equal moles of RNA were delivered, and the dose given was scaled to 1/6 of a human dose for both the mRNA-LNPs and Flulaval vaccine. Post-vaccination sera revealed that the mRNA-LNP vaccines that encoded for NA or HA (as well as the Flulaval control) elicited robust antibody responses against the targeted A/Tasmania/503/2020 protein (Fig. 2b–e). Importantly, mice vaccinated with the NA-F2A-HA mRNA-LNP vaccine elicited a similar, or in the case of HA, better, antibody response to both viral proteins than mice vaccinated with mRNA-LNPs expressing either antigen alone or when the NA mRNA-LNP and HA mRNA-LNP vaccines were mixed (Fig. 2b–e). Thus, although we didn’t monitor protein expression from this mRNA in vivo, it is likely that both HA and NA were appropriately expressed as per our in vitro DNA-based expression experiments.

a Schematic of vaccine groups and vaccination regimen. Created in BioRender. (Heaton, N. (2024) BioRender.com/l47n232). b Cell-based ELISAs against 293T cells transfected with an A/Tasmania/503/2020 NA expression plasmid. c AUC analysis for data shown in b. d Cell-based ELISAs against 293T cells transfected with an A/Tasmania/503/2020 HA expression plasmid. e AUC analysis for data shown in d. f HAI assays performed with the indicated virus. Values below the limit of detection (LOD) (10 HAI units, dotted line) were set to 6 HAI units for inclusion in the plot. g ELLA assays performed with an active N2 enzyme. Values below the LOD (10 units, dotted line) were set to 5 for inclusion in the plot. h MN50 assays using the indicated virus. Values below LOD (1, dotted line) were set to 0.5 for inclusion in the plot. i PRNT50 assays using the indicated virus in MDCK cells. Any values under the LOD (ULD) (100, dotted line) are shown as 50 for inclusion in the plot. j Schematic of vaccine groups and challenge layout. Created in BioRender. (Heaton, N. (2024) BioRender.com/l47n232). k Change in bodyweight over 14 days following lethal challenge with the indicated virus. Dashed line indicates human endpoint, loss of greater than 25% of starting bodyweight. l Percent survival of vaccinated mice following challenge with the indicated virus. If only part of the group survived, that number is indicated in parentheses. For panels f–i, line shown is the mean. For all experiments, n = 5 mice per group and experiments using sera were all performed with sera taken 21 days post vaccination. Mice were vaccinated with doses as follows: BSA, 10 μg; luciferase mRNA-LNPs, 5.1 μg; NA mRNA-LNPs, 2.275 μg; HA mRNA-LNPs, 2.725 μg; NA mRNA-LNP, 2.275 and HA mRNA-LNPs, 2.725 μg; NA-F2A-HA mRNA-LNPs 5.1 μg; Flulaval, 10 μg. For all panels, data are representative of two independent experiments, and are shown as means ± SEM. For all panels including statistics, significance was determined using Kruskal–Wallis test followed by pairwise Wilcoxon rank-sum tests with a Benjamini–Hochberg FDR correction. FDR-corrected p-values are reported above sample groups. Source data are provided as a Source Data file.
Next, we performed a series of experiments to assess if the antibodies elicited were functional. Indeed, the HA mRNA-LNPs, NA + HA mixture mRNA-LNPs, NA-F2A-HA mRNA-LNPs, and Flulaval vaccines elicited hemagglutinin-specific neutralizing antibodies (Fig. 2f), and the NA mRNA-LNP, NA + HA mixture mRNA-LNPs, and NA-F2A-HA mRNA-LNP vaccines also elicited antibodies with detectible neuraminidase inhibition activity above the background level of naïve mouse sera (Fig. 2g). We also performed multicycle microneutralization assays (MN50) and plaque-reduction assays (PRNT50). These assays, which principally measure the activity of HA-directed antibody responses, both revealed that the HA mRNA-LNP, NA + HA mixture mRNA-LNPs, and NA-F2A-HA mRNA-LNP vaccines elicited equally higher levels of neutralizing antibodies than other groups (Fig. 2h, i). Notably, each of these assays showed noninferiority of delivering both antigens through our NA-F2A-HA mRNA construct compared to vaccinating with each antigen alone or via a mixture of HA and NA mRNA-LNPs. Finally, we challenged vaccinated mice with a lethal dose of a mouse-adapted antigenically similar H3N2 virus (A/Switzerland/9175293/2013) to assess protection from disease (Fig. 2j). Luciferase mRNA-LNP-, BSA-, and Flulaval-vaccinated controls experienced severe weight loss and generally died by day 8 post-infection, with some protection mediated by Flulaval where 3/5 vaccinated mice were able to recover from infection. Mice vaccinated with NA mRNA-LNPs, HA mRNA-LNPs, NA + HA mixture mRNA-LNPs, and NA-F2A-HA mRNA-LNPs all survived infection and displayed marked bodyweight loss (Fig. 2k, l). Therefore, the NA-F2A-HA mRNA-LNP vaccine elicited an immune response against both NA and HA similar to that generated following vaccination with mRNA-LNPs encoding for either antigen alone or mixed. Further, vaccination with our NA-F2A-HA mRNA-LNP vaccine is protective against lethal challenge and, at least in this model of disease, provides improved protection relative to current industry standard vaccines.
NA-F2A-HA mRNA-LNP vaccines targeting non-H3N2 viral strains are immunogenic in mice
We next wanted to see if we could apply the NA-F2A-HA mRNA design to other strains of influenza A and influenza B viruses. We therefore generated NA-F2A-HA mRNA-LNP vaccines targeting the remaining three strains included in the 2021–22 influenza vaccine: H1N1 A/Victoria/2570/2019, IBV_V B/Washington/02/2019 and IBV_Y B/Phuket/3073/2013. These vaccines were then independently administered to mice as per our previous experiments with the H3N2-based vaccine (Fig. 3a). Analysis of the post-vaccination sera revealed that the NA-F2A-HA mRNA-LNP vaccines encoding for the H1N1 antigens (Fig. 3b–e), the IBV_V antigens (Fig. 3g–j), and IBV_Y antigens (Fig. 3l–o) all elicited significantly higher antibody responses against their respective NAs and HAs compared to luciferase mRNA-LNPs and Flulaval. Further, these antibodies were found to be neutralizing against antigenically matched (in the case of IBV_Y) or with as antigenically similar viruses as we could acquire and culture in the lab, A/Hawaii/70/2019 for the H1N1 and B/Colorado/06/2017 for the IBV_V (Fig. 3f, k, p).

a Schematic showing vaccine design of H1N1, IBV-Victoria and IBV-Yamagata NA-F2A-HA mRNA-LNPs. Created in BioRender. (Heaton, N. (2024) BioRender.com/h60o989). b Cell-based ELISAs against 293Ts transfected with the indicated NA expression plasmid. c AUC analysis of data in b. d Cell-based ELISAs against 293Ts transfected with the indicated HA expression plasmid. e AUC analysis of data in d. f PRNT50 assays using the indicated virus in MDCK cells. g Cell-based ELISAs against 293Ts transfected with the indicated NA expression vector. h AUC analysis of data in g. i Cell-based ELISAs against 293Ts transfected with the indicated HA expression plasmid. j AUC analysis of data in i. k PRNT50 assay using the indicated virus in MDCK cells. l Cell-based ELISAs against 293Ts transfected with the indicated NA expression plasmid. m AUC analysis of data in l. n Cell-based ELISAs against 293Ts transfected with the indicated HA expression plasmid. o AUC analysis of data in n. p PRNT50 assays using the indicated virus in MDCK cells. For panels f, k, and p, any values under the LOD (ULD) (100, dotted line) are shown as 50 for inclusion in the plot. For all experiments, n = 5 mice per group and experiments using sera were all performed with sera taken 21 days post vaccination. mRNA-LNP vaccinated mice all received 5 μg of their respective vaccine, and Flulaval vaccinated mice received 10 μg of a Flulaval dose. For all panels, data are representative of two independent experiments, and shown as means ± SEM. For all panels including statistics, significance was determined using Kruskal–Wallis test followed by pairwise Wilcoxon rank-sum tests with a Benjamini–Hochberg FDR correction. FDR-corrected p-values are reported above sample groups. Source data are provided as a Source Data file.
A four NA-F2A-HA mRNA-LNP mixture is immunogenic and provides protection from H1N1, H3N2, and IBV-Yamagata challenge
Once we confirmed that NA-F2A-HA mRNA-LNP vaccines encoding for the NAs and HAs of four distinct influenza viruses generated strong immune responses when delivered individually, we next wanted to test if we could protect against all four strains of influenza virus with a single four mRNA-LNP, but “octavalent” vaccination. Mice were vaccinated with a mixture of equal parts of the NA-F2A-HA mRNA-LNP vaccines targeting either the H1N1, H3N2, IBV-Y or IBV-V strains of influenza virus at half the dosage as when they were administered individually (Fig. 4a). This ‘four mRNA-LNP’ vaccine was found to generate strong antibody responses against the NAs and HAs of all four vaccine strains, non-inferiorly to those elicited following Flulaval vaccination (Fig. 4b–e). To test if our vaccine was able to boost pre-existing immunity similar to what would be found in a human population, we performed a study in which pre-immunity was first established via either BSA control or one of two “pre-immune mixes”. “Pre-immune mix 1” consisted of inactivated A/California/04/2009, A/Wyoming/03/2003, B/Malaysia/2506/2004, B/Yamagata/16/1988 viral particles, and “Pre-immune mix 2” was Flulaval. Once these different pre-immune profiles were established (Supplementary Fig. 2A, B), we then vaccinated mice with the four mRNA-LNP vaccine and found that our vaccine was able to successfully boost anti-NA and anti-HA antibody levels in both pre-existing immune backgrounds (Supplementary Fig. 2C–F).

a Schematic of vaccination and challenge experiment. Created in BioRender. (Heaton, N. (2024) BioRender.com/k48b864). b–e AUC analysis using cell-based ELISAs against 293Ts transfected with the NA or HA for the indicated virus strains. f–i PRNT50 assays using the indicated viruses in MDCK cells. Any values under the LOD (ULD) (100, dotted line) are shown as 50 for inclusion in the plot. j–l Change in bodyweight through 14 days following lethal challenge with the indicated viruses. Dashed line indicates human endpoint, loss of greater than 25% of starting bodyweight. m–o Percent survival of vaccinated mice following challenge with the indicated viruses. For all experiments, n = 5 mice per group and experiments using sera were all performed with sera taken 21 days post vaccination. mRNA-LNP vaccinated mice all received 10 μg total of their respective vaccine (2.5 μg of each H1N1, H3N2, IBV-V and IBV-Y NA-F2A-HA mRNA-LNPs for ‘four mRNA-LNP’ group), and Flulaval vaccinated mice received 10 μg of a Flulaval dose. For all panels, data are representative of two independent experiments, and are shown as means ± SEM. For all panels including statistics, significance was determined using Kruskal–Wallis test followed by pairwise Wilcoxon rank-sum tests with a Benjamini–Hochberg FDR correction. FDR-corrected p-values are reported above sample groups. Source data are provided as a Source Data file.
To assess the functional neutralization capability of these antibodies, we performed PRNT50 assays (using the same viruses as in the separate vaccination experiments) and found that the four mRNA-LNP vaccine elicited significantly higher levels of neutralizing H1N1, H3N2, IBV_V and IBV_Y antibodies compared to the luciferase mRNA-LNP controls and generally higher responses than animals that received Flulaval (Fig. 4f–i). Finally, we wanted to determine if the four mRNA-LNP vaccine was protective against lethal challenge. Although we were unable to identify a contemporary IBV_V that caused disease in mice, we challenged the quadrivalent vaccine group (and controls) with a contemporary H1N1 (A/Hawaii/70/2019) (Fig. 4j), H3N2 (A/Switzerland/9175293/2013) virus (Fig. 4k), or matched IBV_Y virus (B/Phuket/3073/2013) (Fig. 4l). All mice vaccinated with the four mRNA-LNP or Flulaval were found to be completely protected against lethal challenge with all three viruses, while luciferase mRNA-LNP-vaccinated mice died between days 7–10 post infection (Fig. 4m–o). We also noted a trend for less bodyweight loss in the four mRNA-LNP-vaccinated group relative to the Flulaval group after the H3N2 and IBV_Y viral challenges.
The four NA-F2A-HA mRNA-LNP vaccine mixture elicits protective immunity in ferrets
Finally, we wanted to confirm that our four mRNA-LNP vaccine would behave similarly in a ferret model to what we had previously observed in mice. We therefore vaccinated ferrets with either luciferase mRNA-LNP, our four mRNA-LNP vaccine, or Flulaval (Fig. 5a). As expected, animals vaccinated with our four mRNA-LNP vaccine elicited robust antibody responses against all four vaccine strain antigens (Fig. 5b–e), with higher responses relative to Flulaval for H1N1, H3N2, and IBV_Y antigens and similar levels as Flulaval for IBV_V antigens. We also found that our four mRNA-LNP vaccine elicited significantly higher levels of neutralizing antibodies post-vaccination, and similar levels compared to Flulaval-vaccinated animals against all four influenza strains (Fig. 5f–i). We then challenged with the contemporary H1N1 strain, A/Hawaii/66/2019 (98.41% A/Hawaii/70/2019 HA sequence homology to A/Victoria/2570/2019 HA) (Fig. 5a). While ferrets are good models of the human clinical course of disease during influenza virus infection, their susceptibility to contemporary influenza viruses is variable61. As expected, we saw little change in bodyweight over the course of infection between groups (Fig. 5j) but were able to see a modest decrease in viral RNA from four mRNA-LNP-vaccinated animals at days 2, 4, and 7 of infection (Fig. 5k). Importantly, viral titer determined from oral swabs indicated that infection was less likely to be established in our four mRNA-LNP vaccinated ferrets, and if they were infected, they were able to clear the virus more efficiently than luciferase mRNA-LNP- or Flulaval-vaccinated ferrets (Fig. 5l). Thus, despite being a sub-optimal challenge model for this contemporary H1N1 strain, the ferret challenge data is consistent with the antibody analyses showing enhanced vaccine responses in the four mRNA-LNP vaccine group.

a Schematic of vaccination and challenge experiment. Created in BioRender. (Heaton, N. (2024) BioRender.com/k70m759). b–e AUC analysis using cell-based ELISAs against 293Ts transfected with the NA or HA for the indicated virus strains. ELISAs are shown as net change in antibody reactivity between pre-vaccination sera (d-2) to post-vaccination (d28) sera. f–i PRNT50 assays using the indicated viruses in MDCK cells. Any values under the LOD (ULD) (100, dotted line) are shown as 50 for inclusion in the plot. Any values over the LOD (OLD) (3200) are shown as 4000 for inclusion in the plot. j Change in bodyweight over 14 days post viral challenge. Dotted line = 0. k qRT-PCR on oral swab samples taken from ferrets at −3-, 2-, 4- or 7-days post infection. l Viral load determined from plaque assays using oral swabs taken from ferrets at −3, 2-, 4-, or 7-days post infection. Any values not detected (LOD = 10 PFU/mL, dotted line) are shown as 5 for inclusion in the plot. m, n Cell-based ELISAs (left) and corresponding AUC analysis (right) against 293Ts transfected with the indicated HA stalk expression plasmid. AUC analysis is shown as net change in antibody reactivity between pre- (dotted line) and post-vaccination (solid line) ferret sera. o Luminex binding assay of group 1 HAs (left), and group 2 HAs (right). Comparisons to the right of each panel are between the four mRNA-LNP- and Flulaval-vaccinated groups of animals. For all panels, n = 4 ferrets per experimental group and the experiment was performed once. mRNA-LNP vaccinated ferrets received 30 μg total of their respective vaccine (7.5 μg of each H1N1, H3N2, IBV-V and IBV-Y NA-F2A-HA LNPs for ‘four mRNA-LNP’ group), and Flulaval vaccinated ferrets received a full 60 μg of a Flulaval dose. Data are shown as means ± SEM. For all panels including statistics, significance was determined using Kruskal–Wallis test followed by pairwise Wilcoxon rank-sum tests with a Benjamini–Hochberg FDR correction. FDR-corrected p-values are reported above sample groups. Source data are provided as a Source Data file.
It has been previously suggested that mRNA-LNP vaccination itself can elicit higher levels of antibodies that recognize the conserved stalk of HA, which can increase protection through non-neutralizing mechanisms50,62,63. Using this knowledge, taken in combination with our findings that four mRNA-LNP-vaccinated ferrets had increased antibody binding but similar viral neutralization activities relative to Flulaval-vaccinated ferrets, we wanted to test if our four mRNA-LNP vaccine elicited non-neutralizing antibodies against epitopes other than the HA head. We therefore performed cell-based ELISAs against the stalk ___domain of the vaccine-matched H1 and H3 HAs using headless, stalk-only HA expression plasmids that were previously validated using conformation-specific antibodies (Supplementary Fig. 3). Interestingly, we found that indeed, our four mRNA-LNP vaccine elicited higher levels of HA stalk-specific antibodies than luciferase mRNA-LNPs or Flulaval in ferrets (Fig. 5m–n). To test if this enhanced antibody response to a more conserved HA ___domain would allow for increased binding to heterologous HAs, we performed a Luminex-based purified HA binding assay using group 1 and 2 HAs from 1933-2021. Within group 1 HAs (Fig. 5o, left) we found that our four mRNA-LNP vaccine elicited higher magnitude responses compared to Flulaval to both older and contemporary H1s and, to a lesser degree, the H2, H5, H6, and H9 HAs. Within the group 2 HAs (Fig. 5o, right), our vaccine elicited similar antibody responses as Flulaval to older H3 HAs, but significantly higher responses to more contemporary H3 and H7 HAs. These findings suggest that our four mRNA-LNP vaccine elicits not only a strong response against vaccine-matched strains, but also a generally more cross-reactive antibody profile against heterologous viruses.
Discussion
While inactivated seasonal influenza vaccines elicit protective immunogenicity, their overall efficacy is limited13,16,18,19,20,21,22. Advancements in mRNA-based technologies in the recent years now make this a practical vaccine modality; however, questions remain regarding the antigen composition and the best configurations to express those antigens. In this study, we present a method of improving seasonal influenza vaccination using a dual antigen-expressing mRNA-LNP strategy. Using only four mRNA-LNPs, we can express eight individual NA and HA proteins by separating the glycoprotein ORFs with furin and 2A cleavage motifs. This NA-F2A-HA mRNA design can be broadly applied to influenza A and influenza B viral glycoproteins, and individual mRNA-LNPs can be mixed to recapitulate the breadth of a typical quadrivalent, virion-based, seasonal vaccine. Further, the four mRNA-LNP vaccine was found to elicit fully protective, and in some cases superior, immune responses to the NA and HA of all four vaccine strains compared to the current inactivated industry standard vaccine in both mice and ferrets.
In addition to its ability to act as an improved seasonal influenza vaccine, our four mRNA-LNP also demonstrated an ability to induce higher responses to the relatively conserved stalk of the HA protein. This ability was likely responsible for the increased binding we observed after mRNA-LNP vaccination to highly drifted HA variants. Although stalk-specific antibodies are non-neutralizing, they have been shown to confer protection through alternate immune pathways, such as the antibody-dependent cytotoxicity (ADCC) pathway62. While these data are promising, future studies will be needed to demonstrate that this increased binding translates to functional activity. Further work will also be required to define which specific epitopes are being uniquely or disproportionately targeted after mRNA-LNP vaccination. mRNA vaccines have also been shown to induce superior cellular immune responses that are critical in viral control and clearance, i.e., antigen-specific CD8 + T-cell responses64,65,66 and potentially improved mucosal IgA responses. We did not formally measure such responses, however, and future work understanding the full breadth of mechanisms that mediate the protection afforded by our vaccine will be required.
Future studies will also be required to understand the relative contributions and mechanisms of NA-mediated immunity after vaccination. Our results, consistent with previous literature42,67,68,69,70, showed that current inactivated vaccines elicit little NA-specific immunity. It is estimated that inactivated vaccines only elicit ~30% seroconversion against NA, and it is suggested this is due to a lack of normalization of NA amounts or NA epitope disruption during vaccine production14,15,42,67,68,69. Thus, while improved HA-directed responses (both in magnitude and breadth) will almost certainly contribute to improved vaccine-mediated protection, the ability of this vaccine to express NA in stochiometric equivalent to HA may also improve protection via elicitation of strong-NA directed responses. Furthermore, it will also be necessary to dissect any potential impacts of expressing NA and HA together through the NA-F2A-HA construct on the immunogenicity of the viral proteins themselves. It has been suggested that for some antigens, removal of sialic acids specifically can increase their antigenicity71. Thus, in addition to simply expressing HA and NA viral proteins simultaneously, the phenomena of NA removing sialic acid from HA-linked glycans52,53 may also modulate antigenicity of the protein itself. Future studies, however, will be required to determine if this is indeed the case.
While the vaccine approach described in this study appears promising, there are several limitations that should be considered. Despite the NA-F2A-HA construct design halving the number of mRNAs to be made, our mRNAs are longer than those encoding for a single antigen. This might leave them more vulnerable to human RNAses upon vaccination, leading to more rapid degradation in vivo compared to their smaller counterparts. Therefore, studies on mRNA half-life and the length of antigen expression in vivo, as well as the consequences of any changes, will be necessary future steps. Furthermore, in addition to the NA-F2A-HA mRNA-LNP simplifying the manufacturing process by reducing production time and cost, we initially hypothesized that coordinated expression of HA and NA could affect antigenicity of the proteins. We ultimately did not perform any studies to directly test this hypothesis in this work, but additional studies looking at differences in the antibody composition elicited by different vaccines will be important. Finally, while we attempted to model pre-existing influenza immunity in mice, the immune history of humans is much more complicated and nuanced. For this reason, our vaccine will need to be carefully assessed for its ability to boost immune responses in other pre-existing immunity animal models and ultimately in humans.
In conclusion, mRNA vaccines have the potential to eliminate many of the limitations that plague current vaccine platforms. By incorporating multiple antigens into a single formulation, our vaccine offers a promising strategy to improve vaccine effectiveness without increasing manufacturing complexity. Moving forward, application of the advances in mRNA technology such as those that we propose here, will allow for improved influenza vaccines which will ultimately help to mitigate the burden of influenza globally. Outside of influenza vaccines, our approach for delivering multiple antigens at once can potentially be applied to other fields of mRNA delivery research, such as for cancer, rare genetic disorder therapies, and other infectious diseases.
Methods
In vitro transcription and mRNA-LNP production
The HA and NA constructs were codon optimized to enhance protein expression and reduce immunogenicity. The codon optimized sequences were gene synthetized at GenScript (New Jersey, USA) and cloned into an in vitro transcription template plasmid containing a T7 promoter, 5′ and 3′UTR regions, and a 100-nucleotide poly (A) tail. mRNA was synthetized and co-transcriptionally capped using the Megascript transcription kit™ (ThermoFisher, Cat# AMB 1334) and the CleanCap™ dinucleotide system (TriLink Biotechnologies), precipitated, and purified using a modified cellulose based chromatography method72. Length and mRNA integrity were assessed using the native agarose gel (1.4%). Removal of double stranded RNA (dsRNA) contaminants was confirmed using dot blot and endotoxin levels were measured using the Genscript Toxisensor chromogenic assay (<0.05 EU/mL). The different HA and NA mRNA were stored frozen (1 mg/mL) at −20 °C in nuclease and pyrogen free water until use. mRNA was encapsulated into lipid nanoparticles as previously described73. A mixture of an ionizable lipid (proprietary to Acuitas Therapeutics), cholesterol, DSPC, and PEG-Lipid in an ethanolic solution was rapidly mixed with an aqueous phase containing the mRNA, dialyzed, concentrated to 1 mg/mL and stored at −80 °C until used. The ionizable lipid and LNP composition are described in patent application WO 2017/004143A1. mRNA-LNP were characterized for their hydrodynamic size, and polydispersity index (PDI) using dynamic light scattering (DLS), and the encapsulation efficiency and concentration measured using the RiboGreen™ RNA Assay (Invitrogen, Cat# R11490).
Cell lines
293T (CRL-3216) and Madin-Darby canine kidney (MDCK, CCL-34) cells were obtained from American Type Culture Collection (ATCC) and grown at 37 °C in 5% CO2. 293 T cells were maintained in Dulbecco’s modified Eagles medium (DMEM, GibcoTM) supplemented with 5% fetal bovine serum (FBS), GlutaMAXTM (GibcoTM), and penicillin-streptomycin (P/S), and MDCKs were maintained in minimal essential medium supplemented with 5% FBS, HEPES, sodium bicarbonate, GlutaMAXTM and penicillin-streptomycin. All media was supplemented with Plasmocin prophylactic (Invitrogen, ANT-MPP) to prevent mycoplasma contamination.
Plasmids
For plasmids used for experiments in Fig. 1 and cell-based ELISAs, all viral protein sequences were obtained through NCBI and cloned into pCAGGs expression plasmids using human-codon optimized gBlocks with an artificial Kozak sequence (IDT) (Protein accession numbers: A/Victoria/2570/2019 NA, WEY08939; A/Victoria/2570/2019 HA, WEY08940; A/Tasmania/503/2020 NA, WMW30851; A/Tasmania/503/2020 HA, WMW30850, T176K, S202R; B/Washington/02/2019 NA, QCG86179; B/Washington/02/2019 HA, QCG86180; B/Phuket/3073/2013 NA, EPI544263; B/Phuket/3073/2013 HA, EPI544264). PCR products were inserted into the pCAGGs expression vector using the EcoRI and NheI restriction sites and HiFi DNA Assembly (New England BioLabs [NEB], Cat #E5520). DNA products were transformed into chemically competent NEB 5-alpha high-efficiency cells (NEB, Cat. C2987H) and purified plasmids were then confirmed using Sanger sequencing.
HA stalk design and expression
Our HA stalk constructs were designed to express the viral stalk ectodomain, anchored by the transmembrane ___domain and cytoplasmic tail. H1N1 A/Victoria/2570/2019 HA (accession: WEY08940) stalk-only design was based on the Gen6 HA-SS design used in Yassine et al.74, and H3N2 A/Tasmania/503/2020 HA (accession: WMW30850: T176K, S202R) stalk-only design was based on the ‘H3ssF_C’ design used in Corbett, K. S., et al.75. For both designs, amino acid (aa) numbering began at the initiator methionine. Briefly, for H1N1 design, the region between aaL49 and L328 was replaced with a GSG linker ___domain, and region between aaM402 and T436 was replaced with a GSGGSG linker. Four additional mutations were added to promote correct folding of the stalk: K394M, Y437D, N438L, E446L. Briefly, for H3N2 stalk-only design, aa66-329, aa407-408, and aa412-436 were deleted. The following mutations were also inserted: aa60-65 (QNSSIG to FPGCGV), K396M, L397V, L400V, G402E, K403L, T404M, N405E, E406Q, H409G, Q410G, I411P, W437D, S438C, N440L, E448L, and N461R. Human codon-optimized gBlocks (IDT) were designed according to the A/Victoria/2570/2019 and A/Tasmania/503/2020 sequences and used to clone into the pCAGGs expression vector using Gibson assembly. These constructs were validated to have the correct trimeric structure using the conformation-specific monoclonal antibody, CR9114 (produced by the Moody laboratory at Duke University) (Supplementary Fig. 3).
Confocal microscopy
293T cells were seeded onto poly-L-lysine treated coverslips in 24-well plates and then transfected with the indicated plasmid in OptiMeM™ (GibcoTM, #31985070) using Trans-IT-LT1 (MIR #2304) transfection reagent and allowed to incubate at 37 °C for 24 h. Cells were then fixed with 4% paraformaldehyde (PFA) and blocked with 3% BSA in phosphate-buffered saline (PBS) (w/v) for two hours at room temperature and stained with 9H10 (10 μg/mL) or anti-N2 antibody (Sino Biological, #40017-RP01-100, 1:1000) in PBS/BSA overnight at 4 °C. Cells were washed with PBS then incubated with secondary antibody (goat anti-mouse AF488 (Invitrogen, #A11001) and goat anti-rabbit AF647 (Invitrogen, # A21245)) diluted 1:1000 in 3% BSA in PBS for 2 h at room temperature. Coverslips were finally washed again, stained for nuclei using Hoechst 33342 (Life Technologies, Cat. #H3570, 1:2500), then mounted onto slides (Prolong Diamond #P36961). Slides were imaged using an inverted IX83 Olympus microscope with a motorized XY-stage (Prior Scientific) and images were processed identically using ImageJ (NIH).
HA fusion assay
293T cells were seeded onto poly-L-lysine treated coverslips in 24-well plates to reach high confluency the next day. Cells were then transfected with the indicated plasmid in OptiMeM™ using LipofectamineTM 3000 (Invitrogen, #L3000001) and incubated at 37 °C for 24 h. Medium was replaced with OptiMeM™ supplemented with 0.35% BSA, 0.01% FBS and 5% P/S with 1 μg/mL TPCK-trypsin for 20 min, and then cells were treated with either DMEM at pH = 7.4 or DMEM acidified using citric acid at pH = 5 for 20 min. All medium was then replaced with fresh DMEM at pH = 7.4 and cells were allowed to recover at 37 °C for 4 h. Cells were then fixed with 2% PFA and stained with 20 μg/mL Wheat Germ Agglutinin (WGA) (Vector Laboratories, #FL-1021-5) fluorescent lectin in PBS for 1 h at room temperature and Hoescht 33342, according to the above described confocal microscopy protocol. Slides were then imaged on a confocal microscope according to the above described protocol.
Vaccine doses
Vaccine doses for mRNA-LNP vaccination were determined based on pre-existing vaccine doses used in Phase 3 clinical trials. Pfizer delivered 30 μg of mRNA-LNP vaccine to humans, so we decided to initially test 1/6th of this dose for mice, ~5 μg, and a similar 1/6th (10 μg) of the human dose of inactivated vaccine, Flulaval76,77. For experiments Fig. 2, mice were vaccinated with doses as follows: BSA, 10 μg; luciferase mRNA-LNP, 5.1 μg; NA mRNA-LNP, 2.275 μg; HA mRNA-LNP, 2.725 μg; NA mRNA-LNP, 2.275 and HA mRNA-LNP, 2.725 μg; NA-F2A-HA mRNA-LNP 5.1 μg; Flulaval, 10 μg. Size of mRNA transcript length were taken into account for the NA and HA proteins and roughly scaled accordingly. For experiments in Fig. 3, mice were vaccinated with doses as follows: luciferase mRNA-LNPs, 5 μg; NA-F2A-HA mRNA-LNP, 5 μg; Flulaval, 10 μg. For experiments in Fig. 4, mice were vaccinated with doses as follows: luciferase mRNA-LNP, 10 μg; four mRNA-LNP, 10 μg (2.5 μg of each H1N1, H3N2, IBV_V and IBV_Y NA-F2A-HA mRNA-LNPs; Flulaval, 10 μg. For ferret experiments in Fig. 5, 6-month old male ferrets (n = 4) were intramuscularly vaccinated with 30 μg of either luciferase mRNA-LNP or the four NA-F2A-HA mRNA-LNP mixture (7.5 μg of each NA-F2A-HA mRNA-LNP) or 60 μg of Flulaval in 500 μL PBS. In Supplementary Fig. 2, mice were first primed with either 10 μg of BSA, 10 μg of inactivated A/California/04/2009, A/Wyoming/03/2003, B/Malaysia/2506/2004, and B/Yamagata/16/1988 (2.5 μg of each virus) mix for ‘Pre-immune mix 1’ group, or 10 μg of Flulaval for ‘Pre-immune mix 2’ group, intramuscularly. Mice were then boosted with either 10 μg of BSA or 10 μg of the four mRNA-LNP mix (2.5 μg of each H1N1, H3N2, IBV-V and IBV-Y NA-F2A-HA mRNA-LNPs). In Supplementary Fig. 4, mice received 10 μg of BSA or 10 μg of Flulaval.
Vaccination and challenge
Six- to ten-week-old female DBA/2J mice from the Jackson Laboratory (#000671) were used for all experiments involving mice (n = 5 for all experiments shown). Mice were vaccinated intradermally (except for prime in Supplementary Fig. 2) with all vaccines to maintain consistency, unless otherwise noted. To confirm that Flulaval did not induce different immunogenicity when administered intradermally, we vaccinated mice both intradermally and intramuscularly with the same dose of Flulaval (10 μg) and saw no difference in antibody levels between groups (Supplementary Fig. 4). Three weeks post-vaccination, mice were bled via a cheek bleed to collect serum. Mice were allowed to recover for 2–3 days and then were challenged with virus. For viral infections, mice were anesthetized with 100 μL of ketamine/xylazine and infected intranasally with 40 μL of virus diluted in pharmaceutical grade PBS (Corning, 21-040-CV). Mice were challenged with the following doses of virus: Fig. 2, H3N2 dose = 1000 PFU; Fig. 4, H1N1 dose = 200 PFU, H3N2 dose = 100 PFU, and IBV_Y dose = 396,000 PFU. After infection, mouse bodyweight was recorded daily for 14 days and mice were sacrificed if bodyweight fell below the predetermined humane endpoint (75% of starting body weight). For ferret experiments, 6-month old male ferrets were first bled three days prior to vaccination, then were intramuscularly vaccinated with the intended vaccines. At 28 days post-vaccination, ferrets were bled again and oral swabs were taken. Ferrets were then infected with 500 μL of virus (intended dose = 1000 PFU of A/Hawaii/66/2019) intranasally after inhalation of isoflurane at 31 days post-vaccination. Ferrets were monitored for change in bodyweight, and oral swabs were obtained at days 2, 4, and 7 post-infection. Animal viral titer was determined using standard plaque assays on MDCK cells using oral swabs in duplicate dilution series. All ferret experiments were conducted in the Duke Regional Biocontainment Laboratory with their independent viral stocks.
Ethics statement
All experiments involving animals were approved by Duke Univeristy IACUC under the protocol numbers A142-21-07 (mouse) and A077-23-03 (ferret). Animals were housed in ambient temperature and humidity with free access to food and water and with a 12 h light/dark cycle.
Viruses
IAV viruses were propagated on MDCK cells or in eggs at 37 °C, and IBV viruses were grown at 33 °C on MDCK cells or in eggs. All viral stocks were then titered using standard plaque assays on MDCK cells. The A/Tasmania/503/2020, B/Phuket/3073/2013, B/Colorado/06/2017 viruses were obtained from BEI resources. To generate the “pre-immune” vaccine, IAV and IBV viruses (A/California/04/2009, A/Wyoming/03/2003, B/Malaysia/2506/2004 and B/Yamagata/16/1988) were propagated on eggs at either 37 °C or 33 °C and then viral stocks were concentrated using a 30% sucrose cushion via ultracentrifugation (27,500 rpm for 1 h). Concentrated viruses were resuspended in pharmaceutical grade PBS (Corning, 21-040-CV) and inactivated with 0.02% formalin at 4 °C for 48 h. Inactivated viruses were then dialyzed using Slide-A-Lyzer cassettes (Thermo Fisher, #66370) and quantified using Pierce Rapid Gold BCA Protein Assay Kit (Thermo Fisher #A53226) and stored at −80 °C for future use. All viral stocks were sequence confirmed using reverse-transcription followed by Sanger sequencing of the HA, NA, and NP or PA proteins. Sequence alignments for HA percent homology were aligned using Clustal Omega v1.2.4. software. For viruses that did not induce robust clinical disease in mice or were not available, similar viral strains with HA sequence homology >95% were chosen to be used (Supplementary Figs. 5–7).
Enzyme-linked immunosorbance assays (ELISAs)
For cell-based ELISAs, 293Ts were plated in poly-L-lysine-treated 96-well plates and then transfected with the indicated plasmid in OptiMeM™ using Trans-IT LT1 and left to incubate for 24 h at 37 °C with 5% CO2. Cells were then fixed with 2% PFA, washed with PBS, and then blocked in 3–5% nonfat milk in PBS (w/v). Serial dilutions of sera in nonfat milk in PBS were then added and allowed to incubate, followed by secondary antibody staining (goat anti-mouse HRP (Invitrogen, #A16072, 1:10,000), goat anti-human HRP (Invitrogen, #A18805, 1:10,000) or goat anti-ferret HRP (Novus Biologicals, #NB7224, 1:10,000). Plates were finally washed again before 1-Step TMB ELISA Substrate Solutions (Fisher Scientific, #PI34028) was added. Signal was allowed to develop until the reaction was stopped with 1 M H2SO4. Absorbance of signal at 450 nm was then read on a Varioskan LUX plate reader. The area under the curve (AUC) for each ELISA was calculated using the average of wells with no primary antibody added as the baseline cutoff, using Prism 9 (GraphPad). For non cell-based ELISAs, ImmunoGrade 96-well plates (BrandTech, #781722) were coated with 0.2 μg/well of the target antigen or 1.0 × 105 PFU/well whole virus in coating buffer (PBS with NaHCO3 and Na2CO3) overnight at 4 °C. Plates were then blocked and signal developed using the same protocol described above. For ELISAs using ferret sera, initial sera dilutions in 3% milk were briefly incubated with 293T cells to reduce background reactivity. Cells were pelleted, then mixed with sera dilutions for 10 min, pelleted again, and the supernatant was then used in the rest of the ELISA protocol as described above.
Hemagglutinination inhibition assays (HAIs)
Sera from vaccinated mice was diluted 1:4 with receptor-destroying enzyme (RDE, Denka Seiken #370013) and incubated for 16–20 h at 37 °C, followed by inactivation of the RDE with incubation at 56 °C for 45 min. Sera was serially diluted in PBS in 96-well V-bottom microplates (Corning, #CLS3897), then mixed with equal volume of standardized virus dilution (2 HA units) and allowed to incubate at room temperature for 15 min. Turkey red blood cells were then diluted 1:40 in PBS and added to each well of sera/virus mixture. Plates were covered and left to incubate at 4 °C overnight before HAI titers were recorded. HAI titers were calculated as the reciprocal of the last dilution of sera that inhibited hemagglutination, indicated by a clear red blood cell pellet.
Enzyme-linked lectin assays (ELLAs) and neuraminidase inhibition assays (NAIs)
For both ELLA assays and NAI assays, immuno-grade 96-well plates were coated with 100 μL 25 mg/mL fetuin (Sigma #F3385) in 1x coating buffer (KPL coating buffer #50-84-10) for at least 24 h at 4 °C before the experiment was conducted. Fetuin-coated plates which were thoroughly washed with PBS-T (0.05% Tween-20). For ELLA assays to detect neuraminidase activity, 293T cells were transfected with plasmids using LipofectamineTM 3000 and left to incubate at 37 °C. At 24 h post transfection, cells were resuspended in OptiMeM™ supplemented with 0.35% BSA, 0.01% FBS and 5% penicillin/streptomycin. Cells were counted and all samples were normalized to the same cell concentration. Samples were added to fetuin-coated plates, firmly sealed, and incubated at 37 °C for 18 h. Plates were thoroughly washed with PBS-T, then peroxidase conjugated-peanut agglutinin-HRPO (1 mg/mL, Sigma #L7759, 1:1000) was diluted in sample buffer and added to each well and left to incubate for 2 h in the dark at room temperature. Plates were washed again with PBS-T then signal was developed as previously described for ELISAs. To assess NAI using ELLAs, RDE-treated sera from vaccinated mice were serially diluted in sample buffer (Dulbecco’s PBS with CaCl2 and MgCl2, Gibco, with 0.5% Tween-20) and added to fetuin-coated plates. Recombinant N2 (Sino Biologicals, #40017-VNAHC-800) was diluted in sample buffer and added to each well (except for a no N2 control). Plates were incubated at 37 °C for 18 h, and signal was developed as previously described for ELLAs. NA activity was calculated as the following percentage: ((serum absorbance-mean background absorbance)/(mean positive control absorbance-mean background absorbance))*100. NAI titer is reported as the reciprocal of the lowest dilution of sera that inhibited greater than or equal to 50% NA activity.
Microneutralization assays (MN50)
RDE-treated sera from vaccinated mice were serially diluted in 96-well microplates in sample diluent (PBS/BSA with 0.1% TPCK-trypsin (Thermo #20233)). Each plate also included virus positive control wells and uninfected negative control wells. Virus suspension was prepared in sample diluent and an equal volume (containing 32,000 PFU A/Tasmania/503/2020 virus) was added to each well containing sera dilutions except the uninfected control wells. The virus-serum mixtures were allowed to incubate at 37 °C with 5% CO2 for 1 h, before an equal volume of MDCK cells (containing 1 × 104 cells) was added to each well on top of the virus-serum mixtures. Plates were incubated for 20 h at 37 °C with 5% CO2, and then were fixed with 4% PFA. Plates were thoroughly washed with wash buffer (PBS + 0.3% Tween-20), then monoclonal human-anti influenza HA-stalk antibody (CR9114) was diluted 1:4000 in antibody diluent (wash buffer + 5% nonfat milk) and added to each well for overnight incubation at 4 °C. Plates were washed with wash buffer then goat-anti human-HRP secondary antibody (1:10,000) was diluted 1:5000 in antibody diluent and added to each well for 1 h at room temperature. Plates were washed with wash buffer a final time, and signal was developed as previously described for ELISAs. The reaction was stopped using 1 M H2SO4 and plates’ absorbances at 450 nm were read using a Varioskan LUX plate reader. In calculating the half-maximal inhibitory titer (MN50), the virus control wells for each plate were set to 100% and the uninfected negative controls were set to 0%. A four-parameter nonlinear regression was then applied to each plate individually using Prism 9 (GraphPad), and the MN50 was interpreted as the log 10 of the lowest dilution of sera that inhibited virus greater than or equal to 50% of control viral infection.
Plaque-reduction neutralization assays (PRNT50)
RDE-treated sera were serially diluted in PBS/BSA, then each dilution was mixed with standardized virus suspension and allowed to incubate for 45 min at room temperature. Confluent 6-well plates of MDCK cells were washed with PBS then infected with the virus/sera mixtures for 1 hour at 37 °C and 5% CO2. Virus/sera mixtures were then removed and replaced with an agar overlay supplemented with TPCK-trypsin. PRNTs were left to incubate for 48–72 h at 37 °C with 5% CO2 until plaques were visible. PRNTs were then fixed for at least 3 hours at room temperature with 4% PFA, before the agar overlay was removed and replaced with CR9114 antibody diluted in antibody dilution buffer (5% nonfat dried milk, 0.05% Tween-20 in PBS, 1:1000) overnight at 4 °C. PRNTs were then washed with PBS and incubated with goat anti-human HRP diluted in antibody dilution buffer for 1 h at room temperature (1:4000). Finally, PRNTs were washed again and stained with KPL TrueBlueTM peroxidase substrate (SeraCare, #95059–168). PRNTs were then dried, and plaques were counted. PRNT50 titer is reported as the reciprocal of the lowest dilution of sera that inhibited greater than or equal to 50% the number of plaques as the no antibody, infected control.
qRT-PCR
Viral RNA was isolated from ferret oral swabs throughout infection using the Qiagen QIAmp Viral RNA Mini Kit (Cat #52904). Purified RNA extracted from 140 μL of oral swab was then stored at −80 °C until further use. For samples with limited oral swab volume, RNA was extracted from 70 μL of sample and then 2x the measured RNA value was used in subsequent analyses. One-step qRT-PCR was then performed using EXPRESS One-Step Superscript qRT-PCR kit (Invitrogen, #11781200) on an Applied Biosystems QuantStudio 3 Real-Time PCR System. Custom TaqMan probes synthesized by IDT were used to target the viral RNA of the A/Hawaii/70/2019 NP segment (Forward primer: 5′-CAGTGAGTACCCTTCCCTTTC-3′,reverse primer:5′-CTGAGAGGATCAGTTGCACATA-3′,Probe:5′-/56-FAM/TGTGTGTAT/ZEN/GGGCTTGCAGTAGCA/3IABkFQ/-3′). Endogenous eukaryotic 18S rRNA (Applied Biosystems, #4319413E) was also targeted as an endogenous control.
Influenza HA production
Recombinant HA (rHA) were purchased from Sino Biological, the IRR, the Duke CIVICs Reagent Core, or produced by GenScript as indicated in Supplementary Table 1. For rHA produced by Genscript, the soluble full-length ectodomains for the HAs of the influenza strains cloned into pFastBac expression constructs containing a T3-fribitin (FoldOn) trimerization ___domain and C-terminal hexa-His tag as described78. Baculoviruses from Sf9-transfected cells were used to infect Sf9 cells cultured in Sf-900TM II SFM medium (Life Technologies, #10902104). Three days post-infection, the rHA were purified by Ni-NTA column.
Multiplex HA binding assays
rHA or BSA (Sigma – negative control) were conjugated to magnetic microspheres (Luminex Corp.) by carbodiimide coupling using the manufacturer’s protocol. rHA- and BSA-conjugated microspheres (1500 of each) were mixed with 10-fold serially-diluted samples diluted 1:100 to 1:10,000,000 in PBS, 1% BSA pH = 7.4 (assay buffer; bioWorld), incubated for 60 min at room temperature on an orbital shaker and then washed in assay buffer. Microspheres were incubated with 10 μg/mL phycoerythrin (PE)-conjugated goat anti-ferret IgG heavy & light chains (Abcam #ab112768 conjugated to PE using a Lightning-Link labeling kit (Abcam)) for 30 min, washed in assay buffer, and the PE median fluorescence intensity (MFI) of each microsphere population measured using an Intelliflex DR-SE bead reader (Luminex). Data were analyzed using Bio-Plex Manager v.6.2. Different HAs exhibited parallelism and dilutional linearity at different sample dilutions, and so for each HA, the raw result was calculated as the average background-corrected MFI from duplicate wells at the first dilution at which the data appeared parallel and linear for all samples. These MFI values were then each normalized to the values of luciferase mRNA-LNP vaccinated animals within each HA, then log transformed to obtain the reported values. These normalized MFI values are shown individually for each animal. HAs for which neither four mRNA-LNP- nor Flulaval-vaccinated animals had a statistically significantly higher normalized MFI value compared to luciferase mRNA-LNP-vaccinated animals are not reported.
Graphing and statistical analyses
Experiments were graphed using Prism 9 (GraphPad Software) and all statistical analysis was performed using R statistical software (R Foundation for Statistical Computing, Vienna, Austria). Initially, comparaisons of more than two groups were performed using a Kruskal–Wallis test. If the resulting p < 0.05, a Wilcoxon rank-sum test was performed to compare each pairs of groups. The Benjamini–Hochberg procedure was applied within each figure panel to control the false discovery rate (FDR) for multiple comparaisons. All tests were two-sided, with an alpha level of 0.05. All experiments were performed in two independent experiments unless otherwise noted. For data in which groups were overlapping over the same values, groups were nudged apart vertically or horizontally to help distinguish groups. For sample groups that yielded values below/above the LOD in their assays, groups were assigned an arbitrary value below the LOD to be included in the graph and in statistical analysis. Schematics featured in figures were created using BioRender.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data for all graphs are provided as a Source Data file. Protein sequences are available using the following accession numbers: A/Victoria/2570/2019 NA, WEY08939; A/Victoria/2570/2019 HA, WEY08940; A/Tasmania/503/2020 NA, WMW30851; A/Tasmania/503/2020 HA, WMW30850 (T176K, S202R) (https://www.ncbi.nlm.nih.gov/protein/WMW30850); B/Washington/02/2019 NA, QCG86179; B/Washington/02/2019 HA, QCG86180; B/Phuket/3073/2013 NA, EPI544263; B/Phuket/3073/2013 HA, EPI544264 Source data are provided with this paper.
References
Thompson, W. W. et al. Influenza-associated hospitalizations in the United States. JAMA 292, 1333–1340 (2004).
Prevention, C.f.D.C.a. Preliminary Estimated Influenza Illnesses, Medical Visits, Hospitalizations, and Deaths in the United States—2022–2023 Influenza Season. 2024 (2023).
Putri, W., Muscatello, D. J., Stockwell, M. S. & Newall, A. T. Economic burden of seasonal influenza in the United States. Vaccine 36, 3960–3966 (2018).
Hobson, D., Curry, R. L., Beare, A. S. & Ward-Gardner, A. The role of serum haemagglutination-inhibiting antibody in protection against challenge infection with influenza A2 and B viruses. J. Hyg. (Lond.) 70, 767–777 (1972).
Chen, Y. Q. et al. Influenza infection in humans induces broadly cross-reactive and protective neuraminidase-reactive antibodies. Cell 173, 417–429.e410 (2018).
Wei, C. J. et al. Next-generation influenza vaccines: opportunities and challenges. Nat. Rev. Drug Discov. 19, 239–252 (2020).
Fleury, D., Wharton, S. A., Skehel, J. J., Knossow, M. & Bizebard, T. Antigen distortion allows influenza virus to escape neutralization. Nat. Struct. Biol. 5, 119–123 (1998).
Guthmiller, J. J. et al. First exposure to the pandemic H1N1 virus induced broadly neutralizing antibodies targeting hemagglutinin head epitopes. Sci. Transl. Med. 13, eabg4535 (2021).
Yamazaki, T., Chiba, J. & Akashi-Takamura, S. Neutralizing anti-hemagglutinin monoclonal antibodies induced by gene-based transfer have prophylactic and therapeutic effects on influenza virus infection. Vaccines (Basel) 6, 35 (2018).
Medina, R. A. et al. Glycosylations in the globular head of the hemagglutinin protein modulate the virulence and antigenic properties of the H1N1 influenza viruses. Sci. Transl. Med. 5, 187ra170 (2013).
Heaton, N. S., Sachs, D., Chen, C. J., Hai, R. & Palese, P. Genome-wide mutagenesis of influenza virus reveals unique plasticity of the hemagglutinin and NS1 proteins. Proc. Natl. Acad. Sci. USA 110, 20248–20253 (2013).
Krammer, F. The human antibody response to influenza A virus infection and vaccination. Nat. Rev. Immunol. 19, 383–397 (2019).
Gouma, S., Anderson, E. M. & Hensley, S. E. Challenges of making effective influenza vaccines. Annu. Rev. Virol. 7, 495–512 (2020).
Krammer, F. et al. NAction! How can neuraminidase-based immunity contribute to better influenza virus vaccines? mBio 9, e02332–17 (2018).
Wu, N. C. & Ellebedy, A. H. Targeting neuraminidase: the next frontier for broadly protective influenza vaccines. Trends Immunol. 45, 11–19 (2024).
Chen, J. R., Liu, Y. M., Tseng, Y. C. & Ma, C. Better influenza vaccines: an industry perspective. J. Biomed. Sci. 27, 33 (2020).
Gouma, S. et al. Nucleoside-modified mRNA-based influenza vaccines circumvent problems associated with H3N2 vaccine strain egg adaptation. J. Virol. 97, e0172322 (2023).
Tricco, A. C. et al. Comparing influenza vaccine efficacy against mismatched and matched strains: a systematic review and meta-analysis. BMC Med 11, 153 (2013).
Zost, S. J. et al. Contemporary H3N2 influenza viruses have a glycosylation site that alters binding of antibodies elicited by egg-adapted vaccine strains. Proc. Natl. Acad. Sci. USA 114, 12578–12583 (2017).
Nayak, J. L. & Caserta, M. T. Cell-culture-based influenza vaccines: don’t put all of your influenza vaccines in the egg basket. Pediatrics 150, e2022058143 (2022).
Chambers, B. S., Parkhouse, K., Ross, T. M., Alby, K. & Hensley, S. E. Identification of hemagglutinin residues responsible for H3N2 antigenic drift during the 2014-2015 influenza season. Cell Rep. 12, 1–6 (2015).
Prevention, C.f.D.C.a. CDC Seasonal Flu Vaccine Effectiveness Studies, https://www.cdc.gov/flu-vaccines-work/php/effectiveness-studies/index.html 2024 (2024).
Goel, R. R. et al. mRNA vaccines induce durable immune memory to SARS-CoV-2 and variants of concern. Science 374, abm0829 (2021).
Goel, R. R. et al. mRNA vaccination induces durable immune memory to SARS-CoV-2 with continued evolution to variants of concern. bioRxiv (2021).
Painter, M. M. et al. Rapid induction of antigen-specific CD4(+) T cells is associated with coordinated humoral and cellular immunity to SARS-CoV-2 mRNA vaccination. Immunity 54, 2133–2142 e2133 (2021).
Chaudhary, N., Weissman, D. & Whitehead, K. A. mRNA vaccines for infectious diseases: principles, delivery and clinical translation. Nat. Rev. Drug Discov. 20, 817–838 (2021).
Zhang, G., Tang, T., Chen, Y., Huang, X. & Liang, T. mRNA vaccines in disease prevention and treatment. Signal Transduct. Target Ther. 8, 365 (2023).
Baden, L. R. et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 384, 403–416 (2021).
Polack, F. P. et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N. Engl. J. Med 383, 2603–2615 (2020).
Wilson, E. et al. Efficacy and safety of an mRNA-Based RSV PreF vaccine in older adults. N. Engl. J. Med 389, 2233–2244 (2023).
Creech, C. B. et al. Evaluation of mRNA-1273 Covid-19 vaccine in children 6 to 11 years of age. N. Engl. J. Med 386, 2011–2023 (2022).
Furer, V. et al. Immunogenicity and safety of the BNT162b2 mRNA COVID-19 vaccine in adult patients with autoimmune inflammatory rheumatic diseases and in the general population: a multicentre study. Ann. Rheum. Dis. 80, 1330–1338 (2021).
Jamous, Y. F. & Alhomoud, D. A. The safety and effectiveness of mRNA vaccines against SARS-CoV-2. Cureus 15, e45602 (2023).
Sahin, U., Kariko, K. & Tureci, O. mRNA-based therapeutics–developing a new class of drugs. Nat. Rev. Drug Discov. 13, 759–780 (2014).
Eichelberger, M. C., Morens, D. M. & Taubenberger, J. K. Neuraminidase as an influenza vaccine antigen: a low hanging fruit, ready for picking to improve vaccine effectiveness. Curr. Opin. Immunol. 53, 38–44 (2018).
Monto, A. S. et al. Antibody to influenza virus neuraminidase: an independent correlate of protection. J. Infect. Dis. 212, 1191–1199 (2015).
Giurgea, L. T., Morens, D. M., Taubenberger, J. K. & Memoli, M. J. Influenza neuraminidase: a neglected protein and its potential for a better influenza vaccine. Vaccines (Basel) 8, 409 (2020).
Hansen, L. et al. Human anti-N1 monoclonal antibodies elicited by pandemic H1N1 virus infection broadly inhibit HxN1 viruses in vitro and in vivo. Immunity 56, 1927–1938.e1928 (2023).
Jiang, H. et al. Structure-based modification of an anti-neuraminidase human antibody restores protection efficacy against the drifted influenza virus. mBio 11, e02315–e02320 (2020).
Kilbourne, E. D., Johansson, B. E. & Grajower, B. Independent and disparate evolution in nature of influenza A virus hemagglutinin and neuraminidase glycoproteins. Proc. Natl. Acad. Sci. USA 87, 786–790 (1990).
Stadlbauer, D. et al. Broadly protective human antibodies that target the active site of influenza virus neuraminidase. Science 366, 499–504 (2019).
Wohlbold, T. J. et al. Vaccination with adjuvanted recombinant neuraminidase induces broad heterologous, but not heterosubtypic, cross-protection against influenza virus infection in mice. mBio 6, e02556 (2015).
McMahon, M. et al. Assessment of a quadrivalent nucleoside-modified mRNA vaccine that protects against group 2 influenza viruses. Proc. Natl. Acad. Sci. USA 119, e2206333119 (2022).
Rcheulishvili, N. et al. Designing multi-epitope mRNA construct as a universal influenza vaccine candidate for future epidemic/pandemic preparedness. Int. J. Biol. Macromol. 226, 885–899 (2023).
Rijnink, W. F. et al. Characterization of non-neutralizing human monoclonal antibodies that target the M1 and NP of influenza A viruses. J. Virol. 97, e0164622 (2023).
Moderna, I. Moderna Announces Interim Phase 3 Safety and Immunogenicity Results for mRNA-1010, a Seasonal Influenza Vaccine Candidate. (2023).
Moderna, I. Moderna Announces First Participants Dosed in Phase 1/2 Study with mRNA-1020 and mRNA-1030 Seasonal Influenza Vaccine Candidates. (2022).
Li, M. et al. Influenza a neuraminidase-based bivalent mRNA vaccine induces Th1-type immune response and provides protective effects in mice. Vaccines (Basel) 12, 300 (2024).
Freyn, A. W. et al. A multi-targeting, nucleoside-modified mRNA influenza virus vaccine provides broad protection in mice. Mol. Ther. 28, 1569–1584 (2020).
Arevalo, C. P. et al. A multivalent nucleoside-modified mRNA vaccine against all known influenza virus subtypes. Science 378, 899–904 (2022).
Pardi, N. et al. Development of a pentavalent broadly protective nucleoside-modified mRNA vaccine against influenza B viruses. Nat. Commun. 13, 4677 (2022).
Basak, S., Tomana, M. & Compans, R. W. Sialic acid is incorporated into influenza hemagglutinin glycoproteins in the absence of viral neuraminidase. Virus Res. 2, 61–68 (1985).
Palese, P., Tobita, K., Ueda, M. & Compans, R. W. Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology 61, 397–410 (1974).
McAuley, J. L., Gilbertson, B. P., Trifkovic, S., Brown, L. E. & McKimm-Breschkin, J. L. Influenza virus neuraminidase structure and functions. Front Microbiol. 10, 39 (2019).
Su, B. et al. Enhancement of the influenza A hemagglutinin (HA)-mediated cell-cell fusion and virus entry by the viral neuraminidase (NA). PLoS One 4, e8495 (2009).
Li, F. et al. Generation of replication-competent recombinant influenza A viruses carrying a reporter gene harbored in the neuraminidase segment. J. Virol. 84, 12075–12081 (2010).
Pan, W. et al. Visualizing influenza virus infection in living mice. Nat. Commun. 4, 2369 (2013).
Fang, J. et al. An antibody delivery system for regulated expression of therapeutic levels of monoclonal antibodies in vivo. Mol. Ther. 15, 1153–1159 (2007).
Harding, A. T., Heaton, B. E., Dumm, R. E. & Heaton, N. S. Rationally designed influenza virus vaccines that are antigenically stable during growth in eggs. mBio 8, e00669–17 (2017).
Alameh, M. G. et al. Lipid nanoparticles enhance the efficacy of mRNA and protein subunit vaccines by inducing robust T follicular helper cell and humoral responses. Immunity 54, 2877–2892 e2877 (2021).
Belser, J. A., Katz, J. M. & Tumpey, T. M. The ferret as a model organism to study influenza A virus infection. Dis. Model Mech. 4, 575–579 (2011).
DiLillo, D. J., Palese, P., Wilson, P. C. & Ravetch, J. V. Broadly neutralizing anti-influenza antibodies require Fc receptor engagement for in vivo protection. J. Clin. Invest 126, 605–610 (2016).
Pardi, N. et al. Nucleoside-modified mRNA immunization elicits influenza virus hemagglutinin stalk-specific antibodies. Nat. Commun. 9, 3361 (2018).
Lim, J. M. E. et al. A comparative characterization of SARS-CoV-2-specific T cells induced by mRNA or inactive virus COVID-19 vaccines. Cell Rep. Med 3, 100793 (2022).
Knudson, C. J. et al. Lipid-nanoparticle-encapsulated mRNA vaccines induce protective memory CD8 T cells against a lethal viral infection. Mol. Ther. 29, 2769–2781 (2021).
Brasu, N. et al. Memory CD8(+) T cell diversity and B cell responses correlate with protection against SARS-CoV-2 following mRNA vaccination. Nat. Immunol. 23, 1445–1456 (2022).
Powers, D. C., Kilbourne, E. D. & Johansson, B. E. Neuraminidase-specific antibody responses to inactivated influenza virus vaccine in young and elderly adults. Clin. Diagn. Lab Immunol. 3, 511–516 (1996).
Couch, R. B. et al. Randomized comparative study of the serum antihemagglutinin and antineuraminidase antibody responses to six licensed trivalent influenza vaccines. Vaccine 31, 190–195 (2012).
Sandbulte, M. R. et al. Discordant antigenic drift of neuraminidase and hemagglutinin in H1N1 and H3N2 influenza viruses. Proc. Natl. Acad. Sci. USA 108, 20748–20753 (2011).
Laguio-Vila, M. R. et al. Comparison of serum hemagglutinin and neuraminidase inhibition antibodies after 2010-2011 trivalent inactivated influenza vaccination in healthcare personnel. Open Forum Infect. Dis. 2, ofu115 (2015).
Schauer, R. Sialic acids as antigenic determinants of complex carbohydrates. Adv. Exp. Med Biol. 228, 47–72 (1988).
Baiersdorfer, M. et al. A facile method for the removal of dsRNA contaminant from in vitro-transcribed mRNA. Mol. Ther. Nucleic Acids 15, 26–35 (2019).
Maier, M. A. et al. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol. Ther. 21, 1570–1578 (2013).
Yassine, H. M. et al. Hemagglutinin-stem nanoparticles generate heterosubtypic influenza protection. Nat. Med 21, 1065–1070 (2015).
Corbett, K. S. et al. Design of nanoparticulate group 2 influenza virus hemagglutinin stem antigens that activate unmutated ancestor B cell receptors of broadly neutralizing antibody lineages. mBio 10, e02810–e02818 (2019).
Park, J. W., Lagniton, P. N. P., Liu, Y. & Xu, R. H. mRNA vaccines for COVID-19: what, why and how. Int. J. Biol. Sci. 17, 1446–1460 (2021).
Vogel, A. B. et al. BNT162b vaccines protect rhesus macaques from SARS-CoV-2. Nature 592, 283–289 (2021).
Whittle, J. R. et al. Broadly neutralizing human antibody that recognizes the receptor-binding pocket of influenza virus hemagglutinin. Proc. Natl. Acad. Sci. USA 108, 14216–14221 (2011).
Acknowledgements
We would like to thank Heather Froggatt and Xinyu Zhu for reagent generation, Emily Troutman for experimental assistance, Zhaochen Luo for helpful discussions, Florian Krammer and Peter Palese at Icahn School of Medicine at Mount Sinai for the kind gifts of the A/Switzerland/9715293/2013 H3N2 virus and the 9H10 antibody, respectively. We would also like to thank the lab of Dr. Carolyn Coyne for experimental support, and the following members of the Duke Regional Biocontainment Laboratory for their help with the ferret experimentation: Herman Staats, Kristina Riebe, Nicholas Blackburn and Jacquelyn DuVall. Multiplex binding assays were performed in the Duke Regional Biocontainment Laboratory, which received partial support for construction from the NIH/NIAID (UC6 AI058607). This research was funded in part with federal funds under a contract from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, contract no. 75N93019C00050. N.S.H. holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund.
Author information
Authors and Affiliations
Contributions
R.A.L. and N.S.H. conceived of and designed the study. R.A.L. performed experimentation with support from K.N.B. and A.N.M.; R.L.S. performed all statistical analyses. Y.T., M.G.A., and D.W. produced the mRNA-LNPs and provided experimental guidance and support. R.A.L. and N.S.H. wrote the manuscript and all authors aided with revisions and editing.
Corresponding author
Ethics declarations
Competing interests
Duke University has filed for protection of intellectual property related to the vaccine approach described in this manuscript on behalf of N.S.H. Y.T. is an officer with Acuitas Therapeutics, a company dedicated to the development of lipid nanoparticulate nucleic acid delivery systems for therapeutic and other applications. Y.T. is named on patents describing uses of modified mRNA LNPs. D.W. is a coinventor on patents that describe the use of nucleoside-modified mRNA as a platform to deliver therapeutic proteins or vaccines. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Bao-Zhong Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Leonard, R.A., Burke, K.N., Spreng, R.L. et al. Improved influenza vaccine responses after expression of multiple viral glycoproteins from a single mRNA. Nat Commun 15, 8712 (2024). https://doi.org/10.1038/s41467-024-52940-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52940-z
This article is cited by
-
Expert consensus on the benefits of neuraminidase in conventional influenza vaccines: a Delphi study
BMC Infectious Diseases (2025)
-
Use standard and enhanced vaccines to prevent influenza, while new classes remain in development
Drugs & Therapy Perspectives (2025)
