
Abstract
In drug development, the substitution of benzene rings in aniline-based drug candidates with saturated bridged bicyclic ring systems often enhances pharmacokinetic properties while preserving biological activity. However, current efforts predominantly focuses on bicyclo[1.1.1]pentylamines, accessing analogs capable of mimicking ortho- and meta-substituted anilines remains challenging due to the lack of a versatile and modular synthetic methods. Herein, we present a modular approach to access a diverse array of saturated bioisosteres of anilines via photoelectrochemical-induced decarboxylative C(sp3)–N Coupling. The success of this reaction hinges on the merging the cooperative ligand-to-metal charge transfer (LMCT) with copper-catalyzed amination. Notably, this net-oxidative C(sp3)–N forming reaction operates under mild electrode potentials and proceeds through hydrogen evolution, eliminating the need for external chemical oxidants. Our research enables the facile decarboxylative amination of a set of sp3-rich small-ring cage carboxylic acids, thus offering a versatile bioisosteric replacement for ortho-, meta-, and para-substituted anilines and di(hetero)aryl amines.
Similar content being viewed by others

Introduction
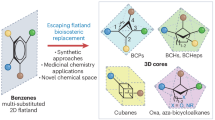
Phenyl rings are ubiquitous in medicinal chemistry, featuring prominently in various biologically relevant molecules1,2. Departing from the concept of ‘flatland’, a range of saturated bridged bicyclic ring systems—such as bicyclo[1.1.1]-pentane (BCP), bicyclo[2.1.1]hexane (BCHex), bicyclo [3.1.1]heptane (BCHep), and bicyclo[2.2.2]octane (BCO)—have been developed to replace phenyl rings, yielding drug molecules with enhanced biological activity and improved physicochemical properties (Fig. 1a)3,4,5,6,7,8,9,10,11. Among the phenyl containing bioactive molecules, aniline stands as a prevalent structural motif, especially in high-throughput screening libraries (Fig. 1b)12,13. However, its prevalence poses substantial challenges in later pharmaceutical development stages, notably due to its tendency to drive compounds towards metabolically derived toxicities or heighten the risk of adverse drug-drug interactions14,15. For instance, Capesaris, a synthetic nonsteroidal estrogen initially developed by GTx, Inc. for advanced prostate cancer treatment, was found to have a high risk of venous thromboembolism in the clinical trials due to the reactive metabolite (RM) formation16,17. One of the approach to tackle this challenge involves leveraging sp3-rich saturated bridged bicyclic ring systems (e.g. [1.1.1]BCP to replace the phenyl ring) (Fig. 1b)1,10,18,19,20,21,22. These isosteric replacements often demonstrate heightened resistance against reactive metabolite formation, along with great opportunities for intellectual property development. However, current synthetic methodologies predominantly focus on bioisosteric analogs of non- or para-substituted anilines (e.g., amino-BCP with substituents at the bridgehead position), necessitating intricate de novo synthesis with lengthy steps and limited modularity22,23,24,25,26,27,28,29. Recently, small-ring cage bioisostere scaffolds like bridgehead-disubstituted [2.1.1]BCHex and [3.1.1]BCHep, and 1,2-disubstituted [2.2.1]BCHep have emerged to mimic meta- and ortho-substituted arenes (Fig. 1c)30,31,32,33,34,35,36. Considering that many drug candidates containing aniline motifs also display meta- or ortho-substitutions, the creation of a versatile synthetic approach with high modularity, capable of universally generating saturated bioisosteres of substituted anilines—encompassing para-, meta-, and ortho-substitutions—holds immense value.

a Examples of the phenyl bioisosteres core appearing in drugs and bioactive compounds. b The world’s best-selling small molecule drugs containing aniline and diaryl amine motifs. The toxicity of Capesaris associated with the metabolism of aniline could be mitigated by BCP isostere replacement. c Representative saturated bioisosteric scaffolds that faithfully replicate the geometry of substituted anilines. d Typical approaches to amino-bioisosteres from small-ring cage carboxylic acids require pre-activation of carboxylic acids with iodomesitylene diacetate. In this work, a photoelectrochemical decarboxylative platform enables modular access to saturated bioisosteres of anilines.
Small-ring cage carboxylic acids such as 3-(methoxycarbonyl)bicyclo[1.1.1]pentane-1-carboxylic acid 1a and 4-(methoxycarbonyl)bicyclo[2.2.2]octane-1-carboxylic acid 1b are readily accessible from commercial sources or synthesized using established methods. This accessibility has sparked our interest in devising a catalytic direct decarboxylative amination reaction. Such a process would enable the streamlined and modular synthesis of saturated bioisosteres of substituted anilines directly from the corresponding carboxylic acids. An elegant example from the MacMillan group showcased the facile decarboxylative amination of certain small-ring cage carboxylic acids with indazole, employing a synergistic combination of photoredox catalysis and copper catalysis29,36. However, the protocol’s requirement for pre-activation of carboxylic acids with iodomesitylene diacetate to form redox-active iodonium carboxylates, crucial for generating key alkyl radical species such as BCP radical, presents inherent limitations within organic synthesis (Fig. 1d, top left). Direct decarboxylative amination offers synthetic appeal as it circumvents the need for preparing and isolating redox-active carboxylic acid derivatives37,38,39,40. Nevertheless, a significant challenge lies in effectively oxidizing carboxylic acids or carboxylates (e.g. hexanoate ion, E1/2red = +1.16 V vs SCE) without affecting the more readily oxidizable amine coupling partners (e.g. p-toluidine, E1/2red = +0.81 V vs SCE)41. Additionally, closing the net-oxidative catalytic cycle without resorting to stoichiometric chemical oxidants like DTBP or copper salts remains another key concern. The use of stoichiometric oxidants can indeed limit the range of substrates, posing challenges for large-scale preparation, and may not be applicable to saturated bridged bicyclic ring systems39,40,42. The interrupted Kolbe reaction allows for the direct anodic oxidation of alkyl carboxylic acids to generate carbocation species43. These species can engage in the subsequent nucleophilic replacement, enabling direct decarboxylative C(sp3)–N bond forming reactions44,45. However, applying this method to small-ring cage skeletons commonly found in saturated bioisosteres has proven challenging due to the highly unstable carbocations formed at the 3o bridgehead position, resulting in rapid ring fragmentation (Fig. 1d, bottom left)44,45,46.
Drawing inspiration from recent advancements in photoelectro-catalytic transformations47,48,49,50,51,52,53, we envisage to invent a modular assembly of aniline bioisosteres through photoelectro-catalytic decarboxylative C(sp3)–N coupling process. Specifically, our approach is centered on generating kinetically stable small-ring cage radical intermediates (e.g., BCP radical) using a photo-induced ligand-to-metal charge transfer (LMCT) process within the solution phase54,55,56,57,58. This strategic design aims to bypass the potential issue of direct electrode overoxidation of radicals into carbocations, consequently mitigating the risk of skeletal rearrangement in small-ring cage bioisosteres. On the other hand, harnessing the synergies of light and electrical energy enables a substantial reduction in the required electrode potential for the crucial redox event, thus offering mild reaction conditions that are suitable for the coupling of easily oxidizable amines. Additionally, employing an electrochemical potential instead of stoichiometric chemical oxidants in the net oxidative coupling process not only provides practical, economic, and sustainable advantages but also has potential to broaden the scope of both reactants42,47. Herein, we report the discovery of a photoelectro-catalysis enabled direct decarboxylative C(sp3)–N coupling reaction (Fig. 1d, right). The chemistry displays a high degree of practicality with a remarkable scope (>65 examples) across various valuable substrates (including many previously inaccessible saturated bioisosteres of meta-, ortho- substituted anilines) using inexpensive carbon-based electrodes. The utility of this approach was demonstrated through the direct conversion of natural products and drug molecules into bioisosteric aniline analogs, as well as in the synthesis of BCP analogs of clinically relevant drugs.
Our proposed mechanism for the direct decarboxylative C(sp3)–N Coupling is depicted in Fig. 2a. We aimed to merge the capacity of LMCT photocatalysis to generate alkyl radicals from carboxylic acids with copper’s established role in reductive elimination for C–N bond formation. Upon light irradiation, the Fe(III) complex A undergoes homolysis of the Fe-O2CR bond (LMCT decarboxylation), leading to CO2 extrusion and yielding Fe(II) photocatalyst B along with an alkyl radical. This alkyl radical could be rapidly captured by the copper(II)-amido complex C, derived from the coordination of the amine coupling partner with a copper(II) precatalyst, forming the crucial copper(III) complex D. Subsequent reductive elimination occurs to give the desired sp3 C–N coupling product and copper(I) catalyst E59,60. The copper(I) species E and Fe(II) species B are expected to undergo a facile anode oxidation to yield the higher valent metals to close the catalytic cycles. Meanwhile, to maintain electrochemical balance, hydrogen evolution occurs through proton reduction at the cathode.

a Proposed catalytic cycles. b Investigation of reaction parameters, standard conditions: 1a (0.6 mmol, 3.0 equiv), 2a (0.2 mmol, 1.0 equiv), Fe(OAc)2 (15 mol%), Cu(acac)2 (15 mol%), Et3N (0.5 equiv), TBABF4 (1.2 equiv), DMF (4.0 mL), carbon felt as the anode, RVC as the cathode, undivided cell, I = 2.5 mA, 6 W LEDs (390 nm), N2, 20 °C, 15 h. bYield determined by 1H NMR. cIsolated yield.
Results
Evaluation of the reaction conditions
Based on this mechanistic understanding, we explored the decarboxylative C(sp3)–N cross-coupling between BCP-derived carboxylic acid 1a and p-toluidine 2a (Fig. 2b). After extensive evaluation of the reaction parameters, we identified the optimal reaction conditions—simultaneous electrolysis and violet light-emitting diode (LED) irradiation (λmax = 390 mm) of the reaction mixture—employing a cooperative iron mediated photocatalysis and copper catalysis. This led to the formation of the BCP-based saturated bioisosteric product 3 in an 83% yield, with 10% of 2a remaining unreacted (Entry 1). One of the major byproducts from 1a is the decarboxylative hydrogenation by-product, specifically methyl bicyclo[1.1.1]pentane-1-carboxylate, as detected by GC-MS analysis. Replacing the Fe(OAc)2 with commonly used LMCT decarboxylation catalysts, such as Ce(OTf)3 or CeCl3, resulted in complete reaction cessation (Entry 2). This observation could be attributed to the inherent higher oxidation potential of cerium(III), which renders it unsuitable for C–N bond formations with easily oxidizable amine coupling partners47,50. Similarly, no reactivity was observed using 4CzIPN as the photocatalyst (Entry 3), underscoring the crucial role of Fe(OAc)2 in this photoelectro-catalytic reaction. Alternative transition metal catalysts like Cu(hfacac)2 and Ferrous oxalate provided the product in moderate yields (Entries 4 and 5). Employing nickel foam as the cathode dramatically decreased the yield to 8% (Entry 6). When using RVC instead of carbon felt as the anode, a moderate yield of 48% was obtained (Entry 7). A shift to 365 nm LED reduced the yield to 20% (Entry 8). Furthermore, 2,4,6-collidine proved to be an effective base, affording 3 in 65% yield (Entry 9). It is worth noting that constant voltage electrolysis (0.8 V) could also deliver the product in 51% yield (Entry 10). Finally, control studies confirmed the indispensable roles of light irradiation, electricity, and Fe, Cu catalysts (Entries 11 − 12).
Scope of amines
We next turned our attention to exploring the scope of this direct decarboxylative C(sp3)–N bond forming reaction. As shown in Fig. 3, an extensive range of aryl and heterocyclic amines were successfully coupled with BCP carboxylic acid 1a under standard conditions, affording the corresponding amino-BCPs as products. Aryl amines bearing electron-donating groups such as Me (3, 6), PhO (7), Boc-protected amine (12) underwent the targeted reaction in moderate to good yields (35-81%). The reaction also displayed compatibility with electron-withdrawing groups including CF3O (8), HCF2O (9), CF3S (11), CF3 (13), CN (14), COOMe (15), SO2Me (16), POMe2 (17). Our reaction’s chemoselectivity was illustrated by its compatibility with various functional groups, including aryl halides (18-21), Bpin (22), ester (23), CN (24), unprotected OH (25), among others. Notably, secondary amines like indoline (27), methyl indoline-2-carboxylate (28) smoothly participated, yielding the desired tertiary amine products in good yields (60%, 66% respectively). Our methodology extended beyond aryl amines, demonstrating success even with pyridine or pyrimidine-containing heterocyclic amines which might otherwise cause catalyst poisoning, highlighting the protocol’s potential attractiveness in drug discovery. In addition, sulfoxide imines (37-38) and imine (39) also demonstrated a high degree of reaction compatibility, with sulfoxide imine 38 being prepared in 75% yield on a gram scale. Furthermore, the reaction exhibited exceptional broad functional-group tolerance, as evidenced by late-stage couplings of complex structures derived from natural products and drug molecules, including 4-amino-L-Phe-Gly (40), aminoglutethimide (41), florfenicol (42), uridine (43), estradiol benzoate (44), and podophyllotoxin (45).

Red indicates newly formed C–N bonds. Isolated yields (given as a percentage) are denoted for each product. Reaction conditions: 1a (0.6 mmol, 3.0 equiv), amine (0.2 mmol), Fe(OAc)2 (15 mol%), Cu(acac)2 (15 mol%), Et3N (0.5 equiv), TBABF4 (1.2 equiv), DMF (4.0 mL), carbon felt as the anode, RVC as the cathode, undivided cell, I = 1–2.5 mA, 6 W LEDs (390 nm), N2, 20 °C, 15–18 h. The current and reaction time vary slightly for each substrate; see Supplementary Information, Section 4 for the specific reaction conditions.
Scope of carboxylic acids
To explore the versatility of our technology in creating saturated bioisosteres of anilines, we evaluated a diverse range of BCP carboxylic acids featuring various bridgehead substitutions (Fig. 4). Encouragingly, functionalities such as CF2H (46), phenyl (47), CN (48), silyl-protected oxygen (49), amides (50, 51), carbamate (52), and ester (53) were well accommodated, yielding the desired bioisosteres products with high efficiency (40-65%). The reactivity could be extended to [2.2.2]BCO (54) and [2.2.1]BCHep (55) scaffolds, offering alternative products suitable as para-substituted aniline bioisosteres. Of particular note, several intricate bicyclic ring systems, recently identified as meta-substituted phenyl bioisostere mimics, were also amenable to our current C(sp3)–N cross-coupling conditions, furnishing aniline bioisosteres that were challenging to obtain (56-58). Moreover, our method allowed for the synthesis of ortho-substituted aniline bioisostere in a synthetically useful yield (59). The successful incorporation of complex units from natural products and drug molecules into BCP-based bioisosteres underlines the potential of our approach in drug discovery (60-62). Beyond bioisosteric replacement scenarios, this newly developed reaction exhibited applicability in the decarboxylative functionalization of secondary and primary alkyl carboxylic acids, providing N-alkyl products in good yields (63-71).

Red indicates newly formed C–N bonds. Isolated yields (given as a percentage) are denoted for each product. Reaction conditions: alkyl carboxylic acid (0.6 mmol, 3.0 equiv), amine (0.2 mmol), Fe(OAc)2 (15 mol%), Cu(acac)2 (15 mol%), Et3N (0.5 equiv), TBABF4 (1.2 equiv), DMF (4.0 mL), carbon felt as the anode, RVC as the cathode, undivided cell, I = 1.0–3.0 mA, 6 W LEDs (390 nm), N2, 20 °C, 15 h. The catalysts loading for primary carboxylic acids are Fe(OAc)2 (20 mol%), Cu(acac)2 (20 mol%). The current vary slightly for each substrate; see Supplementary Information, Section 4 for the comprehensive details.
Synthetic applications
The product derivatization is illustrated in Fig. 5a. Ester hydrolysis under basic conditions yielded 72 in nearly quantitative yield, offering a versatile carboxylate handle for diversification. For example, treatment of 72 with MeLi resulted in the formation of ketone-substituted BCP 73 in 90% yield. The Curtius rearrangement occurred smoothly to form the other bridgehead C(sp3)–N bond (74). Additionally, BH3 reduction of the carboxylate group produced BCP containing primary alcohol (75) in 80% yield. Condensation with Meldrum’s acid, followed by decarboxylation under heating conditions, provided 76 in moderate yield. The oxidation of 27 to indole substituted BCP 77 was easily within reach. The facile transformation of the indole-substituted BCP carboxylic acid 78 into 79 through a photo-induced decarboxylative Heck-type reaction further demonstrated the versatility of our method. We also explored integrating the BCP scaffold into drug molecules. As depicted in Fig. 5b, the BCP analog of the anticancer agent Capesaris was synthesized from decarboxylative amination product 18. It was achieved through a synthetic sequence involving amide formation, Weinreb ketone synthesis, Baeyer–Villiger oxidation, deprotection of the silyl, and hydrolysis (22% yield over 6 steps). This three-dimensional bioisosteric replacement has the potential to enhance absorption, distribution, and metabolism properties, while addressing issues associated with metabolically derived toxicities (vide supra).

a Product derivatization. b This protocol enables the rapid preparation of saturated bioisosteres of anticancer drug Capesaris. c The radical clock and radical inhibition experiments reveal the involvement of alkyl radical intermediate. d The carbocation pathway is excluded by using water as nucleophile. e Experiments investigating the impact of Et3N suggest its potential role as a protector for both the amine substrate and product, shielding them from decomposition. f The reaction proceeds using stoichiometric metal catalysts even in the absence of electricity. g Prussian Blue detection experiment proved the existence of Fe(III) species. h Cyclic voltammetry experiments indicated that TolNH2 promoted the generation of Cu(II) species, while carboxylic acid facilitated the formation of Fe(III) intermediates (compound concentration:10 mM in DMF, reference Electrode: Ag/AgCl electrode, scan rate:100 mV s–1). i, UV−vis absorption experiments provided further evidence for photoexcited Fe(III) intermediate.
Mechanistic studies
Mechanistic studies were conducted to unravel the reaction pathway. As shown in Fig. 5c, radical clock reaction with 2-cyclopropylacetic acid 83 produced the ring-opening product 84 in 30% yield. Introduction of 2.0 equiv of TEMPO in a radical inhibition experiment reduced the yield of 3–26%, and HRMS analysis confirmed the presence of TEMPO adduct product 85. Moreover, replacing amine 2a with H2O or MeOH failed to produce alcohol product 87 or ether product 88 (Fig. 5d). All these findings exclude the carbocation pathway and strongly suggest the participation of an alkyl radical intermediate in this C–N coupling process. The substantial increase in the recovery rate of both 2a and 3 upon adding Et3N suggested a dual role for Et3N, serving not only as a base but also protecting the amine substrate and product from decomposition (Fig. 5e). This stabilization is further corroborated by the detection of diethylamine by HRMS, indicating that Et3N plays a protective role for the reaction components. In another experiment, employing a stoichiometric amount of Fe(III) catalyst [Fe(OAc)2OH] and Cu(acac)2 without electricity, the desired product was obtained in 31% yield (Fig. 5f). In addition, a Prussian Blue detection experiment confirmed the formation of a Fe(III) species through the electrochemical oxidation of the Fe(OAc)2 precatalyst (Fig. 5g)61. This collective evidence indicates the pivotal role of Fe(III) as the active species, initiating LMCT to generate alkyl radicals that participate in the ensuing C–N bond formation. We also conducted cyclic voltammetry experiments to further elucidate the reaction mechanism (Fig. 5h). Cu(acac)2 exhibited no distinct redox features between –0.5 V and 1.0 V. However, the addition of TolNH2 introduced a new anodic event around 0.52 V, corresponding to the oxidation of XCu(I)NH2Tol to XCu(II)NHTol at approximately 0.52 V (vs. Ag/Ag+). An increase in current was observed upon the addition of Et3N, suggesting an interaction between Et3N and XCu(I)NH2Tol. In comparison to Fe(OAc)2, the inclusion of carboxylic acid 1a led to the appearance of a new anodic event with an onset potential around 0.04 V. These observations suggested that TolNH2 promoted the production of Cu(II) species, similar to how carboxylic acid facilitated the formation of Fe(III) intermediates. Additionally, UV−vis absorption experiments clearly showed the emergence of a new ultraviolet absorption peak upon the addition of carboxylic acid 1a, indicating the formation of the Fe(II)-carboxylic acid complex (Fig. 5i). After the electrolysis, the absorption was significantly enhanced, revealing the formation of a Fe(III) intermediate that was photoexcited, displaying LMCT transitions.
Discussion
In summary, we have demonstrated how the combination of photoelectro-catalysis and copper catalysis can be used to overcome the long-standing challenge of the direct decarboxylative C(sp3)–N coupling, providing a versatile and modular approach for the assembly of saturated bioisosteres of aniline-based drug molecules. Noteworthy features of our protocol include its mild reaction conditions, simple experimental operations, broad scope and generality across a wide number of counterparts, enabling the universal synthesis of saturated bioisosteres for various substituted anilines, encompassing para-, meta-, and ortho-substitutions. Given the importance of bioisosteric replacement in drug development, we anticipate that this reaction will significantly propel advancements within pharmaceutical research.
Methods
General procedure for synthesis of saturated anilines bioisosteres
To an ElectraSyn vial was added TBABF4 (0.24 mmol, 1.2 equiv), carboxylic acid (0.4 mmol, 2.0 equiv), Fe(OAc)2 (0.03 mmol, 15 mol%), Cu(acac)2 (0.03 mmol, 15 mol%) and aniline (0.20 mmol, 1.0 equiv). The vial was transferred into a N2-filled glovebox. Degassed DMF (4.0 mL) was added followed by Et3N (0.10 mmol, 50 mol%). The ElectraSyn vial was sealed with a cap which equipped with anode (carbon felt) and cathode (RVC), then removed from the glovebox. The reaction was irradiated with LEDs (6 W, 390 nm) under the vessel and electrolysis was initiated at a constant current of 1.0–2.5 mA. After 10 h at 20 oC, a solution of carboxylic acid (0.20 mmol, 1.0 equiv) in DMF (0.20 mL) was add to the reaction. After 5 h, the photolysis and electrolysis were terminated, the tube cap was removed and electrodes were rinsed with EtOAc, which was combined with the crude mixture. The organic layers were further washed with brine, dried over anhydrous Na2SO4 and concentrated in vacuo and purified by alkaline prepared thin layer chromatography plate.
Data availability
Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Center, under deposition numbers CCDC 2321005 (17). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. Experimental procedures, characterization of new compounds and all other data supporting the findings are available in the Supplementary Information. All data are available from the corresponding author upon request.
References
Subbaiah, M. A. M. & Meanwell, N. A. Bioisosteres of the phenyl ring: Recent strategic applications in lead optimization and drug design. J. Med. Chem. 64, 14046–14128 (2021).
Taylor, R. D., MacCoss, M. & Lawson, A. D. G. Rings in drugs. J. Med. Chem. 57, 5845–5859 (2014).
Lovering, F., Bikker, J. & Humblet, C. Escape from Flatland: Increasing saturation as an approach to improving clinical success. J. Med. Chem. 52, 6752–6756 (2009).
Lovering, F. Escape from Flatland 2: Complexity and promiscuity. Med. Chem. Commun. 4, 515–519 (2013).
Stepan, A. F. et al. Application of the Bicyclo[1.1.1]pentane motif as a nonclassical phenyl ring bioisostere in the design of a potent and orally active γ-secretase inhibitor. J. Med. Chem. 55, 3414–3424 (2012).
Measom, N. D. et al. Investigation of a bicyclo[1.1.1]pentane as a phenyl replacement within an LpPLA2 inhibitor. ACS Med. Chem. Lett. 8, 43–48 (2017).
Goh, Y. L. et al. Toward resolving the resveratrol conundrum: Synthesis and in vivo pharmacokinetic evaluation of BCP–resveratrol. ACS Med. Chem. Lett. 8, 516–520 (2017).
Mykhailiuk, P. K. Saturated bioisosteres of benzene: where to go next? Org. Biomol. Chem. 17, 2839–2849 (2019).
Shire, B. R. & Anderson, E. A. Conquering the synthesis and functionalization of bicyclo[1.1.1]pentanes. JACS Au 3, 1539–1553 (2023).
Pu, Q. et al. Discovery of potent and orally available bicyclo[1.1.1]pentane-derived indoleamine-2,3-dioxygenase 1 (IDO1) inhibitors. ACS Med. Chem. Lett. 11, 1548–1554 (2020).
Denisenko, A. et al. 1,2-Disubstituted bicyclo[2.1.1]hexanes as saturated bioisosteres of ortho-substituted benzene. Chem. Sci. 14, 14092–14099 (2023).
McGrath, N. A., Brichacek, M. & Njardarson, J. T. A graphical journey of innovative organic architectures that have improved our lives. J. Chem. Ed. 87, 1348–1349 (2010).
Kumar, A. et al. Novel anti-campylobacter compounds identified using high throughput screening of a pre-selected enriched small molecules library. Front. Microbiol. 7, 405 (2016).
Walsh, J. S. & Miwa, G. T. Bioactivation of drugs: Risk and drug design. Annu. Rev. Pharmacol. Toxicol. 51, 145–167 (2011).
Kalgutkar, A. S. Should the incorporation of structural alerts be restricted in drug design? an analysis of structure-toxicity trends with aniline-based drugs. Curr. Med. Chem. 22, 438–464 (2014).
James, D. Estrogen receptor ligands and methods of use thereof. WO 2010096801 A1 (2010).
Yu, E. Y. et al. Phase 2 trial of GTx-758, an estrogen receptor alpha agonist, in men with castration-resistant prostate cancer. Clin. Genitourin. Cancer 18, 436–443 (2020).
Staveness, D. et al. Providing a new aniline bioisostere through the photochemical production of 1-aminonorbornanes. Chem 5, 215–226 (2019).
Yang, Y. et al. Programmable late-stage functionalization of bridge-substituted bicyclo[1.1.1]pentane bis-boronates. Nat. Chem. https://doi.org/10.1038/s41557-023-01342-7 (2023).
Aguilar, A. et al. Discovery of 4‑((3′R,4′S,5′R)‑6′′-Chloro-4′-(3-chloro-2-fluorophenyl)-1′-ethyl-2′′-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3′′-indoline]-5′-carboxamido) bicyclo[2.2.2]octane-1-carboxylic Acid (AA-115/APG-115): A potent and orally active murine double minute 2 (MDM2)inhibitor in clinical development. J. Med. Chem. 60, 2819–2839 (2017).
Lars, G. K. et al. 4-(2-fluoro-4-methoxy-5-3-(((1-methylcyclobutyl) methyl) carbamoyl) bicyclo[2.2.1]heptan-2-yl) carbamoyl)phenoxy) 1-methylcyclohexane-1-carboxylic acid derivatives and similar compounds as RXFP1 modulators for the treatment of heart failure. WO 2022122773 A1 (2022).
Zhang, X. et al. Copper-mediated synthesis of drug-like bicyclopentanes. Nature 580, 220–226 (2020).
Alvarez, E. M. et al. O-, N- and C-bicyclopentylation using thianthrenium reagents. Nat. Synth. 2, 548–556 (2023).
Gianatassio, R. et al. Strain-release amination. Science 351, 241–246 (2016).
Pickford, H. D. et al. Twofold radical-based synthesis of N,C-difunctionalized bicyclo[1.1.1]pentanes. J. Am. Chem. Soc. 143, 9729–9736 (2021).
Kim, J. H., Ruffoni, A., Al-Faiyz, Y. S. S., Sheikh, N. S. & Leonori, D. Divergent strain-release amino-functionalization of [1.1.1]propelling with electrophilic nitrogen-radicals. Angew. Chem. Int. Ed. 59, 8225–8231 (2020).
Livesley, S. et al. Electrophilic activation of [1.1.1]propellane for the synthesis of nitrogen-substituted bicyclo[1.1.1]pentanes. Angew. Chem. Int. Ed. 61, e202111291 (2022).
Kanazawa, J., Maeda, K. & Uchiyama, M. Radical multicomponent carboamination of [1.1.1]propellane. J. Am. Chem. Soc. 139, 17791–17794 (2017).
Liang, Y., Zhang, X. & MacMillan, D. W. C. Decarboxylative sp3 C–N coupling via dual copper and photoredox catalysis. Nature 559, 83–88 (2018).
Rigotti, T. & Bach, T. Bicyclo[2.1.1]hexanes by visible light-driven intramolecular crossed [2+2] photocycloadditions. Org. Lett. 24, 8821–8825 (2022).
Frank, N. et al. Synthesis of meta-substituted arene bioisosteres from [3.1.1]propellane. Nature 611, 721–726 (2022).
Garry, O. L. et al. Rapid access to 2-substituted bicyclo[1.1.1]pentanes. J. Am. Chem. Soc. 145, 3092–3100 (2023).
Zheng, Y. et al. Photochemical intermolecular [3σ+2σ]-cycloaddition for the construction of aminobicyclo[3.1.1]heptanes. J. Am. Chem. Soc. 144, 23685–23690 (2022).
Denisenko, A. et al. 2-Oxabicyclo[2.1.1]hexanes as saturated bioisosteres of the ortho-substituted phenyl ring. Nat. Chem. 15, 1155–1163 (2023).
Denisenko, A., Garbuz, P., Shishkina, S. V., Voloshchuk, N. M. & Mykhailiuk, P. K. Saturated bioisosteres of ortho-substituted benzenes. Angew. Chem. Int. Ed. 59, 20515–20521 (2020).
Wiesenfeldt, M. P. et al. General access to cubanes as benzene bioisosteres. Nature 618, 513–518 (2023).
Nguyen, V. T. et al. Visible-light-enabled direct decarboxylative N-alkylation. Angew. Chem. Int. Ed. 59, 7921–7927 (2020).
Xiong, N., Li, Y. & Zeng, R. Merging photoinduced iron-catalyzed decarboxylation with copper catalysis for C-N and C-C couplings. ACS Catal. 13, 1678–1685 (2023).
Li, Q. Y. et al. Decarboxylative cross-nucleophile coupling via ligand-to-metal charge transfer photoexcitation of Cu(II) carboxylates. Nat. Chem. 14, 94–99 (2022).
Lutovsky, G. A., Gockel, S. N., Bundesmann, M. W., Bagley, S. W. & Yoon, T. P. Iron-mediated modular decarboxylative cross-nucleophile coupling. Chem 9, 1610–1621 (2023).
Roth, H. G., Romero, N. A. & Nicewicz, D. A. Experimental and calculated electrochemical potentials of common organic molecules for applications to single-electron redox chemistry. Synlett 27, 714–723 (2016).
Zhang, W., Carpenter, K. L. & Lin, S. Electrochemistry broadens the scope of flavin photocatalysis: Photoelectrocatalytic oxidation of unactivated alcohols. Angew. Chem. Int. Ed. 59, 409–417 (2020).
Hofer, H. & Moest, M. Mittheilung aus dem elektrochemischen Laboratorium der Königl, Technischen Hochschule zu München. Justus Liebigs Ann. Chem. 323, 284–323 (1902).
Xiang, J. et al. Hindered dialkyl ether synthesis with electrogenerated carbocations. Nature 573, 398–402 (2019).
Sheng, T. et al. Electrochemical decarboxylative N-alkylation of heterocycles. Org. Lett. 22, 7594–7598 (2020).
Applequist, D. E., Renken, T. L. & Wheeler, J. W. Polar substituent effects in 1,3-disubstituted bicyclo[1.1.1]pentanes. J. Org. Chem. 47, 4985–4995 (1982).
Huang, H., Steiniger, K. A. & Lambert, T. H. Electrophotocatalysis: Combining light and electricity to catalyze reactions. J. Am. Chem. Soc. 144, 12567–12583 (2022).
Lai, X.-L., Chen, M., Wang, Y., Song, J. & Xu, H.-C. Photoelectrochemical asymmetric catalysis enables direct and enantioselective decarboxylative cyanation. J. Am. Chem. Soc. 144, 20201–20206 (2022).
Liu, J., Lu, L., Wood, D. & Lin, S. New redox strategies in organic synthesis by means of electrochemistry and photochemistry. ACS Cent. Sci. 6, 1317–1340 (2020).
Wang, F. & Stahl, S. S. Merging photochemistry with electrochemistry: Functional-group tolerant electrochemical amination of C(sp3)−H bonds. Angew. Chem. Int. Ed. 58, 6385–6390 (2019).
Lu, J., Yao, Y., Li, L. & Fu, N. Dual transition metal electrocatalysis: Direct decarboxylative alkenylation of aliphatic carboxylic acids. J. Am. Chem. Soc. 145, 26774–26782 (2023).
Fan, W. et al. Electrophotocatalytic decoupled radical relay enables highly efficient and enantioselective benzylic C−H functionalization. J. Am. Chem. Soc. 144, 21674–21682 (2022).
Yang, Z. et al. Electrophotochemical Ce-catalyzed ring-opening functionalization of cycloalkanols under redox-neutral conditions: Scope and mechanism. J. Am. Chem. Soc. 144, 13895–13902 (2022).
Juliá, F. Ligand-to-metal charge transfer (LMCT) photochemistry at 3d-metal complexes: An emerging tool for sustainable organic synthesis. ChemCatChem 14, e202200916 (2022).
Bian, K.-J. et al. Photocatalytic hydrofluoroalkylation of alkenes with carboxylic acids. Nat. Chem. 15, 1683–1692 (2023).
Kao, S.-C. et al. Photochemical iron-catalyzed decarboxylative azidation via the merger of ligand-to-metal charge transfer and radical ligand transfer catalysis. Chem. Catal. 3, 100603 (2023).
Abderrazak, Y., Bhattacharyya, A. & Reiser, O. Visible-light-induced homolysis of earth-abundant metal-substrate complexes: A complementary activation strategy in photoredox catalysis. Angew. Chem. Int. Ed. 60, 21100–21115 (2021).
Sugimori, A. & Yamadam, T. Visible light- and radiation-induced alkylation of pyridine ring with alkanoic acid. Effective alkylation in the presence of iron(III) sulfate. Bull. Chem. Soc. Jpn. 59, 3911–3915 (1986).
Mao, R., Frey, A., Balon, J. & Hu, X. Decarboxylative C(sp3)–N cross-coupling via synergetic photoredox and copper catalysis. Nat. Catal. 1, 120–126 (2018).
Barzanò, G., Mao, R., Garreau, M., Waser, J. & Hu, X. Tandem photoredox and copper-catalyzed decarboxylative C(sp3)–N coupling of anilines and imines using an organic photocatalyst. Org. Lett. 22, 5412–5416 (2020).
Ludi, A. Prussian blue, an inorganic evergreen. J. Chem. Educ. 58, 1013 (1981).
Acknowledgements
We thank the Instrumental Analysis Center of SJTU for characterization experiments and Financial support for this work was provided by National Natural Science Foundation of China (Grant No. 22101171, 22471163).
Author information
Authors and Affiliations
Contributions
M.S. conceived the research concept and directed the project. M.S. and K.-N.Y. designed the experiments and analyzed the data. K.-N.Y., H.Z., J.W., Y.S. and H.-Q.Y. performed the experiments. M.S. wrote the manuscript. K.-N.Y., M.-H.L. and L.-L.Xu. assisted in writing and editing the manuscript.
Corresponding author
Ethics declarations
Competing interests
A Chinese patent application on this work has been filed by Shanghai Jiao Tong University (application no. 202311813231X), with M.S., K.-N.Y., J.W., M. H.L. listed as inventors. The patent covers the discovery of this new reaction, the scope of both reactants and their applications in the drug development. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hai-Chao Xu and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yuan, KN., Zhuang, H., Wei, J. et al. Modular access to saturated bioisosteres of anilines via photoelectrochemical decarboxylative C(sp3)–N coupling. Nat Commun 16, 920 (2025). https://doi.org/10.1038/s41467-024-54648-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-54648-6
