
Abstract
The asymmetric Büchner reaction and related arene cyclopropanations represent one type of the powerful methods for enantioselective dearomatization. However, examples of asymmetric Büchner reactions via a non-diazo approach are quite scarce, and the related arene cyclopropanation based on alkynes has not been reported. Herein, we disclose an asymmetric Büchner reaction and the related arene cyclopropanation by copper-catalyzed controllable cyclization of N-propargyl ynamides via vinyl cation intermediates, leading to chiral tricycle-fused cycloheptatrienes and benzonorcaradienes in high yields and enantioselectivities. Importantly, this protocol represents an asymmetric arene cyclopropanation reaction of alkynes and an asymmetric Büchner reaction based on vinyl cations.
Similar content being viewed by others

Introduction
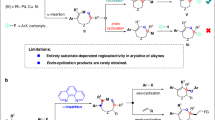
Catalytic asymmetric dearomatization (CADA) has proven to be one of the most attractive synthetic strategies to transform aromatic compounds to three dimensional molecules1,2,3,4,5,6,7,8. In the past decades, a wide range of efficient CADA reactions have been developed with a focus on the electron-rich aromatic rings, such as indoles and phenols1,2,3,4,5,6,7,8. Conversely, the CADA reactions of simple benzenes and naphthalenes have been rarely reported9,10, because of their intrinsic aromatic stability. Büchner reaction, as a unique type of expansive dearomatization of unactivated arenes, has become a practical strategy for the straightforward assembly of valuable functionalized cycloheptatrienes11,12,13,14,15,16,17,18,19,20,21,22,23. In recent years, the asymmetric Büchner reaction has received extensive attention and represents a significant advance in CADA reactions, offering the great potential to build valuable chiral cycloheptatrienes (CHTs)24,25,26,27,28,29,30,31,32,33,34,35,36. Moreover, the arene cyclopropanation product (norcaradiene, NCD), as an intermediate in the Büchner reaction, has also been widely used to prepare versatile polycyclic compounds13,37,38,39,40,41,42. In 1990, McKervey and co-workers achieved an asymmetric Büchner reaction by rhodium catalysis24,25, and since then, this chiral rhodium-catalyzed Büchner reaction was extensively studied by Xu and Doyle26 and others27,28,29. In addition, chiral copper- and ruthenium-catalyzed Büchner reactions were also nicely explored by Maguire30,31,32,33 and Iwassa34, respectively. Despite these significant advances (Fig. 1a), these protocols have to rely on the use of diazo compounds as the carbene precursors. Particularly, compared to the classical Büchner reaction, the related asymmetric arene cyclopropanation reaction is highly challenging and has been scarcely reported37,38,39,42. Firstly, the generated norcaradienes are susceptible to isomerize into the more stable cycloheptatrienes11,12,13,28,37,38,39,40,41,42. Secondly, the interrupted cyclopropanation products could undergo easy racemization11,12,13,26 and suffer from low regiocontrol11,12,13,39. This also increases the difficulty in achieving the diversification of asymmetric arene Büchner reaction and cyclopropanation reaction. In 2021, Nemoto and Harada demonstrated an example of asymmetric Büchner reaction based on alkynes via a non-diazo approach (Fig. 1b)35. Very recently, an asymmetric Büchner reaction by chiral rhodium-catalyzed enynone cycloisomerization43 via the donor-donor Rh carbenes was elegantly studied by Zhu and co-workers36. However, to our best knowledge, the asymmetric arene cyclopropanation reaction based on alkynes remains unrealized. Furthermore, direct divergent synthesis of chiral cycloheptatriene and benzonorcaradiene products have not yet been explored.

a Traditional asymmetric Büchner and arene cyclopropanation reactions. b Asymmetric Büchner and arene cyclopropanation reactions of alkynes. c This work: Cu-catalyzed asymmetric Büchner and arene cyclopropanation of alkynes.
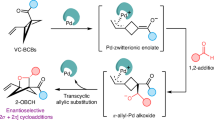
As a versatile intermediate in organic synthesis, vinyl cations have gained particular attention for their unique carbene-like reactivity in the past decade44,45. However, the exploitation of an asymmetric catalysis based on vinyl cation intermediates remains elusive but highly desirable46. In the past several years, our group has developed a facile copper-catalyzed diyne cyclization for the generation of vinyl cations. By using this strategy, a variety of useful asymmetric transformations have been established via a remote control of enantioselectivity47,48,49,50,51,52,53,54, including intramolecular aromatic C(sp2)–H functionalization47, vinylic C(sp2)–H functionalization49, unactivated C(sp3)–H functionalization54, cyclopropanation47, and [1,2]-Stevens-type rearrangement50, and intermolecular annulations with styrenes48 and ketones51, and atroposelective cyclization52,53. Inspired by these results and by our recent studies on the ynamide chemistry for N-heterocycle synthesis55,56,57,58,59,60,61, we envisaged that intramolecular arene moieties might capture the vinyl cations generated from the diyne cyclization and eventually lead to the dearomatized products (Fig. 1c).
Herein, we report the successful implementation of this mechanistic design to a highly enantioselective synthesis of a wide range of tricycle-fused cycloheptatrienes by Büchner reaction via the copper-catalyzed cyclization of phenyl-substituted N-propargyl ynamides. Interestingly, such a copper-catalyzed cyclization of naphthyl-substituted N-propargyl ynamides allows the formation of chiral benzonorcaradienes by interrupted arene cyclopropanation. Thus, by utilizing alkynes as precursors, this method leads to practical and divergent synthesis of enantioenriched cycloheptatrienes and benzonorcaradienes. Importantly, this protocol not only represents an asymmetric arene cyclopropanation reaction of alkynes, but also constitutes an asymmetric Büchner-type reaction based on vinyl cations. Of note, chiral fused bicyclo[5,4,0] rings and benzonorcaradienes are important structural motifs found in various natural products and bioactive molecules (Fig. 2)36,62,63,64,65,66,67,68,69,70.

Some of representative molecules are listed.
Results
Screening of conditions on the asymmetric Büchner reaction
At the outset, N-propargyl ynamide 1a bearing an electronically deactivated arene moiety was used as the model substrate35,36 to explore the asymmetric Büchner reaction based on our previous copper-catalyzed diyne cyclization47,48,49,50,51,52,53,54, and selected results are summarized in Table 1 (see the Supplementary Materials, Table S1). In the presence of 10 mol % Cu(MeCN)4PF6 as the catalyst and 12 mol % NaBArF4 as the additive in DCM at 35 °C, we were pleased to find that the expected chiral pyrrole-fused cycloheptatriene 2a was obtained in high yields ( > 80%) with moderate enantioselectivities by using bisphosphine ligands L1 and L2 as chiral ligands, and no background aromatic C − H insertion product was observed47 (Table 1, entries 1 and 2). Then, various bisoxazoline (BOX) ligands L3–L9 (12 mol %) were screened. It was found that poor enantioselectivities were achieved when typical BOX ligands such as L3 and L4 were employed as chiral ligands (Table 1, entries 3 and 4). In addition, the use of BOX ligand L5 also led to low enantioselectivity (Table 1, entry 5, 42% ee). Subsequently, Tang’s side-armed bisoxazoline (SaBOX) ligands71 were investigated on the basis of our previous studies47,48,49,50,51,52,53,54. Gratifyingly, the desired chiral product 2a could be obtained in 94% yield with 79% ee by using the dibenzyl-substituted SaBOX ligand L6 (Table 1, entry 6). Further screening of other SaBOX ligands L7–L9 (Table 1, entries 7–9) revealed that the use of L9 allowed the formation of the desired 2a in 95% yield with 81% ee (Table 1, entry 9). Next, we screened some other typical solvents such as DCE, toluene and THF (Table 1, entries 10–12), and found that the use of toluene as solvent could further improve the enantioselectivity (Table 1, entry 11). To our delight, a significant temperature effect was observed (Table 1, entries 13 and 14), and the chiral product 2a was formed in 95% yield with 96% ee when the temperature was lowered to −20 °C (Table 1, entry 14).
Reaction scope study on the asymmetric Büchner reaction
After establishing the optimal reaction conditions (Table 1, entry 14), we then exploited the generality of this catalytic asymmetric Büchner reaction. As illustrated in Fig. 3, the Büchner reactions of various N-protected ynamides 1a–1 f were first carried out to afford the desired chiral pyrrole-fused cycloheptatrienes 2a–2 f in 95–99% yields with excellent enantioselectivities (91–96% ees). Then, N-propargyl ynamides 1 g and 1 h with para-halogen-substituents (R2) on the aromatic ring were found to be suitable substrates, furnishing the corresponding products 2 g and 2 h in excellent yields and enantioselectivities. However, the use of diyne 1i possessing a meta-halogen-substituent at the biaryl moiety led to the desired 2i with excellent enantioselectivity (98% ee) but in decreased yield35, and significant formation of arene cyclopropanation product (49%) was observed (see the Supplementary Materials, Figs. S1, S2). Besides, the variation of aryl substituents of N-propargyl ynamides was studied, such as substrates 1j–1p containing different substituents at the 4-position of the aromatic ring, and the expected products 2j–2p were formed in 85–99% yields with 80–96% ees. We also examined diynes with the piperonyl group (1q) and disubstituted aromatic group (1r), and found that the reaction could efficiently generate the corresponding chiral cycloheptatrienes 2q (99%, 92% ee) and 2r (99%, 81% ee), respectively. In addition to the aryl- substituted diynes, the heteroaryl-substituted N-propargyl ynamide 1 s was also suitable for this reaction, and the desired product was obtained in 95% yield and 95% ee. Next, a wide range of ynamides containing different R1 substituents were screened, leading to products 2t–2 y in excellent yields (91–99%) and enantioselectivities (90–95% ees). Notably, the reaction was also extended to diyne substrates bearing no electron-withdrawing groups to produce the desired 2z (98%, 91% ee) and 2aa (99%, 83% ee) at 30 °C in DCM. Interestingly, the reaction also proceeded smoothly with the cyclohexenyl-linked aryl-diyne, yielding the corresponding product 2ab in 85% yield with 96% ee. Of note, in cases of the diyne substrates 1ac–1ad with non-electron-rich groups, higher temperature was required (50 °C) and low enantioselectivities ( < 10% ees) were observed (see the Supplementary Materials, Fig. S3), which is similar to the previous protocols47,48,49,50,51,52,53,54. Our attempts to extend the reaction to Cy-substituted diyne 1ae failed to obtain the corresponding Büchner product, and instead a hydroarylation product was formed in 69% yield (see the Supplementary Materials, Fig. S4). In addition, this asymmetric Büchner reaction could proceed smoothly with the heterocycle-linked diynes 1af–1ag and the alkyl-linked aryl diynes 1ah–1ai, but only gave the corresponding cycloheptatriene products 2af–2ai with moderate enantioselectivities (40–53% ees) under the optimized reaction conditions (see the Supplementary Materials, Fig. S5). Attempts to extend the reaction to the cyclopropyl-linked aryl diyne 1aj only led to the formation of the corresponding cyclopropane 4aj in 38% yield with 20% ee (see the Supplementary Materials, Fig. S6). Importantly, almost no arene cyclopropanation product (except substrate 1i) and no C − H insertion product were detected in all these cases. The absolute configuration of product 2b was confirmed by X-ray crystallographic analysis. Thus, this protocol constitutes a chiral copper-catalyzed asymmetric Büchner reaction of alkynes.

Reaction conditions: 1 (0.1 mmol), Cu(MeCN)4PF6 (0.01 mmol), L9 (0.012 mmol), NaBArF4 (0.012 mmol), toluene (2 mL), −20 °C, in vials; yields are those for the isolated products; ees are determined by HPLC analysis. aDCM (2 mL), 20 °C; bDCM (2 mL), 30 °C. Ts = p-toluenesulfonyl, Mbs = 4-methoxybenzenesulfonyl, Mts = 2-mesitylenesulfonyl, Bs = 4-bromobenzenesulfonyl, PMP = 4-methoxyphenyl.
Screening of conditions on the asymmetric cyclopropanation
Interestingly, during the substrate scope study of the above Büchner reaction, it was found that when the naphthyl-substituted diyne was employed as substrate, the corresponding arene cyclopropanation product was obtained instead and no formation of the seven-membered Büchner cyclization product was observed. Inspired by this dearomatized cyclopropanation reaction, we then chose the naphthalene ring-substituted N-propargyl ynamide 3a as the model substrate to investigate this asymmetric arene cyclopropanation, and some of the results are displayed in Table 2 (see the Supplementary Materials, Table S2). In the presence of Cu(MeCN)4PF6 (10 mol %), NaBArF4 (12 mol %) and bisphosphine ligand L1 or L2 as chiral ligand in DCM at 35 °C, we were delighted to find that the desired tricycle-fused benzonorcaradiene 4a bearing three chiral centers could be generated in excellent yields with moderate enantioselectivities (Table 2, entries 1 and 2). Further screening of various BOX ligands L3–L6 and L10–L12 (Table 2, entries 3–9) revealed that the use of SaBOX ligand L12 led to the expected chiral benzonorcaradiene 4a in 93% yield with 92% ee (Table 2, entry 9). Subsequently, the effect of solvent was explored (Table 2, entries 10–12), and slightly improved yield and enantioselectivity could be achieved by using 2-MeTHF as the solvent (Table 2, entry 12). Finally, it was found that lowering the reaction temperature to 0 °C allowed the formation of the desired cyclopropane product 4a in 95% yield with 96% ee (Table 2, entry 13).
Reaction scope study on the asymmetric cyclopropanation
With the optimal reaction conditions in hand (Table 2, entry 13), the substrate scope of this asymmetric arene cyclopropanation was investigated. As shown in Fig. 4, diynes with various N-protecting groups were first explored to provide the expected chiral tricycle-fused benzonorcaradienes 4a–4e in generally excellent yields with high enantioselectivities (90–96% ees). We then examined the substitutions of the parent ring of substrates 3f–3 m with different electron-donating and -withdrawing substituents in 4- and 5-positions, and found that the desired benzonorcaradienes 4f–4 m were furnished in 80–98% yields with 92–96% ees. In addition, diynes containing different naphthalene moieties were appropriate substrates to afford the corresponding enantioenriched cyclopropanes 4n–4w in high yields with the ees of 85–96%. Moreover, this cyclopropanation reaction could also proceed smoothly in case of diynes bearing different nitrogen-substituted aromatic rings (3x and 3 y). Similarly, the cyclohexenyl-linked N-propargyl ynamide 3z was also suitable for this reaction, yielding the desired chiral cyclopropane product 4z in 90% yield and 95% ee. However, the reaction of 1-naphthyl- and PMP-substituted diynes 3aa–3ab afforded the desired products 4aa (64% ee) and 4ab (66% ee) with moderate enantioselectivities, and the formation of the Büchner cyclization product was observed in the latter case (see the Supplementary Materials, Figs. S7, S8). We speculate that the moderate enantiomeric excess value in the former case may be attributed to the steric hindrance between the bridging benzene ring and the dearomatized naphthalene ring of the obtained product. Interestingly, this arene cyclopropanation proceeded smoothly with the heterocycle-linked diynes 3ac and 3ad, furnishing the corresponding cyclopropanes 4ac (99%, 74% ee) and 4ad (96%, 93% ee), respectively. Finally, it was found that the use of the alkyl-linked naphthyl-diyne 3ae only led to the desired 4ae in 90% yield with 42% ee under the optimized reaction conditions (see the Supplementary Materials, Fig. S9). Of note, neither Büchner cyclization product nor C − H insertion product was observed in all cases. Importantly, four rings containing three stereocenters with high stereospecificity are assembled in one step under mild conditions. The absolute configuration of product 4 l was confirmed by X-ray crystallographic analysis.

Reaction conditions: 3 (0.15 mmol), Cu(MeCN)4PF6 (0.015 mmol), L12 (0.018 mmol), NaBArF4 (0.018 mmol), 2-MeTHF (3 mL), 0 °C, in vials; yields are those for the isolated products; ees are determined by HPLC analysis.
Synthetic applications
To showcase the synthetic utility of this method, the gram-scale reactions and further product elaborations were explored (Fig. 5). The gram-scale synthesis of chiral cycloheptatriene 2a was first explored, and the desired product was obtained in 97% yield with a slightly decreased enantioselectivity (94% ee) under 5 mol % of chiral copper catalyst, as shown in Fig. 5a. Next, selective hydrogenation of the double bond of cycloheptatriene moiety of 2a with Pd/C generated the debromination product 5a in 98% yield with 12:1 dr. Further hydrogenation of the pyrrole moiety with Pd(OH)2/C under a H2 atmosphere (8 MPa) could lead to the formation of pyrrolidine- fused product 5b in 50% yield with excellent dr. In addition, further transformation of cycloheptatriene 2 l, which was synthesized on a preparative-scale in 99% yield with 96% ee, was also investigated. It was found that facile Sonagashira coupling and Suzuki coupling afforded the corresponding products 5c (98%, 95% ee) and 5d (71%, 96% ee), respectively. Subsequently, the gram-scale synthesis and synthetic applications of the cyclopropane products 4 were demonstrated, as depicted in Fig. 5b. In the presence of 5 mol % of chiral copper catalyst, the preparative reaction of 3a resulted in the formation of the desired product 4a in 93% yield and 96% ee. Interestingly, the Ms protecting group was readily removed by treating with KOH and protected again by the Boc group, furnishing the corresponding 5e in 95% yield with 95% ee (two steps). Additionally, the two-step reduction reactions of 4a with NaBH3CN and Pd/C/H2, respectively, could lead to the dihydropyrrole product 5 f in 51% yield (two steps). Then, the Diels-Alder reaction was also tested by the use of DMAD reagent, and the unexpected ring-expansion product 5 g was formed in 90% yield and 96% ee. The relative configuration of the product 5 g was confirmed by X-ray crystallographic analysis. Moreover, the NMe2 group of the cyclopropane product 4x could be further converted into the aryl group by Pd-catalyzed cross-coupling with an aryl Grignard reagent, delivering the corresponding 5 h in 75% yield. Significantly, almost no erosion of the enantiopurity of the compounds was observed in all these elaborations.

a Preparative-scale reaction of 1a and synthetic applications. b Preparative-scale reaction of 3a and synthetic applications. Reagents and conditions: (i) Cu(MeCN)4PF6 (5 mol %), NaBArF4 (6 mol %), L9 (6 mol %), toluene, −20 °C, 9 d. (ii) Pd/C (10 mol %), H2 (1 atm), MeOH, rt, 12 h. (iii) Pd(OH)2/C (10 mol %), H2 (8 MPa), AcOH:EA = 1:1, 80 °C, 72 h. (iv) Cu(MeCN)4PF6 (10 mol %), NaBArF4 (12 mol %), L9 (12 mol %), DCM, 20 °C, 4 d. (v) Phenylacetylene (4 equiv), Pd(PPh3)4 (5 mol %), CuI (10 mol %), THF:Et3N = 3:1, 50 °C, 1 h. (vi) PhB(OH)2 (3.5 equiv), Pd(PPh3)4 (10 mol %), CsF (2.5 equiv), DME, 50 °C, 8 h. (vii) Cu(MeCN)4PF6 (5 mol %), NaBArF4 (6 mol %), L12 (6 mol %), 2-MeTHF, 0 °C, 51 h. (viii) KOH (10 equiv), THF:MeOH = 1:1, 50 °C, 1.5 h, then DMAP (20 mol %), (Boc)2O (3 equiv), Et3N (4 equiv), DCM, rt, 2 h. (ix) NaBH3CN (5 equiv), DCM:TFA = 10:1, rt, 1 h; Pd/C (10 mol %), H2 (1 atm), AcOH, 60 °C, 12 h. (x) DMAD (20 equiv), toluene, 60 °C, 12 h. (xi) MeOTf (10 equiv), Et2O, 0 °C to rt, 2 h; Pd(PPh3)2Cl2 (5 mol %), PhMgBr (2 equiv), THF, rt, 2 h.
Mechanistic investigations
On the basis of the aforementioned experimental observations, our previous studies47,48,49,50,51,52,53,54, and comprehensive computational analysis (see the Supplementary Materials, Tables S6–9), a plausible vinyl cation-involved mechanism from 1a to 2a and 3x to 4x is exhibited in Fig. 6. The reaction is initialized via a preferential coordination of the CuI catalyst to activate the electron-richer amide-tethered C ≡ C bond of 1a and 3x to produce the precursor A1 and A2, followed by an intramolecular cyclization to afford the vinyl cation intermediate B1 and B2 with a free energy barrier of 8.9 kcal/mol and 11.1 kcal/mol, respectively. Nevertheless, the reaction deviates upon reaching the intermediate B. Upon the generation of the vinyl cation intermediate B1, the vinyl cation undergoes an electrophilic addition to the aryl group connected to the side chain, conquering a free energy barrier of 5.6 kcal/mol, forming a dearomatized carbon cation intermediate C1. Then, within intermediate C1, an electrophilic addition and cyclopropanation process occurs to afford the cyclopropane-tethered copper carbenoid intermediate D1, undergoing TSC1 with a slight barrier height of only 4.0 kcal/mol. Subsequently, the cyclopropane structure undergoes a ring-expansion process, resulting in Büchner-type copper carbenoid intermediate E1. Finally, the Büchner-type product 2a is obtained through a rate-determining Lewis base (1a)-assisted 1,4-H migration process similar to our previous studies49,50,51,52,53,54, with a barrier height of 13.8 kcal/mol (Fig. 6a). While in the reaction pathway starting from intermediate B2, after going through the same process and forming a cyclopropane-type copper carbenoid intermediate D2, instead of the same ring-expansion process as is mentioned above, which undergoes a higher free energy barrier in the subsequent steps, the cyclopropane-type product 4x is much more thermodynamically favorable right after the same rate-determining Lewis base (3x)-assisted 1,4-H migration process from intermediate D2, undergoing a free energy barrier of 20.1 kcal/mol (Fig. 6b). All above, under the provided reaction conditions, this reaction can proceed smoothly, with the Lewis base-assisted 1,4-H migration process being the rate-determining step.

Relative free energies (ΔG, in kcal/mol) were computed at: a (PCM, solvent = toluene)-PBE0-D3/6-311 + + G(d,p)-SDD//B3LYP-D3/6-31 G(d)-LANL2DZ level of theory. b (PCM, solvent = 2-MeTHF)-PBE0-D3/6-311 + + G(d,p)-SDD//B3LYP-D3/6-31 G(d)-LANL2DZ level of theory.
The enantio-determining step in the synthesis of chiral products 2a and 4x was also computationally investigated employing the chiral ligand L9 and L12 coordinated to the CuI center in the irreversible enantio-determining electrophilic addition step (Fig. 7). Upon further observation of structures of these enantio-determining transition states, it is found that in CuL9-(S)-TSB1, there is a significant π\(\cdots\)π interaction between the substrate and the branched phenyl group of L9, which stabilizes the transition state. Above factor results in the free energy difference of 4.8 kcal/mol between the two enantio-determining transition states, ultimately leading to the enantioselectivity of the Büchner-type product. Similarly, in CuL12-(S)-TSB2, there is a fairly strong C-H\(\cdots\)π interaction between the substrate and the branched phenyl group of L12, stabilizing the transition state, accounting for the energy difference of 4.0 kcal/mol between the two enantio-determining transition states, finally leading to the enantioselectivity of the cyclopropane-type product.

All hydrogen atoms are omitted for clarity except for those involved in critical interactions. Relative free energies (ΔΔG, in kcal/mol) were computed at the PCM(toluene)-PBE0-D3/6-311 + + G(d,p)-SDD//B3LYP-D3/6-31 G(d)-LANL2DZ level of theory and PCM(2-MeTHF)-PBE0-D3/6-311 + + G(d,p)-SDD//B3LYP-D3/6-31 G(d)-LANL2DZ level of theory. Color code: red = O; white = H; gray = C; yellow = S; blue = N; brown = Cu.
Discussion
In summary, we have developed an asymmetric Büchner reaction and the related arene cyclopropanation through copper-catalyzed controllable cyclization of N-propargyl ynamides via vinyl cations, enabling divergent and atom-economic synthesis of a wide range of chiral tricycle-fused cycloheptatrienes and benzonorcaradienes in generally excellent yields and enantioselectivities. Significantly, this protocol not only represents an asymmetric arene cyclopropanation reaction of alkynes, but also constitutes an asymmetric Büchner-type reaction based on vinyl cations. Moreover, theoretical calculations further support the mechanism of vinyl cation-involved dearomatized cyclization and elucidate the origin of enantioselectivity. We believe that these findings will offer further perspectives and explorations in the field of asymmetric catalysis based on dearomatization reaction and vinyl cation chemistry.
Methods
General
For 1H, 13C, and 19F nuclear magnetic resonance (NMR) spectra of compounds in this manuscript and details of the synthetic procedures as well as more reaction condition screening, see Supplementary Information.
General procedure for the synthesis of chiral cycloheptatrienes 2
To an oven-dried Schlenk tube with a stir bar were sequentially added Cu(MeCN)4PF6 (0.01 mmol, 3.8 mg), L9 (0.012 mmol, 8.8 mg) and NaBArF4 (0.012 mmol, 10.6 mg, white crystal) under argon atmosphere. After injecting toluene (1 mL) into the Schlenk tube, the mixture was stirred at 25 °C for 2 h. Then the mixture reaction was cooled to −20 °C, and N-propargyl ynamide 1 (0.1 mmol) in toluene (1 mL) was added into the reaction mixture dropwise. The progress of the reaction was monitored by TLC. Upon completion, the reaction mixture was directly purified by column chromatography on silica gel (eluent: PE/EtOAc) to afford the desired chiral cycloheptatriene 2.
General procedure for the synthesis of chiral tetracyclopropanes 4
To an oven-dried Schlenk tube with a stir bar were sequentially added Cu(MeCN)4PF6 (0.015 mmol, 5.7 mg), L12 (0.018 mmol, 15.0 mg) and NaBArF4 (0.0018 mmol, 16.2 mg, white crystal) under argon atmosphere. After injecting 2-MeTHF (1.5 mL) into the Schlenk tube, the mixture was stirred at 25 °C for 2 h. Then the mixture reaction was cooled to 0 °C, and N-propargyl ynamide 3 (0.15 mmol) in 2-MeTHF (1.5 mL) was added into the reaction mixture dropwise. The progress of the reaction was monitored by TLC. Upon completion, the reaction mixture was concentrated under reduced pressure and purified by column chromatography on silica gel (eluent: PE/EtOAc) to afford the desired chiral benzonorcaradiene 4.
Data availability
Data for the crystal structures reported in this paper have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under the deposition numbers 2301623 (2b), 2301624 (for 4 l) and 2301659 (for 5 g). Copies of these data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif. All other data supporting the findings of this study, including experimental procedures and compound characterization, are available within the paper and its Supplementary Information files or from the corresponding authors on request. The coordinates of the optimized structures in this study are provided in the Source Data file. Source data are provided with this paper.
References
Liu, Y.-Z., Song, H., Zheng, C. & You, S.-L. Cascade asymmetric dearomative cyclization reactions via transition-metal catalysis. Nat. Synth. 1, 203–216 (2022).
Zheng, C. & You, S.-L. Advances in catalytic asymmetric dearomatization. ACS Cent. Sci. 7, 432–444 (2021).
Huck, C. J. & Sarlah, D. Shaping molecular landscapes: recent advances, opportunities, and challenges in dearomatization. Chem 6, 1589–1603 (2020).
Xia, Z.-L., Xu-Xu, Q.-F., Zheng, C. & You, S.-L. Chiral phosphoric acid-catalyzed asymmetric dearomatization reactions. Chem. Soc. Rev. 49, 286–300 (2020).
Sheng, F.-T., Wang, J.-Y., Tan, W., Zhang, Y.-C. & Shi, F. Progresses in organocatalytic asymmetric dearomatization reactions of indole derivatives. Org. Chem. Front. 7, 3967–3998 (2020).
Zheng, C. & You, S.-L. Catalytic asymmetric dearomatization by transition-metal catalysis: a method for transformations of aromatic compounds. Chem 1, 830–857 (2016).
Wu, W.-T., Zhang, L. & You, S.-L. Catalytic asymmetric dearomatization (CADA) reactions of phenol and aniline derivatives. Chem. Soc. Rev. 45, 1570–1580 (2016).
Zhuo, C.-X., Zheng, C. & You, S.-L. Transition-metal-catalyzed asymmetric allylic dearomatization reactions. Acc. Chem. Res. 47, 2558–2573 (2014).
Wertjes, W. C., Southgate, E. H. & Sarlah, D. Recent advances in chemical dearomatization of nonactivated arenes. Chem. Soc. Rev. 47, 7996–8017 (2018).
Pape, A. R., Kaliappan, K. P. & Kündig, E. P. Transition-metal-mediated dearomatization reactions. Chem. Rev. 100, 2917–2940 (2000).
Shi, C.-Y., Zhu, G.-Y., Xu, Y., Teng, M.-Y. & Ye, L.-W. Recent advances in catalytic asymmetric Büchner reaction. Chin. Chem. Lett. 34, 108441 (2023).
Shi, C.-Y. et al. Recent advances in transition-metal-catalyzed Büchner reaction of alkynes. Org. Biomol. Chem. 21, 5150–5157 (2023).
Reisman, S. E., Nani, R. R. & Levin, S. Büchner and beyond: arene cyclopropanation as applied to natural product total synthesis. Synlett 17, 2437–2442 (2011).
Yuan, D.-F. et al. Hypervalent iodine promoted the synthesis of cycloheptatrienes and cyclopropanes. Chem. Sci. 13, 478–485 (2022).
Xia, J., Liu, J., Yu, Y., Zhang, J. & Huang, X. Divergent access to polycyclic N-heterocyclic compounds through Büchner-type dearomatization enabled cycloisomerization of diynamides under gold catalysis. Org. Lett. 24, 4298–4303 (2022).
Zeng, Q. et al. Divergent construction of macrocyclic alkynes via catalytic metal carbene C(sp2)–H insertion and the Büchner reaction. ACS Catal. 9, 10773–10779 (2019).
Claus, V. et al. Gold-catalyzed dimerization of diarylalkynes: direct access to azulenes. Angew. Chem., Int. Ed. 57, 12966–12970 (2018).
Nakayama, H., Harada, S., Kono, M. & Nemoto, T. Chemoselective asymmetric intramolecular dearomatization of phenols with α-diazoacetamides catalyzed by silver phosphate. J. Am. Chem. Soc. 139, 10188–10191 (2017).
Wang, H., Zhou, C.-Y. & Che, C.-M. Cobalt-porphyrin-catalyzed intramolecular Büchner reaction and arene cyclopropanation of in situ generated alkyl diazomethanes. Adv. Synth. Catal. 359, 2253–2258 (2017).
Liu, Z. et al. Wang, Transition-metal-free intramolecular carbene aromatic substitution/Büchner reaction: synthesis of fluorenes and [6,5,7]benzo-fused rings. Angew. Chem. Int. Ed. 54, 3056–3060 (2015).
Wang, X., Abrahams, Q. M., Zavalij, P. Y. & Doyle, M. P. Highly regio- and stereoselective dirhodium vinylcarbene induced nitrone cycloaddition with subsequent cascade carbenoid aromatic cycloaddition/N=O cleavage and rearrangement. Angew. Chem. Int. Ed. 51, 5907–5910 (2012).
Panne, P. & Fox, J. M. Rh-catalyzed intermolecular reactions of alkynes with α-diazoesters that possess β-hydrogens: ligand-based control over divergent pathways. J. Am. Chem. Soc. 129, 22–23 (2007).
Galan, B. R., Gembicky, M., Dominiak, P. M., Keister, J. B. & Diver, S. T. Carbon monoxide-promoted carbene insertion into the aryl substituent of an N-heterocyclic carbene ligand: Büchner reaction in a ruthenium carbene complex. J. Am. Chem. Soc. 127, 15702–15703 (2005).
Kennedy, M., McKervey, M. A., Maguire, A. R. & Roos, G. H. P. Asymmetric synthesis in carbon-carbon bond forming reactions of α-diazoketones catalyzed by homochiral rhodium(II) carboxylates. J. Chem. Soc., Chem. Commun. 361–362 (1990).
McCarthy, N. et al. A new rhodium(II) phosphate catalyst for diazocarbonyl reactions including asymmetric synthesis. Tetrahedron Lett. 33, 5983–5986 (1992).
Xu, X., Wang, X., Zavalij, P. Z. & Doyle, M. P. Straightforward access to the [3.2.2]nonatriene structural framework via intramolecular cyclopropenation/Büchner reaction/Cope rearrangement cascade. Org. Lett. 17, 790–793 (2015).
Hoshi, T., Ota, E., Inokuma, Y. & Yamaguchi, J. Asymmetric synthesis of a 5,7-fused ring system enabled by an intramolecular Büchner reaction with chiral rhodium catalyst. Org. Lett. 21, 10081–10084 (2019).
Darses, B., Maldivi, P., Philouze, C., Dauban, P. & Poisson, J.-F. Asymmetric intramolecular Büchner reaction: from high stereoselectivity to coexistence of norcaradiene, cycloheptatriene, and an intermediate form in the solid state. Org. Lett. 23, 300–304 (2021).
Ly, D., Bacsa, J. & Davies, H. M. L. Rhodium(II)-catalyzed asymmetric cyclopropanation and desymmetrization of [2.2]paracyclophanes. ACS Catal. 14, 6423–6431 (2024).
O’Keeffe, S., Harrington, F. & Maguire, A. R. Enantioselective intramolecular Büchner reaction of α-diazoketones. Synlett 15, 2367–2370 (2007).
O’Neil, S., O’Keeffe, S., Harrington, F. & Maguire, A. R. Enhancement of enantioselection in the copper-catalyzed intramolecular Büchner reaction by variation of the counterion. Synlett 14, 2312–2314 (2009).
Slattery, C. N. et al. Investigation of additive effects in enantioselective copper-catalyzed C−H insertion and aromatic addition reactions of α-diazocarbonyl compounds. Synlett 23, 765–767 (2012).
Crowley, D. C., Lynch, D. & Maguire, A. R. Copper-mediated, heterogeneous, enantioselective intramolecular Büchner reactions of α-diazoketones using continuous flow processing. J. Org. Chem. 83, 3794–3805 (2018).
Thanh, N. P. T., Tone, M., Inoue, H., Fujisawa, I. & Iwasa, S. Highly stereoselective intramolecular Büchner reaction of diazoacetamides catalyzed by a Ru(II)-pheox complex. Chem. Commun. 55, 13398–13401 (2019).
Ito, T. et al. Asymmetric intramolecular dearomatization of nonactivated arenes with ynamides for rapid assembly of fused ring system under silver catalysis. J. Am. Chem. Soc. 143, 604–611 (2021).
Zhu, D., Cao, T., Chen, K. & Zhu, S. Rh2(II)-Catalyzed enantioselective intramolecular Büchner reaction and aromatic substitution of donor-donor carbenes. Chem. Sci. 13, 1992–2000 (2022).
Guan, F. et al. Asymmetric dearomative cyclopropanation of naphthalenes to construct polycyclic compounds. Chem. Sci. 13, 13015–13019 (2022).
Otog, N., Gantogos, B., Fujisawa, I. & Iwasa, S. Highly enantioselective synthesis of norcaradiene derivatives from naphthyl diazoacetamides using a Ru(II)-pheox complex. Chem. Commun. 58, 12325–12328 (2022).
Smith, K. L., Padgett, C. L., Mackay, W. D. & Johnson, J. S. Catalytic, asymmetric dearomative synthesis of complex cyclohexanes via a highly regio- and stereoselective arene cyclopropanation using α-cyanodiazoacetates. J. Am. Chem. Soc. 142, 6449–6455 (2020).
Guo, Y., Nguyen, T. V. & Koenigs, R. M. Norcaradiene synthesis via visible-light-mediated cyclopropanation reactions of arenes. Org. Lett. 21, 8814–8818 (2019).
Nani, R. R. & Reisman, S. E. α-Diazo-β-ketonitriles: uniquely reactive substrates for arene and alkene cyclopropanation. J. Am. Chem. Soc. 135, 7304–7311 (2013).
Doyle, M. P., Ene, D. G., Forbes, D. C. & Pillow, T. H. Chemoselectivity and enantiocontrol in catalytic intramolecular metal carbene reactions of diazo acetates linked to reactive functional groups by naphthalene-1,8-dimethanol. Chem. Commun. 1691–1692 (1999).
Zhu, D., Chen, L., Fan, H., Yao, Q. & Zhu, S. Recent progress on donor and donor–donor carbenes. Chem. Soc. Rev. 49, 908–950 (2020).
Liu, X.-J., Xu, Y., Tang, C., Qian, P.-C. & Ye, L.-W. Unactivated C(sp3)−H functionalization via vinyl cations. Sci. China Chem. 65, 20–30 (2022).
Niggemann, M. & Gao, S. Are vinyl cations finally coming of age? Angew. Chem. Int. Ed. 57, 16942–16944 (2018).
Nistanaki, S. K. et al. Catalytic asymmetric C–H insertion reactions of vinyl carbocations. Science 378, 1085–1091 (2022).
Hong, F.-L. et al. Generation of donor/donor copper carbenes through copper-catalyzed diyne cyclization: enantioselective and divergent synthesis of chiral polycyclic pyrroles. J. Am. Chem. Soc. 141, 16961–16970 (2019).
Hong, F.-L. et al. Copper-catalyzed asymmetric reaction of alkenyl diynes with styrenes by formal [3 + 2] cycloaddition via Cu-containing all-carbon 1,3-dipoles: access to chiral pyrrole-fused bridged[2.2.1] skeletons. J. Am. Chem. Soc. 142, 7618–7626 (2020).
Zhu, X.-Q. et al. Copper-catalyzed asymmetric cyclization of alkenyl diynes: method development and new mechanistic insights. Chem. Sci. 12, 9466–9474 (2021).
Hong, F.-L. et al. Copper-catalyzed asymmetric diyne cyclization via [1,2]-Stevens-type rearrangement for the synthesis of chiral chromeno[3,4-c] pyrroles. Angew. Chem. Int. Ed. 61, e202115554 (2022).
Qi, L.-J. et al. Enantioselective copper-catalyzed formal [2 + 1] and [4 + 1] annulations of diynes with ketones via carbonyl ylides. Angew. Chem. Int. Ed. 61, e202210637 (2022).
Chen, Y.-B. et al. Construction of axially chiral arylpyrroles via atroposelective diyne cyclization. Angew. Chem. Int. Ed. 62, e202303670 (2023).
Li, C.-T. et al. Asymmetric formal C–C bond insertion into aldehydes via copper-catalyzed diyne cyclization. Nat. Commun. 14, 7058 (2023).
Chen, Y.-B. et al. Enantioselective functionalization of unactivated C(sp3)–H bonds through copper-catalyzed diyne cyclization by kinetic resolution. Nat. Commun. 15, 2232 (2024).
Hu, Y.-C., Zhao, Y., Wan, B. & Chen, Q.-A. Reactivity of ynamides in catalytic intermolecular annulations. Chem. Soc. Rev. 50, 2582–2625 (2021).
Lynch, C. C., Sripada, A. & Wolf, C. Asymmetric synthesis with ynamides: unique reaction control, chemical diversity and applications. Chem. Soc. Rev. 49, 8543–8583 (2020).
Chen, Y.-B., Qian, P.-C. & Ye, L.-W. Brønsted acid-mediated reactions of ynamides. Chem. Soc. Rev. 49, 8897–8909 (2020).
Hong, F.-L. & Ye, L.-W. Transition metal-catalyzed tandem reactions of ynamides for divergent N-heterocycle synthesis. Acc. Chem. Res. 53, 2003–2019 (2020).
Luo, J. et al. Exploiting remarkable reactivities of ynamides: opportunities in designing catalytic enantioselective reactions. ACS Catal. 10, 13978–13992 (2020).
Zhou, B., Tan, T.-D., Zhu, X.-Q., Shang, M. & Ye, L.-W. Reversal of regioselectivity in ynamide chemistry. ACS Catal. 9, 6393–6406 (2019).
Wang, X.-N. et al. Ynamides in ring forming transformations. Acc. Chem. Res. 47, 560–578 (2014).
McDowell, P. A., Foley, D. A., O’Leary, P., Ford, A. & Maguire, A. R. Asymmetric synthesis of cis-7-methoxycalamenene via the intramolecular Büchner reaction of an α-diazoketone. J. Org. Chem. 77, 2035–2040 (2012).
Chung, S. et al. Synthesis, activity, and structural analysis of novel α-hydroxytropolone inhibitors of human immunodeficiency virus reverse transcriptase-associated ribonuclease H. J. Med. Chem. 54, 4462–4473 (2011).
Michalak, K., Michalak, M. & Wicha, J. Construction of the tricyclic 5-7-6 scaffold of fungi-derived diterpenoids. Total synthesis of (±)-heptemerone G and an approach to Danishefsky’s intermediate for guanacastepene a synthesis. J. Org. Chem. 75, 8337–8350 (2010).
Trost, B. M., Hu, Y. & Horne, D. B. Total synthesis of (+)-frondosin A. Application of the Ru-catalyzed [5 + 2] cycloaddition. J. Am. Chem. Soc. 129, 11781–11790 (2007).
Wright, D. L., Whitehead, C. R., Sessions, E. H., Ghiviriga, I. & Frey, D. A. Studies on inducers of nerve growth factor: Synthesis of the cyathin core. Org. Lett. 1, 1535–1538 (1999).
Levin, S., Nani, R. R. & Reisman, S. E. Rapid assembly of the salvileucalin B norcaradiene core. Org. Lett. 12, 780–783 (2010).
King, G. R., Mander, L. N., Monck, N. J. T., Morris, J. C. & Zhang, H. A new and efficient strategy for the total synthesis of polycyclic diterpenoids: the preparation of gibberellins (±)-GA103 and (±)-GA73. J. Am. Chem. Soc. 119, 3828–3829 (1997).
Mitrenga, M. & Hartmann, R. N-oxide formation causes loss of aromatase inhibitory activity of pyridyl-substituted tetrahydronaphthalenes. Eur. J. Med. Chem. 30, 241–244 (1995).
Hanessian, S. & Schutze, G. Notes. Synthetic penicillins derived from benznorcaradienecarboxylic acids. J. Med. Chem. 12, 529–531 (1969).
Liao, S., Sun, X.-L. & Tang, Y. Side arm strategy for catalyst design: modifying bisoxazolines for remote control of enantioselection and related. Acc. Chem. Res. 47, 2260–2272 (2014).
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (22125108, 22121001 and 22331004; 22122109 and 22271253, X.H.), the National Key R&D Program of China (2022YFA1504301, X.H.), the Zhejiang Provincial Natural Science Foundation of China (LDQ23B020002, X.H.), the Starry Night Science Fund of Zhejiang University Shanghai Institute for Advanced Study (SNZJU-SIAS-006, X.H.), Beijing National Laboratory for Molecular Sciences (BNLMS202102, X.H.), the CAS Youth Interdisciplinary Team (JCTD−2021-11, X.H.), the Fundamental Research Funds for the Central Universities (226−2022-00140, 226−2022-00224 and 226-2023-00115, X.H), the State Key Laboratory of Physical Chemistry of Solid Surfaces (202210, X.H.), the Leading Innovation Team grant from the Department of Science and Technology of Zhejiang Province (2022R01005, X.H.), the Natural Science Foundation of Jiangsu Province (BK20211059), and NFFTBS (J1310024). Dedicated to Professor Yong Tang at Shanghai Institute of Organic Chemistry on the occasion of his 60th birthday.
Author information
Authors and Affiliations
Contributions
Y.X.Z., T.Q.H., X.Li. and B.Z. performed experiments. X.H. and X.Lu. designed the DFT calculations. L.G.L. performed the DFT calculations. L.W.Y. and Z.X. conceived and directed the project and wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ruihan Wang, Yiming Wang, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zheng, YX., Liu, LG., Hu, TQ. et al. Asymmetric Büchner reaction and arene cyclopropanation via copper-catalyzed controllable cyclization of diynes. Nat Commun 15, 9227 (2024). https://doi.org/10.1038/s41467-024-53605-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-53605-7
This article is cited by
-
Ligand-controlled divergent asymmetric C(sp3)−H and C(sp3)−O insertion via vinyl cations
Nature Communications (2025)
