
Abstract
The RNA-dependent RNA polymerase (RdRp), 3C-like protease (3CLpro), and papain-like protease (PLpro) are pivotal components in the viral life cycle of SARS-CoV-2, presenting as promising therapeutic targets. Currently, all FDA-approved antiviral drugs against SARS-CoV-2 are RdRp or 3CLpro inhibitors. However, the mutations causing drug resistance have been observed in RdRp and 3CLpro from SARS-CoV-2, which makes it necessary to develop antivirals with novel mechanisms. Through the application of a structure-based drug design (SBDD) approach, we discover a series of novel potent non-covalent PLpro inhibitors with remarkable in vitro potency and in vivo PK properties. The co-crystal structures of PLpro with lead compounds reveal that the residues D164 and Q269 around the S2 site are critical for improving the inhibitor’s potency. The lead compound GZNL-P36 not only inhibits SARS-CoV-2 and its variants at the cellular level with EC50 ranging from 58.2 nM to 306.2 nM, but also inhibits HCoV-NL63 and HCoV-229E with EC50 of 81.6 nM and 2.66 μM, respectively. Oral administration of the GZNL-P36 results in significantly improved survival and notable reductions in lung viral loads and lesions in SARS-CoV-2 infection mouse model, consistent with RNA-seq data analysis. Our results indicate that PLpro inhibitors represent a promising SARS-CoV-2 therapy.
Similar content being viewed by others

Introduction
Over 670 million people have been infected and over 6.8 million people have died in the worldwide pandemic caused by the SARS-CoV-2 virus according to data from Johns Hopkins University. Despite the efficacy demonstrated by vaccines and targeted small molecule drugs in preventing and treating COVID-19, the ongoing emergence of viral mutations, such as Alpha, Beta, Gamma, Delta and Omicron presents escalating challenges1,2,3,4,5,6. The high mutation frequency of spike protein is responsible for the escape of SARS-CoV-2 from the vaccines. Unlike spike protein, the non-structural proteins, including 3CLpro (nsp5) and PLpro (nsp3), remain conserved among coronavirus functional proteins and show much lower mutation frequency in natural SARS-CoV-2 variants7,8. Their high conservativeness makes 3CLpro and PLpro attractive drug targets. At present, there are several clinically available small-molecule anti-SARS-CoV-2 drugs. Among these drugs, remdesivir, molnupiravir, and VV116 target RNA-dependent RNA polymerase (RdRp)9,10,11, nirmatrelvir, ensitrelvir, atilotrelvir, leritrelvir, and simnotrelvir target chymotrypsin-like protease (3CLpro, also referred as Mpro)12,13,14,15,16. Unfortunately, the clinical efficacy of remdesivir is controversial17, and multiple reported cases have already outlined an increasing observed resistance to remdesivir in immuno-compromised patients undergoing treatment with the drug18,19,20. Molnupiravir is not authorized to be used for patients under the age of 18 due to its bone and cartilage toxicity, and also not applicable for pregnant patients due to the potential risk of major birth defects and miscarriage21. In order to increase the half-life and the in vivo concentration of nirmatrelvir, ritonavir is included as a boosting agent to inhibit the activity of cytochrome P450 3A4 (CYP3A4)22. Ensitrelvir, a novel selective 3CL pro inhibitor, showed favorable clinical antiviral efficacy, albeit having potent CYP3A4 inhibitory activity13,23,24. Although 3CLpro is known as a well-conserved protein, Duan et al. reported several potential mutant sites by which SARS-CoV-2 might evolve the resistance to nirmatrelvir25.
PLpro, a major functional ___domain in SARS-CoV and SARS-CoV-2 non-structural protein 3 (nsp3), is an essential enzyme involved in viral replication and immune evasion26,27,28. PLpro plays an important role in viral transcription and replication by cleaving the peptide bonds in the viral polyprotein, while the deubiquitinating and deISGylating activity of PLpro is related to the immune evasion by antagonizing the host’s innate immune response upon viral infection27. PLpro-mediated deubiquitination of STING disrupted the STING-IKKε-IRF3 complex by removing the K63-linked polyubiquitin chain from LYS289 of STING28. Subsequently, the IFN-I signal pathway was inhibited. These data indicate PLpro is a promising drug target against SARS-CoV-2.
GRL0617, a SARS-CoV PLpro inhibitor, also shows inhibition activity for SARS-CoV-2 PLpro29,30,31. In addition, several other SARS-CoV-2 PLpro inhibitors (Supplementary Table 2) have been reported32,33,34,35. GRL0617 prevents the substrate binding by inducing a conformation change of Y268 on the BL2 loop, which closes the BL2 loop and narrows the binding cleft36. However, GRL0167 displays low potency, from μM to sub-μM, in inhibiting PLpro enzymatic activity. Analogous of GRL0617 have been reported with moderate enzymatic and cellular activities, such as Jun9-84-337, XR8-2434, Jun1127338 and the covalent inhibitors CP739. Exceptionally, Shen et al. substituted the BL2 group in GRL0617 with several other aromatic ring systems, but these modifications did not increase bio-activity. The most promising compound XR-24 has a 2-phenylthiophene group to imitate the interaction of naphthalene at the BL2 groove. This compound has an IC50 of 0.56 μM for PLpro inhibition and an EC50 of 1.2 μM for antiviral effects, which is comparable to GRL061734. Recently, Tan et al. reported a potent PLpro inhibitor Jun12682 with an enzymatic activity IC50 of 106.8 nM and antiviral activity EC50 against different SARS-CoV-2 variants ranging from 0.44 to 2.02 μM40. PF-07957472 was another potent, selective, and orally available PLpro inhibitor with antiviral activity EC50 of 147 nM against SARS-CoV-241. However, there are no approved PLpro inhibitor drugs in clinical treatment. In this study, we synthesized a series of novel PLpro inhibitors and evaluated their activities. The lead compound (GZNL-P36) showed excellent in vitro potency, decent oral in vivo pharmacokinetic (PK) properties, and more importantly displays similar in vivo antiviral efficacy in SARS-CoV-2 infected mice with ensitrelvir.
Results
Structure-based discovery and optimization of novel PLpro inhibitors
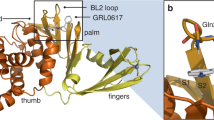
The co-crystal structure of GRL0617 reveals the compound bound at a shallow pocket on the protein surface (Fig. 1a), which has been recognized as the substrate binding site, contacting various hydrophobic and hydrophilic residues (Fig. 1b). The naphthalene ring sandwiched in the BL2 groove (i.e., S1 site) is a critical group for the binding of GRL0617. In previous studies, most PLpro inhibitors keep the naphthalene ring as the group that binds to the BL2 groove42, and replacing this group with alternative aromatic ring systems can somewhat maintain but does not increase bio-activity exampled by the study of XR-2434. We hypothesized that compound binding affinity can be enhanced through the substitution of naphthalene with a bulkier substitute, as the naphthalene ring does not fully occupy the entire surface of the groove, in particular the area that includes residue P247. We first introduced a tricyclic structure, i.e., the 1,2-dihydroacenaphthylene (DHAN) group as a replacement of naphthalene ring to expand the hydrophobic contact surface with P247, and GZNL-P1 was synthesized (Fig. 1c). Encouragingly, its inhibitory activity (IC50 = 2.83 μM) is better than GRL0617 (IC50 = 4.82 μM) (Fig. 1d, Supplementary Table 3), indicating the beneficial effect of introducing a bulkier substitute in the BL2 groove. This modification shares similarity with the design of the recently reported highly potent inhibitor, Jun12682, which extends the interaction in the BL2 groove by an aryl substitution40. Furthermore, we found that compound potency can be improved more than 10-fold when we simultaneously changed the substituent in the linking group (L) from methyl to cyclopropyl, resulting in GZNL-P3 (IC50 = 185.80 nM) (Fig. 1c and d, Supplementary Table 3). Notably, the cyclopropyl group is a much better substituent than the di-methyl, as the IC50 of GZNL-P2 is 6.90 μM. This suggests that the linker part is also very important for increasing activity, which is just confirmed by the companion study that reported a potent PLpro inhibitor PF-07957472 with the same cyclopropyl linker41. Therefore, we reasoned that GZNL-P3 serves as a good starting point for further lead optimization.

a Analysis of potential ligand binding sites S1 and S2 (PDB: 7JRN). The critical hydrogen bonds between GRL0617 and PLpro are shown as marine dash lines. GRL0617 and the key residues of the ligand binding pocket are shown as cyan sticks and wheat sticks, respectively. b Strategies of structure-guided compound design. The optimized groups are shown as colored circles, bright orange for hydrophobicity, and split pea for hydrophilicity. c Procedures of activity optimization and in vitro PK optimization indicated by the representative compounds. d The inhibition activity on SARS-CoV-2 PLpro of the representative compounds. Data are presented as mean of two (GZNL-P1, GZNL-P3, GZNL-P17, GZNL-P35, and GZNL-P36) or three (GRL0617 and GZNL-P4) technical replicates. Three independent experiments were tested and shown in Supplementary Table 3, only one representative is shown here. e The in vivo PK profiling of GZNL-P35 and GZNL-P36 at 10 mg/kg and 20 mg/kg (n = 3 per group). f The in vitro stability in liver microsome of the representative compounds. g Summary of in vivo PK, metabolism, distribution, and toxicity properties of GZNL-P36. Source data of (d) and (e) are provided as a Source Data file.
Previous work done by Shen et al. has shown that it is possible to engage positively charged amine groups on the benzene of GRL0617 to interact with residue E167 at the S2 site34. This interaction could enhance activity by forming salt bridge and hydrogen bonding. A docking calculation-based library design was carried out to explore R1 groups (Fig. 1b, c) that can form a salt bridge with E167. Forty-one library compounds were selected for synthesis based on docking score, and it was found that piperazine derivatives generally exhibit strong inhibition on PLpro. Meanwhile, compounds with 3-substituted azetidines also showed excellent activity against PLpro. Remarkably, GZNL-P17 is the most potent compound in this series with an IC50 of 2.91 nM, which is more potent than the best piperazine derivative, i.e., GZNL-P4 (IC50 = 36.29 nM). However, modifying the methyl group on the ortho position of the benzene ring (i.e., R2 group in Fig. 1b), which extends to the recently identified important residue L16243, leads to a decrease in antiviral activity. At this stage, we successfully achieved potent enzymatic activity that is hundreds of times improved compared to the parent compound GRL0617. The most active compounds, GZNL-P4 and GZNL-P17, were then selected to measure their antiviral activity against both wild-type SARS-CoV-2 and its two epidemic variants (Supplementary Fig. 4, Supplementary Table 6) in infected VeroE6 cells and sub-micromolar anti-viral potency was achieved, which is much improved comparing with GRL0617 (EC50 = 23.64 μM37). To evaluate their ADME properties, in vitro liver stability of GZNL-P4 and GZNL-P17 (Fig. 1f) was measured. In rat liver microsomes, the half-life time (T1/2) of both compounds is lower than 15 minutes and their intrinsic clearance rate (Clint) is very high. Moreover, they have worse stability in human liver microsomes. Metabolite identification work of GZNL-P4 indicates that compound instability could partially be attributed to the oxidation of the DHAN ring (see metabolites analysis in Supplementary Fig. 1). To address the liver stability problem, we changed the DHAN ring to the benzoindolone ring. To our delight, for GZNL-P35 and 36, the liver stability was considerably improved (Fig. 1f), while their enzymatic potencies were maintained at the same level, 8.15 nM and 6.45 nM, respectively (Fig. 1d, Supplementary Table 3). The inhibitors can stabilize PLpro, hence, they increase the unfolding or melting temperature (Tm) of SARS-CoV-2 PLpro (Supplementary Fig. 3h). The cellular antiviral activity for benzoindolone compounds were examined, where GZNL-P31, 35, and 36 exhibited strong antiviral activity against different SARS-CoV-2 variants, for example, against XBB.1 strains with the EC50 of 41.6 nM, 43.3 nM and 58.2 nM, respectively (Fig. 3e, Supplementary Fig. 4c, Supplementary Table 5). These benzoindolone compounds also showed low toxicity to normal cell HEK293T with CC50 of 157.4 μM, 67.67 μM, and 88.41 μM, respectively (Table 1). Overall, GZNL-P35 and 36 demonstrate the most favorable profile in terms of potency and liver metabolic stability. The overall workflow for lead optimization is shown in Fig. 1b, c. Bioactivity data of selected compounds are listed in Fig. 1d and Table 1.
An in vivo PK study was carried out for GZNL-P35 and 36 using 3 male CD1 mice (SPF level) per group with a dosage of 10 mg/kg, both compounds can reach the maximum plasma concentration at 1.58 and 1.67 h (Tmax), respectively with a peak plasma concentration (Cmax) of 227 and 549 ng/mL (Fig. 1e). However, the clearance of GZNL-P35 (T 1/2 = 0.96 h) is much faster than GZNL-P36 (T 1/2 = 1.45 h). This results in an enhancement of the performance of GZNL-P36 on drug blood exposure. Particularly, the bioavailability (F%) of GZNL-P36 is much higher than GZNL-P35. Further profiling of PK properties demonstrated that GZNL-P36 exists significantly weaker inhibition on major metabolic enzymes (CYP 1A2, 2C9, 2C19, 2D6, and 3A4) in the liver than GZNL-P35 (Fig. 1g, Supplementary Table 4). Additionally, its moderate hERG inhibition should be taken with care in the future following studies. In summary, the strong in vitro activity and good PK properties of GZNL-P36 make it suitable to move forward to in vivo efficacy study.
X-ray crystal structures of SARS-CoV-2 PLpro with inhibitor
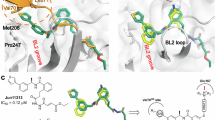
To clarify the binding mechanism of inhibitors, the co-crystal structures of SARS-CoV-2 PLpro with GZNL-P4, 28, 31, and 35 were determined (resolution range of 1.7 to 2.6 Å; Fig. 2, Supplementary Table 1). These compounds have similar binding patterns, and the unbiased electron density for these inhibitors are unambiguous. The amide structures of GZNL-P4, 28, and 31 form two hydrogen bonds with residues Q269 and D164, whereas GZNL-P35 forms hydrogen bonds between two amide groups with D164 and E167. Compared to the naphthalene ring of GRL0617, the characteristic tricyclic group (DHAN or benzoindolone group) at the BL2 groove makes a similar π-π stacking interaction with Y268 and further expands its contact surface with P247 and P248 (Fig. 2). For these compounds, the substitution with methyl-substituted piperazine at R1 makes additional hydrogen bond and salt bridge with the residue E167 and Q269 that is responsible for the significant potency enhancement (Fig. 2). Based on the remarkable enhancement of enzymatic activity and the structure comparison (Table 1) of GZNL-P1 and 3, it is clear that the cyclopropyl group is critical for the enzymatic inhibition activity improvement. Comparison among co-crystal structures of PLpro with GRL0617 (PDB: 7JRN), GZNL-P4, and 35 shows that the cyclopropyl group makes the plane of amido bond rotate 42.7 degrees for GZNL-P4 and 34.2 degrees for GZNL-P35 (Supplementary Fig. 2j, k) comparing to that of GRL0617. This conformational change enables GZNL-P4 and GZNL-P35 to form a hydrogen bond with the residue Y264 (Supplementary Fig. 2d-f). In addition, the CH-π interaction between the cyclopropyl group and the aromatic side chain of Y264 is another favourable factor.

X-ray co-crystal structure of SARS-CoV-2 PLpro with GZNL-P4 (a), GZNL-P28 (b), GZNL-P31 (c), and GZNL-P35(d). The residues interacting with the ligand are shown as yellow sticks, GZNL-P4 (PDB: 8YX2), GZNL-P28 (PDB: 8YX3), GZNL-P31 (PDB: 8YX4), and GZNL-P35 (PDB: 8YX5) are shown as brown sticks. Hydrogen bonds are shown as black dashed lines and the water molecules are shown as small red spheres. The distances of hydrogen bonds and the residues are labeled. The omitted electron densities (2Fo-Fc) of GZNL-P4, 28, 31, and 35 were shown at 1 σ level. The values of IC50 are presented as mean of two (GZNL-P28, GZNL-P31, and GZNL-P35) or three (GZNL-P4) technical replicates.
The mechanism of GZNL-P36 in inhibiting SARS-CoV-2 PLpro
PLpro plays a key role in the proteolytic processing of viral polyproteins and the dysregulation of the host immune response. The deubiquitylation and de-ISGylation activity of PLpro is related with the host innate immune pathways and the innate immune evasion of SARS-CoV-227. To characterize the enzymatic inhibition of the designed compounds, we performed a PLpro enzymatic assay using the labeled peptide substrate RLRGG-AMC (GLPBIO, GA23715). The final selected candidate compound GZNL-P36 showed potent enzymatic inhibition with IC50 value of 6.45 nM, compared to 4.82 μM for reference compound GRL0617 and 36.29 nM for the lead compound GZNL-P4 (Fig. 1d, Supplementary Table 3). To investigate the thermodynamic profile of the binding between ligands and PLpro, we performed isothermal titration calorimetry (ITC) experiments. The measured binding affinities of GRL0617, GZNL-P35 and GZNL-P36 with PLpro are 1.82 μM, 5.83 nM and 24.6 nM, respectively (Supplementary Fig. 3a-d). The structure optimization from GRL0617 to GZNL-P35 and GZNL-P36 is driven by favorable changes in enthalpy (ΔH) and entropy ( − TΔS). The improvement of Gibbs free energy (ΔG) from GRL0617 to GZNL-P4/GZNL-P17 and GZNL-P35/GZNL-P36 is benefited from the substitution of naphthalene by the tricyclic group of DHAN or benzoindozolone and the additional N-methyl-substituted piperazine group (GZNL-P4) or bridged piperazine (GZNL-P35) that contribute bigger interaction area with PLpro (Supplementary Fig. 2a–c). In addition, the piperazine group also forms a hydrogen bond with residue E167 (Fig. 2). The increase of van der Waals interactions and the additional hydrogen bond formation are responsible for the decrease of enthalpy in the structure optimization44. To investigate the binding kinetic properties between ligand and receptor, we performed biolayer interferometry (BLI) to obtained the binding association constant (Kon), dissociation constant (Koff), and equilibrium dissociation constant (Kd) of GRL0617, GZNL-P4, GZNL-P36 binding to PLpro. The Kd values obtained by BLI are consistent with those from ITC, with values of 25.0 μM, 282 nM and 7.36 nM for GRL0617, GZNL-P4 and GZNL-P36, respectively (Supplementary Fig. 3e-g). The change of Kon from GRL0617 to GZNL-P4 and GZNL-P36 are mainly due to the enhance of hydrophobic interaction through the substitution of naphthalene by a bulkier tricyclic acenaphthylene group and the additional of piperazine containing group, increasing the polar contact due to the formation of hydrogen bond between the piperazine group of GZNL-P4/GZNL-P36 and the residue E16744. The thermal unfolding (Tm) of PLpro determined by differential scanning fluorimetry (DSF) assay indicated that PLpro is significantly stabilized by the ligand binding, the ΔTm of PLpro with or without incubation of GRL0617, GZNL-P4, GZNL-P17, GZNL-35, and GZNL-P36 increased from 6.3 to 19.5 °C (Supplementary Fig. 3h). Comparison of the binding conformations of PLpro with substrate RLRGG (PDB: 6YVA) and inhibitors (Supplementary Fig. 2g–i) suggests that enzymatic inhibition activity correlates with the blocking of substrate binding to PLpro by inhibitors. Specifically, it has been demonstrated that the binding of GRL0617 to PLpro impedes residue L73 of the substrate (72RLRGG76), while the piperazine group of GZNL-P4 and GZNL-P35 also obstructs residue R72 of the substrate.
Evaluation of in vitro antiviral activity cross coronavirus family
To test whether GZNL-P36 could effectively inhibit PLpro across multiple coronavirus subtypes, we performed a fluorescence resonance energy transfer (FRET) inhibition assay against PLpro proteins from different species coronaviruses from genera alpha-, beta-, gamma-, and delta-coronaviruses (Fig. 3a, b, Supplementary Fig. 5). GZNL-P36 exhibited broad maximum inhibition efficacy against PLpro derived from all coronaviruses tested (Fig. 3a). The cellular antiviral activity of GZNL-P36 was examined by a cell protection assay. In this assay, the cytopathic effect (CPE) of SARS-CoV-2-infected Vero E6 cells with or without treatment by the compounds was assessed using Celigo Image Cytometer45. The cells were challenged with WT SARS-CoV-2 and two other variants Omicron BA.5 and XBB.1. GZNL-P36 dose-dependently protected cells from death with 50% effective concentration (EC50) values for wild type (WT), Omicron BA.5 and XBB.1 is 111.0 nM, 306.2 nM and 58.2 nM, respectively. Compared with S-217622 (Ensitrelvir), GZNL-P36 possessed similar effective anti-viral activity for SARS-CoV-2 and its variants (Fig. 3c–e, Supplementary Table 5). Besides SARS-CoV-2, GZNL-P36 also illustrated anti-viral activity for the other coronaviruses, such as HCoV-NL63 (EC50: 81.6 nM) and HCoV-229E (EC50: 2.66 μM) (Fig. 3f, Supplementary Table 5). Together, our data demonstrated that GZNL-P36 rendered superb cross-protection against SARS-CoV-2, HCoV-229E, and HCoV-NL63, exhibiting potent broad-spectrum anti-coronaviral efficacy.

a Enzymatic maximum inhibition efficacy of GZNL-P36 against coronaviruses PLpro. b The phylogenetic tree of coronaviruses PLpro used in this experiment (Shang, J. (2024) BioRender.com/b84m836). Antiviral activity of GZNL-P36 against SARS-CoV-2 wild type (WT) (c), variants Omicron BA.5 (d), and XBB.1 (e). Vero E6 cells were pre-treated with indicated compounds with different concentrations for 1 h and then infected with SARS-CoV-2 wild type (WT), variants Omicron BA.5, and XBB.1 at an MOI of 0.01. The EC50 was assessed after being cultured for three days. f The representative inhibition curves of GZNL-P36 against HCoV-229E and HCoV-OC43 in Huh-7 cells. The EC50 was assessed after being cultured for two days. Data are presented as mean of two (HCoV-NL63 in f) or mean ± s.d. of three (a, c, d, e, and HCoV-OC43 and HCoV-OC229E in f) technical replicates. The error bars are mean ± s.d. Three independent experiments were tested and shown in Supplementary Table 5, only one representative is shown here. Source data are provided as a Source Data file.
In vivo antiviral efficacy of GZNL-P36
To assess the in vivo anti-viral activity of GZNL-P36, we treated the model mice infected with SARS-CoV-2 XBB.1 by oral administration (Fig. 4a). K18-hACE2 transgenic mice aged 8 weeks were used as our mouse model, forty-eight female hACE2 transgenic mice were divided into six groups with eight mice in each group to evaluate the efficacy of mock, vehicle, positive comparator S-217622 of 25 milligrams per kilograms (mpk), and GZNL-P36 of 25 mpk, 50 mpk and 100 mpk in the therapeutic treatment. The weight loss plot shows the about 15% loss of the vehicle group, but the weight loss is less than 10% for the treated groups (Fig. 4b). The lung live viral titers cannot be detected (Fig. 4c) for the group-treated with GZNL-P36 at 100 mpk. The groups treated by GZNL-P36 at the dose of 25 mpk or 50 mpk also showed significant viral titer decrease at 2 days post-infection. The anti-viral efficiency of GZNL-P36 is slightly weaker than the same dose of positive drug S-217622. Immunohistochemistry assays with SARS-CoV-2 nucleocaspid protein antibody revealed that abundant expression of viral antigen was identified in the lung of vehicle-treated mice at 4 days post-infection (Fig. 4d). In contrast, GZNL-P36 treatment, even when administered after the virus challenge, markedly suppressed viral nucleocapsid protein expression in the lung (Fig. 4d). As the concentration of GZNL-P36 increased, the amount of stained viral N protein decreased, and in the group treated with 100 mg/kg of GZNL-P36, it became undetectable. The amount of viral N protein in the 25 mg/kg of S-217622 treatment group was comparable to that in the 25 mg/kg and 50 mg/kg of GZNL-P36 treatment group. Next, haematoxylin and eosin (H&E) stained lungs indicate that the lung was significantly protected from the GZNL-P36 treatment (Fig. 4e). Compared to the vehicle group, all treated groups showed significant improvement in the lung lesion. Among them, the groups treated with 25 mg/kg and 50 mg/kg of GZNL-P36 showed slightly better improvement than the group treated with 25 mg/kg of S-217622, with the 100 mg/kg of GZNL-P36 treatment group exhibiting the greatest improvement.

(a) Experimental design for the 4-day experiment in K18-ACE2 mouse (Shang, J. (2024) BioRender.com/p67u335). (b) Body weight loss of mice from different groups (n = 5 per group). (c) Live viral titers in lungs collected at 2 d.p.i. (n = 5 per group) (d) and (e) Lungs collected at 4 d.p.i. from different groups were immunostained with SARS-CoV-2 nucleocaspid protein antibody (d) or stained with haematoxylin and eosin (H&E) (e) (n = 5 per group). The data are representative of at least two experiments. The error bars are mean ± s.d. Statistical differences were determined by Ordinary one-way ANOVA with Dunnett multiple comparisons test for the body weight on day 4 in (b) and the live viral titer in c. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; ns, not significant. Source data of (b) and (c) are provided as a Source Data file.
PLpro can dysregulate the host inflammation and antiviral response due to its deubiquitinating activity. The transcription levels of inflammatory genes, including Cxcl10, IFNB, and IFN-g, were determined with the lungs of the mice collected at 2 d.p.i. Compared to the vehicle group, the transcription level of these pro-inflammatories in GZNL-P36 treated groups was significantly decreased. Furthermore, the transcription level of Cxcl10, IFNB, and IFN-g was tested by qRT-PCR. Compared to the vehicle group, the transcription level of Cxcl10 and IFN-g in both S-217622 and GZNL-P36 treated groups were significantly decreased (Fig. 5). The transcription level of Cxcl10 and IFN-g in the S-217622 treatment group (25 mg/kg) were comparable to those in the 25 mg/kg of GZNL-P36 treatment group, and were significantly lower in the 50 mg/kg and 100 mg/kg of GZNL-P36 treatment groups (Fig. 5a–c). These results indicated that PLpro inhibitors may provide more benefits on the anti-inflammation properties than S-217622.

Relative mRNA expression of Cxcl10 (a), IFNB (b), and IFN-g (c) of the lungs collected at 2 d.p.i. Three technical replicates were tested for each mouse sample. Each dot represents one mouse at the indicated time point. Source data are provided as a Source Data file.
We further performed bulk RNA sequencing on lung samples of all SARS-CoV-2 infected mice. GSVA analysis results revealed that GZNL-P36 successfully reversed most of the SARS-CoV-2-induced changes, identical to the 3CL pro inhibitor S-217622 (Fig. 6). More importantly, high dose of GZNL-P36 greatly reversed the GSVA scores of both WP_FOXP3_IN_COVID19 and WP_PATHOGENESIS_OF SARSCOV2_MEDIATED_BY_NSPINSP10_COMPLEX genesets, while S-217622 failed to reverse these genes, suggesting our GZNL-P36 might possess synergistic effect on the recovery of SARS-CoV-2 infected mice comparing with the 3CL pro inhibitor S-217622 (Supplementary Fig. 6).

GSVA scores of selected genesets on bulk RNAseq data of GZNL-P36 in SARS-CoV-2 infected Mice (n = 5 per group). Genesets were obtained from MSigDB and curated based on a criterion of including keywords such as “SARS” or “COVID”. The data were then scaled, GSVA scores ranging from -1 (blue, down-regulated) to 1 (red, up-regulated). Source data are provided as a Source Data file.
Discussion
Small molecule antiviral drugs targeting the conserved viral proteases are particularly important therapeutic modalities, as vaccines and neutralizing antibodies cannot provide complete protection against the continuously emerging variants of SARS-CoV-2. At present, there are several clinically available drugs targeting RdRp and 3CLpro. Unfortunately, the resistance mutations of RdRp19,20 and 3CL pro 25,46,47,48 have been reported. PLpro is another important potential SARS-CoV-2 antiviral drug target, yet no drug targeting PLpro has been reported. Starting from a weak PLpro inhibitor GRL0617, a series of novel benzoindolone PLpro inhibitors was discovered through the utilization of docking-based library design. Our lead compound GZNL-P36 shows excellent PLpro inhibitory potency and decent pharmacokinetics and in vitro safety profile.
To investigate the broad-spectrum antiviral activity against various coronaviruses, we tested the inhibitory activity of GZNL-P36 by enzymatic assay and antiviral cell models. The substrate peptide used in our enzymatic assay is highly potent for SARS-CoV and SARS-CoV-2 PLpro, but much lower for others, which makes it challenging to measure inhibition IC50. For comparison, we calculated the maximum inhibition rate of GZNL-P36 for different coronavirus PLpros at the concentration of 20 μM for SARS-CoV and SARS-VCo-2, 100 μM for the other (Fig. 3a, Supplementary Fig. 5). Thus, the broad-spectrum activity against PLpros of various coronaviruses is not very significant due to the low substrate efficiency. The cellular antiviral activity of GZNL-P36 against HCoV-NL63 and HCoV-229E is much better than that of enzymatic inhibition activity (Fig. 3c-f, Supplementary Fig. 5). This result implies that the low efficiency of enzymatic inhibition activity is most likely due to the low substrate activity. It is certainly worthwhile to develop highly potent substrate peptides of other coronavirus PLpros for the investigation of broad-spectrum activity. Considering that the antiviral efficiency of GZNL-P36 (Fig. 3f) is much higher than its enzyme inhibition activity (Supplementary Fig. 5), the possibility for off-target effects of the compound in cells cannot be excluded. To further validate the broad-spectrum antiviral activity of GZNL-P36, additional studies on PLpro from various coronaviruses is needed in the future.
To investigate the in vivo anti-viral efficacy, we evaluated the in vivo anti-viral efficacy of GZNL-P36 against SARS-CoV-2 XBB.1 variants in the established H11-K18-hACE2 transgenic mouse model. In comparison to other small-animal models for COVID-19, the H11-K18-hACE2 mouse model allows for robust SARS-CoV-2 replication with lethal outcomes and significant body weight loss. This model effectively mimics the human severe COVID-19 phenotype, which makes it an ideal model for evaluating the drug candidates in ameliorating severe SARS-CoV-2 infections15,45. In the 100 mg/kg GZNL-P36 treated group, we noticed a significant decrease in the body weight of some mice compared to those of the vehicle group. In order to confirm whether the weight loss of mice was due to the toxicity of GZNL-P36, we carried out an acute toxicity experiment at doses of 300 mg/kg and 600 mg/kg, and the body weight did not decrease after 4 days of administration (Supplementary Fig. 7a, b). No apparent lesions were observed in the heart from the GZNL-P36 treated groups (Supplementary Fig. 7c). Furthermore, on the fourth day, the weight decrease of the mice in GZNL-P36 treated group is still less than that of the vehicle group and several clinically available COVID-19 drugs, such as PF-07321332, RAY1216 etc, also demonstrated the same pattern of body weight loss on the fourth day in their animal studies15. So, the observed weight change of mice in the treatment group with GZNL-P36 is unlikely due to compound toxicity. At the same time, we predicted the possible metabolites of GZNL-P36 using the online server GLORY49,50, the acute toxicity results suggested that GZNL-P36 and its metabolites (Supplementary Table 7) did not lead to acute adverse effects on the body.
Furthermore, this compound shows strong in vivo anti-viral efficacy in antiviral mice model suggesting its potential as a COVID-19 therapy candidate. Interestingly, compared to S-217622, GZNL-P36 treatment showed lower expression of the pro-inflammatory genes. Our results demonstrate that PLpro is an attractive druggable antiviral target and PLpro inhibitor is a class of promising antiviral drug with dual-effect on antiviral and anti-inflammation.
Methods
Our research complies with all relevant ethical regulations. Animal Ethics Committee at Guangzhou Customs Inspection and Quarantine Technology Center (IQTC2023015) and Pharmaron’s Institutional Animal Care and Use Committee (PK-M-07182023, PK-D-06012023, 24-165) approved the study protocols.
Docking method
Molecular docking was carried out using the Glide docking algorithm integrated in the software of Schrödinger (version 2020). The co-crystal structure of SARS-CoV-2 PLpro with GRL0617 (PDB code: 7JRN) was selected as the receptor model for docking. The protein structure was preprocessed using the Protein Preparation Wizard in the software. Bond orders were assigned to all bonds in the structure properly. Amino acids with missing side chains were also repaired. In addition, all crystal waters were removed. The Epik algorithm was chosen to generate ionization and tautomeric states at the pH of 7.0, and the structure was minimized using the OPLS3 force field until the heavy atoms converged to 0.3 Å root mean square deviation to the original coordinates. After this, the minimized structure was then used to prepare the receptor grid for Glide docking, which defined a box space centered on the gravity of the crystal ligand as the docking site. On the other hand, ligands for docking were prepared using the LigPrep module in Schrödinger. Similar to protein preprocessing, the ligands were protonated using the Epik algorithm. Tautomers and stereoisomers were also enumerated. One lowest energy conformation per compound was generated using the OPLS3 force field. The prepared ligands were then docked to the predefined docking site. Standard docking precision was employed, where flexible mode was adopted for ligand conformation sampling. At most 10 poses per ligand were eventually reserved after docking and they were ranked by the Glidescore, which is a scoring function for pose measurement. The poses which did not map to the binding mode of GZNL-P3, including the π-π interaction within the BL2 groove and the two hydrogen bonds with Q269 and D164 mediated by the amide group were removed and the rest went through further analysis.
Plasmid Construction, Protein Expression and Purification
The papain-like protease ___domain sequence was obtained from the SARS-CoV-2 complete genome (NCBI gene databank, severe acute respiratory syndrome coronavirus 2, Gene ID: 43730578). The PLpro Ubl ___domain protein sequence (amino acids, 746-1060) of Nsp3 protein from SARS-CoV-2 (Nsp3, YP_009742610.1) was codon optimized, synthesized, and cloned into pET28a with BamHI and XhoI (Tsingke). The TEV protease recognition sequence was inserted between the N-terminal His6-tag and PLpro sequence. The mutant of PLpro for C111S (PLproC111S) was generated by polymerase chain reaction (PCR). BL21(DE3) Escherichia coli competent cells were used for protein expression. The competent cells transformed with PLpro expression plasmid were inoculated in LB medium and grown to an OD600 of 0.6-0.8, 0.5 mM of isopropyl-D-thiogalactopyranoside (IPTG), and 1 mM zinc chloride (ZnCl2) were used to induce protein production at 16°C for 18 hours. Cell pellets were collected by centrifugation at 3000 × g for 30 minutes and then stored at −80 °C for use. The cell pellets were thawed and resuspended with buffer A [50 mM Tris-HCl, 150 mM NaCl, 10 mM imidazole, and 1 mM TCEP (pH 7.4)] supplemented with lysozyme, deoxyribonuclease, and cOmpleteTM protease inhibitor cocktail (Roche). After lysed by sonication, the cell lysates were separated by centrifugation at 18,000 × g for 35 minutes twice at 4 °C. The supernatant was further filtered using a 0.22 μm pore-size membrane. His-tagged proteins were loaded onto a HisTrap HP column (5 ml; GE) equilibrated with buffer A for 10 column volumes. The unspecific binding proteins were washed with buffer A for 16 column volumes and the target protein was eluted with buffer B [50 mM Tris-HCl, 150 mM NaCl, 250 mM imidazole, and 1 mM TCEP (pH 7.4)] for 20 column volumes by linear gradient. The His-tag was removed by TEV protease during dialysis. Then, the protein cleaved His-tag was concentrated to 2 ml and loaded onto Superdex 75 column (120 ml; GE) equilibrated with buffer C [20 mM Tris-HCl, 100 mM NaCl, and 1 mM TCEP (pH 7.4)] for 1 column volumes.
The recombinant expression plasmids of other PLpros were constructed following the same procedure of SARS-CoV-2 PLpro. The protein expression and purification of these PLpros are also following the similar protocol of SARS-CoV-2 PLpro with minus changes. The PLpros tested in this study are from four different subfamily coronaviruses, including alpha-coronavirus (TEGV-PLpro, Mink-PLpro, Feline-PLpro, HCoV-NL63-PLpro, HCoV-229E-PLpro), beta-coronavirus (HCoV-SARS-PLpro, HCoV-SARS-2-PLpro, Equine-PLpro,), delat-coronavirus (pigeon-PLpro, PDCoV-PLpro, quail-PLpro), and gamma- coronavirus (Avian-PLpro).
PLpro enzymatic assay
The activity of PLpro was measured using the labeled peptide Z-RLRGG-AMC (GLPBIO, GA23715) as substrate in a black 384-well plate (GREINER, 784076), using wavelengths of 340 nm and 460 nm for excitation and emission, respectively. The assay buffer contains 50 mM HEPES, 10 mM DTT, 0.1 mM EDTA, 0.005% tween 20, pH7.2. GRL0617 (TargetMol, Shanghai, China) was used as the positive control. The gradient-diluted compounds were added into PLpro solution diluted with assay buffer and incubated for 10 minutes. The substrate peptide diluted with assay buffer was added into PLpro and inhibitor mix solution and incubated for 1 hour at 37 °C. The final concentrations are 10 nM and 20 μM for PLpro and substrate, respectively. Fluorescence intensity was monitored with a BioTek Neo2 multimode plate reader (Agilent). The results were plotted as dose inhibition curves using nonlinear regression to determine the IC50 values of inhibitor compounds using GraphPad Prism 9.0.
For the representative compounds, three independent experiments were performed and one representative is shown in Fig. 1d. The values of IC50 from 3 independent experiments are shown in Supplementary Table 3.
The enzymatic inhibition efficacy of GZNL-P36 against different coronavirus PLpros was calculated:
Where Top and Bottom are taken from the corresponding terms in the nonlinear fitting formula,
Then, all the efficacies of PLpros are normalized to that of SARS-CoV-2 PLpro.
Crystallization, data collection, and structure determination
Purified SARS-CoV-2 PLproC111S was incubated with inhibitors (GZNL-P4, GZNL-P28, GZNL-P31, and GZNL-P35) at a 1:3 protein/ligand molar ratio in buffer overnight and concentrated to 8 mg/ml. Protein-ligand complex crystals were obtained after 7-10 days at 4 °C by sitting-drop vapor diffusion against 40 μL of well solution using 96-well crystallization plates. The crystallization drops contained 0.75 μL of protein complex sample mixed with 0.75 μL of reservoir solution. The crystals with well diffraction ability were obtained in conditions containing 0.1 M HEPES pH 7.5, 0.2 M Lithium sulfate monohydrate, and 25% (w/v) Polyethylene glycol 3350 for GZNL-P4; 0.03 M Citric acid, 0.07 M BIS-TRIS propane pH 7.6, 20% w/v Polyethylene glycol 3,350 for GZNL-P28; 5 mM CoCl2-6H2O, 5 mM NiCl2-6H2O, 5 mM CdCl2-H2O, 5 mM MgCl2-6H2O, 0.1 M HEPES pH 7.5, 12% w/v PEG3,350 for GZNL-P31; 0.1 M Bis-Tris pH 6.5, 0.2 M Magnesium chloride hexahydrate, and 25% (w/v) Polyethylene glycol 3350 for GZNL-P35.
X-ray diffraction data were collected at beamline BL19U1 and BL02U1 at the Shanghai Synchrotron Radiation Facility and processed with the program autoPROC1.051 and Aquarium52. The structures were solved by molecular replacement using the program Phaser53 integrated in PHENIX54 package using the previously published PLpro structure (PDB code: 7CJD)36 as the search model. The structures were refined using PHENIX with several cycles of manually interactive model rebuilding in Coot55. The ligand constraints were generated using Phenix.elbow56 and the ligand models were built according to the omit map. The data collection and refinement statistics are summarized in Supplementary Table 1.
Bio-layer interferometry (BLI)
BLI assay was carried out at 25 °C using Octet R8 (Sartorius) with phosphate buffer solution (PBS) containing 0.005% tween-20 (PBST). Biotinylation SARS-CoV-2 PLpro protein was produced using a commercially available kit (G-MM-IGT, Genemore). The PLpro protein (50 μg/mL) was immobilized onto Super Streptavidin SSA capture biosensors (18-5070, Sartorius) pre-equilibrated with PBST. The biosensors were then exposed to different concentrations of the compound prepared with PBST for association, and followed by the dissociation in PBST. Data was analyzed using the Octet BLI Analysis software.
Isothermal titration calorimetry (ITC)
ITC assays were carried out at 25 °C with 19 injections, including the first pre-injection of 0.4 μL and 18 injections of 2 μL using a MicroCal PEAQ-ITC instrument (Malvern Panalytical). Reference power was set to 5 μcal/s, and the compound solution was titrated at 150 s. The PLpro proteins and compounds were diluted with a buffer (pH 7.4) containing 20 mM Tris-HCl, 100 mM NaCl, 1 mM TECP, and 0.5% (v/v) DMSO. All samples were centrifugated at 18,000× g for 10 minutes to remove precipitation and bubbles before titration. The PLpro proteins (20 μM for GRL0617, and 10 μM for GZNL-P35 or GZNL-P36) were injected into the sample cell, and compounds (200 μM for GRL0617, and 100 μM for GZNL-P35 or GZNL-P36) were loaded into the syringe cell, whereas deionized water was injected into the reference cell as a heat balance control. Data were fitted into a one-site model, as well as Kd, N, ΔG, ΔH, and −TΔS values were calculated using MicroCal PEAQ-ITC analysis software.
Differential scanning fluorimetry (DSF)
DSF experiment was carried out on a CFX384 Touch Real-Time PCR Detection System (Bio-Rad) in PBS buffer containing 0.005% Tween-20, and 1 mM TCEP. The reaction system contained 5 × SYPRO Orange dye (Sigam-Aldrich, S5692), 10 μM of SARS-CoV-2 PLpro in the presence or the absence of compounds at the concentration of 10 μM. Fluorescence was monitored when the temperature was gradually raised from 30 to 80 °C in 0.2 °C increments at 5-second intervals. Melt curve data were plotted using the Boltzmann model to obtain the melt temperature (Tm, the midpoint of unfolding of the protein) using GraphPad Prism 9.0.
Cell culture and cell viability assay
Vero E6 (ATCC, CRL-1586), Huh-7 (JCRB, 0403), HEK293T (ATCC CRL-3216) and HCT-8 (ATCC, CCL-244) cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 IU/mL penicillin and 100 μg/mL streptomycin. The cells were cultured at 37 °C in a fully humidified atmosphere containing 5% CO2, and have been tested negative for mycoplasma infection.
Cell viability was evaluated using a Cell Titer-Glo 2.0 Cell Viability Assay (G9242, Promega) according to the manufacturer’s instructions. In brief, 4 ×103 cells in 20 μL culture medium were seeded into opaque-walled 384-well plates and incubated for 12 hours for adherence. After being treated with compounds for 48 h, 20 μL of Cell Titer-Glo reagent was added into each well. After 2 min shaking and 10 min incubation, luminescence was measured by BioTek Synergy H1 multimode reader (Agilent).
Virus preparation and titrations
SARS-CoV-2 wild-type strain (WT, GenBank: MT123291), Omicron BA.5 variant (Omicron BA.5, GDPCC-303), and XBB.1 variant (XBB.1, IQTC-1596943) were propagated in Vero E6 cells and stored at −80 °C. Virus titers were determined with 10-fold serial dilutions in confluent Vero E6 cells in 96-well microtiter plates. Three days after inoculation, a cytopathic effect (CPE) was scored, and the Reed-Muench formula was used to calculate the TCID50. All of the infection experiments were performed at BSL-3 in Guangzhou Customs Inspection and Quarantine Technology Center (IQTC).
HCoV-229E strain (VR-740) was propagated in Huh-7 cells and stored at −80 °C. Virus titers were determined with 10-fold serial dilutions in confluent Huh-7 cells in 96-well microtiter plates. Three days after inoculation, a cytopathic effect (CPE) was scored, and the Reed-Muench formula was used to calculate the TCID50. All of the infection experiments were performed at BSL-2 in the Guangzhou Laboratory.
HCoV-OC43 strain (VR-1558) and HCoV-NL63 strain (NR-470) were propagated in HCT-8 and Huh7-hACE2 cells and stored at −80 °C, respectively. Virus titers were determined with an indirect immunofluorescence assay. In brief, the cells were fixed in 4% paraformaldehyde for 15 minutes at 24 hours post-infection and permeabilized with 0.3% Triton X-100 for 15 minutes at room temperature. HCoV-OC43 and HCoV-NL63 were probed using anti-HCoV-OC43 Nucleoprotein antibody (Abcam, ab309964, 1:100) and anti-HCoV-NL63 Nucleoprotein antibody (SinoBiological, 40641-T62, 1:500) over 1 hour incubation at room temperature and followed by Alexa Fluor488-labelled secondary antibody (Jackson), respectively. All the cells were stained with 4,6-diamidino-2-phenylindole (DAPI, Sigma, USA) for nuclear visualization. Fluorescent images were acquired using an Evos M5000 Cell Imaging System (Thermo Fisher Scientific, Massachusetts, USA) at 4×magnification. Fluorescence dots were quantified and normalized to the total nuclear count using Image J. All of the infection experiments were performed at BSL-2 in the Guangzhou Laboratory.
In vitro antiviral activity assay
2 × 104 Vero E6 cells were seeded in a 96-well plate for 24 hours. The compounds with different dilution concentrations were mixed with SARS-CoV-2 (MOI = 0.01), and 200 μL mixtures were inoculated onto monolayer Vero E6 cells. Seventy-two hours after inoculation, CPE was scored by Celigo Image Cytometer. The inhibition of compounds and the value of EC50 is calculated from SARS-CoV-2 ‘s CPE rates. Three independent experiments were performed with eight concentration gradients, each with triplicate wells, and one representative is shown.
2 × 104 Huh-7 cells were seeded in a 96-well plate for 24 h. GZNL-P36 with different dilution concentrations were mixed with HCoV-229E (MOI = 0.3), and 200 μL mixtures were inoculated onto monolayer Huh-7 cells. Forty-eight hours after inoculation, CPE was scored by Celigo Image Cytometer. The inhibition of compounds and the value of EC50 are calculated from HCoV-229E’s CPE rates. Three independent experiments were performed with eight concentration gradients, each with triplicate wells, and one representative is shown.
1 × 105 Huh-7 (for HCoV-OC43) or Huh7-hACE2 (for HCoV-NL63) cells were seeded in a 24-well plate for 24 h, respectively. GZNL-P36 with different dilution concentrations were mixed with HCoV-OC43 (MOI = 0.3) or HCoV-NL63 (MOI = 0.3), and 200 μL mixtures were inoculated onto monolayer cells. Forty-eight hours (for HCoV-OC43) or ninety-six hours (for HCoV-NL63) after inoculation, the intracellular total RNA was collected by Trizol reagent. The viral RNA level was determined using qRT-PCR. For HCoV-OC43, the primer (F/R, 5’-3’) targets N gene: GCTCAGGAAGGTCTGCTCC/TCCTGCACTAGAGGCTCTGC. For HCoV-NL63, the primer (F/R, 5’-3’) targets N gene: AGGACCTTAAATTCAGACAACGTTCT/GATTACGTTTGCGATTACCAAGACT, probe: 5’-FAM-TAACAGTTTTAGCACCTTCCTTAGCAACCCAAACA-3’-BHQ1. The inhibition rates were calculated as the percentage of the viral RNA level relative to the control.
Three independent experiments were performed, each with triplicate wells, and one representative is shown in Fig. 3 and Supplementary Fig. 4. The values of IC50 from 3 independent experiments are shown in Supplementary Table 5, 6.
Fluorescence focus assay
Confluent monolayers of Vero E6 cells were incubated with 50 μL pulmonary tissue homogenate of mice with threefold serial dilutions for 2 h at 37 °C, 5% CO2, in triplicate per condition. The inoculum was removed and cells were overlayed with a virus growth medium containing 2% carboxymethylcellulose sodium. At 24 hours post-infection, cells were fixed in 4% paraformaldehyde and permeabilized with 0.2% Triton-X-100, and virus plaques were visualized by immunostaining using an anti-nucleocapsid antibody and action of HRP on a tetramethylbenzidine-based substrate.
SARS-CoV-2 infection in K18-hACE2 mice
K18-hACE2 transgenic mice aged 8 weeks were obtained from the GemPharmatech. The use of K18-hACE2 transgenic mice has received ethical approval from the Animal Ethics Committee at Guangzhou Customs Inspection and Quarantine Technology Center (IQTC2023015). Forty-eight female hACE2 transgenic mice were divided into six groups with eight mice in each group to evaluate the efficacy of GZNL-P36 in the therapeutic treatment. On the day of infection, the hACE2 mice were intranasally inoculated with either 5×104 TCID50 XBB.1, pre-diluted in 50 μL DMEM. Treatment was delayed until 2 hours post-infection (h.p.i.). K18-hACE2 transgenic mice were orally administered a dose of 25 mg/kg S-217622 (MedChemExpress, HY-143216), GZNL-P36 (25, 50 or 100 mg/kg) diluted in 200 μL 5% DMSO/20% hydroxypropyl-beta-cyclodextrin for the treatment group or vehicle solution only for the control group. Mice were killed at the designated time points and organ tissues were sampled for virological and histopathological analysis.
RNAseq analysis of lung tissue from the SARS-COV-2 infected mice
Lung tissue samples were collected and processed in P3 laboratory for safety issues according to the regulation. Total RNA was then extracted and used for the construction of cDNA libraries by an Illumina TruseqTM RNA sample prep Kit. Sequence-by-synthesis single reads of 54-base-length using the Hiseq2000 Truseq SBS Kit (v3-HS, Illumina) were generated on the HiSeq X system. Raw data were collected, and Salmon was used for the quantification of the count and TPM matrix. Subsequently, the tidyverse, clusterProfiler, and GSVA packages of R were used for the downstream analysis. Briefly, the GSVA scores were calculated based on geneset information provided by the msigdbr packages, and the GSVA score for SARS or COVID-containing genesets were selected, and the core enriched genes were then extracted as well. All heatmaps or dot plots were created by the pheatmap or ggplot2 packages.
RNA extraction and qRT-PCR
Tissue samples were lysed and extracted with the Trizol (Invitrogen) reagent according to the manufacturer’s protocols. After RNA extraction, complementary DNA was synthesized through reverse transcription using the Evo M-MLV RT Mix Kit with gDNA Clean reagent for qPCR (AG11705, Accurate Biology). Real-time PCR was carried out using 2 × SYBR Green Pro Taq HS Master Mix qPCR Kit (AG11701, Accurate Biology) on a CFX384 Touch Real-Time PCR Detection System (Bio-Rad). The setting procedures were as follows: 95 °C for 30 seconds, 95 °C for 5 seconds, 60 °C for 30 seconds, a total of 40 cycles. Glyceraldehyde-3- phosphate dehydrogenase (GAPDH) was used as an internal control. The relative expression level of the target gene was calculated by the 2−ΔΔCT method. The specific primers of immune-related genes used for Real-time PCR are listed as follows:
GAPDH sense (5’- AATGAAGGGGTCATTGATGG -3’) and antisense (5’- AAGGTGAAGGTCGGAGTCAA -3’),
IFNB sense sense (5’- AGCTCCAAGAAAGGACGAACA-3’) and antisense (5’-GCCCTGTAGGTGAGGTTGAT-3’),
Cxcl10 sense (5’-CCAAGTGCTGCCGTCATTTTC -3’) and antisense (5’-GGCTCGCAGGGATGATTTCAA-3’),
IFN-g sense (5’-GCCACGGCACAGTCATTGA-3’) and antisense (5’-TGCTGATGGCCTGATTGTCTT-3’).
LogD and solubility
To test the partition coefficient between the aqueous and organic phase (LogD) of GZNL-P36, 15 µL of stock solutions (10 mM) of each sample were placed into their proper 96-well rack, 500 µL of PBS-saturated 1-octanol was added into each vial of the cap-less LogD plate followed by the addition of 500 µL of 1-octanol saturated PBS (pH 7.4). One stir stick was added to each vial and a molded PTFE/Silicone plug was used to seal each vial. Then the LogD plate was transferred to the Eppendorf Thermomixer Comfort plate shaker and shaken at 25 °C with 1,100 rpm for 1 hour. After completion of 1 hour, plugs were removed and the stir sticks were removed using a big magnet. The samples were then centrifuged at 25 °C at 20,000 × g for 20 min to separate the phases, and a pipette and syringe were used to remove the upper (1-octanol) and lower (buffer) phases to the empty tubes, respectively. Aliquots of 5 µL were taken from the upper phases followed by an addition of 495 µL of a mixture of H2O and acetonitrile (1:1 in v/v). Vortex for 1 minute, and then aliquots of 50 µL were taken from the diluent followed by an addition of 450 µL of a mixture of H2O and acetonitrile (1:1 in v/v). Then, aliquots of 50 µL were taken from lower phases followed by an addition of 450 µL of a mixture of H2O and acetonitrile (1:1 in v/v). 200 μL of diluent was transferred to a new 96-well plate for LC-MS/MS analysis.
To test the solubility of GZNL-P36, a proper amount of compounds was weighed out and placed into a 96-well rack. Based on the amount, the proper volume of PBS pH 7.4 was added to each vial of the solubility sample plate which was transferred to the Eppendorf Thermomixer Comfort plate shaker and shaken at 25 °C and 1,100 rpm for 24 h. After completion of 24 h, the samples were transferred from the Solubility Sample plate into a filter plate for removing the un-soluble ingredient using the vacuum manifold. The filtrate was diluted by 1000-fold ((An aliquot of 5 µL was taken from the filtrate followed by the addition of 5 µL DMSO and 490 µL of a mixture of H2O and acetonitrile (1:1 in v/v), vortex well and then aliquot of 50 µL was taken from the diluent followed by addition of 450 µL of a mixture of H2O and acetonitrile (1:1 in v/v). 200 μL of diluent was transferred to a new 96-well plate for LC-MS/MS analysis.
Analysis of compound metabolites
The in vitro metabolites analysis of GZNL-P4 was evaluated using rat liver cells. The compound was diluted to 20 μM using William’s E medium as a test solution. Rat liver cell suspension was prepared at the density of 1 × 106 cells/mL. The test solution and cell suspension were incubated in a constant temperature oscillation incubator at 37 °C with 5% CO2 for 5 min. For the time point 0 min, 600 μL of acetonitrile and 100 μL of cell suspension were mixed by vortex oscillation for 5 min, then followed by 100 μL of the test solution and another vortex oscillation for 5 min. For the time point 20 min, 100 μL of test solution was added into 100 μL of cell suspension to start the reaction. After incubating in a constant temperature oscillation incubator at 37 °C with 5% CO2 for 20 min, 600 μL of acetonitrile was added into the sample and oscillated by vortex for 5 min. All samples were centrifuged at 18,000 × g for 10 min, and the supernatant was transferred to fresh tubes for use. 700 μL of supernatant was dried under nitrogen airflow at 37 °C. The dried sample was re-dissolved by 200 μL of 75% acetonitrile (v/v) for LC-UV-HRMS analysis (Vanquish Flex, Q Exactive HF-X, Thermo Scientific).
Metabolic stability in liver microsome
The metabolism in human liver microsomes was used to evaluate the in vitro metabolic stability of GZNL-P4, GZNL-P17, GZNL-P29, GZNL-P31, GZNL-P35, and GZNL-P36. 1.5 μM of compounds were incubated with rat or human liver microsomes (0.75 mg/mL) in a volume of 0.5 mL PBS, respectively. Assay plates dispensed 30 µL of 1.5 µM compound solution containing 0.75 mg/mL microsomes were designated for different time points (0, 5, 15, 30, and 45 min). NADPH (final concentration at 2 mM) was used to start the reaction at 37 °C and acetonitrile solution containing IS (100 nM alprazolam, 200 nM labetalol, 200 nM caffeine and 2 µM ketoprofen) was used to terminate the reaction at different time points. The plates were centrifuged at 6000× g for 15 min after shaking for 10 min at 600 rpm. Precipitated protein was removed and 80 μL supernatant was transferred to 96-well plate containing 140 μL pure water for LC/MS (Shimadzu Nexera LC-40 & SCIEX TQ-6500 + ) analysis. Intrinsic clearance by substrate depletion was calculated taking into account mg of microsomes per liver weight and grams of liver per Kg body weight.
Metabolic stability in hepatocyte
The cryopreserved hepatocytes were thawed in a 37 °C water bath and gently shaking the vials for 2 min. The hepatocytes were transferred into a 50 mL conical tube containing thawing medium. The thawing medium was removed after centrifuged at 100× g for 10 minutes and the hepatocytes were resuspended in enough incubation medium to yield 1.5 × 106 cells/mL. 1 μM compound was incubated with pre-warmed hepatocytes in an incubation medium at 0.5 × 106 live cells/mL. The 25 μL sample taken at time points of 0, 15, 30, 60, 90, and 120 minutes, were mixed with 6 volumes (150 μL) of acetonitrile containing internal standards (IS: 100 nM alprazolam, 200 nM labetalol, 200 nM caffeine, and 2 µM ketoprofen) to terminate the reaction. After centrifugation for 20 min at 3,220 × g, an aliquot of 100 µL of the supernatant was mixed with 100 µL of ultra-pure water for LC-MS/MS analysis. Boiled cells were used as control. The in vitro half-life (in vitro T1/2) was determined from the slope value by equation (1):
where k = rate constant (-slope value).
The intrinsic clearance (CLint, in µL/min/106cells) was calculated using the equation (2):
where V = incubation volume (0.2 mL);
N = number of hepatocytes per well (1 × 105 cells)
Inhibition of hERG
To evaluate the inhibitory effects of GZNL-P36 on the hERG, the current on human embryonic kidney cells stably expressing hERG channels (hERG-HEK293 cells, Creacell, A-0320) was monitored using a patch clamp system that the core components were bought from Sutter Instrument (USA). The cells were clamped using the patch clamp system to perform a whole-cell voltage clamp mode, the hERG currents were elicited with corresponding voltages. The cells were treated with serially diluted cisapride (sigma, positive control) and GZNL-P36. Tail currents of hERG channels were recorded to obtain the peak tail current at each concentration using Patchcontrol HT and Patchmaster. The current recorded at non-compound containing solution was used as the control for each cell. The current was recorded twice independently for each cell, and at least 2 cells were recorded for each concentration.
Inhibition of CYP isoforms
In this study, test compounds were added into the liver microsomes solution pre-warmed at 37 °C for 5 min. Subsequently, 20 µL of pre-warmed (37 °C) 10 mM NADPH solution was added into 180 µL of liver microsomes solution treated by compounds to initiate the enzyme reaction. The assay plate was incubated at 37 °C until the designated time when 300 µL of termination solution (35 ng/mL ketoprofen, 7.5 ng/mL carbamazepine, 5 ng/mL diphenhydramine, 10 ng/mL tolbutamide) was added to quench the reaction. Then the samples were centrifuged at 3,220 × g for 40 min. The supernatant was transferred to an analysis plate containing a proper volume of ultra-pure water for LC-MS/ MS analysis. The inhibition IC50 was determined for the CYP isoforms 1A2, 2C9, 2C19, 2D6, and 3A4.
Plasma protein binding assay
Firstly, a basic solution was prepared by dissolving 14.2 g/L Na2HPO4 and 8.77 g/L NaCl in deionized water and the solution could be stored at 4 °C for up to 7 days. An acidic solution was prepared by dissolving 12.0 g/L NaH2PO4 and 8.77 g/L NaCl in deionized water and the solution could be stored at 4 °C for up to 7 days. Then, the working buffer was prepared by titrating the basic solution with the acidic solution to pH 7.4 and store at 4 °C for up to 7 days. pH was checked on the day of the experiment and was adjusted if outside the specification of 7.4 ± 0.1. The plasma stored at −80 °C was thawed and centrifuged at 3220× g for 10 min to remove clots and the supernatant was collected into a fresh tube. Compounds were added into the plasma to achieve a final concentration of 1 μM (0.5% DMSO). The dialysis membranes were soaked in ultra-pure water for 60 min to separate strips, then in 20% ethanol for 20 minutes, and finally in dialysis buffer for 20 min. 50 µL of the spiked plasma samples were transferred to separate wells of a 96-well plate and incubated at 37 °C with 5% CO2 for 6 hours. At the end of incubation, samples from both buffer and plasma chambers were transferred to wells of a 96-well plate. A room-temperature quench solution (acetonitrile containing 200 nM labetalol, 100 nM tolbutamide, and 100 nM ketoprofen) was added to precipitate protein. Samples in the plate were vortexed and centrifuged at 3220× g for 30 min at 4 °C. And 100 µL of supernatant was transferred to a new 96-well plate with 100 µL of water for LC-MS/MS analysis.
In vivo PK and acute toxicity experiments
In this study, mouse and dog pharmacokinetic and acute toxicity studies were done at Pharmaron (Beijing, PRC). The PK studies were approved by Pharmaron’s Institutional Animal Care and Use Committee (IACUC) with the ethical approval number PK-M-07182023 for mice and PK-D-06012023 for dogs. The acute toxicity study was approved by Pharmaron TSP NB IACUC with the ethical approval number 24-165. Male CD1 mice (n = 3) and male beagle dogs (n = 3) were treated with intravenous or oral administration. Blood samples were collected at 0.25 h, 0.5 h, 1 h, 2 h, 4 h, 8 h, 24 h after administration and analyzed using liquid chromatography tandem mass spectrometry (LC-MS/MS). The pharmacokinetic parameters AUC0-t、Cmax、Tmax、T1/2 were calculated using Phoenix WinNonlin 7.0. Three male and three female C57BL/6 J mice were used for the acute toxicity study at the dose of 300 mg/kg and 600 mg/kg, respectively. Four days after oral administration of GZNL-P36, the body weight was recorded, and the heart was harvested and stained with haematoxylin and eosin (H&E) for analysis.
Histology and immunohistochemistry
Hematoxylin/eosin (HE) staining of lung samples followed standard HE staining procedures. The lung tissue was cut off and fixed in a 4% polymethylaldehyde solution for 24 hours. Briefly, the sections (3-4 μm-thick) were stained with hematoxylin for 3 minutes and then soaked in the acidic liquid alcohol differentiation for 30 s. After staining with eosin for 15 s and dehydrated by ethanol, the sections were finally cleared by xylene and mounted. The images of per slide were captured using a microscope (LEICA Aperio Versa 8, Germany) and analyzed by the Image J software.
The lung tissue was dewaxed in xylene and hydrated in ethanol. The endogenous peroxidase was blocked using a 3% H2O2 solution. The antigen was retrieved by boiling in a citric acid solution (pH = 6.0), and non-specific binding sites were blocked with 3% BSA at room temperature for 30 minutes. The tissues were then incubated with anti-SARS-CoV-2 (COVID-19) Nucleocapsid primary antibodies (1:1,000 dilution) at 4 °C overnight. After washing, the sections were conjugated with a horseradish peroxidase (HRP) antibody (1:200 dilution; Servicebio) at room temperature for 50 minutes. The tissues were developed using 3,3’-diaminobenzidine (DAB) reagent, counterstained with hematoxylin, dehydrated, and mounted.
Statistical analysis
The data in the figures represent mean ± SD. All data were analyzed using GraphPad Prism 9.0 software. Statistical comparison between different groups was performed using corresponding statistical analysis labeled in figure legends combining several experiments. P‐values were calculated, and statistical significance was reported as highly significant with ***P < 0.001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Crystal structures generated during the current study are available in the Protein Data Bank (PDB) under accession codes 8YX2 (PLpro bound to GZNL-P4), 8YX3 (PLpro bound to GZNL-P28), 8YX4 (PLpro bound to GZNL-P31) and 8YX5 (PLpro bound to GZNL-P35). The RNA sequencing (RNA-Seq) datasets of lung tissues have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences under accession code CRA017011 that are publicly accessible at https://ngdc.cncb.ac.cn/gsa. Source data are provided with this paper. The Supplementary note for synthetic compounds can be found in Supplementary Information. Source data are provided with this paper.
Change history
21 January 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41467-025-56341-8
References
Planas, D. et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 596, 276–280 (2021).
Lopez Bernal, J. et al. Effectiveness of Covid-19 Vaccines against the B.1.617.2 (Delta) Variant. N. Engl. J. Med. 385, 585–594 (2021).
Tuekprakhon, A. et al. Antibody escape of SARS-CoV-2 Omicron BA.4 and BA.5 from vaccine and BA.1 serum. Cell 185, 2422–2433 e2413 (2022).
Cao, Y. et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature 608, 593–602 (2022).
Yao, L. et al. Omicron subvariants escape antibodies elicited by vaccination and BA.2.2 infection. Lancet Infect. Dis. 22, 1116–1117 (2022).
Andrews, N. et al. Covid-19 Vaccine Effectiveness against the Omicron (B.1.1.529) Variant. N. Engl. J. Med. 386, 1532–1546 (2022).
Wang, H., Xue, S., Yang, H. & Chen, C. Recent progress in the discovery of inhibitors targeting coronavirus proteases. Virol. Sin. 31, 24–30 (2016).
Li, X. & Song, Y. Targeting SARS-CoV-2 nonstructural protein 3: Function, structure, inhibition, and perspective in drug discovery. Drug Discov. Today 29, 103832 (2024).
Kokic, G. et al. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat. Commun. 12, 279 (2021).
Jayk Bernal, A. et al. Molnupiravir for Oral Treatment of Covid-19 in Nonhospitalized Patients. N. Engl. J. Med. 386, 509–520 (2022).
Cao, Z. et al. VV116 versus Nirmatrelvir-Ritonavir for Oral Treatment of Covid-19. N. Engl. J. Med. 388, 406–417 (2023).
Arbel, R. et al. Nirmatrelvir Use and Severe Covid-19 Outcomes during the Omicron Surge. N. Engl. J. Med. 387, 790–798 (2022).
Mukae, H. et al. Efficacy and Safety of Ensitrelvir in Patients With Mild-to-Moderate Coronavirus Disease 2019: The Phase 2b Part of a Randomized, Placebo-Controlled, Phase 2/3 Study. Clin. Infect. Dis. 76, 1403–1411 (2023).
Zhang, H. et al. Phase I study, and dosing regimen selection for a pivotal COVID-19 trial of GST-HG171. Antimicrob. Agents Chemother. 68, e0111523 (2024).
Chen, X. et al. Preclinical evaluation of the SARS-CoV-2 M(pro) inhibitor RAY1216 shows improved pharmacokinetics compared with nirmatrelvir. Nat. Microbiol 9, 1075–1088 (2024).
Jiang, X. et al. Structure-based development and preclinical evaluation of the SARS-CoV-2 3C-like protease inhibitor simnotrelvir. Nat. Commun. 14, 6463 (2023).
Consortium, W. H. O. S. T. Remdesivir and three other drugs for hospitalised patients with COVID-19: final results of the WHO Solidarity randomised trial and updated meta-analyses. Lancet 399, 1941–1953 (2022).
Heyer, A. et al. Remdesivir-induced emergence of SARS-CoV2 variants in patients with prolonged infection. Cell Rep. Med 3, 100735 (2022).
Hogan, J. I. et al. Remdesivir Resistance in Transplant Recipients With Persistent Coronavirus Disease 2019. Clin. Infect. Dis. 76, 342–345 (2023).
Gandhi, S. et al. De novo emergence of a remdesivir resistance mutation during treatment of persistent SARS-CoV-2 infection in an immunocompromised patient: a case report. Nat. Commun. 13, 1547 (2022).
Saravolatz, L. D., Depcinski, S. & Sharma, M. Molnupiravir and Nirmatrelvir-Ritonavir: Oral Coronavirus Disease 2019 Antiviral Drugs. Clin. Infect. Dis. 76, 165–171 (2023).
Tan, H., Hu, Y., Jadhav, P., Tan, B. & Wang, J. Progress and Challenges in Targeting the SARS-CoV-2 Papain-like Protease. J. Med Chem. 65, 7561–7580 (2022).
Shimizu, R. et al. Evaluation of the Drug-Drug Interaction Potential of Ensitrelvir Fumaric Acid with Cytochrome P450 3A Substrates in Healthy Japanese Adults. Clin. Drug Investig. 43, 335–346 (2023).
Shimizu, R. et al. Safety, Tolerability, and Pharmacokinetics of the Novel Antiviral Agent Ensitrelvir Fumaric Acid, a SARS-CoV-2 3CL Protease Inhibitor, in Healthy Adults. Antimicrob. Agents Chemother. 66, e0063222 (2022).
Duan, Y. et al. Molecular mechanisms of SARS-CoV-2 resistance to nirmatrelvir. Nature 622, 376–382 (2023).
Báez-Santos, Y. M., St John, S. E. & Mesecar, A. D. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antivir. Res. 115, 21–38 (2015).
Shin, D. et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 587, 657–662 (2020).
Cao, D. et al. The SARS-CoV-2 papain-like protease suppresses type I interferon responses by deubiquitinating STING. Sci. Signal 16, eadd0082 (2023).
Ratia, K. et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl Acad. Sci. USA 105, 16119–16124 (2008).
Yan, H. et al. A robust high-throughput fluorescence polarization assay for rapid screening of SARS-CoV-2 papain-like protease inhibitors. Virology 574, 18–24 (2022).
Osipiuk, J. et al. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 12, 743 (2021).
Zang, Y. et al. High-throughput screening of SARS-CoV-2 main and papain-like protease inhibitors. Protein Cell 14, 17–27 (2023).
Wang, Q. et al. Structure-Based Design of Potent Peptidomimetic Inhibitors Covalently Targeting SARS-CoV-2 Papain-like Protease. Int. J. Mol. Sci. 24, https://doi.org/10.3390/ijms24108633 (2023).
Shen, Z. et al. Design of SARS-CoV-2 PLpro Inhibitors for COVID-19 Antiviral Therapy Leveraging Binding Cooperativity. J. Med Chem. 65, 2940–2955 (2022).
Yuan, S. et al. Targeting papain-like protease for broad-spectrum coronavirus inhibition. Protein Cell 13, 940–953 (2022).
Gao, X. et al. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B 11, 237–245 (2021).
Ma, C. et al. Discovery of SARS-CoV-2 Papain-like Protease Inhibitors through a Combination of High-Throughput Screening and a FlipGFP-Based Reporter Assay. ACS Cent. Sci. 7, 1245–1260 (2021).
Jadhav, P. et al. Structure-based design of SARS-CoV-2 papain-like protease inhibitors. Eur. J. Med Chem. 264, 116011 (2024).
Sanders, B. C. et al. Potent and selective covalent inhibition of the papain-like protease from SARS-CoV-2. Nat. Commun. 14, 1733 (2023).
Tan, B. et al. Design of a SARS-CoV-2 papain-like protease inhibitor with antiviral efficacy in a mouse model. Science 383, 1434–1440 (2024).
Garnsey, M. R. et al. Discovery of SARS-CoV-2 papain-like protease (PLpro) inhibitors with efficacy in a murine infection model. bioRxiv (2024).
Calleja, D. J., Lessene, G. & Komander, D. Inhibitors of SARS-CoV-2 PLpro. Front Chem. 10, 876212 (2022).
Shiraishi, Y. & Shimada, I. NMR Characterization of the Papain-like Protease from SARS-CoV-2 Identifies the Conformational Heterogeneity in Its Inhibitor-Binding Site. J. Am. Chem. Soc. 145, 16669–16677 (2023).
Liu, W., Jiang, J., Lin, Y., You, Q. & Wang, L. Insight into Thermodynamic and Kinetic Profiles in Small-Molecule Optimization. J. Med Chem. 65, 10809–10847 (2022).
Guo, Y. et al. Discovery and characterization of potent pan-variant SARS-CoV-2 neutralizing antibodies from individuals with Omicron breakthrough infection. Nat. Commun. 14, 3537 (2023).
Focosi, D. et al. Nirmatrelvir and COVID-19: development, pharmacokinetics, clinical efficacy, resistance, relapse, and pharmacoeconomics. Int J. Antimicrob. Agents 61, 106708 (2023).
Iketani, S. et al. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature 613, 558–564 (2023).
Kiso, M. et al. In vitro and in vivo characterization of SARS-CoV-2 resistance to ensitrelvir. Nat. Commun. 14, 4231 (2023).
de Bruyn Kops, C. et al. GLORY: Generator of the Structures of Likely Cytochrome P450 Metabolites Based on Predicted Sites of Metabolism. Front Chem. 7, 402 (2019).
Stork, C. et al. NERDD: a web portal providing access to in silico tools for drug discovery. Bioinformatics 36, 1291–1292 (2020).
Vonrhein, C. et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr D. Biol. Crystallogr 67, 293–302 (2011).
Yu, F. et al. Aquarium: an automatic data-processing and experiment information management system for biological macromolecular crystallography beamlines. J. Appl. Crystallogr. 52, 472–477 (2019).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl Crystallogr 40, 658–674 (2007).
Adams, P. D. et al. The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106 (2011).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. Biol. Crystallogr 60, 2126–2132 (2004).
Moriarty, N. W., Grosse-Kunstleve, R. W. & Adams, P. D. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr D. Biol. Crystallogr 65, 1074–1080 (2009).
Acknowledgements
We thank Prof. Douglas J. Kojetin, Vanderbilt University, for critical reading and discussions. We are grateful for support from Shanghai Synchrotron Radiation Facility (SSRF) beamlines (BL19U1 and BL02U1) and Guangzhou Laboratory Core Facility and Animal Center. This study was supported by grants from the National Natural Science Foundation of China (82170473 to J.S.), the startup and R&D Program of Guangzhou National Laboratory (GZNL2024A01022 to J.S.; YW-YWYM0202 to J.S.; GZNL2023A02012 to J.S.; GZNL2023A01008 to J.S., W.K.Z., H.C. and D.G.; GZNL2024A01005 to H.C.; SRPG22-002 to J.S. and X.C.; SRPG22-003 to J.S.), and the Guangdong Natural Science Foundation (2021QN020451 to J.S.; 2021CX020227 to H.C.), and the Basic and Applied Basic Research Projects of Guangzhou Basic Research Program (2023A04J0161 to Q.Y.).
Author information
Authors and Affiliations
Contributions
Y.L., Q.Y., W.L., M.D., J.T., J.C., Jiy.Z., A.Z., S.Y., D.G., K.W., and X.W. performed the cellular and biochemical assays. Y.L. and J.S. performed crystallography. G.Z., P.Z., H.C., P.H., Jinp.Z. and T.R. performed the medicinal chemistry. Q.Y., W.L., B.X., and J.T. performed the animal models. T.R., M.T., and H.C. performed the computational studies. Y.L., W.L., M.D., J.C., Jiy.Z., K.W., and X.W. performed cloning and/or purified proteins. Y.L., W.L., J.S., and W.K.Z. performed the transcriptome study. Y.L., Q.Y., T.R. X.C., H.C., and J.S. conceived the experiments and wrote the manuscript with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Chamandi Dampalla, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lu, Y., Yang, Q., Ran, T. et al. Discovery of orally bioavailable SARS-CoV-2 papain-like protease inhibitor as a potential treatment for COVID-19. Nat Commun 15, 10169 (2024). https://doi.org/10.1038/s41467-024-54462-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-54462-0
This article is cited by
-
Identification of naturally occurring drug-resistant mutations of SARS-CoV-2 papain-like protease
Nature Communications (2025)
