
Abstract
Acute myeloid leukemia (AML) featuring retinoic acid receptor-gamma (RARG) rearrangements exhibits morphological features resembling those of acute promyelocytic leukemia but is associated with drug resistance and poor clinical outcomes. However, the mechanisms underlying the role of RARG fusions in leukemogenesis remain elusive. Here, we show that RARG fusions disrupt myeloid differentiation and promote proliferation and self-renewal of hematopoietic stem and progenitor cells (HSPCs) by upregulating BCL2 and ATF3. RARG fusions overexpression leads to preleukemic phenotypes but fails to induce oncogenic transformation. However, the co-occurrence of RARG fusions and heterozygous Wt1 loss induce fully penetrant AML by activating MYC and HOXA9/MEIS1 targets. Leveraging Connectivity Map resources and high-throughput screening, we identify venetoclax, homoharringtonine, and daunorubicin as potential therapeutic options for RARG-AML. Overall, our findings provide pivotal insights into the molecular mechanisms governed by RARG fusions and enhanced by WT1 loss in AML development and propose a rational therapeutic strategy for RARG-AML.
Similar content being viewed by others


Introduction
AML is a hematological malignancy characterized by abnormal expansion of hematopoietic precursors that are blocked at different stages of differentiation1. Chromosomal translocations in AML lead to the synthesis of chimeric proteins that interfere with cell signaling pathways involved in growth, differentiation, and survival2. Next-generation sequencing has revolutionized cancer genomics, and its application in AML has led to the discovery of an increasing number of molecular abnormalities, which in turn has expanded the classification of AML subtypes3. In 2011, the first case of AML harboring a RARG was reported4. AML with RARG rearrangements, including NUP98::RARG, PML::RARG, CPSF6::RARG, NPM1::RARG::NPM1, and HNRNPC::RARG, has been identified as a subtype of AML5. The method to diagnose patients with AML with RARG rearrangement involves the multiplex quantitative polymerase chain reaction (qPCR) using primers covering the 5 RARG fusions or fluorescence in situ hybridization (FISH) analysis with RARG specific fluorescence in situ hybridization probes from BAC (bacterial artificial chromosome) clones. The morphological features of leukemia blasts from patients with AML with RARG rearrangement exhibited purple cytoplasm granules, kidney-shaped, bilobed or irregularly shaped nuclei. These cases share clinical, morphological, and immunophenotypic features similar to those of classical acute promyelocytic leukemia (APL), but they are unresponsive to standard APL treatments (ATRA and/or ATO) and have poor prognosis2,4,5,6,7,8,9,10,11,12,13,14,15,16. However, the mechanism of action of RARG fusions and therapeutic approaches for RARG-AML remain largely unknown.
Retinoic acid receptors (RARs) are nuclear receptors that act as transcription factors and are involved in the regulation of gene expression during the development, differentiation, and maintenance of tissue homeostasis17. RAR fusions have been repeatedly found in AML, particularly in the APL subtype, which is characterized mainly by the presence of PML::RARA resulting from t(15;17) chromosomal translocation18,19. PML-RARA acts as a transcriptional repressor of RARA signaling to interfere with the gene expression programs involved in both progenitor self-renewal and terminal myeloid cell differentiation18,20. RARG fusions differ from PML-RARA primarily in their N-terminal region. Although RARG shares 90% homology with RARA and RARB21, the transcriptional regulatory ability of RARG fusions remains poorly understood. Importantly, RARG-AML patients does not respond to standard APL treatments, implying that RARG-AML has a distinct etiology and requires distinct therapeutic strategies.
Wilms’ tumor 1 (WT1) acts as a transcription factor that suppresses tumor growth and promotes stem cell quiescence22,23. Concurrent mutations in WT1 have been observed in 58.3% of patients with RARG rearrangements5. However, whether WT1 mutations cooperate with RARG fusions in AML development and pathology remains unclear. Here, we found that RARG fusions exhibited effects on proliferation, self-renewal, and disruption of myeloid differentiation similar to those of PMLRARA. We also found that RARG fusions overexpression, in combination with WT1 deficiency, contributes to AML formation in mouse models. To elucidate the molecular mechanisms underlying the differences between RARG fusions and PML-RARA, we integrated RNA-seq analysis and ChIP-seq data to identify BCL2 and ATF3 as critical factors involved in the development of AML. Additionally, we used the Connectivity Map resource and high-throughput screening to identify potential therapeutic options for AML with RARG rearrangement. Finally, we validated the potential efficacy of these therapeutic approaches in transplantation mouse models and patient-derived xenograft (PDX) models.
Results
RARG fusions have molecular functions similar to those of PML-RARA
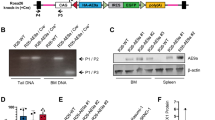
To investigate the role of RARG fusions in AML, we generated a schematic overview of RARG fusions and full-length RARG fusion genes (Fig. 1a). Furthermore, the RARG fusions were subcloned into retroviral vectors and used to infect human CD34+ (hCD34+) cells (Fig. 1b). After infection, the percentage of GFP+ cells was determined by flow cytometry (Supplementary Fig. 1a) and the expression of RARG fusions was confirmed by immunoblotting (Fig. 1c). After sorting for GFP+ cells, these cells were cultured in a liquid medium system, allowing long-term expansion of HSPCs24. Cells overexpressing RARG fusions grew much faster than those infected with control retrovirus (Fig. 1d). In addition, CFU assays revealed that overexpression of RARG fusions, as well as PML-RARA, in hCD34+ cells increased their self-renewal ability (Fig. 1e). Overexpression of RARG fusions or PML-RARA promoted the expansion of immature myeloid cells (CD45+CD33+CD34+), reduced the percentage of mature granulocytes (CD45+CD33+CD66b+), and inhibited myeloid cell differentiation (CD45+CD33+Cb11b+)25 (Fig. 1f–h). These results showed that similar to PML-RARA, RARG fusions increased the proliferation and self-renewal abilities of primary hCD34+ cells while disrupting myeloid differentiation.

a Schematic representation of the main domains of RARG fusion genes. b Approaches to investigate the effect of RARG fusions on the transformation of hCD34+ hematopoietic stem and progenitor cells. c The expression of RARG fusions in hCD34+ cells after sorting for GFP+ cells were measured by western blotting (n = 3 independent experiments). d Growth curves of hCD34+ cells with or without RARG fusions overexpression. e Colony-forming capacity of hCD34+ cells with or without RARG fusions overexpression from the 1st and 2nd rounds of cell plating in methylcellulose and culture for 14 days. f–h Flow cytometric analysis of the effect of RARG fusions overexpression on hCD34+ cell myeloid differentiation. The percentages of immature myeloid cells (f), granulocytes (g) and differentiation of myeloid cells (h) are shown. The gating strategy is shown in Supplementary Fig. 7a. i The effect of RARG fusions on the transcriptional activity of PU.1, CEBPE and GFI1. The luciferase reporter plasmids expressing the CEBPE, PU.1, or GFI1 promoter were cotransfected with empty vector or RARG fusions plasmids into U937 cells. Luciferase activity was measured 24 h after transduction. j The homodimerization of CPSF6-RARG or NUP98-RARG was evaluated by CO-IP assays (n = 3 independent experiments). k The heterodimerization of CPSF6-RARG or NUP98-RARG was evaluated by CO-IP assays (n = 3 independent experiments). l The interactions of CPSF6-RARG or NUP98-RARG with endogenous transcriptional corepressors were evaluated by CO-IP assays. Leukemia cells were isolated from Wt1F/+CreMx-1;CPSF6::RARG mice and Wt1F/+CreMx-1;NUP98::RARG mice and cell extracts were subjected to IP with an anti-HA antibody (n = 3 independent experiments). m, n Real-time PCR analysis of the effect of overexpression of RARG fusions or PML-RARA in hCD34+ cells on the mRNA level of CD38, SCUBE1, RARG, POSTN, or DEFA1. d–i, m, n n = 3 technical replicates. Results are representative of three independent experiments. Statistical significance was calculated by (d–i, m, n) one-way ANOVA with Tukey’s multiple comparison tests; Data are presented as means ± S.E.M. In each of the immunoblots j–l, all samples derive from the same experiments but different gels for each antibody were processed in parallel. Source data are provided as a Source Data file.
PML-RARA has long been considered as a transcriptional repressor that blocks the differentiation of myeloid cells by repressing the target genes CEBPE and PU.126,27,28,29. Recently, PML-RARA was shown to be a transcriptional activator that promotes the expression of genes essential for APL pathogenesis such as GFI130. We found that RARG fusions exerted transrepressing effects on PU.1 and CEBPE and transactivating effects on GFI1 (Fig. 1i). RARA fusions have been reported to form homodimers that bind corepressors and DNA sequences, which mediate transcriptional deregulation and leukemogenesis31,32. As shown in Fig. 1j and Supplementary Fig. 1b, Flag-tagged RARG fusions pulled down HA-tagged RARG fusions, indicating the formation of homodimers. Additionally, PML-RARA forms a heterodimer with RXR, which enhances the binding of PML-RARA to DNA and is required for RA-induced APL cell differentiation33. Consistent with this observation, RARG fusions formed heterodimers with RXRA (Fig. 1k and Supplementary Fig. 1c), and interacted with nuclear receptor corepressors such as NCOR2, Sin3A, and HDAC3 (Fig. 1l and Supplementary Fig. 1d). To provide further evidence of the transcriptional effects of RARG fusions, we examined the cellular localization of RARG fusions. We found that RARG fusions were localized predominantly in nucleus, with a lower distribution in cytoplasm (Supplementary Fig. 1e). To further define the specific ___domain within the RARG fusion protein responsible for its intracellular localization, we selected CPSF6-RARG and constructed six CPSF6-RARG truncations and two CPSF6-RARG mutants in key residues M624 and T62733,34. Interestingly, we found that ΔDBD, ΔRRM, ΔRARG1, CPSF6, and RARG had an intracellular distribution similar to that of CPSF6-RARG (Supplementary Fig. 1f). However, the ΔLBD and M624R/T627R mutants exhibited a loss of the diffuse intranuclear pattern (Supplementary Fig. 1f). Additionally, we confirmed the interaction between CPSF6-RARG and RXRA and found that LBD ___domain and residues M624 and T627 were critical for mediating this interaction (Supplementary Fig. 1g, h). Previous study has shown that the most significant differentially expressed genes in RARG-AML patients, in comparison to PML::RARA-positive APL, include CD38, SCUBE1, HOXA6, RARG, POSTN, DEFA1, among others5. As shown in Fig. 1m, n, the mRNA levels of CD38 and SCUBE1 were decreased, while the mRNA levels of RARG, POSTN, and DEFA1 were increased in hCD34+ cells with RARG fusions overexpression. Taken together, these data imply that RARG fusions, similar to PML-RARA, not only promote cell proliferation and self-renewal and block myeloid cell differentiation but also contribute to the formation of transcriptional complexes that regulate target gene transcription.
RARG fusions transcriptionally activate ATF3 and BCL2
To understand how RARG fusions regulate their target genes in the hematopoietic system, we overexpressed fusions in hCD34+ cells and identified target genes that are regulated by RARG fusions (Fig. 2a). A total of 4556 CPSF6-RARG-binding sites, 4852 NUP98-RARG-binding sites, 2075 PML-RARG-binding sites, 3304 HNRNPC-RARG-binding sites, 2684 NPM1-RARG-binding sites, and 2785 PML-RARA-binding sites were identified (Fig. 2b). Notably, RARG fusions shared 423 common binding sites (Fig. 2c), with the binding sites being highly enriched at promoter regions (Supplementary Fig. 2a). Additionally, there were 318 common binding sites and 105 unique binding sites between RARG fusions and PML-RARA (Fig. 2d). By integrating ChIP-seq and RNA-seq data, we further created an atlas of direct target genes of RARG fusions, including 537 activated genes and 583 repressed genes for CPSF6-RARG, 402 activated genes and 355 repressed genes for NUP98-RARG, 68 activated genes and 36 repressed genes for HNRNPC-RARG, 53 activated genes and 49 repressed genes for NPM1-RARG, 166 activated genes and 119 repressed genes for PML-RARG, and 113 activated genes and 89 repressed genes for PML-RARA (Supplementary Fig. 2b).

a Schematic illustration of the experimental design. b Density heatmap of HA-X-RARG fusion genes binding peaks. The heatmap encompasses a region extending from −5 kb to +5 kb centered on TSS enriched by the anti-HA antibody. c Venn diagram indicating the common binding peaks of RARG fusions identified by ChIP-seq. d Venn diagram indicating the common binding peaks in both RARG fusions and PML::RARA identified by ChIP-seq. e Venn diagram indicating the genes uniquely activated and repressed by RARG fusions compared to PML-RARA based on integrated analysis of ChIP-seq and RNA-seq data. f Motif enrichment at RARG fusions binding sites by HOMER analysis. g Real-time PCR analysis of the effect of CPSF6-RARG and NUP98-RARG overexpression in hCD34+ cells on the expression of activated genes and repressed gene. h Genome browser tracks showing the binding patterns of RARG fusions and PML::RARA on ATF3 and BCL2. i hCD34+ cells overexpressing RARG fusions were analyzed by ChIP with an anti-HA antibody or IgG. Immunoprecipitated DNA was quantified using qPCR for the anti-HA antibody- or IgG- bound BCL2 and ATF3 promoter and enhancer regions. j Effect of RARG fusions on the transcriptional activity of ATF3 and BCL2. k Heat maps and density plots of ATAC-seq peaks aligned to human hg38 genome around the TSS ± 5 kb. l, m Heatmaps and histograms shown the abundant CPSF6-RARG, NUP98-RARG, H3K27ac, H3K4me1, H3K4me3, and ATAC-seq occupancy on RARG fusions binding sites. A 5 kb window is shown with the summit of RARG fusions binding in the center of each panel. n Real-time PCR analysis of the effect of p300 inhibitor C646 on the expression of BCL2 and ATF3 in hCD34+ cells with or without CPSF6-RARG or NUP98-RARG overexpression. g, i, j, n n = 3 technical replicates. Results are representative of three independent experiments. Statistical significance was calculated by (i, j) one-way ANOVA with Tukey’s multiple comparison tests; (g, n) two-tailed Student’s t test; Data are presented as means ± S.E.M. Source data are provided as a Source Data file.
More importantly, we identified eight uniquely activated genes and two repressed genes by RARG fusions compared to PML-RARA through the integrated analysis of ChIP-seq and RNA-seq data (Fig. 2e). Motif analysis revealed that RARG fusions binding sites were enriched for motifs of transcription factors KLF1, KLF10, E2F2, SP1, CEBPG, and SMAD4 (Fig. 2f). Overexpression of RARG fusions led to increased transcript levels of activated genes (EREG, SYLT3, REXO1, RASGEF1B, INPP5A, NCAPG2, ATF3, and BCL2) and decreased transcript levels of repressed gene (EIF1 and RPS8). Notably, ATF3 and BCL2 exhibited the highest upregulation in most hCD34+ cells overexpressing RARG fusions, while the mRNA levels of EREG, SYLT3, REXO1, RASGEF1B, INPP5A, and NCAPG2 were increased in some of these cells (Fig. 2g and Supplementary Fig. 2c, d). Additionally, there was a slight increase in the mRNA levels of EREG, RASGEF1B, ATF3, and BCL2 in hCD34+ cells overexpressing PML::RARA (Supplementary Fig. 2e). Previous studies have shown that ATF3 sustained the cycling and survival of leukemia cells and contributed to the differentiation arrest35. BCL2, a well-known anti-apoptotic gene, has been implicated in various cancers, including AML36. The upregulation of ATF3 and BCL2 in RARG-fusion AML suggests their potential role in inhibiting differentiation and maintaining survival of leukemia cells.
In addition, the binding signals of RARG-fusions were more abundant in the promoter and enhancer regions of ATF3 and BCL2, as well as the promoter region of EIF1 compared to PML::RARA (Fig. 2h and Supplementary Fig. 2f). Furthermore, ChIP‒qPCR confirmed the enrichment of these regions of ATF3, BCL2, and EIF1 in RARG fusions targets (Fig. 2i and Supplementary Fig. 2g). RARG fusions and PML::RARA could enrich the promoter and enhancer regions of ATF3 and BCL2, and there was a more abundant enrichment of RARG-fusions on the promoter (peak 1) and enhancer (peak 2) regions of ATF3 and BCL2 compared to PML::RARA (Fig. 2i). We then constructed the promoter (peak 1) region of ATF3, BCL2, and EIF1 into luciferase reporter plasmid and detected the effect of RARG fusions on their transcriptional regulation. The luciferase reporter assays indicated that RARG fusions transcriptionally activated ATF3 and BCL2 while repressing EIF1 (Fig. 2j and Supplementary Fig. 2h). To further confirm the effects of RARG fusions on the enhancer region of ATF3 and BCL2, constructs containing the enhancer (peak 1) and promoter (peak 2) regions of these genes were generated. The results showed an increase in luciferase activity compared to the promoter construct of ATF3 or BCL2 alone (Fig. 2j). Moreover, PML::RARA could transcriptionally activate ATF3 and BCL2, but the transcriptional activities of ATF3 and BCL2 were higher in RARG fusions overexpression compared to PML::RARA overexpression (Fig. 2j). These findings elucidate the specific alterations in gene expression associated with different RARG fusion variants, highlighting the roles of these fusions in hematopoiesis and their potential contributions to the development and treatment of AML.
We defined the chromatin accessibility using ATAC-seq assay in human CD34+ (hCD34+) cells with or without CPSF6::RARG or NUP98::RARG overexpression. ATAC-seq peaks were identified at 50,676 regions in hCD34+ cells, 80,002 regions in hCD34+ cells with CPSF6::RARG, and 76,322 regions in NUP98::RARG overexpression (Fig. 2k, and Supplementary Data 1). The chromatin accessibility was increased in hCD34+ cells with CPSF6::RARG or NUP98::RARG overexpression compared to hCD34+ cells (Fig. 2k). The ATAC-seq peaks were mainly enriched at promoters, intron, and intergenic regions (Supplementary Fig. 2i). Subsequently, differential chromatin accessibility was analyzed based on sites with FDR ≤ 0.05 and the average absolute log2 (fold change) ≥ 1.0 in CPSF6::RARG and NUP98::RARG overexpressing hCD34+ cells compared to hCD34+ cells. Using this threshold, we identified 1145 differential regions in CPSF6::RARG overexpressing hCD34+ cells and 6008 in NUP98::RARG overexpressing hCD34+ cells compared to hCD34+ cells (Supplementary Fig. 2j). Of these regions, CPSF6::RARG overexpressing hCD34+ cells exhibited 1135 regions with increased chromatin accessibility and 10 regions with decreased accessibility. In NUP98::RARG overexpressing hCD34+ cells, 5952 regions showed increased accessibility, while 56 regions displayed decreased accessibility (Supplementary Fig. 2j, k). Moreover, there were 625 differentially accessible regions in CPSF6::RARG and NUP98::RARG overexpressing hCD34+ cells (both increased and decreased) compared to hCD34+ cells (Supplementary Fig. 2k). Notably, these differentially accessible regions were primarily enriched at promoter, intron, and intergenic regions (Supplementary Fig. 2l). These data suggest that RARG fusions increased DNA accessibility across numerous genomic regions.
We further performed ChIP-seq assay to investigate the epigenetic changes (H3K27ac, H3K4me1, and H3K4me3) associated with RARG fusions binding in hCD34+ cells overexpressing CPSF6::RARG and NUP98::RARG. Our analysis revealed that regions bound by RARG fusions exhibited elevated levels of ATAC-seq, H3K27ac, H3K4me3, and H3K4me1 signals compared to hCD34+ cells without RARG fusions overexpression (Fig. 2l, m), indicating a potential role of RARG fusions in regulating promoter and enhancer. In hCD34+ cells with CPSF6::RARG and NUP98::RARG overexpression, the promoter and enhancer sites of ATF3 and BCL2 showed significant enrichment of ATAC-seq, H3K27ac, H3K4me1, and H3K4me3 signals compared to hCD34+ cells without RARG fusions overexpression (Supplementary Fig. 2m). Previous reports have highlighted the activating functions of PML-RARA through its interaction with coactivators. Our previous work established that PML-RARA interacts with coactivator P300 to activate specific target genes30. To further investigate the interaction between P300 and RARG fusions and its impact on gene activation, we initially performed a CO-IP assay to examine the binding of P300 with CPSF6-RARG or NUP98-RARG. The CO-IP assay results showed that P300 indeed interacted with both CPSF6-RARG and NUP98-RARG (Supplementary Fig. 2n). Subsequently, we treated hCD34+ cells overexpressing CPSF6::RARG or NUP98::RARG with the P300 inhibitor C646 to detect the effect on downstream targets BCL2 and ATF3. The expression of BCL2 and ATF3 were decreased in hCD34+ cells overexpressing CPSF6::RARG or NUP98::RARG following treatment with C646 (Fig. 2n). This suggests that P300 plays a critical role in mediating the activation of downstream targets such as BCL2 and ATF3 by RARG fusions.
BCL2 and ATF3 are crucial for the maintenance of RARG-AML
Next, we investigated the clinical relevance of BCL2 and ATF3 in RARG-AML. Gene set enrichment analysis (GSEA) using RNA-seq data5 from RARG-AML patients revealed that ATF3 target genes were enriched in RARG-AML, whereas the apoptosis pathway was enriched in APL (Fig. 3a). We then detected the mRNA and protein levels of BCL2, ATF3, and ATF3 target genes (TNFA1P6 and ITGA4) in U937 cells stably overexpressing RARG fusions, PML::RARA or control vector. Our data show a slight increase in the mRNA levels of BCL2, ATF3, and ATF3 target genes, as well as protein levels of BCL2 family proteins in U937 cells overexpressing PML::RARA compared to control U937 cells (Fig. 3b–d). Importantly, we found a significant upregulation of these proteins in U937 cells overexpressing RARG fusions compared to those overexpressing PML::RARA (Fig. 3b–d). Moreover, other proteins relating anti-apoptotic pathways were also upregulated in U937 cells overexpressing RARG fusions (Fig. 3d). The IGV tracks showed that there were binding signals in the promoter regions of BCL2L1, BCL2L2, and MCL1 (Supplementary Fig. 3a). We also found that the mRNA levels of BCL2L1, BCL2L2, or MCL1 were increased in hCD34+ cells with X-RARG overexpression (Supplementary Fig. 3b). These data suggested that the upregulation of BCL2 family protein BCL2L1, BCL2L2, and MCL1 by X-RARG may be mediated by its transactivation activities.

a GSEA results showing the enrichment of apoptosis pathways and ATF3 target genes in RARG-AML patients compared with APL patients. Statistical significance was determined by a one-sided permutation test, and statistical adjustments were made for multiple comparisons. b, c Real-time PCR analysis of the effect of stable overexpression of RARG fusions or PML-RARA in U937 cells on the mRNA level of BCL2 (b), ATF3 and ATF3 target genes TNFA1P6 and ITGA4 (c). d The protein levels of apoptosis pathway proteins and ATF3 in U937 cells with or without RARG fusions or PML-RARA overexpression were measured by Western blotting (n = 3 independent experiments). The samples derive from the same experiments but different gels for BCL2L1, BCL2, BCL2L2, MCL1, ATF3, HA, and Actin were processed in parallel. e, f Flow cytometric analysis of the effect of RARG fusions overexpression on differentiation (e) and apoptosis (f) in hCD34+ cells with or without ATF3 or BCL2 knockdown. e CON-shRNA VS ATF3-shRNA p value: 0.0009; CON-shRNA VS BCL2-shRNA, p value: 0.0011. The gating strategy is shown in Supplementary Fig. 7b. f CON-shRNA VS ATF3-shRNA, p value: 0.0086; CON-shRNA VS BCL2-shRNA, p value: 0.0016. The gating strategy is shown in Supplementary Fig. 7c. g The effect of RARG fusions or PML-RARA overexpression on the colony-forming capacity of hCD34+ cells with or without ATF3 or BCL2 knockdown. CON-shRNA VS ATF3-shRNA, p value: 0.0041; CON-shRNA VS BCL2-shRNA, p value: 0.0061. h, i The effect of ATF3 or BCL2 knockdown on the apoptosis and differentiation of hCD34+ cells and primary NUP98-RARG patient cells. The relative change in apoptosis and differentiation cells (Fold of CON-shRNA group) were calculated. j Long-term culture-initiating cell (LTC-IC) assays were performed from hCD34+ cells or primary NUP98-RARG patient cells with or without ATF3 or BCL2 knockout. LTC-IC colonies were counted as a percentage of colonies from CON-shRNA groups of hCD34+ cells or primary NUP98-RARG patient cells. b, c, e–j n = 3 technical replicates. Results are representative of three independent experiments. Statistical significance was calculated by (b, c, e–j) one-way ANOVA with Tukey’s multiple comparison tests; Data are presented as means ± S.E.M. Source data are provided as a Source Data file.
Moreover, we also detected the protein levels of BCL2 and ATF3 in human primary NUP98::RARG primary blasts, primary PML::RARA blasts, and healthy donors, and found that BCL2 and ATF3 seemed to have elevated tendency in NUP98::RARG primary blasts compared to PML::RARA blasts (Supplementary Fig. 3c). Additionally, we found that RARG fusions overexpression in U937 cells decreased the percentage of apoptotic and differentiated leukemia cells and increased the colony-forming ability compared to control U937 cells. Knockout of ATF3 or BCL2 in U937 cells resulted in increased apoptosis and differentiation, as well as a decrease in colony-forming ability. However, the effects of RARG fusions overexpression on apoptosis, differentiation, and colony-formation were diminished in ATF3- or BCL2- knockout U937 cells (Supplementary Fig. 3d–h). These data suggested that ATF3 and BCL2 are crucial in maintaining apoptosis, differentiation, and colony-forming ability of RARG fusions overexpressing U937 cells.
We also used human CD34+ cells model to investigate the role of BCL2 and ATF3 in the maintenance of RARG-AML. Overexpression of RARG fusions or PML-RARA in hCD34+ cells reduced the percentage of apoptotic cells and differentiated myeloid cells (CD33+CD11b+) and increased the CFU numbers compared to hCD34+ cells transduced with control vector (Fig. 3e–g, black columns). ATF3 or BCL2 knockdown in hCD34+ cells increased the percentage of apoptotic and differentiated cells and decreased the CFU numbers. However, the effects of apoptosis and differentiation arrest and increase of colony-forming ability induced by RARG fusions overexpression in hCD34+ cells were diminished in BCL2- or ATF3- knockdown hCD34+ cells (Fig. 3e–g, red and pink columns). Moreover, we found that PML-RARA overexpression in hCD34+ cells also induced the apoptosis and differentiation arrest and increase of colony-forming ability. Notably, these effects were still observed even when PML-RARA was overexpressed in hCD34+ cells with knockdown of BCL2 or ATF3 (Fig. 3e–g). Overall, these results reveal that the apoptosis and differentiation arrest and increase of colony-forming ability induced by RARG fusions were mediated by transactivating BCL2 and ATF3.
In addition, BCL2 or ATF3 knockdown in hCD34+ cells had minimal effect on the apoptosis and differentiation (Fig. 3h, i). However, BCL2 or ATF3 knockdown in primary NUP98-RARG patient cells increased the percentage of apoptotic and differentiated leukemia cells (Fig. 3h, i). Moreover, we also performed long-term culture-initiating cells (LTC-ICs) assay to evaluate the effect BCL2 or ATF3 knockdown on the leukemia stem cells (LSCs) function of primary NUP98-RARG leukemia cells. Primary NUP98-RARG leukemia cells infected with or without BCL2- or ATF3- shRNA lentivirus were cultured for 2 days before being subjected to LTC-ICs assays for 3 weeks, followed by colony-forming assay. The results showed that BCL2 or ATF3 knockdown in primary NUP98-RARG leukemia cells significantly reduced the colony formation compared to cells without the knockdown (Fig. 3j). These results indicate that BCL2 and ATF3 are crucial for the maintenance of RARG-AML.
RARG fusions drives the expansion of HSPCs
To examine the function of RARG fusions in hematopoietic development and leukemogenesis, we generated a mouse model by knock-in Cpsf6-rarg cDNA in exon 3 of Rarg (Supplementary Fig. 4a). The Cpsf6rargF/F mice were crossed with the CreVav strain to induce Cpsf6::rarg overexpression in the HSCs (Fig. 4a). The expression of Cpsf6-rarg in the BM of Cpsf6-rargcKI mice was validated by western blotting (Supplementary Fig. 4b). Hematological phenotype analysis indicated that white blood cell counts, platelet counts, and hemoglobin levels were similar between Cpsf6::rargcKI and CreVav mice throughout their lifespans (Supplementary Fig. 4c). The proportions of B, T, and myeloid cells in the PB and spleen of 6-month-old Cpsf6::rargcKI mice did not differ significantly from those in CreVav controls (Supplementary Fig. 4d). The number of BM cells in 6-month-old Cpsf6::rargcKI mice was similar to that in CreVav controls (Supplementary Fig. 4e). This finding suggests that Cpsf6-rarg overexpression alone does not cause any overt changes in hematopoiesis.

a Schematic strategy used to establish the mouse model of HSC-specific overexpression of Cpsf6::rarg. b, c Representative images of Wright-Giemsa-stained PB smears, BM smears (b), spleens (c, left) and statistical analysis of the spleen weights (c, right) from 12-month-old Crevav mice and Cpsf6::rargCKI mice (n = 5 mice per group). d, e Flow cytometric analysis of PB, spleen cells (d) and BM cells (e) isolated from 12-month-old Crevav mice and Cpsf6::rargCKI mice (n = 5 mice per group). The gating strategy is shown in Supplementary Fig. 7e. f–j Representative FACS plots and quantification of the percentage of granulocytes (CD11b+Gr-1+), immature myeloid cells (CD11b+c-kit+) (f) and myeloid precursor cells (Gr-1+c-kit +) (g), Lin- cells, LSK cells, HPCs (h), MPPs, ST-HSCs, LT-HSCs (i), MEPs, GMPs, and CMPs (j) in BM from 12-month-old Crevav mice and Cpsf6::rargCKI mice. (n = 5 mice per group). k Schematic strategy to investigate the effect of transplantation of BM cells isolated from Cpsf6::rargCKI mice or Crevav mice on AML development. l Representative images of spleens (top) and statistical analysis of the spleen weight (bottom) in recipient mice 2 months after transplantation of BM cells isolated from 12-month-old Crevav mice or Cpsf6::rargCKI mice (n = 5 mice per group). m–o Flow cytometric analysis of BM cells isolated from recipient mice 2 months after transplantation of BM cells isolated from 12-month-old Crevav mice and Cpsf6::rargCKI mice (n = 5 mice per group). p Real-time PCR analysis of the mRNA levels of Atf3 (top) and Bcl2 (bottom) in myeloid cells sorted from the BM of Crevav mice and Cpsf6::rargCKI mice (n = 3 mice per group). The strategy for isolating myeloid cells from BM is shown in Supplementary Fig. 7f. Statistical significance was calculated by (c–j, l–p) two-tailed Student’s t-test; Data are presented as means ± S.E.M. Source data are provided as a Source Data file.
Although there were no significant differences in blood cell differentials or splenomegaly at the age of 12 months (Fig. 4b, c), the proportion of B cells was decreased, while that of myeloid cells was increased in the PB and spleen of 12-month-old Cpsf6::rargcKI mice (Fig. 4d). A more detailed analysis of the BM showed that the proportions of B cells and T cells were decreased in Cpsf6::rargcKI mice, while those of myeloid cells, granulocytes (CD11b+Gr-1+), and immature myeloid cells (CD11b+c-kit+) were increased (Fig. 4e, f and Supplementary Fig. 4f, g). Moreover, the percentage of myeloid precursors (Gr-1+c-kit+) in the BM of 12-month-old Cpsf6::rargcKI mice was higher than that in BM of CreVav mice (Fig. 4g).
We then further analyzed the impact of Cpsf6::rarg on HSPCs, which are phenotypically defined as Lin−Sca1+c-kit+ (LSK) and Lin−Sca1−c-kit+ (HPC) respectively. Interestingly, the proportions of lineage-negative (Lin-) cells in the BM and LSK and HPC cells in the Lin- cell population were increased in the BM of both 6 and 12-month-old Cpsf6-rargcKI mice (Fig. 4h and Supplementary Fig. 4h). Furthermore, the proportions of multipotent progenitor cells (MPPs) and short-term repopulating HSCs (ST-HSCs) among LSK cells, as well as granulocyte-monocyte progenitors (GMPs) among HPC cells, were increased in the BM of these mice (Fig. 4i, j, and Supplementary Fig. 4i). In contrast, the proportions of megakaryocyte-erythroid progenitor cells (MEPs) and common myeloid progenitors (CMPs) among the HPCs were decreased in these mice (Fig. 4j). Taken together, these results imply that the CPSF6-RARG drives aberrant expansion of HSPCs, which may lead to a preleukemic phenotype.
To investigate whether the preleukemia phenotype originated from transformed HSPCs harboring CPSF6-RARG, we conducted secondary BM transplantation experiments. BM cells from 12-monthold Cpsf6::rargcKI mice were transplanted into lethally irradiated recipient mice (Fig. 4k). Similar to the observations in the donor mice, there were no detectable differences in PB cell counts of recipient mice (Supplementary Fig. 4j). As expected, the reproduction of the preleukemia phenotypes was observed. These phenotypes included spleen enlargement (Fig. 4l), as well as a decrease in the proportion of B cells and increase in the proportion of myeloid cells in the PB and spleen of Cpsf6::rargcKI-transplanted recipient mice (Supplementary Fig. 4k, l). Furthermore, aberrant expansion of myeloid cells, granulocytes, and immature myeloid cells, as well as LSK and HPC populations, was observed in the BM of Cpsf6::rargcKI-transplanted recipient mice (Fig. 4m, n). The percentages of MPPs and GMPs also increased in the BM of recipient mice (Fig. 4o). We next measured Bcl2 and Atf3 expression in Cpsf6::rargcKI mice. The mRNA levels of Bcl2 and Atf3 were significantly increased in the myeloid cells isolated from the BM of Cpsf6::rargcKI mice compared to those in control mice (Fig. 4p). These results showed that these abnormal cell populations originated from abnormal BM cells in Cpsf6::rargcKI donor mice. However, overt AML development upon transplantation in this mouse model suggests that additional events or genetic alterations may be required to fully promote hematopoietic transformation and leukemia development.
Wt1 haploinsufficiency cooperates with RARG fusions to induce AML
Based on clinical observations that monoallelic germline Wt1 mutations frequently co-occur with RARG-AML5, we investigated whether partial Wt1 deficiency cooperates with RARG fusions to induce AML. We first crossed CreVav mice with Wt1F/F mice to generate mice with Wt1 deletion in hematopoietic system. Unfortunately, CreVavWt1F/+ mice did not survive longer than 2–3 weeks after birth, possibly due to hematopoietic cell dysfunction. Subsequently, we generated an inducible hematopoiesis-specific conditional Wt1-knockout mouse model by crossing Wt1F/F mice with CreMx-1 mice (Supplementary Fig. 5a, b). Lin- cells were isolated from the BM of 8-month-old CreMx-1 and Wt1F/+CreMx1 mice, transduced with X-RARG-fusions-GFP retrovirus, and transplanted into lethally irradiated recipient mice (Fig. 5a). Eight to nine weeks after transplantation, all Wt1F/+CreMx-1; X-RARG transplanted mice developed aggressive AML with significantly enlarge spleen (Fig. 5b). Wright staining and HE staining confirmed the infiltration of leukemic blasts into the PB, BM, and spleen (Fig. 5c and Supplementary Fig. 5c). FACS analysis showed that the GFP-positive cells from the BM and spleen of Wt1F/+CreMx-1;CPSF6::RARG-transplanted mice were myeloid cells (Fig. 5d). Furthermore, mice transplanted with Wt1F/+CreMx-1 Lin- cells expressing RARG fusions exhibited a median latency of 60 days (Fig. 5e).

a Schematic model of the procedure used to study the effect of heterozygous Wt1 knockout in combination with RARG fusions overexpression on AML development. b Representative images of spleens (left) and statistical analysis of the spleen weights (right) in recipient mice from the indicated groups (n = 5 mice per group). c Representative images of BM smears and H&E staining of the spleen tissues from recipient mice in the indicated groups (n = 5 mice per group). d Representative FACS plots of GFP positive cells in the BM and spleen of CreMx-1 CPSF6::RARG mice or Wt1F/+CreMx-1 CPSF6::RARG mice. e Kaplan–Meier survival curves for recipient mice with primary transplantation of Lin- cells isolated from CreMx-1 mice (left) or Wt1F/+CreMx-1 mice (right) and infected with the control retrovirus or RARG fusions retroviruses (n = 5 mice per group). f Kaplan–Meier survival curves for recipient mice with secondary transplantation of spleen cells isolated from primary Wt1F/+CreMx-1;X::RARG mice (leukemic) or X::RARG mice (n = 5 mice per group). g Enrichment analysis of the top enriched pathways related to the upregulated genes (left) and downregulated genes (right) in leukemia cells isolated from the spleen of Wt1F/+CreMx-1;CPSF6::RARG mice compared to CreMx-1;CPSF6::RARG mice. Statistical analysis was conducted with Metascape (https://metascape.org/gp/index.html), using the two-sided cumulative hypergeometric distribution. h GSEA showing the enrichment of indicated gene sets in the Wt1F/+CreMx-1;CPSF6::RARG group compared with the CreMx-1;CPSF6::RARG group. The P value was determined by a one-sided permutation test. Statistical adjustments were made for multiple comparisons. i The protein levels of BCL2 and ATF3 in the BM of PML::RARA-driven APL mice and Wt1F/+CreMx-1;X::RARG-driven AML mice were detected by western blotting (n = 3 independent experiments). The samples derive from the same experiments but different gels for ATF3, BCL2, and GAPDH were processed in parallel. j Kaplan–Meier survival curves for recipient mice with secondary transplantation of spleen cells isolated from primary Wt1F/+CreMx-1;CPSF6::RARG mice (leukemic) and infected with control, Atf3 or Bcl2 shRNA lentivirus particles (n = 8 mice per group). Statistical significance was calculated by (b) two-tailed Student’s t-test; (e, f, j) two-sided log-rank test. Data are presented as means ± S.E.M. Source data are provided as a Source Data file.
However, similar to the mice transplanted with CreMx-1 Lin- cells, most mice transplanted with CreMx1 Lin- cells with RARG fusions overexpression exhibited normal spleen sizes, PB, BM, and spleen staining, a long survival time, and good hematopoietic reconstruction capacity (Fig. 5b–e and Supplementary Fig. 5c). Only one CreMx-1;CPSF6::RARG mouse died after transplantation and succumbed after approximately four months, with an enlarged spleen. Moreover, Wt1F/+CreMx-1 transplanted mice did not develop an overt leukemic phenotype and had life spans comparable to those of CreMx-1 transplanted mice (Fig. 5b–d). All Wt1F/+CreMx-1;X::RARG mice with secondary transplantation died of AML within 2 months, whereas in most mice, X-RARG fusion overexpression alone did not result in leukemia-related death during the three-month observation period after secondary transplantation (Fig. 5f).
To investigate the mechanism by which Wt1 haploinsufficiency cooperates with RARG fusions overexpression to induce AML, we analyzed the gene expression profiles of leukemia cells isolated from CreMx-1;CPSF6::RARG and Wt1F/+CreMx-1;CPSF6::RARG mice. Functional enrichment analysis showed that the upregulated genes in leukemia cells isolated from Wt1F/+CreMx-1;CPSF6::RARG mice were mainly enriched in transcriptional regulation by RUNX1, myeloid leukocyte activation, HATs acetylate histones, and cell cycle pathways (Fig. 5g left). Furthermore, the downregulated genes in leukemia cells isolated from Wt1F/+CreMx-1;CPSF6::RARG mice were mainly enriched in the myeloid cell differentiation, T-cell differentiation and activation, and hemopoiesis pathways (Fig. 5g right). GSEA showed that MYC and HOXA9/MEIS1 targets were significantly enriched in Wt1F/+CreMx-1;CPSF6::RARG mice (Fig. 5h). These results imply that MYC- and HOXA9/MEIS1 mediated transcriptional regulation may contribute to AML pathogenesis induced by Wt1 haploinsufficiency in cooperation with RARG fusions.
Furthermore, we determined the roles of Bcl2 and Atf3 in RARG-AML using a CPSF6::RARG transplantation mouse model. The protein levels of ATF3 and BCL2 and mRNA levels of Atf3 and Bcl2 were significantly increased in Wt1F/+CreMx-1;X::RARG-driven AML mice compared to those in control mice and PML::RARA-driven APL mice (Fig. 5i and Supplementary Fig. 5f). Moreover, knockdown of either Bcl2 or Atf3 in leukemia cells significantly prolonged the survival of CPSF6::RARG AML cells engrafted mice (Supplementary Fig. 5g and Fig. 5j). These results indicate that BCL2 and ATF3 are crucial for the maintenance of AML with RARG rearrangements and could be targeted therapeutically.
Identifying potential therapeutic approaches for RARG-AML
Similar to clinical observations5, both ATRA and ATO showed weak effects on the proliferation of U937 cells overexpressing CPSF6-RARG (Supplementary Fig. 6a). In addition, we detected the effects of ATRA and ATO in primary leukemia cell derived from primary NUP98-RARG patient blasts and Wt1F/+CreMx1;CPSF6::RARG mice and found that these leukemia cells showed resistance to ATRA and ATO in the induced differentiation and apoptosis (Supplementary Fig. 6b, c). To identify potential therapeutic approaches for RARG-AML, we performed HTS in U937 cells overexpressing CPSF6-RARG (Fig. 6a). Four small-molecule drugs, topotecan hydrochloride, mitoxantrone, HHT, and venetoclax (BCL2 inhibitor), showed strong inhibitory activity against CPSF6-RARG-overexpressing U937 cells (Fig. 6b). We used C-MAP analysis to screen for potential drug candidates by integrating RNA-seq data from RARG-AML and APL patients5. C-MAP analysis identified six drugs, idelalisib, lopinavir, prilocaine, oxfendazole, mitoxantrone, and dasabuvir, as potential therapeutic candidates for RARG-AML (Fig. 6c). Subsequently, U937 cells overexpressing CPSF6-RARG were sensitive to chemotherapeutic drugs (mitoxantrone, HHT, and daunorubicin) as well as to targeted drugs (lopinavir and venetoclax), and the IC50 values of these drugs were lower in U937 cells overexpressing RARG fusions compared to parental U937 cells (Fig. 6d). However, they were tolerant to comparatively high concentrations of dasabuvir, oxfendazole, prilocaine, and idelalisib (Supplementary Fig. 6d). This suggested that U937 cells with RARG fusions are more sensitive to daunorubicin, mitoxantrone, HHT, and venetoclax. Based on our previous data showing the activation of BCL2 by RARG fusions in RARG-AML cells, the BCL2 inhibitor could be an attractive targeted agent for RARG-AML.

a Schematic illustration of the procedure for HTS of small molecule drugs against RARG-AML. An FDA-approved drug library containing 10,000 small molecule drugs was used to treat CPSF6-RARG-overexpressing U937 cells. Hits in the top 2% were reconfirmed by cell viability testing. b Volcano plot showing the hits in the top 2% (left). The viability of CPSF6-RARG-overexpressing U937 cells treated with the indicated small molecule drugs is also shown. c The top 10 targets and potential drug candidates identified by C-MAP analysis are shown in the table. d Effects of HHT, mitoxantrone, daunorubicin, or venetoclax on the growth of U937 cells with or without CPSF6-RARG overexpression. The data are a summary of the IC50 values. e, f Effects of HHT, mitoxantrone, venetoclax, or daunorubicin on the growth of NPM1-RARG, NUP98-RARG, PML-RARG, and HNRNPC-RARG overexpressing U937 cells. The data are summary of the IC50 values. g Effects of HHT, venetoclax, or daunorubicin on the growth of hCD34+ cells with or without CPSF6-RARG, NUP98-RARG, HNRNPC-RARG, NPM1-RARG, and PML-RARG overexpression. The data are summary of the IC50 values. h Flow cytometric analysis of the effect of different concentrations of daunorubicin, HHT or venetoclax on the apoptosis of hCD34+ cells, primary AML cells (AML-M2), or primary NUP98-RARG leukemia cells. hCD34+ cells, primary AML cells (AML-M2), or primary NUP98-RARG leukemia cells were treated with different concentrations of daunorubicin, HHT or venetoclax for 48 h, and the percentage of Annexin V+ cells were calculated with FlowJo software. The gating strategy is shown in Supplementary Fig. 7c. The relative change in apoptosis and differentiation cells (Fold of CON group) were calculated. d–h n = 3 technical replicates. Results are representative of three independent experiments. Statistical significance was determined by (h) one-way ANOVA with Tukey’s multiple comparison tests. Data are presented as means ± S.E.M. Source data are provided as a Source Data file.
Furthermore, U937 cells overexpressing RARG fusions exhibited sensitivity to all four drugs in a concentration-dependent manner, indicating the potential therapeutic efficacy of these drugs in RARG-AML (Fig. 6e, f). Because daunorubicin and mitoxantrone are both chemotherapeutic agents, and daunorubicin is the first-line therapeutic drug for AML treatment in clinic37, we did not verify the effect of mitoxantrone on RARG-AML in further studies. Moreover, we also detected the dose-response effect of daunorubicin, HHT, and venetoclax on hCD34+ cells with or without X-RARG fusions overexpression by the CCK-8 assay. Our results demonstrated that hCD34+ cells overexpressing RARG fusions exhibited more sensitivity to daunorubicin, HHT, and venetoclax in a concentration-dependent manner (Fig. 6g). These findings suggest the potential therapeutic efficacy of these drugs in RARG-AML.
We then evaluated and compared the therapeutic effects of low concentrated daunorubicin, HHT, and venetoclax in primary NUP98::RARG leukemia cells, as well as other type of primary AML cells (AML-M2) and hCD34+ cells. We found that low concentrations of HHT (0.1 nM and 1 nM), daunorubicin (0.1 nM and 1 nM), and venetoclax (100 nM) could induce the apoptosis of primary NUP98-RARG patient cells. However, these low concentrations of the drugs did not have obvious effect on normal hCD34+ cells or primary AML-M2 cells (Fig. 6h). These data suggest that daunorubicin, HHT, and venetoclax show some level of specificity towards X-RARG.
To assess the anti-AML effects of daunorubicin, HHT, and venetoclax in vivo, we used a transplantation mouse model harboring the Wt1F/+CreMx-1;CPSF6::RARG (Fig. 7a). After 3 weeks of administration, the mice in the daunorubicin, HHT, and venetoclax treatment groups exhibited dramatic reductions in spleen size and weight and (Fig. 7b, c). Additionally, these treatments decreased the population of immature blast cells, increased the proportion of apoptotic leukemia cells in the BM and spleen, and prolonged the survival of recipient mice (Fig. 7d–f).

a Strategy for investigating the anti-AML effects of daunorubicin, HHT, and venetoclax in a Wt1F/+CreMx-1;CPSF6::RARG transplantation mouse model in vivo. b, c Representative images of spleens (b) and statistical analysis of the spleen weights (c) in Wt1F/+CreMx-1;CPSF6::RARG transplanted recipient mice treated with the indicated agents (n = 4 mice per group). d, e Percentages of CD11b+c-kit+ cells (d) and apoptotic cells (e) in the BM and spleens of Wt1F/+CreMx-1;CPSF6::RARG transplanted recipient mice treated with the indicated agents (n = 5 mice per group). The gating strategy is shown in Supplementary Fig. 7g and Supplementary Fig. 7c. f Kaplan–Meier survival curves for Wt1F/+CreMx-1;CPSF6::RARG transplanted recipient mice treated with the indicated agents (n = 8 mice per group). g Strategy for investigating the anti-AML effects of daunorubicin, HHT, and venetoclax in the PDX model established with CPSF6::RARG patient cells in vivo. h Representative images of spleens (left) and statistical analysis of the spleen weight (right) in the indicated PDX model (n = 5 mice per group). i The percentages of human leukemia cells in the spleens and BM of CPSF6-RARG PDX-bearing mice with the indicated treatments were determined by flow cytometry (n = 5 mice per group). The gating strategy is shown in Supplementary Fig. 7h. j Kaplan–Meier survival curves for CPSF6-RARG PDX-bearing mice with the indicated treatment (n = 8 mice per group). k Kaplan–Meier survival curves for CPSF6-RARG PDX-bearing mice with 1 mg/kg HHT and 1 mg/kg daunorubicin for 1 week (three times a week) treatment (n = 3 mice per group). l The percentages of human leukemia cells in the spleens were determined by flow cytometry (n = 3 mice per group). m Schematic diagram illustrating the molecular mechanisms governed by RARG fusions and propose a rational therapeutic strategy for RARG-AML. Statistical significance was determined by (c–e, h, i, l) one-way ANOVA with Tukey’s multiple comparison tests; (f, j, k) two-sided log-rank test. Data are presented as means ± S.E.M. Source data are provided as a Source Data file.
In addition, we examined the therapeutic effects of daunorubicin, HHT, and venetoclax in an established PDX model of RARG-AML derived from CPSF6::RARG patients (Fig. 7g). Daunorubicin, HHT, and venetoclax reduced the spleen size and weight of PDX mice (Fig. 7h). Moreover, these treatments decreased the percentage of human CD45+ cells in the spleen and BM, and prolonged overall survival of PDX mice (Fig. 7i, j). The survival data presented in Fig. 7j suggests a weak response to HHT and daunorubicin, indicating that the immunodeficient NOD-SCID mice may not have been tolerant to these chemotherapy drugs. We then reduced treatment regimen and treated CPSF6::RARG PDX mice with 1 mg/kg HHT or 1 mg/kg daunorubicin for one week (three times a week). The survival data indicated that HHT and daunorubicin treatment prolonged the survival time of CPSF6::RARG PDX mice (Fig. 7k). Furthermore, the percentage of human CD45+ cells in the spleen and BM of CPSF6::RARG PDX mice was remarkably decreased (Fig. 7l). These findings underscored the potential efficacy of HHT and daunorubicin in treating RARG-AML in vivo. Taken together, these results support the therapeutic potential of daunorubicin, HHT, and venetoclax for the treatment of RARG-AML (Fig. 7m).
Discussion
In our previous study, we identified AML with RARG rearrangements as a distinct AML entity. However, AML with RARG rearrangements is insensitive to ATRA and ATO, and has poor prognosis, leading to a mortality rate of over 60%. Due to the similarities in morphological features between RARG-AML and APL, it is expected that ATRA or ATO treatment would be effective in treating RARG-AML patients. However, these treatments have proven to be ineffective in RARG-AML patients, putting them at a high risk of early death5. Therefore, there is an urgent and unmet need for combinatorial approaches that involve molecular targeted therapies in combination with current chemotherapies to prevent early death and subsequent relapse in RARG-AML patients. In this study, we demonstrated that RARG genes play a role similar to that of PML-RARA in supporting cell proliferation and self-renewal, disrupting myeloid differentiation, and regulating transcription. RARG-AML also has distinct differences from PML::RARA-positive APL, such as lack of CD38 expression5. Our RNA-seq and CUT&Tag data revealed that the upregulation of ATF3- and BCL2- mediated apoptotic signaling and the reduced EIF1 level in RARG-AML contributed to the expansion of leukemogenic cells, distinct from the mechanism observed in PML::RARA-positive APL. Using HTS, we identified the BCL2 inhibitor as a potential targeted treatment agent for RARG-AML. Our study has identified promising therapeutic options for RARG-AML treatment in clinical settings, including the use of daunorubicin, HHT, and venetoclax as treatment agents. Our work for the first time offers not only crucial insights into its pathogenesis but also proposes potential therapeutic options for RARG-AML.
ATRA signaling relies on retinoic acid receptors (RARA, RARB, and RARG) and their heterodimeric partners RXR to bind DNA response elements to activate the transcription of target genes, which have been implicated as key regulators of normal and transformed blood cells38. Among the RARs, RARA and RARG have distinct functions, particularly in the field of hematopoiesis. RARA, a low-efficacy receptor for ATRA, primarily exerts ATRA reversible basal repressive functions. On the other hand, RARG, a high-efficacy receptor for ATRA, acts as a potent ligand-dependent transcriptional activator39. Interestingly, overexpression of RARA but not RARG is sufficient to induce myeloid progenitor immortalization40. In leukemia, activation of RARA signaling by ligand promotes myeloid differentiation and loss of self-renewal, whereas ATRA activation of RARG may enhance self-renewal41,42. The role of RARG in AML cells differentiation and clonogenic activity requires further investigation. AML with RARG rearrangements exhibits distinct differences in immunophenotypes, such as the lack of CD38 expression (0% positive) compared to PML::RARA-positive APL, which shows 88% positive expression5. The involvement of RARG-RXR-RARE axis in CD38 downregulation of RARG fusions needs to be explored in the future.
The WT1 gene is frequently mutated in T-ALL (10%), non-APL AML (10%), and typical APL with PML::RARA (19%), and RARG-AML (58.3%)43,44,45,46. Alterations in WT1 are predominantly heterozygous frameshift mutations in exon 7, but also include mutations in exons 1 and 9. These mutations often lead to the synthesis of truncated proteins lacking C-terminal DBD or loss of WT1 function due to nonsense-mediated RNA decay45. In some cases, monoallelic or subclonal deletions have been observed in association with mutations44. However, it is difficult to replicate the diverse mutation status of WT1 in the context of RARG fusions with a single-site mutation. Here, we found that WT1 R462Q mutant overexpression did not produce synergistic leukemic effects with CPSF6::RARG (Fig. S5E). Instead, we used mice with heterozygous loss of Wt1 to demonstrate that overexpression of RARG fusions, coupled with partial loss of Wt1, resulted in AML transformation. A previous study showed that Wt1-haploinsufficient hematopoietic cells progressed to leukemic transformation, whereas Wt1-deficient cells did not47. Furthermore, RARG-AML has been associated with low frequencies of KRAS, NRAS, TP53, and GATA mutations5. Further exploration is needed to understand the status of these mutations and their contributions to the pathogenesis of RARG-AML.
Daunorubicin and HHT are chemotherapeutic drugs, which are also sensitive to AML. BCL2 inhibitor venetoclax is also used for AML treatment. We verified Daunorubicin, HHT and venetoclax are also sensitive to RARG-AML on RARG-AML PDX model and RARG-AML mouse model. Our studies provided potential therapeutic options for RARG-AML treatment, which is blank in clinical. It is urgent for screening therapeutic agents and verifying the therapeutic efficiency specially for RARG fusions in pre-clinical studies. Moreover, there are discrepancy in survival curves between daunorubicin treated immunodeficient NOD-SCID xenograft model (human primary RARG-AML PDX model) and C57B6/L transplantation mouse model harboring the Wt1F/+CreMx-1;CPSF6::RARG. We speculate that immunodeficient NOD-SCID mice are intolerant and low dose of daunorubicin treatment had side-effect on immunodeficient NOD-SCID mice, but low dose of daunorubicin treatment had little side-effect on immunocompetent mice.
The limitations of this study include the small size of primary RARG-AML patient samples, lack of other primary RARG-AML patient samples except for CPSF6::RARG and NUP98::RARG blasts, the lack of experiments in RARG-AML cell lines and the use of AML cell lines overexpressing CPSF6::RARG for confirmation and drug screening experiments. It would be valuable to verify the mechanism on primary RARG-AML patient samples. Furthermore, it would be valuable to use AML cell lines specifically harboring RARG rearrangements to mimic the genetic context of RARG-AML better. Additionally, the absence of experiments with an ATF3 inhibitor and the need for more robust investigations into the mechanism of RARG fusions combined with Wt1 inactivation in AML development are noted limitations.
Overall, our study provides valuable insights into the molecular mechanisms of RARG fusions in AML development, highlighting the similarities between RARG fusions and PML-RARA and potential therapeutic targets. The identification of BCL2 and ATF3 as targets in RARG fusions-driven AML suggests that inhibiting BCL2, such as venetoclax, could be a rational therapeutic strategy for RARG-AML. Prospective clinical trials in RARG-AML patients are warranted as proof of concept.
Methods
Study approval
Human-mobilized peripheral blood collected from healthy donors, APL patient specimens and RARG-AML patient specimens were obtained from the Beijing Chao-Yang Hospital. Informed consent was obtained from all the participants in accordance with the Declaration of Helsinki. Patients received no financial compensation for study participation. The study protocol was approved by the Institutional Review Board of the Ethics Committee of the Beijing Chao-Yang Hospital. All participants provided written informed consent to publish the information that identified the individuals. Our study is compliant with the ‘Guidance of the Ministry of Science and Technology (MOST) for the Review and Approval of Human Genetic Resources, which requires formal approval for the export of human genetic material or data from China. The patient-related information is identified in Supplementary Data 2.
Animal studies
NOD-scid IL2Rgnull (NSG) mice (6–8 weeks old, male) were purchased from Beijing Biocytogen Co., Ltd. (Beijing, China). C57BL/6 mice (6–8 weeks old, male or female) were purchased from Beijing Hua Fu Kang Bioscience Co. Ltd. (Beijing, China). CreVav (B6. Cg-Commd10Tg(VAV1-icre/A2Kio/J) mice (5–6 weeks old, one male and two females) (The Jackson Laboratory, 008610) were obtained from Cyagen Biosciences Inc. (Suzhou, China). Cpsf6-rarg knock-in mice (Cpsf6::rarg loxP/loxP, Cpsf6::rarg F/F) were generated by the National Human Disease Animal Model Resource Center (Beijing, China). Cpsf6::rarg overexpression in hematopoietic stem cells (Cpsf6::rarg cKI mice) was performed by breeding CreVav mice with Cpsf6::rarg F/F mice. CreMx-1(B6.Cg-Tg(Mx1-cre) 1Cgn/J/) mice (5–6 weeks old, two males and two females) (The Jackson Laboratory, 003556) and Wt1F/F (C57BL/6JSmoc-Wt1tm3(flox)Smoc) mice (5–6 weeks old, two males and two females) (NM-CKO-200041) were obtained from Shanghai Research Center for Model Organisms (Shanghai, China). Inducible Wt1-knockout mice were generated by breeding Wt1F/F mice with CreMx-1 mice to generate CreMx-1Wt1F/+ mice, which were treated with polyinosinic–polycytidylic acid (poly I:C) to induce Wt1 heterozygous knockout. To induce Wt1 in CreMx-1Wt1F/+ mice, 8-month-old CreMx-1Wt1F/+ mice were treated with poly I:C (Sigma-Aldrich, 528906) at a dose of 20 μg/kg i.p. for five days, followed by transplantation. If mice manifested symptoms such as failure to thrive, weight loss > 10% of total body weight, hunching, activity decrease (stationary unless stimulated or hind limb paralysis), bleeding, infection, and/or fatigue, they were killed immediately, which was approved by the Animal Experimentation Ethics Committee of the Chinese Academy of Medical Sciences. The criteria of diagnosing AML in mice includes the detection of massive splenomegaly with myeloid infiltration, BM infiltration with high blast counts, and expansion of c-Kit-high myeloid cells in the BM and spleen. Animals were housed in groups of 4–6 mice per individually ventilated cage under a 12 h light/dark cycle with controlled room temperature (23 ± 2 °C) and relative humidity (40–50%).
All mice were maintained in the animal facility at the Institute of Medicinal Biotechnology under specific pathogen-free (SPF) conditions. For animal studies, mice were earmarked before grouping and then randomly separated into groups by an independent person; however, no particular method of randomization was used. The sample size was predetermined according to previous experience using the same strains and treatments. In general, we used ≥5 mice per genotype and condition. We ensured that the experimental groups were balanced in terms of age and weight. All animal procedures were conducted in accordance with the guidelines of the Institutional Committee for the Ethics of Animal Care and Treatment in Biomedical Research of the Chinese Academy of Medical Sciences and Peking Union Medical College (PUMC). Animal studies were performed according to ARRIVE guidelines.
Cell culture
U937 (Acute myeloid leukemia) cells, and HEK 293 T cells were purchased from the Cell Resource Center, Peking Union Medical College. lenti-X293T cells were purchased from Takara Bio (Cat. #632180). These cells were recently authenticated by short tandem repeat (STR) profiling and characterized by mycoplasma and cell vitality detection. U937cells were cultured and maintained in 1640 medium supplemented with 10% fetal bovine serum (FBS; Invitrogen, CA, USA) and 1% penicillin streptomycin. HEK 293T and lenti-X293T cells were cultured and maintained in DMEM supplemented with 10% FBS. All cell lines were verified to be negative for mycoplasma using the MycoAlertTM Mycoplasma Detection Kit (Lonza, LT07-318). To generate U937 cells stably overexpressing X-RARG fusions, X-RARG-HA-IRES-GFP or MIGR1-HA-IRES-GFP retrovirus particles were infected into U937 cells. After 48 h of infection, GFP-positive U937 cells were sorted using FACS Aria III (BD Biosciences) and cultured for experiments without cloning. To generate U937 cells stably expressing AFT3Cas9 or BCL2Cas9, AFT3Cas9 or BCL2 Cas9 lentiviral plasmids were purchased from Wengene Biosciences Co., Ltd. (Beijing, China). The gRNA sequence targeting AFT3 was 5’- TGTCAGCGACAGACCCCTCGGGG -3′; 5′- TTTGTGATGGACACCCCGAGGGG -3′; and the gRNA sequence targeting BCL2 was 5′- CATTATAAGCTGTCGCAGAG GGG-3′; 5′- GGAGAACAGGGTACGATAAC CGG -3′; After 24 h of infection, stable cells were selected in medium containing 4 μg/mL puromycin (Gibco) for 14 days. After 2–3 passages in the presence of puromycin, cultured cells were used for experiments without cloning.
Plasmid construction
Human RARG-untagged (HG18253-UT), Human NUP98-untagged (HG17778-UT), Human RXRA (HG10731-CF), and Human CPSF6-Flag-tagged (HG11458-CF) proteins were purchased from Sino Biological Inc. (Beijing, China). Human PML::RARA-Myc-tagged proteins were generated as previously described 56. The full-length fusion transcript of CPSF6::RARG spanning from the start codon to exon 4 (519 nt) of CPSF6 cDNA (NM001300947) was fused to the RARG 5′-UTR from exon2 to exon3 (210 nt) plus the RARG coding region (NG029822) from the start codon to the stop codon, forming a 2094-bp in-frame fusion transcript (GenBank MG715502), which is predicted to encode a 698-aa fusion protein. The full-length fusion transcript of HNRNPC::RARG, spanning from the start codon to the 5′-region, encodes an RNA recognition motif (RRM) of HNRNPC cDNA (NM_031314) that was fused to the RARG coding region (NG029822) from the DNA binding ___domain to stop codon forming a1422-bp in-frame fusion transcript, which is predicted to encode a 474-aa fusion protein. The full-length fusion transcript of PML::RARG spanning from the start codon to exon 3 (1183 nt) of PML cDNA (NM_002675.3) was fused to the RARG 5′-UTR from exon2 to exon3 (209 nt) plus the RARG coding region (NG029822) from the start codon to the stop codon, forming a 2754-bp in-frame fusion transcript, which is predicted to encode a 918-aa fusion protein. The full-length fusion transcript of NUP98::RARG spanning from start exon 4 to the stop codon (1409 nt) of NUP98 cDNA (NM_016320) was fused to the RARG coding region (NG029822) from the start codon to the stop codon to form a 2586-bp in-frame fusion transcript, which is predicted to encode an 862aa fusion protein. The full-length fusion transcript of NPM1::RARG spanning exon 1 to exon 4 (352 nt) of NPM1 cDNA (NM_002520) was fused to the RARG coding region (NG029822) from partial exon 4 to exon 9 plus NPM1 exon 11 to form a 1389-bp in-frame fusion transcript, which is predicted to encode a 463-aa fusion protein. Full-length cDNA sequences of NPM1::RARG and HNRNPC::RARG were synthesized by Ruibiotech (Beijing, China). Full-length cDNA sequences of CPSF6::RARG, NUP98::RARG, and PML::RARG were constructed by standard subcloning. The CPSF6::RARG, NUP98::RARG, PML::RARG, NPM1::RARG, HNRNPC::RARG and PML::RARAHA-IRES-GFP plasmids were cloned into the MIGR1 vector (Addgene, #27490) by standard subcloning, and an HA-tag or Flag-tag was added. The Myc-tagged X-RARG fusion plasmid was cloned into the pcDNA3.1-Myc-his vector using standard subcloning. The truncations of CPSF6::RARG (amino acids 1-698), CPSF6 (amino acids 1-173), RARG (amino acids 243-698), CPSF6::RARG-△-RARG1 (amino acids 1-173 and 243-698), CPSF6::RARG-△-RRM (amino acids 1-81 and 173-698), CPSF6::RARG-△-DBD (amino acids 1-328 and 408-698), CPSF6::RARG-△-LBD (amino acids 1431 and 661-698), CPSF6::RARG-M624R and CPSF6::RARG-T627R were inserted into the pcDNA3.1-Myc-his vector by standard subcloning. shRNAs specifically targeting murine Bcl2 and Atf3 or human BCL2 and ATF3 were constructed into the ZsGreen1-tagged lentiviral vector pLVX-shRNA2 (Clontech, Palo Alto, CA, USA) according to the Lenti-X shRNA expression system user manual. The shRNA sequence targeting murine Atf3 was 5′- AGAAGGAACATTGCAGAGCTA-3′; and the shRNA sequence targeting murine Bcl2 was 5′- CGTGATGAAGTACATACATTA-3′; The shRNA sequence targeting human ATF3 was 5′- AGGCGACGAGAAAGAAAT-3′; and the shRNA sequence targeting human BCL2 was 5′-AACATCGCCCTGTGGATGACT-3′; For the construction of luciferase reporter plasmids of BCL2, we cloned the Chr18: 63318000-63319000 promoter sequences of BCL2 into pGL3-basic vector, or Chr18: 63318000-63319000 promoter sequences plus Chr18: 63366500-63367700 enhancer sequences into pGL3-basic vector. For the construction of luciferase reporter plasmids of ATF3, we cloned the Chr1: 212566200-212567300 promoter sequences of ATF3 into pGL3-basic vector, or Chr1: 212566200-212567300 promoter sequences plus Chr1: 212607500-212609000 enhancer sequences into pGL3-basic vector (Promega, Madison, WI, USA). For the construction of luciferase reporter plasmids of EIF1, we cloned the Chr17: 41688400-41689700 promoter sequences of ATF3 into pGL3-basic vector. PGL3-CEBPE, pGL3-PU.1, and pGL3-GFI1 were gifts from the Kankan Wang Lab.
Retrovirus or lentivirus production
Lenti-X-293T cells were seeded in a 15 cm culture dish (Corning Inc., #430599) for retrovirus particle or lentivirus particle production. For retrovirus particle production, 15 μg of retrovirus plasmids (MIGR1 or X::RARG-HA-IRES-GFP plasmid) and 9 μg of pCL-Ecopack plasmid (Addgene, 12371) were transfected into lenti-X-293T cells in 150 mm culture using polyethylenimine (PEI, Polysciences Inc., #23966). For lentivirus particle production, 2.7 μg of pMD2.G (Addgene, #12259), 5.4 μg of psPAX2 (Addgene, #12260), 10.7 μg of lentivirus target plasmid (Atf3-shRNA, Bcl2-shRNA, ATF3-shRNA, BCL2-shRNA, AFT3Cas9 or BCL2Cas9 lentiviral plasmids) were transfected into lenti-X-293T cells, the virus containing supernatant was collected, filtered with a 0.45 μm PVDF filter (Millipore), and concentrated with Retro-XTM Concentrator (Takara Bio, 631455) or lenti-PacTM Lentivirus Concentration Solution (GeneCopoeia, LT007) at 48 h after transfection. The retrovirus or lentivirus particles were aliquoted and stored at −80 °C freezer until use.
Isolation, culture, and retroviral infection of human CD34+ primary cells
Human CD34+ cells were isolated from mobilized peripheral blood using a human CD34 MicroBead Kit (Miltenyi Biotec,130-046-702). hCD34+ cells were cultured in StemSpanTM SFEM II (Stemcell, 09655) medium supplemented with 2 mM Glutamine, 100 units/ml penicillin/streptomycin (PS), 100 ng/mL human SCF (Peprotech, 300-07-10), 100 ng/mL human TPO (Peprotech, AF-300-18-10), 20 ng/mL human GM-CSF (Peprotech, 300-03-5), 100 ng/mL human Flt3L (Peprotech, 300-19-10), 50 ng/mL human IL-6 (Peprotech, 200-06-20), and 50 ng/mL human IL-3 (Peprotech, 200-03-10) or supplemented with 1x StemSpanTM CD34+ expansion supplement (Stemcell, 02691), and UM729 (Stemcell, 72332). For retroviral infection of human CD34+ cells, hCD34+ cells were infected with MIGR1 or X-RARG-HA-IRES-GFP retrovirus particles supplemented with 8 μg/ml polybrene and then were centrifuged at 1200 × g, 37 °C for 1.5 h. After 48 h of infection, GFP-positive hCD34+ cells were sorted using FACS Aria III (BD Biosciences) and cultured for experiments without cloning.
Flow cytometry
For BM, spleen, and PBMC of Cpsf6-rargcKI mice or recipient mice for primary or secondary transplantation analysis, cell suspensions from the BM, spleen, and PBMC were directly labeled. Fluorescently labeled antibodies against the following surface proteins were used for mouse cells staining: PE anti-mouse B220 antibody (Biolegend, 103207, 1:100), APC/Cyanine7 anti-mouse CD3 antibody (Biolegend, 100221, 1:100), APC anti-mouse Gr-1 antibody (Biolegend, 108411, 1:100), PerCP/Cyanine5.5 anti-mouse/human CD11b antibody (Biolegend, 101227, 1:100), PE/Cyanine7 anti-mouse c-kit antibody (Biolegend, 135111, 1:100), PE anti-mouse Lin- antibody (Biolegend, 13303, 1:100), FITC anti-mouse Lineage cocktail antibody (Biolegend, 133302, 1:100), PerCP/Cyanine5.5 anti-mouse Ly6A/E (Sca-1) antibody (Biolegend, 108121,1:100), APC/Cyanine7 anti-mouse CD48 antibody (Biolegend, 103431, 1:100), APC/Cyanine7 anti-mouse CD16/32 antibody (Biolegend, 101327, 1:100), APC/Cyanine7 anti-mouse CD34 antibody (Biolegend, 128621, 1:100), APC anti-mouse CD150 antibody (Biolegend, 162604, 1:100), APC anti-mouse CD34 antibody (Biolegend, 128611, 1:100), APC anti-mouse CD16/32 antibody (Biolegend, 101325, 1:100). For human CD34+ cell differentiation analysis, cell suspensions from hCD34+ cells overexpressing RARG fusions for seven days were directly labeled. Fluorescently labeled antibodies against the following surface proteins were used for human cell staining: APC anti-human CD33 antibody (BioLegend, 366605, 1:100), PE anti-human CD66b antibody (BioLegend, 305105, 1:100), PE/Cyanine7 anti-human CD45 antibody (BioLegend, 368531, 1:100), PE anti-human CD45 antibody (BioLegend, 304008, 1:100), PE antihuman CD34 antibody (BioLegend, 343506, 1:100), Annexin V-APC (BioLegend, 640920,1:100), and 50 μg/mL propidium iodide (PI) (BD Biosciences, 550825). For human CD34+ cell differentiation and apoptosis analysis, cell suspensions from hCD34+ cells or hCD34+ cells overexpressing RARG fusions with or without ATF3 or BCL2 knockdown cultured for 10 days were directly labeled. Fluorescently labeled antibodies against the following surface proteins were used for human cell staining: APC anti-human CD33 antibody (BioLegend, 366605, 1:100), PE/ Cyanine7 anti-human CD11b antibody (BioLegend, 982608, 1:100), PE antihuman CD34 antibody (BioLegend, 343506, 1:100), Annexin V-APC (BioLegend, 640920, 1:100), and 50 μg/mL propidium iodide (PI) (BD Biosciences, 550825). Cells were then washed, and data were acquired using a CytoFLEX flow cytometer and analyzed using FlowJo and CytExpert software.
Luciferase reporter assays
U937 cells were seeded in 12-well plates and transfected with the indicated luciferase reporter gene plasmids using the Cell Line Nucleofector Kit V (LONZA, VCA-1003) according to the manufacturer’s instructions. pTK-Renilla was used as the internal control. After 24 h of transfection, the cells were harvested with lysis buffer and luciferase activity was measured using a dual-luciferase assay (Promega, Madison, WI, USA, E1910).
Immunoprecipitation, immunoblotting, and immunostaining
Co-IP was performed as previously described. The cells were harvested and lysed for 30 min in CoIP buffer supplemented with a complete protease inhibitor (Cell Signaling Technology, 5817) on ice. Centrifugation was performed to obtain the supernatant, which was then incubated first with the indicated antibodies at 4 °C overnight and then with Protein A/G Plus-Agarose (Santa Cruz Biotechnology, TX, USA) at 4 °C for 2 h. Soluble lysates were incubated with the indicated anti-Myc magnetic beads (Bimake.com, B26302) and anti-HA magnetic beads (Bimake.com, B26202) at 4 °C overnight. The complexes were eluted from the beads and boiled for 10 min. The precipitated proteins were subjected to SDS-PAGE and immunoblotting with the corresponding antibodies.
For immunoblotting, whole-cell extracts were prepared using RIPA lysis buffer. SDS-PAGE was performed with equal amounts of protein per sample and transferred to PVDF membranes. The following antibodies were used for immunoblotting: anti-Bcl-2 (CST, #4223, 1:1000), anti-Bcl-xL (CST, #2764, 1:1000), anti-Mcl-1 (CST, #5453, 1:1000), anti-Bcl-w (CST, #2724, 1:1000), anti-ATF3 (Abcam, ab207434, 1:1000), anti-GAPDH (ZSGB-BIO TA-08, 1:2000), anti-Retinoic Acid Receptor gamma (Abcam, ab187159, 1:1000), anti-RXRA (Abcam, ab125001, 1:1000), anti-NCOR2 (Abcam, ab24551, 1:1000), anti-HDAC3 (CST, #3949, 1:1000), anti-mSin3A (Abcam, ab307197, 1:1000), anti-CPSF6 (Abcam, ab175237, 1:1000), anti-P300 (CST, #54062, 1:1000) anti-HA (CST, #3724, 1:1000), anti-Flag (CST, #14793, 1:1000), anti-GFP (Abcam, ab290, 1:1000), anti-Myc-tag (MBL, #562, 1:1000), anti-DDDDK-tag (MBL, PM020, 1:1000), anti-HA-tag (MBL, #561, 1:1000), and anti-His-tag (MBL, PM032, 1:1000).
For immunofluorescence and colocalization assays, cells were seeded on coverslips and processed according to the experimental requirements. Next, the cells were briefly washed with PBS, fixed with 4% formaldehyde in PBS for 10 min, washed thrice with PBS, and permeabilized with 0.5% Triton X-100 for 15 min. Then, cells were washed with PBS three times and blocked with 3% bovine serum albumin (BSA) for 1 h at 37 °C. samples were incubated with primary antibodies overnight at 4 °C and with secondary antibodies for 2 h. Nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI) in blocking buffer. Images were acquired using a confocal microscope (Olympus Microsystems).
RNA-Seq library preparation and sequencing
Human CD34+ cells infected with control or RARG fusions retroviruses were sorted into GFP-positive cells. Total RNA was extracted from hCD34+ cells with or without RARG fusions overexpression or spleen cells isolated from the spleens of Wt1F/+CreMx-1;CPSF6::RARG mice and CreMx1;CPSF6::RARG mice using the RNA-QuickTM MicroPrep Kit (ZYMO RESEARCH, R1055). RNA purity was evaluated using the Kaiao K5500® spectrophotometer (Kaiao, Beijing, China), and RNA integrity and concentration were assessed using the RNA Nano 6000 Assay Kit and Bioanalyzer 2100 system (Agilent Technologies, CA, USA). An RNA integrity number of 7.5 was required for all samples. Sequencing libraries were generated using the NEBNext® UltraTM RNA Library Prep Kit for Illumina® (#E7530L, NEB, USA), according to the manufacturer’s recommendations, and index codes were added to attribute sequences to each sample. Briefly, mRNA was purified from 2 µg of each total RNA sample using poly T oligo-attached magnetic beads. Fragmentation was performed using divalent cations at an elevated temperature in NEB Next First Strand Synthesis Reaction Buffer (5X). First-strand cDNA was synthesized using random hexamer primers and RNase H. Secondstrand cDNA synthesis was subsequently performed using a buffer, dNTPs, DNA polymerase I, and RNase H. The library fragments were purified using QiaQuick PCR kits and eluted with EB buffer, and terminal repair, A-tailing, and adapter addition were implemented. The intended products were retrieved and PCR was performed, at which point the library construction was complete.
ChIP-seq, ChIP-qPCR and CUT&Tag assay
For ChIP-seq, ChIP assays were performed according to the manufacturer’s protocol using the SimpleChIP® Plus Sonication Chromatin IP Kit #56383 (Cell Signaling Technology, Danvers, MA, USA). Briefly, hCD34+ cells with CPSF6::RARG and NUP98::RARG overexpression were fixated with 1% formaldehyde for 10 min, incubated with glycine (50 mM final) 10 min and washed three times with PBS. After cell lysis and chromatin extraction, chromatin was sonicated to 100–500 bp using a Q800R3 Sonicator Chromatin and DNA Shearing System (Qsonica, USA), followed by centrifugation at 16,000 g for 10 min at 4 °C. Then the lysates were incubated overnight at 4 °C with ChIP grade antibodies: anti-HA tag-ChIP grade (abcam, ab9110, 1:100), anti-Histone H3 (mono methylK4 ChIP grade (abcam, ab8895, 10 μg/sample), anti-Histone H3 (tri methylK4 ChIP grade (abcam, ab213224, 10 μg/sample), and anti-Histone H3 (acetyl K27ChIP grade) (abcam, ab4729, 10 μg/sample), which were then coupled to magnetic beads for 4 h. The Precipitated material was eluted (input chromatin was used as control), the crosslinking was reverted, and DNA was purified by chloroform/phenol extraction and resuspended in DNA elution buffer. Species-matched IgG (Cell Signaling Technology, Danvers, MA, USA) was used as the control for all ChIP-qPCR experiments. Then the purified DNA was used to prepare ChIP-seq DNA libraries for Illumina sequencing using NEBNext Ultra II DNA Library Prep Kit (E7645). Library DNA was sequenced with paired-end 150 reads using the Illumina NovaSeq X plus platform.
For ChIP-qPCR experiments, for BCL2, the ChIP-qPCR regions include the promoter region (peak 1) Chr18: 63318200-63318500 and the enhancer region (peak 2) Chr18: 63367000-63367300. For ATF3, ChIP-qPCR regions include the promoter region (peak 1) Chr1:212566400-212566700 and the enhancer region (peak 2) Chr1:212608000-212608300. The qPCR primer sequences were as follows: hBCL2 (peak 1) forward, 5′ - AACTCAAAGAAGGCCACAATCC -3′; hBCL2 (peak 1) reverse, 5′ - CGCCCCATCCAGCCGCATCCCG -3′; hBCL2 (peak 2) forward, 5′ - AGGCCACTCACCACCACATGCG -3′; hBCL2 (peak 2) reverse, 5′ - CTGGGTGGCGCGCGCGCCGGCT -3′; hATF3 (peak 1) forward, 5′ - ATTTTCCCATTTATAAAATATG -3′; hATF3 (peak 1) reverse, 5′ - GCGTCATTTATAACCTCTCAACT -3′; hATF3 (peak 2) forward, 5′ - CGGGGCTGCGGGCTCGGGCGCAC -3′; hATF3 (peak 2) reverse, 5′ - CCCAGACTAGGTGAGGCTGGGA -3′; hEIF1 forward, 5′ - AGAGGACGCCCTCCTGCCGCAGA -3′; hEIF1 reverse, 5′ - GAAGGCGGCGGCGGCGGTGGCG -3′.
For CUT&Tag, the CUT&Tag assay was performed according to the manufacturer’s protocol using the Hyperactive Universal CUT&Tag Assay Kit for Illumina (Vazyme, Nanjing, China, #TD904). In brief, hCD34+ cells with CPSF6::RARG, NUP98::RARG, PML::RARG, NPM1::RARG, HNRNPC::RARG and PML::RARA overexpression were harvested, counted and centrifuged for 5 min at 600 × g at room temperature, 100,000 cells per sample. The cells were washed with 500 μL of Wash Buffer supplemented with 1× protease inhibitor cocktail and then resuspended in 100 μL of Wash Buffer. ConA Beads pro were prepared with binding buffer, and 10 µL of activated beads were added per sample, inverted to mix, and incubated at room temperature for 10 min. Placed The tube on a magnetic stand and then discard the supernatant, the primary antibody (anti-HA (CST, #3724, 1:100), anti-Flag tag-ChIP grade (CST, #14793, 1:100) incubation was incubated on a rotating platform overnight at 4 °C. The beads were incubated with goat anti-rabbit secondary antibody for 1 h at a dilution of 1:100. After incubation, the beads were washed three times with Dig-wash Buffer. Cells were incubated with a hyperactive pA/G-transposon (pA/G-Tnp) at RT for 1 h and washed three times in Dig-300 buffer to remove unbound pA/G-Tnp. Next, cells were re-suspended in 50 μL Trueprep Tagment Buffer L (TTBL) buffer and incubated at 37 °C for 1 h. The DNA fragments were extracted by adding 2 μL 10% SDS at 55 °C for 10 min. Tubes were placed on a magnet stand to clear, and the liquid was carefully removed to new tubes. DNA fragments was extracted by adding 50 μL prepared DNA Extract Beads Pro at room temperature for 20 min, and the tubes were placed on a magnet stand to clear, and the liquid was carefully withdrawn. Beads were washed twice with 200 μL 1 X BW buffer without disturbing the beads. After drying for approximately 5 min, 15 µL of ddH2O was added and the resuspended beads were used for amplifying libraries.
To amplify the libraries, 15 µL DNA was mixed with 25 µL 2×CUT&Tag amplification mix, 5 µL universal N5, and 5 µL uniquely barcoded N7 primer, using a different barcode for each sample. The sample was placed in a thermocycler with a heated lid using the following cycling conditions: 72 °C for 3 min, 95 °C for 30 min, 14 cycles of 98 °C for 10 s and 60 °C for 5 s, final extension at 72 °C for 1 min, and holding at 4 °C. Post-PCR clean-up was performed by adding 100 µL VAHTS DNA Clean Beads (Vazyme, #N411), and libraries were incubated with beads for 5 min at RT, washed twice gently in 80% ethanol, and eluted in 20 µL ddH2O for sequencing.
For CUT&Tag sequencing, the size distribution of libraries was determined using an Agilent 2100 bioanalyzer, and the libraries were sequenced on the Illumina NovaSeq X plus platform in paired-end reads, following the manufacturer’s instructions.
ATAC-seq
The ATAC-seq assay was performed according to the manufacturer’s protocol using the Hyperactive ATAC-seq Library Prep Kit for Illumina (Vazyme, Nanjing, China, #TD711). In brief, 10,0000 hCD34+ cells with or without CPSF6::RARG or NUP98::RARG overexpression were harvested, centrifuged for 5 min at 500 × g at 4 °C. The cells were washed with 50 μL TW wash buffer twice, resuspended with 50 μL lysis buffer, incubated in lysis buffer on ice for 5 min to isolate the nuclei. Then nuclei were spun at 500 × g for 10 min at 4 °C, and resuspended in 50 μL Fragment Mix buffer. The fragmentation reaction was incubated at 37 °C for 30 min. DNA fragments were extracted by adding 100 μL DNA Extract Beads at room temperature for 5 min, and the tubes were placed on a magnet stand to clear, and the liquid was carefully withdrawn. Beads were washed twice with 80% ethanol, without disturbing the beads. After drying for approximately 5 min, 26 µL of ddH2O was added. The tubes were vortexed, spun quickly, and allowed to sit for 5 min. The tubes were then placed on a magnet stand, and the liquid was withdrawn to a fresh tube.
To amplify the libraries, 20 µL fragmented DNA was mixed with 30 µL 2× CAM buffer, 5 µL universal N5, and 5 µL uniquely barcoded N7 primer, using a different barcode for each sample. The sample was placed in a thermocycler with a heated lid using the following cycling conditions: 72 °C for 3 min, 95 °C for 30 min, 15 cycles of 98 °C for 10 s and 60 °C for 5 s, final extension at 72 °C for 1 min, and holding at 4 °C. Post-PCR clean-up was performed by adding 33 µL ATAC DNA Clean Beads and libraries were incubated with beads for 5 min at RT, the tubes were placed on a magnet stand to clear, and the liquid was removed to a fresh tube. Then the amplified DNA was added 72 µL ATAC DNA Clean Beads and incubated with beads for 5 min at RT, and libraries were incubated with beads for 5 min at RT, washed twice gently in 80% ethanol, and eluted in 20 µL ddH2O for sequencing. For ATAC-seq sequencing, the size distribution of libraries was determined using an Agilent 2100 bioanalyzer, and the libraries were sequenced on the Illumina NovaSeq X plus platform in paired-end reads, following the manufacturer’s instructions.
ChIP-seq, CUT&Tag and ATAC-seq analysis
For ChIP-seq, CUT&Tag and ATAC-seq, reads were trimmed using Cutadapt (version 4.1).and then were aligned to the human genome (hg38) using Bowtie 2 (version 2.2.9) to map the unique reads. Duplicated reads from PCR amplification were further removed with Picard MarkDuplicates (version 1.119). The sam files were converted to bam files and sorted with Samtools (version 1.3). The bam files were converted to BW files by using Deeptools (version 3.5.1). MACS2 (version 2.2.7.1) was used to call peaks48. The Mapped reads were visualized using Integrative Genomics Viewer (IGV, version 2.17.4).
For CUT&Tag, the RARG fusions or PML-RARA binding peaks were identified using MACS2 (version 2.2.6) by comparing differences between those peaks identified in hCD34+ cells with RARG fusions or PML-RARA overexpression with an IgG control. The common peaks in RARG fusions to or unique peaks in RARG fusions compared to PML-RARA were identified by Bedtools intersect. The enrichment motifs in RARG fusions were analyzed using HOMER motif analysis. The ChIPseeker was used for the peak annotations with the default settings. Diffbind49 was used to call differential peaks between CPSF6::RARG or NUP98::RARG overexpressing hCD34+ cells and hCD34+ cells using a threshold of FDR ≤ 0.05 and the average absolute log 2 (fold change) ≥1.0 by DESeq2.
Real-time PCR
RNA was isolated using the RNA-Quick Purification Kit (ES science, RN001). Total RNA (1 mg of total RNA was reverse-transcribed using the TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix (Transgen Biotech, AT311-02). PCR was performed using a MyCycler thermal cycler and analyzed using qTOWER. The PCR primer sequences were as follows: hGAPDH forward, 5′- GTCTCCTCTGACTTCAACAGCG-3′, hGAPDH reverse, 5′- ACCACCCTGTTGCTGTAGCCAA-3′; hATF3 forward, 5′- CGCTGGAATCAGTCACTGTCAG -3′; hATF3 reverse, 5′- CTTGTTTCGGCACTTTGCAGCTG-3′; hBCL2 forward, 5′- ATCGCCCTGTGGATGACTGAGT-3′; hBCL2 reverse, 5′- GCCAGGAGAAATCAAACAGAGGC -3′; hEREG forward, 5′- CTTATCACAGTCGTCGGTTCCAC -3′; hEREG reverse, 5′- GCCATTCAGACTTGCGGCAACT -3′; hSYTL3 forward, 5′- GCCTGTAAGAACCTTGCCTATGG -3′; hSYTL3 reverse, 5′- ACGGTGTTCCTTTGGACTCCAG -3′; hREXO1 forward, 5′- AACGAGTCCACCAGCGTCAAGA -3′; hREXO1 reverse, 5′- CCTTGCTTGGAAAGGTGGGAGA -3′; hRASGEF1B forward, 5′- TTCAGGCGTTCGTGCAGAAGGA -3′; hRASGEF1B reverse, 5′- AGCAACCAAGTAGCTGAGGCGA -3′; hINPP5A forward, 5′- AGCGAGGAGAAGGTTGTCACCT -3′; hINPP5A reverse, 5′- CATGGGCATGAGGTTTACCTGC -3′; hNCAPG2 forward, 5′- GGAACTGGCATTTGACACGAGC -3′; hNCAPG2 reverse, 5′- GCTGCTCTAACAATGGGTGGCT -3′; hEIF1 forward, 5′- ACTGTCCAAGGGATCGCTGATG -3′; hEIF1 reverse, 5′- TCTTGCGTTGGTCACCCTGTAG -3′; hRPS8 forward, 5′- CCGCAAGAAGGGAGCCAAGCT -3′; hRPS8 reverse, 5′- CCAGGAGACTGCTGATTTTGGC -3′; mAtf3 forward, 5′- GAAGATGAGAGGAAAAGGAGGCG -3′; mAtf3 reverse, 5′- GCTCAGCATTCACACTCTCCAG -3′; mBcl2 forward, 5′- CCTGTGGATGACTGAGTACCTG -3′; mBcl2 reverse, 5′- AGCCAGGAGAAATCAAACAGAGG -3′; hBCL2L1 forward, 5′- GCCACTTACCTGAATGACCACC -3′; hBCL2L1 reverse, 5′- AACCAGCGGTTGAAGCGTTCCT -3′; hBCL2L2 forward, 5′- CAAGGAGATGGAACCACTGGTG -3′; h BCL2L2 reverse, 5′- CCGTATAGAGCTGTGAACTCCG -3′; hMCL1 forward, 5′- CCAAGAAAGCTGCATCGAACCAT -3′; hMCL1 reverse, 5′- CAGCACATTCCTGATGCCACCT -3′; hRARG forward, 5′- CATCACCAAGGTCAGCAAAGCC -3′; hRARG reverse, 5′- GCTCACTGAACTTGTCCCACAG -3′; hCD38 forward, 5′- TCTTGCCCAGACTGGAGAAAGG -3′; hCD38 reverse, 5′- TGGACCACATCACAGGCAGCTT -3′; hSCUBE1 forward, 5′- ATGTGGACGAGTGCAGCATGAG -3′; hSCUBE1 reverse, 5′- GCGAGAAAGACACTTGCCTGTC -3′; hPOSTN forward, 5′- CAGCAAACCACCTTCACGGATC -3′; hPOSTN reverse, 5′- TTAAGGAGGCGCTGAACCATGC -3′; hDEFA1 forward, 5′- CCAGAAGTGGTTGTTTCCCTTGC -3′; hDEFA1 reverse, 5′- GGTAGATGCAGGTTCCATAGCG -3′;
Wright-Giemsa staining
The PMBC or BM cells were harvested, washed with PBS, and 1 × 104 cells were centrifuged into glass slides by cytospin at 600 rpm for 5 min. Glass slides were stained with Wright-Giemsa staining solution (E607315, Sangon Biotech, Shanghai, China) according to the manufacturer’s instructions.
Mouse bone marrow transplantation (BMT)
For BMT of Cpsf6::rargCKI mice, 1 × 106 freshly harvested BM cells from 12-month-old Crevav mice and Cpsf6-rargCKI mice were transplanted via tail vein injection into lethally (9 Gy, X-rays, RS2000)irradiated 8-week-old C57BL/6 recipient mice. For Wt1F/+CreMx-1, X-RARG mouse model establishment, lineage-negative (lin-) cells were enriched from 8-month-old CreMx-1or Wt1F/+CreMx1mice (poly I:C-induction) using the ImunoSep Mouse hematopoietic lineage negative cell enrichment kit (Nuowei Biotechnology, 729120), according to the manufacturer’s instructions. Lineage-negative cells were cultured in StemSpanTM SFEM (Stemcell, 09650) supplement with 10 ng/ml recombinant mouse IL-3 (Peprotech, 213-13), 10 ng/ml recombinant human IL-6 (Peprotech, 200-06-20) and 100 ng/ml recombinant mouse SCF (Peprotech, 250-03), 55 mM BME, 1% HEPES, and 1% penicillin/streptomycin (PS) for 24 h. Then Lineage negative cells were infected with MIGR1 or X-RARG-HA-IRES-GFP retrovirus particles supplemented with 8 μg/ml polybrene and then were centrifuged at 1200 g, 37 °C for 1.5 h. After two rounds of infection with retrovirus particles, 1 × 106 lineage-negative cells were transplanted via tail vein injection into lethally (9 Gy, X-rays, RS2000)-irradiated 8-week-old C57BL/6 recipient mice. For secondary transplantation, spleen cells from primary Wt1F/+CreMx-1;X-RARG mice (leukemic) or X-RARG mice were transplanted into sublethally (4.5 Gy) irradiated 8-week-old C57BL/6 recipient mice. Peripheral blood (PB) was collected for peripheral blood cell counts using an ABX Pentra 60 Hematology Analyzer. Portions of the spleen from the leukemic mice were collected at the time of sacrifice, fixed in formalin, and embedded in paraffin. Tissue samples were then sectioned and stained with hematoxylin and eosin (H&E).
PDX mouse models and treatment in vivo
For the PDX model, 6-week-old male NSG mice were implanted with intravenous injections of 5 × 106 leukemia cells from AML patients with CPSF6::RARG and NUP98::RARG rearrangement after 1 Gy irradiation. Approximately 3–4 weeks following implantation, the first-generation mice (G1) were sacrificed, and spleen and BM cells were collected. Leukemia cells were tested for human CD45 expression, and the remaining cells 3 × 106 into 8 NSG mice as the second generation (G2). After stable passage of the third generation (G3), 1 × 106 leukemia cells of the fourth generation (G4) were transplanted into 30–40 NSG mice for in vivo treatment (8 mice per group, dependent on the experimental drug treatment). After confirmation of human leukemia engraftment in murine peripheral blood (>5% human CD45+ cells), mice were treated with vehicle, Venetoclax (ABT-199) (Selleck, S8048) (100 mg/kg/day, p.o.), homoharringtonine (Selleck, S9015) (0.5 mg/kg, i.p. twice a week), or daunorubicin HCl (Selleck, S3035) (0.5 mg/kg, i.p. twice a week) for three weeks. During and after treatment with the vehicle control or the indicated agents, the ratio of human CD45+ cells and survival were evaluated to determine therapeutic efficacy in the PDX models. Mice were sacrificed post-treatment, and the peripheral blood, bone marrow, and spleen were analyzed by flow cytometry.
In vivo study of RARG-AML mice model
To assess the anti-AML effects of HHT, Daunorubicin, and Venetoclax on RARG-AML in vivo, AML like spleen cells from primary transplantation mice (Wt1F/+CreMx-1; CPSF6:RARG mice, leukemic) were intravenously injected into sublethally (4.5 Gy) irradiated 8-week-old C57BL/6 recipient mice (1 × 106 cells per mouse). Mice implanted with leukemic cells were randomly assigned to one of two treatment groups. Three days after transplantation, Venetoclax (ABT-199) (Selleck, S8048) (100 mg/kg/day, p.o.), homoharringtonine (Selleck, S9015) (0.5 mg/kg, i.p. three times a week), or daunorubicin HCl (Selleck, S3035) (0.5 mg/kg, i.p. three times a week) for three weeks. Mice were sacrificed post-treatment, and the peripheral blood, bone marrow, and spleen were analyzed by flow cytometry. To assess the role of Atf3 or Bcl2 knockout on the progression of RARG-AML, mouse AML-like spleen cells from primary transplantation mice (Wt1F/+CreMx-1;NUP98::RARG mice, leukemic) infected with con or Bcl2 and Atf3 shRNA lentiviral particles, and 1 × 106 cells were intravenously injected into sublethally (4.5 Gy) irradiated 8-week-old C57BL/6 recipient mice. The survival rates in the indicated groups were evaluated.
Colony formation assay
Approximately 10,000 human CD34+ cells with or without X-RARG fusion overexpression were seeded in semi-solid medium (MethoCult H4536; Stemcell Technologies, Cambridge, MA, USA) according to the manufacturer’s instructions. Colony formation and colony number were calculated after 14 days. For 2nd replating, the cells from 1st CFU were collected, washed, resuspended, and replated in semi-solid medium. The number were counted after 14 d.
Cell proliferation assay
For the CCK-8 assay, U937 or hCD34+ cells with or without X-RARG overexpression were seeded in 96-well plates at a concentration of 10,000 cells per well in triplicate at 37 °C and 5% CO2. The cells were then treated with prilocaine (AbMole, M5897), oxfendazole (AbMole, M3209), lopinavir (AbMole, M2158), mitoxantrone (AbMole, M3493), dasabuvir (ABT-333) (AbMole, M2333), CAL-101 (Idelalisib) (AbMole, M1945), daunorubicin HCl (Selleck, S3035), venetoclax (ABT-199) (Selleck, S8048), ABT199 (Venetoclax) (AbMole, M2017), and homoharringtonine (Selleck, S9015) at different concentrations for 48 h. Subsequently, 10 μl of CCK-8 solution (Dojindo, CK04) was added to each well, according to the manufacturer’s instructions. Finally, the absorbance was measured at 450 nm wavelength. The IC50 values of the indicated agents were calculated using the GraphPad Prism software (version 9.3.1).
Gene Set Enrichment Analysis (GSEA)
For the GSEA analysis, the gene list was pre-ranked based on log2 fold-change values of gene expression in primary RARG-AML patients vs primary APL patients. Pre-ranked GSEA analysis was performed using an apoptotic pathway gene set or ATF3 target gene sets (ATF3_Q6. v2023.1. Hs). The MisigDB database used in this analysis was originated from our previous study5.
Statistics and reproducibility
Data are expressed as mean ± standard error of the mean (SEM). Comparisons between two groups were performed using an unpaired Student’s t test and one-way ANOVA. Survival rates were analyzed using the Kaplan–Meier method. Sample number (n) indicates the number of independent biological samples in each experiment. The sample numbers and experimental replicates are indicated in the figures and figure legends. All experiments were performed with ≥3 biological replicates. Statistical significance was set at p < 0.05. Analyses were performed using the GraphPad Prism software (version 9.3.1).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq, CUT&Tag, ChIP-seq, and ATAC-seq data have been deposited in the NCBI Gene Expression Omnibus (GEO) database under accession numbers GSE243438, GSE275303, and GSE275304. The uncropped blot figures and original data underlying Figs. 1–7 and Supplementary Figs. 1–6 are provided as a Source Data file. The remaining data are available within the Article, Supplementary Information or Source Data file. Source data are provided in this paper. Source data are provided with this paper.
References
Olsson, I., Bergh, G., Ehinger, M. & Gullberg, U. Cell differentiation in acute myeloid leukemia. Eur. J. Haematol. 57, 1–16 (1996).
Chen, X. et al. A novel NPM1-RARG-NPM1 chimeric fusion in acute myeloid leukaemia resembling acute promyelocytic leukaemia but resistant to all-trans retinoic acid and arsenic trioxide. Br. J. Cancer 120, 1023–1025 (2019).
Duncavage, E. J. et al. Genome sequencing as an alternative to cytogenetic analysis in myeloid cancers. N. Engl. J. Med. 384, 924–935 (2021).
Such, E. et al. A novel NUP98/RARG gene fusion in acute myeloid leukemia resembling acute promyelocytic leukemia. Blood 117, 242–245 (2011).
Zhu H. et al. A global study for acute myeloid leukemia with RARG rearrangement. Blood Adv. 7, 2972–2982 (2023).
Ha, J. S. et al. Identification of a novel PML-RARG fusion in acute promyelocytic leukemia. Leukemia 31, 1992–1995 (2017).
Liu, T. et al. Identification of novel recurrent CPSF6-RARG fusions in acute myeloid leukemia resembling acute promyelocytic leukemia. Blood 131, 1870–1873 (2018).
Miller, C. A. et al. A case of acute myeloid leukemia with promyelocytic features characterized by expression of a novel RARG-CPSF6 fusion. Blood Adv. 2, 1295–1299 (2018).
Qin, Y. Z., Huang, X. J. & Zhu, H. H. Identification of a novel CPSF6-RARG fusion transcript in acute myeloid leukemia resembling acute promyelocytic leukemia. Leukemia 32, 2285–2287 (2018).
Luo, H. et al. A novel entity of acute myeloid leukaemia with recurrent RARG-rearrangement resembling acute promyelocytic leukaemia. Leuk. Res. 77, 14–16 (2019).
Zhang, X. et al. RARgamma-rearrangements resemble acute promyelocytic leukemia and benefit from 3 + 7 regimen. Leuk. Lymphoma 60, 1831–1834 (2019).
Zhao, J. et al. The genetics and clinical characteristics of children morphologically diagnosed as acute promyelocytic leukemia. Leukemia 33, 1387–1399 (2019).
Su, Z. et al. Novel reciprocal fusion genes involving HNRNPC and RARG in acute promyelocytic leukemia lacking RARA rearrangement. Haematologica 105, e376–e378 (2020).
Tao, S. et al. Acute myeloid leukemia with NUP98-RARG gene fusion similar to acute promyelocytic leukemia: case report and literature review. Onco Targets Ther. 13, 10559–10566 (2020).
Wei, W. et al. Alkaloid-based regimen is beneficial for acute myeloid leukemia resembling acute promyelocytic leukemia with NUP98/RARG fusion and RUNX1 mutation: a case report. Medicine 99, e22488 (2020).
Han, X. et al. Acute myeloid leukemia with CPSF6-RARG fusion resembling acute promyelocytic leukemia with extramedullary infiltration. Ther. Adv. Hematol. 12, 2040620720976984 (2021).
Qiu, J. J. et al. Critical role of retinoid/rexinoid signaling in mediating transformation and therapeutic response of NUP98-RARG leukemia. Leukemia 29, 1153–1162 (2015).
de The, H. & Chen, Z. Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nat. Rev. Cancer 10, 775–783 (2010).
di Masi, A. et al. Retinoic acid receptors: from molecular mechanisms to cancer therapy. Mol. Asp. Med. 41, 1–115 (2015).
Sell, S. Leukemia: stem cells, maturation arrest, and differentiation therapy. Stem Cell Rev. 1, 197–205 (2005).
Conserva, M. R. et al. RARG gene dysregulation in acute myeloid leukemia. Front. Mol. Biosci. 6, 114 (2019).
Carlesso, EllisenL. W., Cheng, N., Scadden, T. & Haber, D. T. DA. The Wilms tumor suppressor WT1 directs stage-specific quiescence and differentiation of human hematopoietic progenitor cells. EMBO J. 20, 1897–1909 (2001).
Wang, Y. et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol. Cell 57, 662–673 (2015).
Sarma, N. J., Takeda, A. & Yaseen, N. R. Colony forming cell (CFC) assay for human hematopoietic cells. J. Vis. Exp. 46, 2195 (2010).
Yassin, E. R., Abdul-Nabi, A. M., Takeda, A. & Yaseen, N. R. Effects of the NUP98-DDX10 oncogene on primary human CD34+ cells: role of a conserved helicase motif. Leukemia 24, 1001–1011 (2010).
Truong, B. T. et al. CCAAT/Enhancer binding proteins repress the leukemic phenotype of acute myeloid leukemia. Blood 101, 1141–1148 (2003).
Wang, K. et al. PML/RARalpha targets promoter regions containing PU.1 consensus and RARE half sites in acute promyelocytic leukemia. Cancer Cell 17, 186–197 (2010).
Hu, Z. & Saunthararajah, Y. CEBPE activation in PML-RARA cells by arsenic. Blood 119, 2177–2179 (2012).
Yang, X. W. et al. Coordinated regulation of the immunoproteasome subunits by PML/RARalpha and PU.1 in acute promyelocytic leukemia. Oncogene 33, 2700–2708 (2014).
Tan, Y. et al. A PML/RARalpha direct target atlas redefines transcriptional deregulation in acute promyelocytic leukemia. Blood 137, 1503–1516 (2021).
Kwok, C., Zeisig, B. B., Dong, S. & So, C. W. Forced homo-oligomerization of RARalpha leads to transformation of primary hematopoietic cells. Cancer Cell 9, 95–108 (2006).
Kwok, C., Zeisig, B. B., Qiu, J., Dong, S. & So, C. W. Transforming activity of AML1-ETO is independent of CBFbeta and ETO interaction but requires formation of homo-oligomeric complexes. Proc. Natl Acad. Sci. USA 106, 2853–2858 (2009).
Zhu, J. et al. RXR is an essential component of the oncogenic PML/RARA complex in vivo. Cancer Cell 12, 23–35 (2007).
Qiu, J. J. et al. Leukemic transformation by the APL fusion protein PRKAR1A-RARalpha critically depends on recruitment of RXRalpha. Blood 115, 643–652 (2010).
Di Marcantonio, D. et al. ATF3 coordinates serine and nucleotide metabolism to drive cell cycle progression in acute myeloid leukemia. Mol. Cell 81, 2752–2764.e2756 (2021).
Zhang, L., Lu, Z. & Zhao, X. Targeting Bcl-2 for cancer therapy. Biochim. Biophys. Acta Rev. Cancer 1876, 188569 (2021).
Kantarjian, H. et al. Acute myeloid leukemia: current progress and future directions. Blood Cancer J. 11, 41 (2021).
Geoffroy M. C., de The H. Classic and variants APLs, as viewed from a therapy response. Cancers 12, 967 (2020).
Geoffroy, M. C., Esnault, C. & de, TheH. Retinoids in hematology: a timely revival? Blood 137, 2429–2437 (2021).
Du, C., Redner, R. L., Cooke, M. P. & Lavau, C. Overexpression of wild-type retinoic acid receptor alpha (RARalpha) recapitulates retinoic acid-sensitive transformation of primary myeloid progenitors by acute promyelocytic leukemia RARalpha-fusion genes. Blood 94, 793–802 (1999).
Walkley, C. R. et al. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell 129, 1097–1110 (2007).
Ghiaur, G. et al. Regulation of human hematopoietic stem cell self-renewal by the microenvironment’s control of retinoic acid signaling. Proc. Natl Acad. Sci. USA 110, 16121–16126 (2013).
King-Underwood, L. & Pritchard-Jones, K. Wilms’ tumor (WT1) gene mutations occur mainly in acute myeloid leukemia and may confer drug resistance. Blood 91, 2961–2968 (1998).
Tosello, V. et al. WT1 mutations in T-ALL. Blood 114, 1038–1045 (2009).
Abbas, S., Erpelinck-Verschueren, C. A., Goudswaard, C. S., Lowenberg, B. & Valk, P. J. Mutant Wilms’ tumor 1 (WT1) mRNA with premature termination codons in acute myeloid leukemia (AML) is sensitive to nonsense-mediated RNA decay (NMD). Leukemia 24, 660–663 (2010).
Zhang, J. et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481, 157–163 (2012).
Pronier, E. et al. Genetic and epigenetic evolution as a contributor to WT1-mutant leukemogenesis. Blood 132, 1265–1278 (2018).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Wu, D. Y., Bittencourt, D., Stallcup, M. R. & Siegmund, K. D. Identifying differential transcription factor binding in ChIP-seq. Front. Genet. 6, 169 (2015).
Acknowledgements
This work was supported by grants from the National Key R&D Program of China (2022YFA1106100 to K.L. and 2023YFA1800401 to K.K.W.), National Natural Science Foundation of China (82222070, 82273973 to K.L., 82450101, 82370169 to H.H.Z., and 82350710226, 82370178 to K.K.W.), CAMS Innovation Fund for Medical Sciences (2021I2M-1-030 to K.L.; 2021-I2M-1-070 to T.Z.), CAMS Innovation Engineering Platform Fund for Medical Sciences (2022-I2M-2-002 to K.L.), and Fundamental Research Funds for Central Universities (3332022149 to F.W.).
Author information
Authors and Affiliations
Contributions
K.L., H.H.Z., K.K.W., and Y.Z.Q. conceptualized the study and participated in the overall design, supervision, and coordination of the study. F.W., L.Y.Z., and Y.T. designed and performed most experiments. X.F.C., H.G., H.M.J., Y.L., and Y.X.L. performed molecular and cellular biological experiments. T.T.Z., C.X.Z., T.S., G.L.X., and C.R.W. performed animal experiments. J.H. and X.L. provided scientific guidance for the study. K.L., F.W., and L.Y.Z. prepared the manuscript. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Chi Wai Eric So, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, F., Zhao, L., Tan, Y. et al. Oncogenic role of RARG rearrangements in acute myeloid leukemia resembling acute promyelocytic leukemia. Nat Commun 16, 617 (2025). https://doi.org/10.1038/s41467-024-55047-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55047-7
