
Abstract
Compared with widely established monovalent-ion batteries, aqueous multivalent-ion batteries promise higher capacity release by achieving multiple electron-transfer events per ion intercalation in the host material. Despite plausibility, this high-capacity dream is untenable with the total tolerable redox charge-transfer limit of the host material for all carrier species equally, which is historically assumed to depend on the material rather than the guest carrier itself, and the kinetic hysteresis induced by larger charge/radius ratios induced kinetic hysteresis further enlarges the divide. Herein, we report that copper carrier redox in vanadium sulfide (VS2) exceeds the intrinsic intercalation capacity boundary, with the highest capacity release as 675 mAh g-1 at 0.4 A g-1 among all VS2 cathodes previously reported. Operando X-ray absorption spectroscopy, operando synchrotron X-ray diffraction and composite ex situ characterization jointly demonstrated that intercalated divalent copper is preferentially involved in redox afforded extra electron transfer to form reversible monovalent copper pillars, thus not only ensuring stable topological de/intercalation with high capacity but also sustaining fast migration kinetic paths through reconfigurable pillar effects. Intercalated carrier redox reported here emphasizes the interlayer variable valence advantage of multivalent ions, providing insights into high-performance multivalent-ion storage chemistry in aqueous batteries.
Similar content being viewed by others

Introduction
Rechargeable batteries are indispensable for intermittent energy storage towards renewable energy sources, allowing ions to be reversibly stored in layers of material to provide conversion of chemical and electrical energy1,2,3,4. Monovalent (alkaline) ions represented by lithium ion are currently dominant especially in mobile electronics and electric vehicles1,5, but rising safety and cost buzz has prompted the emergence of aqueous multivalent-ion batteries (AMBs) as a complement to large-scale energy storage, which can plausibly utilize accessible metal anodes such as zinc6,7 or aluminium8 and nonflammable aqueous electrolytes9,10,11. However, the large charge/radius ratios of multivalent ions present a critical challenge to the cathode in AMBs, as the inherent high surface charge density of carriers and predictably tough kinetics in solid-phase diffusion hinder capacity release and reversibility in intercalation chemistry3,12,13. With such consideration, the design of cathode materials and intercalation chemistries is particularly essential, and more efforts are required to enable high-capacity and stable cathodes for multivalent-ion intercalation in AMBs.
Conventionally, it has been theoretically expected that multivalent-ion chemistry can provide greater capacity release to cathodes than monovalent-ion intercalations, based on the fact that multivalent ions can provide multiple electron transfer events in each carrier intercalation at first glance14,15,16,17. In contrast, monovalent ions provide only one electron transfer per carrier intercalation18,19. However, this perspective fundamentally relies on the shaky assumption that the host material can hold the same number of carriers in the interlayer regardless of the type of carrier. Palacín20 and Yao12 contend that, in practical mechanistic considerations, the theoretical capacity boundary of any intercalated compound actually depends on the total number of electrons that can be transferred to the variable-valence redox centres of the host, which is the material endowment property, implying that over-expectations for multivalent-ion intercalation are not feasible. Therefore, in the framework of the intercalation mechanism, the theoretical capacity release boundary of the cathode material in the multivalent-ion system would not be better—if not worse—than that in the monovalent-ion system, especially considering the poor kinetics19,21. As a typical case, layered vanadium sulfide (VS2) has been demonstrated to provide only 180 mAh g-1 in zinc batteries21, which is much lower than that reported in sodium batteries (∼500 mAh g-1) at the same specific current5. Exploring novel cathode chemistries to truly take advantage of the multiple charge carriers of multivalent ions shuttling in the electrolyte remains desirable but challenging.
Beyond established experimental exploration and insights into multivalent-ion chemistry, the potential of multivalent-ion carriers in the valence state22,23, i.e., the redox capability, has long been neglected in intercalated electrodes, although preliminary investigations have been conducted in liquid-flow batteries (electrolyte redox)24,25,26,27 and elemental electrodes (conversion)28; for example, Wu et al. proposed that copper ions can be used as redox charge carriers in sulfur conversion to achieve high capacity29, and the phenomenon was extended to other conversions11,30,31,32,33. Exploration of electroplating/stripping and redox utilizing electrolytes is also in progress22,34. It can be expected that, compared to monovalent carriers acting only as charge carriers in intercalation, the high valence state of intercalated multivalent carriers in host as redox centres endows the possibility to afford extra charge transfer, permitting a fundamental breakthrough in the intrinsic capacity boundary of the electrode material. Moreover, the redox of intercalated multivalent carriers inside the host considerably affects the electronic structure and interlayer ion transport dynamics of the host material7,21, which thus contributes to the deep intercalation of multivalent ions by overcoming kinetic disadvantages and the inherent trade-off between stability and capacity. Predictably, intercalated carrier redox offers apparent benefits in multivalent-ion storage and will undoubtedly advance in-depth research to exceed the established mechanism framework, yet such research is still incipient.
Herein, we report intercalated multivalent-ion redox reaction in VS2 cathode using divalent copper-ion carriers to exceed the theoretically reversible capacity boundary. Operando/ex situ X-ray absorption fine structure spectroscopy (o-XAFS), operando synchrotron X-ray diffraction (o-SXRD), and composite ex situ characterization jointly confirmed that copper carriers were stored as monovalent forms through redox in interlayers at the early stage of intercalation, thus not only doubling the reversible capacity release to 675 mAh g-1 but also realizing the predicted sustainable kinetic advantages with stable operation for 7000 cycles at 10 A g-1 through monovalent copper-ion pillar effects which can be reconfigured by copper-ion de/intercalation. Employing in zinc batteries with decoupled configurations, the VS2 cathode with copper carriers delivers an energy density of 773 Wh g-1 (based on the mass of the VS2 cathode) and the ability to operate for 1000 cycles, providing a substantial boost in stability and energy supply. High-load (7.03 mg cm-2) and scaled-up watt-hour level cells (1.1 Wh) are also built to provide a total charge storage of 0.91 Ah. Initiating carrier redox provides insights into aqueous battery chemistry and represents a kind of high-performance intercalated electrodes.
Intercalated carrier redox mechanism
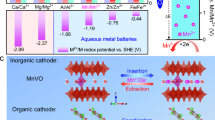
Conceptually formalized VS2 (f-VS2) secondary particles were synthesized via a one-step hydrothermal method and assembled from micrometre-sized hierarchical nanosheets (Supplementary Figs. 1–3)11,35, which were well-indexed to hexagonal-stacked 2H-VS2 (PDF#89-1640) belonging to the P-3m1(164) space group (Fig. 1a, Supplementary Figs. 4–6),36 and the elemental components were as expected (Supplementary Figs. 7–12)4,37,38,39. Structural investigations of several ion intercalations revealed that, unlike the multivalent ion carriers that have been disclosed such as magnesium and zinc, the Cu-S bonds formed by intercalated copper-ions have shorter bond lengths compared to the V-S bonds in VS2 host, implying that stronger interactions may enable the capture of additional electrons from the host to trigger the intercalated carriers redox (Fig. 1b). Previous reports have shown that VS2 delivers an intercalation capacity release of <200 mAh g-1 as cathode in multivalent-ion batteries40,41. Realizing copper-ion intercalation with carrier redox is expected to break this long-standing inherent limitation42,43. Thus, when copper ions are used as carriers, the f-VS2 cathode has a capacity of up to 675 mAh g-1 at 0.4 A g-1 (Fig. 1c, d). This capacity record considerably exceeds all VS2 cathodes revealed in ion batteries to date (Fig. 1e, Supplementary Table 1)6,8,18,21,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60, suggesting the possibility of novel redox mechanisms that have yet to be identified, permitting stable operation for 7000 cycles at high specific current of 10 A g-1 with a Coulombic efficiency (CE) of ∼100% and capacity retention of 82.4% (Fig. 1f). Therefore, the valence evolution of the vanadium centre of f-VS2 obtained via o-XAFS analysis was first investigated (Fig. 1g). An operando approach was taken to capture the XAFS data in fluorescence mode with minimized X-ray attenuation. The near-edge structure of the normalized V K-edge showed a distinct shift in the V state at 5482 ∼ 5485 eV towards lower energy during intercalation, suggesting a decrease in the vanadium valence state during redox (Fig. 1h). The V state in full discharge f-VS2 remains far from the standard V foil (V0), ruling out conversion reactions (Supplementary Fig. 13). When the current is reversed, the edge reversibly moves to higher energy and returns to the pristine state at full charge (Fig. 1i), indicating the complete retrieval of intercalated ions. The Fourier transformed (FT) K-edge extended X-ray absorption fine structure (EXAFS) spectra of the V K-edge were further analysed to clarify the local structure around V during intercalation (Fig. 1j). The strongest main peak corresponded to the nearest-neighbour V–S coordination shell located at ∼1.9 Å (no phase correction)61. Insignificant V‒S bond elongation (<0.12 Å) occurs during the discharge process and vice versa, implying a robust in-plane structure during intercalation. Changes in V–S coordination can also be identified from the peak profiles of ex situ tender-energy S K-edge X-ray absorption near edge structure (XANES) in the range of 2465–2475 eV62, but no extra anionic redox was found since no offset of the main sulfur peak was observed (Supplementary Fig. 14)63,64.

a XRD pattern of VS2. b Structural investigations of ion intercalations, including magnesium, zinc, and copper. c Galvanostatic charge‒discharge (GCD) curve and (d), cycling performance of f-VS2 electrodes with copper intercalation at 0.4 A g-1. e Comparison with the capacity of reported VS2 cathodes to date. VS2 cathodes for monovalent-ion batteries and multivalent-ion batteries are distinguished by blue and red colour blocks, respectively. f Cycling performance of f-VS2 electrodes with copper intercalation at 10 A g−1. g GCD curves and corresponding V K-edge o-XAFS data in (h), deintercalation (charging) and (i), intercalation (discharging). The right-side images show the magnified XANES region. j EXAFS evolution inferred from o-XAFS data. The red and blue dashed lines mark the V‒S peak positions for the fully discharged and fully charged states, respectively.
Nevertheless, from theoretical electron transfer number calculations based on VS2, vanadium centre evolution alone, which does not involve conversion reactions, cannot provide the observed high discharge capacity. The reasonable inference of carrier redox prompted further investigation of the valence evolution of copper during intercalation. Eighteen f-VS2 electrodes were extracted from one cycle with different charge/discharge states, and their copper content was quantified (Fig. 2a), with duplicates for the fully discharged state. The apparent emergence of Cu 2p peaks in X-ray photoelectron spectroscopy (XPS) clearly indicates copper intercalation (Supplementary Fig. 15, Supplementary Fig. 16), and the maximum copper content (fully discharged state) is close to 1.0 Cu per VS2. The corresponding XPS Cu 2p core spectra are shown in Fig. 2b and Fig. 2c. The Cu 2p3/2 peak and the Cu 2p1/2 peak increased in intensity as the discharge process proceeded. Notably, during the first half of intercalation (before the sixth discharge spectrum, a copper content of ∼0.75 per VS2), only isolated Cu 2p3/2 (932.7 eV)/Cu 2p1/2 (952.6 eV) peaks were identified, implying the presence of monovalent copper species. As the depth of intercalation proceeded, additional Cu2+ peaks with signature satellite peaks (located at 943/962 eV) emerged, indicating the presence of Cu2+ species. Similar observations for bulk VS2 corroborate the conclusions (Supplementary Fig. 17). Therefore, considering that Cu2+ species dominate the type of copper carriers in the electrolyte, carrier redox should occur at the early stage of intercalation, i.e., Cu2+ carriers are first redox-converted to Cu+ species after intercalation to remain in the interlayer, whereas Cu2+ carriers intercalate and migrate at the later intercalation stage, and vice versa. When the current is reversed during charging, similar inverse processes are observed, and copper ions are almost completely extracted from the interlayer, confirming the reversibility of the proposed mechanism.

a GCD curves, evolution of the copper content quantified from the XPS spectra, and corresponding Cu 2p XPS data in (b), intercalation (discharging) and (c), deintercalation (charging). d XAFS data for specific discharge states and corresponding XANES region amplification. e Optical images of the CuII and CuI electrolytes and GCD curves of f-VS2 in the corresponding systems. See Methods for the configuration of the CuI electrolyte. f Cycling performance of f-VS2 using the CuI electrolyte. g Comparison of the maximum capacity and cycling lifespan of f-VS2 in the CuII and CuI electrolyte systems. h Schematic of the valence distribution of the intercalated copper ions in the discharge. Monovalent copper resulting from carrier redox appears at the beginning of intercalation, whereas divalent copper appears at the final stage of intercalation. i Schematic of the intercalation process of monovalent-ion, multivalent-ion, and multivalent-ion redox carriers. Intercalation of multivalent-ion redox carriers can improve the charge transfer capacity while occupying a limit number of transition metal (TM) redox sites.
The collection of XAFS spectra for specific discharge states (states I, II, III, and IV) demonstrated similar results (Fig. 2d). In states I and II, the copper species in VS2 have almost unchanged overlapping edge positions, indicating that a low copper valence state coincides with the monovalent copper position (CuI, inferred from Cu2S). Upon further discharging (states III and IV), a shift of the absorption edge towards higher energies (CuII, inferred from CuS) is observed, indicating an increase in the average valence state of the intercalated copper ions. Finally, the average copper valence state reached 1.27. The redox of copper carriers in the interlayer was hence indeed triggered at the beginning of intercalation65, which is consistent with the XPS evidence. For comparison, a monovalent copper-ion (CuI) electrolyte was prepared on the basis of the stability of the Cu+ ions in acetonitrile to exclude the effect of the redox of the copper carriers (Fig. 2e). At the same specific current, f-VS2 in the CuI electrolyte provides only 97 mAh g-1 capacity (rate performance shown in Supplementary Fig. 18), which is ∼14% of the capacity of f-VS2 in the CuII electrolyte. In addition, a 41.6% capacity was obtained after 1000 cycles, which is far inferior to that of f-VS2 in the CuII electrolyte (Fig. 2f), highlighting the composite enhancement of stability and capacity by intercalated carrier redox (Fig. 2g). Therefore, it can be concluded that the copper carriers and vanadium centres involved in the redox process are plausibly jointly contributing to the high reversible capacity of f-VS2 (Fig. 2h), which implies that interlayer carrier redox may optimize the intercalation kinetics and reversibility in host materials. The FT-EXAFS analysis also revealed stable bond structures and an increase in the Cu-Cu ligand content (Supplementary Fig. 19), data that coincide with the insights extracted from the V K-edge. This first-revealed redox of intercalated carriers could bring intrinsic advantages to multivalent-ion batteries (Fig. 2i). Compared with monovalent carriers, multivalent carriers are generally believed to possess a greater potential capacity because they can carry multiple charges at once for redox charge transfer with host materials. However, this expectation is actually strictly constrained by the total number of electron transfers that can be provided by the redox centre in host materials, which theoretically treats all types of carriers equally. Therefore, multivalent carriers would instead be mediocre by occupying more redox centres. In this regard, the use of intercalated multivalent copper ions as redox centres can afford extra electron transfer to form monovalent copper ions, thus increasing the capacity based on limited redox centres in the host, which is difficult to achieve in monovalent-ion batteries. The evolution of intercalated multivalent carriers (CuII, ionic radius = 57 pm) to monovalent carriers (CuI, ionic radius = 60 pm) also leads to an increase in the ion size, thus maintaining sound layer spacing, and the effect of such endogenous carrier-dependent interlayer interactions on the intercalation capability was subsequently revealed via structural research.
Reversible pillar effects in structural evolution
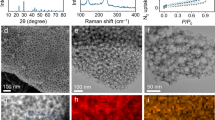
The structural evolution caused by the reversible storage of ions in f-VS2 was further investigated through o-SXRD. The evolution of several major characteristic peaks of f-VS2 during two continuous charging and discharging processes (Fig. 3a), including the (011), (002), and (110) planes (Fig. 3b) is shown. The peak shifts towards smaller angles are considerable in the initial stages of copper-ion intercalation, inducing simultaneous in-plane and out-of-plane lattice expansions that subsequently become moderate. When the electron transfer number reaches ∼2.2 e-/VS2, the SXRD pattern can plausibly be indexed to the Cu0.75VS2 species (Supplementary Fig. 20), indicating the presence of an extra redox centre providing more electron transfer processes, which is consistent with the results of the spectroscopic analysis. Until the fully discharged state was reached, a maximum of 3.08% in the (002) plane and 2.77% in the (110) plane was observed, corresponding to a volume expansion of less than 10%, which coincided with the macroscopic cross-sectional view (Supplementary Fig. 21). The characteristic peaks move similarly but in opposite trends during charging, eventually returning to the pristine peak position at the completion of charging. No additional peak generation/vanishing or tendency breaks were recognized, confirming a reliable intercalation mechanism, and the structure of VS2 fully recovered after deintercalation. Further o-SXRD was performed after 50 cycles (Fig. 3c), and overlapping de/intercalation processes were noted (Fig. 3d). Quantification of the expansion of the characteristic lattice for both o-SXRD measurements reveals that f-VS2 maintains a stable reversible intercalation mechanism (Fig. 3e). Notably, the most significant lattice expansion ( ∼ 2% according to the (002) plane) occurs at the beginning of intercalation, when the copper ions are transformed from divalent copper (CuII) to monovalent copper (CuI) in the interlayer. The larger monovalent copper ions act as pillars to stabilize the interlayer structure and reduce the degree of lattice expansion following ion intercalation (<1% according to the (002) plane) (Fig. 3f). Previous reports have shown that intercalated interlayer pillars can mitigate lattice distortion and retain plausible ion diffusion paths that favour intercalation kinetics through the so-called pillar effect; however, the demonstrated pillars are usually retained in the interlayer and do not deintercalated, thus permanently altering the material structure. In contrast, copper ions with intercalated carrier redox not only ensure reversible stable intercalation with promising decent kinetics but also contribute to desirable capacity enhancement, allowing reconfiguration in layers via reversible de/intercalation. Transmission electron microscopy (TEM; Fig. 3g) and scanning electron microscopy (SEM) observations (Supplementary Fig. 22) of the fully discharged products were performed, and the ultrathin nanosheet morphology with a ten-micrometre horizontal size was well preserved. The uniform elemental distribution with copper intercalation (Fig. 3h–k, Supplementary Fig. 23) and the inherited microstructure (Fig. 3l) similarly support the stable de/intercalation mechanism with reversibility.

a GCD curves and (b) corresponding o-SXRD patterns of typical characteristic Bragg peaks in the initial two cycles. c GCD curves and (d) corresponding o-SXRD patterns of typical characteristic Bragg peaks in the two cycles after cycling. e Quantized lattice expansion during the measured cycles. f Schematic of the pillar effects of copper carrier redox during the intercalation process. g TEM observation, (h) HAADF image, and the corresponding (i), Cu, (j), S, (k), V elemental mapping of full-discharged product. l, HRTEM image and SAED pattern of full-discharged product.
The pillar effects of copper carrier redox during the intercalation process are illustrated via density functional theory (DFT) analysis. Calculations of density of states (DOS) distributions for species in initial (VS2, Fig. 4a), intermediate (Cu0.75VS2, Fig. 4b), and fully intercalated (Cu1.0VS2, Fig. 4c) states show that a continuous conduction band without energy gaps is maintained near the Fermi energy level (Ef), suggesting that the high conductivity of materials is guaranteed by intercalated copper ions. The highest and sub-highest electron-filling intensities near Ef are occupied by V and Cu in the intermediate state, and the sulfur intensity edge is far from the profile, which is consistent with the observation that no anion redox occurs in the experiments. Thermodynamic analysis on the basis of the Gibbs free energy further revealed the feasibility of intercalation (Supplementary Fig. 24). The absorption and migration of copper at the VS2 interface were also examined via first-principles calculations (Fig. 4d). All the possible adsorption sites with optimized structures for copper adsorbed on VS2 and Cu0.75VS2 were considered (Supplementary Fig. 25, Supplementary Table 2). A comparison of the copper adsorption energies for the top (-2.71 eV) and hollow (-2.53 eV) potential sites indicated that copper was susceptible to capture at the interface. Nonetheless, the energy difference across sites is not considerable, and both sites can act as adsorption sites to initiate redox processes. At the intermediate state of intercalation (Cu0.75VS2), the hollow site becomes the dominant site (top -2.11 eV, hollow -2.69 eV (II)/-2.38 eV (I), bridge -2.39 eV; Fig. 4e). The intercalated copper pillar does not considerably change the adsorption energy, maintaining the appreciable interfacial adsorption kinetics of VS2 during intercalation. Migration barriers as low as 0.29 eV were obtained when copper diffused along the top–top path, which enabled unimpeded access of copper to all possible kinetic storage sites (Fig. 4f). The migration barriers of the intermediate state of the intercalation also remain at a low hindering level of 0.63 eV (Fig. 4g). The galvanostatic intermittent titration technique (GITT) provides experimental evidence. Generally, ions that have been intercalated will occupy favourable sites leading to slow kinetics of subsequent intercalation, as shown by the rapid decrease in diffusion coefficients for CuI (Supplementary Fig. 26) or zinc-ion intercalation without redox. The f-VS2 with copper carrier redox provides an average ion diffusion coefficient Dions ∼ 10-10.62 cm2 s-1 (Fig. 4h), which is comparable to the recognized high-speed intercalation cathode Zn0.25V2O5 in aqueous batteries and an order of magnitude greater than the diffusion of zinc carriers without redox in f-VS2 (Supplementary Fig. 27, Supplementary Fig. 28). Therefore, benefitting from the reversible pillar effects of copper carrier redox in the intercalation process, the advantage of fast and reliable kinetics is maintained throughout the de/intercalation process, which is important for obtaining high performance.

a DOS in different intercalated states: pristine state VS2, (b) intermediate state Cu0.75VS2 and (c) fully intercalated state Cu1.0VS2. The dotted lines mark the positions of the Fermi energy levels. d Adsorption energy of copper at accessible top sites and hollow sites of VS2. e Adsorption energy of copper at accessible top sites, hollow sites (multiple), and bridge sites of Cu0.75VS2. The blue, orange, and red atoms represent the vanadium, sulfur, and copper elements, respectively. f Migrated energy barriers on the VS2 and (g) Cu0.75VS2 surfaces corresponding to plausible diffusion paths. h Ion diffusion coefficients of f-VS2 with copper intercalation. CuI in acetonitrile (grey) as reference.
Demonstration of high-performance VS2-Cu | |Zn cell with carrier redox
Aqueous zinc batteries with decoupled configurations and f-VS2 cathodes were established to take advantage of the copper carrier redox mechanisms (Fig. 5a). The CuI electrolyte is not suitable for such systems, thus cells with zinc carriers without carrier redox were also established for comparison. When f-VS2 cycles in the VS2-Zn | |Zn cell (Fig. 5b) - note that here using VS2-M | | N to define the specific battery system, where M is the carrier involved in redox at the cathode and N is the anode - results in a capacity lower than 125 mAh g-1 (Fig. 5c). In contrast, when copper ions are used as carriers (VS2-Cu | |Zn), the performance is drastically improved, as shown in the cyclic voltammetry (CV) curve (Fig. 5b). At 0.2 A g-1, f-VS2 in the VS2-Cu | |Zn cell provides a capacity release of 623 mAh g-1 and an energy supply of 773 Wh kg-1, which is 10 times greater than that of the cell using zinc carriers at the same specific current (all calculated on the basis of the VS2 mass), as shown in the GCD curves. Even if the specific current is increased to 0.5, 1, 2, 4, and 6 A g-1, capacities of 600, 567, 547, 508, and 423 mAh g-1 can be obtained, respectively (Fig. 5d). The energy efficiency ranged from 80% ∼ 96%. When the specific current is returned to 0.2 A g-1, a capacity of 617 mAh g-1 is recovered, demonstrating favourable robustness to current abuse (Supplementary Fig. 29), which is far superior to that of f-VS2 in the VS2-Zn | |Zn cell (Supplementary Fig. 30). Compared with all the VS2 cathodes used in zinc batteries reported to date, the f-VS2 cathode in the VS2-Cu | |Zn cell offers more than twice the energy density advantage (Fig. 5e)6,21,43,55,56,57,58,59,60. Further long-term cycling measurements were performed at 6 A g-1 (Fig. 5f). The capacity retention of 88.7% (compared with the pristine cell) confirms the stability of f-VS2 with a CE of ∼100%. With facile optimization of the zinc anode, the cell lifespan can be further extended to 1800 cycles (Supplementary Fig. 31). After long cycles, retained flower-like clusters are observed at the electrodes (Supplementary Fig. 32), which corroborates the reliable stability. In stark contrast, f-VS2 using zinc as a carrier is constrained by slow kinetics and rapidly degrades to a capacity below 15 mAh g-1 within 10 cycles, two orders of magnitude lower than that of f-VS2 in the VS2-Cu | |Zn cell in terms of energy density and lifespan (Supplementary Fig. 33). The established advantages have driven the build-up of a high-load (7.03 mg cm-2) and scale-up 1.1 Wh cell (Fig. 5g), which can deliver 581 mAh g-1 capacity and 690 Wh kg-1 energy density (calculated on the basis of f-VS2 loading), enabling 0.91 Ah reversible charge storage and an energy efficiency of 81.7% (Supplementary Fig. 34).

a Schematic of the established decoupled cell configurations with f-VS2 cathodes and copper carrier redox mechanisms. b CV curves at 0.2 mV s−1 and (c) GCD curves at 0.2 A g–1 of f-VS2 in the VS2-Cu | |Zn cell and VS2-Zn | |Zn cell. d Rate performance of f-VS2 in the VS2-Cu | |Zn cell and VS2-Zn | |Zn cell. e Comparison of the record reported here with the performance of all known VS2 cathodes in zinc batteries. f Long-term cycling of f-VS2 in a VS2-Cu | |Zn cell at 6 A g–1. g GCD curves of the scale-up 1.1 Wh cell at 50 mA g–1.
Discussion
Driven by resource sustainability and reserve advantages in the Earth’s crust, interest in multivalent-ion batteries, which cover several ion carriers from zinc to aluminium. The taken-for-granted anticipation of the prospect of multivalent ion batteries promises high-capacity availability, which is invoked by the fact that single ion can carry multiple charges: Multivalent ions such as zinc enable more than one charge equivalents transfer per mole of the metal carriers, leading to a higher specific capacity than that of monovalent metal-ion batteries. However, this motivation is untenable for host electrodes employed in ion storage such as intercalation-type VS2 cathodes, due to the limitation of the total number of redox centers. Beyond the upper limit of the total number of transferable electrons per mole of material, the bond–lattice structure of the material irretrievably collapses, depending not on the type of carrier but on the number of redox sites in the material itself. Given these considerations, the most important advantage of multivalent ion carriers may be the variable valence property, whereby the intercalated carriers act as redox centres to undertake electron transfer to break through the upper limit of the total number of electrons that can be transferred in the material itself, which is clearly beyond the reach of monovalent carrier systems. Inspired by the mixed valence of copper minerals in nature, the redox reaction of divalent copper has been developed and applied in many different battery systems. The exploitation of copper elements as anchored redox centers in electrodes has been demonstrated in lithium and zinc batteries66,67, and initiating the deposition of copper ions from the electrolyte to the electrode interface or liquid-flow-type redox as well as conversion in sulfur group element electrodes have shown in several novel battery prototypes22,29,32,68. These established deposition/conversion reactions clearly result in copper carrier redox, however the possibility and mechanism of carrier redox within the intercalated host material is long neglected. Apparently, for the capacity enigma that continues to loom over multivalent-ion intercalation chemistry, realizing the redox of intercalated multivalent-ions with insights into the electronic structure and interlayer ion transport dynamics of the host material in reaction is a desirable starting point. In this work, we report carrier redox in an intercalation-type f-VS2 cathode, which provides the highest recorded capacity among VS2 cathodes of 675 mAh g-1 and enables stable operation for 7000 cycles at 10 A g-1. Composite operando/ex situ characterization and first-principles calculations revealed that the preferential redox of intercalated copper ions in the interlayer not only provides extra capacity for f-VS2 but also maintains a fast kinetic advantage via the reversible pillar effect, conferring a 773 Wh kg-1 energy supply in aqueous zinc-decoupling batteries. We also found that the similar intercalated carrier redox mechanism is present in niobium disulfide (Supplementary Fig. 35). Carrier redox during intercalation provides a potential pathway to truly utilize the multicharge-carrying advantages of multivalent carriers, which is promising for stimulating fervour for research on multivalent-ion batteries.
Methods
Materials
To prepare f-VS2, first, 2 mmol of NH4VO3 was added to a mixture of 30 mL of deionized water and 2 mL of ammonium hydroxide. Then, 20 mmol of thioacetamide was added to the above mixture and stirred for 4 h at room temperature in an atmosphere. The autoclave was sealed and kept at 180 °C for 20 h and then cooled to room temperature. The product was washed with deionized water and ethanol several times and then dried at 60 °C overnight in a vacuum oven. CuSO4·5H2O (99.9%), ZnSO4·5H2O (99.9%), copper foil (99%), and zinc foil (99%) were purchased from Sinopharm Chemical Reagent Co., Ltd. NbS2 were purchased from Nanjing MKNANO Tech. Co., Ltd. and were used directly without further purification.
Electrodes and electrolytes
f-VS2 was mixed with carbon black and polyvinylidene fluoride at a weight ratio of 7:2:1 in 1-methyl-2-pyrrolidinone (i.e., active material as 70 w.t.%), and then, the slurry was pasted on carbon paper and dried under vacuum at 60 °C for 10 h. Typical thickness of the electrodes is 150 μm (including the collector) for a 6 mm diameter disc (area 28.3 mm2). The typical mass loading of the active materials was 1–1.5 mg cm-2. Preparation of NbS2 electrodes followed the same protocol. All metal electrodes were polished with 800 grit silicon carbide sandpaper and cleaned with ethanol before use. 1 M CuSO4 in H2O (for the VS2-Cu | |Zn cell) and 1 M ZnSO4 in H2O (for the VS2-Zn | |Zn cell and VS2-Cu | |Zn cell) were prepared or obtained in stoichiometric ratios. In addition, a CuI electrolyte was used: 0.2 M Cu(ClO4)2 was dissolved in anhydrous acetonitrile, the cleaned copper foils were incubated overnight, and the solution eventually became colourless because of the reduction of Cu2+ in the solution.
Electrochemical evaluation
Electrochemical energy storage tests are carried out in an air-conditioned room with the temperature of the AC set at 27 °C, affected by the ambient air temperature with a variation of 2 °C. All the specific energy/energy density values and the specific power/power density values reported in the manuscript are calculated based on the mass of VS2. For all cells, significantly excessive metal anodes are used ( ∼ 80 mg cm-2, N:P > 10). As previously defined, the cells are named VS2-M | |Zn, where f-VS2 is the cathode material and M is the carrier in the cathode chamber reacting with f-VS2, i.e., Cu or Zn. Specifically, for the VS2-Cu | |Zn cell, an ion-selective membrane (anion exchange membrane, AEM typed as FAB-PK-130 and commercially obtained from FUMATECH BWT GmbH) is used to divide the complete cell into two chambers, referred to as the cathode chamber and the anode chamber. Since AEMs can block cations (e.g., Zn2+ and Cu2+) from crossing over but allow anions (SO42-) to traverse freely, the cathode and anode chambers can be filled with aqueous sulfate solutions containing different cations without worrying about electrolyte mixing (normally 2 mL due to cell size). In the case of f-VS2-Cu | |Zn, for example, the cathode chamber is filled with a 1 M CuSO4 solution, and f-VS2 coated on carbon paper is used as the cathode submerged in the solution. Similarly, the anode chamber was filled with an equal amount of 1 M ZnSO4 solution, and zinc metal was used as the anode. Owing to the confinement of the AEM, Zn ions do not diffuse to the cathode side to disturb the de/intercalation of Cu ions, and SO42- ions are allowed to shuttle between the two chambers to balance the charge. All energy densities in the manuscript are calculated on the basis of the VS2 material in the cathode. The GCD and GITT tests were performed with a LANHE CT2001A (Wuhan Land Electronic Co., Ltd.) battery testing system, with cycling in the voltage range of 1.1 − 1.6 V vs. Zn2+/Zn (for the VS2-Cu | |Zn cell) and 0.4 − 0.8 V vs. Zn2+/Zn (for the VS2-Zn | |Zn cell), and were precycled at a low specific current (0.5 A g-1) for 5 cycles to activate the electrodes before formal testing. When copper foil is used as an anode, a 1 M CuSO4 solution (CuII) or a CuI electrolyte is used as the electrolyte without an AEM. Electrochemical evaluation of NbS2 electrodes followed the same protocol. CV tests were performed with a CHI 760E (Chenhua Instrument Company, Shanghai, China) electrochemical workstation, and the voltage range was consistent with that used in the GCD cycling.
Characterization
The VS2 samples/electrodes were characterized via SEM (Carl Zeiss Gemini SEM 300), XPS (Kratos Analytical Axis UltraDLD), TEM (JEM-F200), and Raman spectroscopy (RENISHAW inVia Basis; 532 nm). The XRD experiments were performed on beamline BL02U2 (X-ray wavelength of 0.6887 Å) at the Shanghai Synchrotron Radiation Facility (SSRF). The S XAFS experiments were performed on beamline BL16U1 at the SSRF, and the Cu XAFS experiments were performed on beamline BL13SSID at the SSRF. The position of the absorption edge was calibrated with S powder or Cu foil, and the obtained data were analysed with the ATHENA software package. For ex situ measurements of the VS2 cathodes, the batteries were first charged/discharged to the specific potential with a LAND workstation and opened in air to collect the cathodes. The prepared cathodes were washed with deionized water and anhydrous ethanol three times and dried at room temperature for 10 min for further measurements. o-SXRD experiments were performed on beamline BL02U2 (X-ray wavelength of 0.6887 Å or 0.695 Å) at the SSRF. The beam size was confined by horizontal and vertical slits to ∼0.3 × 0.3 mm2. The two-dimensional XRD signal was obtained by a PILATUS3S 2 M detector and integrated with Fit2D software. In addition, a special coin cell was designed for the o-SXRD study. There were two observation holes with radii of ∼2 mm in the middle of both sides of the cell, guaranteeing that X-rays penetrated through the active material during operation. The special operando cell observation holes were sealed with polyimide tape. The same cell but with an observation hole (5 mm) was also used to perform V o-XAFS measurements at beamline BL17B1 at the SSRF. The position of the absorption edge was calibrated with V foil, and the obtained data were analysed with the ATHENA software package. During the test, the batteries were cycled at 0.3 A g-1.
Calculation
Calculations were conducted using spin-polarized DFT within the Vienna ab initio simulation package (VASP). Projector-augmented wave (PAW) pseudopotentials were employed to describe the core–valence interactions, whereas the exchange–correlation functional was based on the generalized gradient approximation (GGA) within the PBE framework. The cut-off energy for the plane wave basis was set to 520 eV. All the bulk structures (VS2, Cu0.75VS2, and Cu1.0VS2) were optimized. The k-points were set to 8 × 8 × 1 for the VS2 bulk structure and 3 × 3 × 1 for the Cu0.75VS2 and Cu1.0VS2 bulk structures in the Gibbs free energy calculations. The convergence criteria for energy and force were set to 1 × 10-5 eV/atom and 0.02 eV/Å, respectively. A total of 4 × 4 supercells with 3 atomic layers were selected for the VS2 slabs, whereas a 2 × 2 unit-cell with four atomic layers was selected for the Cu0.75VS2 surface. A 2 × 2 × 1 k-points were set for all optimized slabs. To avoid interactions between periodic images, a vacuum layer over 15 Å was set in the z direction. The climbing image nudged elastic band (CI-NEB) method with the quasi-Newton algorithm was employed to calculate the diffusion barrier of a Cu atom on the surface of a monolayer VS2. The vibrational frequencies of the optimized structures were computed via the finite difference method with a step size of 0.02 Å. Three types of sites were considered for the adsorption of Cu on a single-layer VS2/Cu0.75VS2, including the top, bridge and hollow sites. The energy for the Cu-chemisorbed state was calculated via the formula Eads = ECu-VS2 or Cu-Cu0.75VS2 - ECu - EVS2 or Cu0.75VS2. ECu-VS2 or Cu-Cu0.75VS2 is the energy of the Cu atom adsorbed on the VS2 or Cu0.75VS2 surface, respectively. EVS2 or Cu0.75VS2 represents the energy of single-layer VS2 or Cu0.75VS2. ECu is the energy of the isolated Cu atom.
Data availability
The authors declare that all data supporting the findings of this study are available within the paper and its supplementary information files.
References
Wang, Q. et al. Chemical short-range disorder in lithium oxide cathodes. Nature 629, 341–347 (2024).
Zhao, Y. et al. Ultrastable Cu2+ intercalation chemistry based on a niobium sulfide nanosheet cathode for advanced aqueous storage devices. ACS Nano 17, 6497–6506 (2023).
Sun, Y. et al. Proton-dominated reversible aqueous zinc batteries with an ultraflat long discharge plateau. ACS Nano 15, 14766–14775 (2021).
Ren, Z. et al. Metallic V5S8 microparticles with tunnel-like structure for high-rate and stable zinc-ion energy storage. Energy Storage Mater. 42, 786–793 (2021).
Zhou, J. et al. Hierarchical VS2 nanosheet assemblies: a universal host material for the reversible storage of alkali metal ions. Adv. Mater. 29, 1702061 (2017).
Zhang, L. et al. MXene-stabilized VS2 nanostructures for high-performance aqueous zinc ion storage. Adv. Sci. 11, 2401252 (2024).
Yao, Z. et al. A volume self-regulation MoS2 superstructure cathode for stable and high mass-loaded Zn-ion storage. ACS Nano 16, 12095–12106 (2022).
Wu, L. et al. A rechargeable aluminum-ion battery based on a VS2 nanosheet cathode. Phys. Chem. Chem. Phys. 20, 22563–22568 (2018).
Sun, Y. et al. Ultrahigh-speed aqueous copper electrodes stabilized by phosphorylated interphase. Adv. Mater. 35, 2305087 (2023).
Si, J. et al. Deep multiphase conversion derived from NiTe2 nanosheets with preferred kinetics for highly reversible mild aqueous zinc–tellurium batteries. Adv. Energy Mater. 14, 2303982 (2024).
Sun, Y. et al. Initiating reversible aqueous copper–tellurium conversion reaction with high volumetric capacity through electrolyte engineering. Adv. Mater. 35, 2209322 (2023).
Liang, Y., Dong, H., Aurbach, D. & Yao, Y. Current status and future directions of multivalent metal-ion batteries. Nat. Energy 5, 646–656 (2020).
Ren, Z. et al. Accumulative delocalized Mo 4d electrons to bound the volume expansion and accelerate kinetics in Mo6S8 cathode for high-performance aqueous Cu2+ storage. ACS Nano 17, 19144–19154 (2023).
Liu, Y. et al. Cathode design for aqueous rechargeable multivalent ion batteries: challenges and opportunities. Adv. Funct. Mater. 31, 2010445 (2021).
Manalastas, W. Jr et al. Water in rechargeable multivalent-ion batteries: an electrochemical pandora’s box. ChemSusChem 12, 379–396 (2019).
Fu, N. et al. Electrode materials for aqueous multivalent metal-ion batteries: current status and future prospect. J. Energy Chem. 67, 563–584 (2022).
Liu, Z. et al. Voltage issue of aqueous rechargeable metal-ion batteries. Chem. Soc. Rev. 49, 180–232 (2020).
Zhao, J. et al. Molybdenum atom engineered vanadium disulfide for boosted high-capacity Li-ion storage. Small 19, 2301738 (2023).
Murphy, D. W., Cros, C., Di Salvo, F. J. & Waszczak, J. V. Preparation and properties of LixVS2 (0≤x≤1). Inorg. Chem. 16, 3027–3031 (1977).
Ponrouch, A. et al. Multivalent rechargeable batteries. Energy Storage Mater. 20, 253–262 (2019).
Tan, Y. et al. Unexpected role of the interlayer “dead Zn2+” in strengthening the nanostructures of VS2 cathodes for high-performance aqueous Zn-ion storage. Adv. Energy Mater. 12, 2104001 (2022).
Duan, Z. et al. An aqueous copper battery enabled by Cu2+/Cu+ and Cu3+/Cu2+ redox conversion chemistry. Chem. Commun. 58, 10076–10079 (2022).
Xu, C. et al. Unravelling rechargeable zinc-copper batteries by a chloride shuttle in a biphasic electrolyte. Nat. Commun. 14, 2349 (2023).
Xi, D. et al. Mild pH-decoupling aqueous flow battery with practical pH recovery. Nat. Energy 9, 479–490 (2024).
Carrington, M. E. et al. Associative pyridinium electrolytes for air-tolerant redox flow batteries. Nature 623, 949–955 (2023).
Ma, W. et al. A twelve-electron conversion iodine cathode enabled by interhalogen chemistry in aqueous solution. Nat. Commun. 14, 5508 (2023).
Xie, C. et al. Reversible multielectron transfer I−/IO3− cathode enabled by a hetero-halogen electrolyte for high-energy-density aqueous batteries. Nat. Energy 9, 714–721 (2024).
Bi, S., Zhang, Y., Wang, H., Tian, J. & Niu, Z. High-energy aqueous/organic hybrid batteries enabled by Cu2+ redox charge carriers. Angew. Chem. Int. Ed. 62, e202312172 (2023).
Wu, X. et al. A four-electron sulfur electrode hosting a Cu2+/Cu+ redox charge carrier. Angew. Chem. Int. Ed. 58, 12640–12645 (2019).
Dai, C. et al. Enabling fast-charging selenium-based aqueous batteries via conversion reaction with copper ions. Nat. Commun. 13, 1863 (2022).
Wang, Y., Chao, D., Wang, Z., Ni, J. & Li, L. An energetic CuS–Cu battery system based on CuS nanosheet arrays. ACS Nano 15, 5420–5427 (2021).
Bi, S. et al. Six-electron-redox iodine electrodes for high-energy aqueous batteries. Angew. Chem. Int. Ed. 62, e202312982 (2023).
Zhang, J. et al. Dual synergistic effects assisting Cu-SeS2 electrochemistry for energy storage. Proc. Natl Acad. Sci. USA 120, e2220792120 (2023).
Wang, H. et al. A shuttle-free solid-state Cu−Li battery based on a sandwich-structured electrolyte. Angew. Chem. Int. Ed. 62, e202214117 (2023).
Sun, Y. et al. Facile renewable synthesis of nitrogen/oxygen co-doped graphene-like carbon nanocages as general lithium-ion and potassium-ion batteries anode. Carbon 167, 685–695 (2020).
Wiegers, G. A. Physical properties of first-row transition metal dichalcogenides and their intercalates. Phys. B+C. 99, 151–165 (1980).
Ou, X. et al. Exfoliated V5S8/graphite nanosheet with excellent electrochemical performance for enhanced lithium storage. Chem. Eng. J. 320, 485–493 (2017).
Mohan, P., Yang, J., Jena, A. & Suk Shin, H. VS2/rGO hybrid nanosheets prepared by annealing of VS4/rGO. J. Solid State Chem. 224, 82–87 (2015).
Kumar, G. M. et al. Ultrathin VS2 nanodiscs for highly stable electro catalytic hydrogen evolution reaction. Int. J. Energy Res. 44, 811–820 (2020).
Zhao, R. et al. Lanthanum nitrate as aqueous electrolyte additive for favourable zinc metal electrodeposition. Nat. Commun. 13, 3252 (2022).
Naveed, A. et al. A highly reversible Zn anode with intrinsically safe organic electrolyte for long-cycle-life batteries. Adv. Mater. 31, 1900668 (2019).
Hu, E., Li, H., Zhang, Y., Wang, X.,Liu, Z. Recent progresses on vanadium sulfide cathodes for aqueous zinc-ion batteries. Energies 16, 917 (2023).
Mao, Y. et al. Magneto-electrochemistry driven ultralong-life Zn-VS2 aqueous zinc-ion batteries. Mater. Horiz. 10, 3162–3173 (2023).
Cai, L. et al. Highly crystalline layered VS2 nanosheets for all-solid-state lithium batteries with enhanced electrochemical performances. ACS Appl. Mater. Interfaces 10, 10053–10063 (2018).
Liang, X. et al. Unveiling the solid-solution charge storage mechanism in 1T vanadium disulfide nanoarray cathodes. J. Mater. Chem. A 8, 9068–9076 (2020).
Zhang, X. et al. Insights into the storage mechanism of layered VS2 cathode in alkali metal-ion batteries. Adv. Energy Mater. 10, 1904118 (2020).
Li, L. et al. Vanadium disulfide flakes with nanolayered titanium disulfide coating as cathode materials in lithium-ion batteries. Nat. Commun. 10, 1764 (2019).
Fang, W. et al. Facile hydrothermal synthesis of VS2/graphene nanocomposites with superior high-rate capability as lithium-ion battery cathodes. ACS Appl. Mater. Interfaces 7, 13044–13052 (2015).
Li, W. et al. VS2 nanoarchitectures assembled by single-crystal nanosheets for enhanced sodium storage properties. Electrochim. Acta 286, 131–138 (2018).
Miao, Y. et al. Interlayer engineering of VS2 nanosheets via in situ aniline intercalative polymerization toward long-cycling magnesium-ion batteries. ACS Appl. Mater. Interfaces 15, 57079–57087 (2023).
Jing, P., Lu, H., Yang, W. & Cao, Y. Interlayer-expanded and binder-free VS2 nanosheets assemblies for enhanced Mg2+ and Li+/Mg2+ hybrid ion storage. Electrochim. Acta 330, 135263 (2020).
Zhao, Y. et al. Superior Mg2+ storage properties of VS2 nanosheets by using an APC-PP14Cl/THF electrolyte. Energy Storage Mater. 23, 749–756 (2019).
Xue, X. et al. One-step synthesis of 2-ethylhexylamine pillared vanadium disulfide nanoflowers with ultralarge interlayer spacing for high-performance magnesium storage. Adv. Energy Mater. 9, 1900145 (2019).
Sun, R. et al. High-rate and long-life VS2 cathodes for hybrid magnesium-based battery. Energy Storage Mater. 12, 61–68 (2018).
Mao, Y. et al. Two birds with one stone: V4C3 MXene synergistically promoted VS2 cathode and zinc anode for high-performance aqueous zinc-ion batteries. Small 20, 2306615 (2024).
Mao, Y. et al. Carbon foam-supported VS2 cathode for high-performance flexible self-healing quasi-solid-state zinc-ion batteries. Small 19, 2207998 (2023).
Sun, H. et al. Rose-like VS2 nanosheets chemically anchored on carbon nanotubes for flexible zinc-ion batteries with enhanced properties. ACS Appl. Mater. Interfaces 14, 40247–40256 (2022).
Jiao, T. et al. Binder-free hierarchical VS2 electrodes for high-performance aqueous Zn ion batteries towards commercial level mass loading. J. Mater. Chem. A 7, 16330–16338 (2019).
He, P. et al. Layered VS2 nanosheet-based aqueous Zn ion battery cathode. Adv. Energy Mater. 7, 1601920 (2017).
Liu, J., Peng, W., Li, Y., Zhang, F. & Fan, X. A VS2@N-doped carbon hybrid with strong interfacial interaction for high-performance rechargeable aqueous Zn-ion batteries. J. Mater. Chem. C. 9, 6308–6315 (2021).
Sun, X. et al. In situ unravelling structural modulation across the charge-density-wave transition in vanadium disulfide. Phys. Chem. Chem. Phys. 17, 13333–13339 (2015).
Pascal, T. A. et al. X-ray absorption spectra of dissolved polysulfides in lithium–sulfur batteries from first-principles. J. Phys. Chem. Lett. 5, 1547–1551 (2014).
Cao, D. et al. Understanding electrochemical reaction mechanisms of sulfur in all-solid-state batteries through operando and theoretical studies. Angew. Chem. Int. Ed. 62, e202302363 (2023).
Lin, C.-H. et al. Operando structural and chemical evolutions of TiS2 in Na-ion batteries. J. Mater. Chem. A 8, 12339–12350 (2020).
Le Nagard, N., Collin, G. & Gorochov, O. Structure cristalline et proprietes physiques de Cu0.75VS2. Mater. Res. Bull. 12, 975–982 (1977).
Li, W. & Wang, D. Conversion-type cathode materials for aqueous Zn metal batteries in nonalkaline aqueous electrolytes: progress, challenges, and solutions. Adv. Mater. n/a, 2304983 (2023).
Chen, Y. et al. Hollow CuS nanoboxes as Li-free cathode for high-rate and long-life lithium metal batteries. Adv. Energy Mater. 10, 1903401 (2020).
Sandstrom, S. K. et al. Reversible electrochemical conversion from selenium to cuprous selenide. Chem. Commun. 57, 10703–10706 (2021).
Acknowledgements
Y.S., R.Q., and Q.L. contributed equally to this work. D.Z. acknowledges supports from National Key Research and Development Programme of China (No. 2022YFA1605400, D.Z.), National Natural Science Foundation of China (No. 12275342, D.Z., 12005286, D.Z., U2032204, D.Z.), Youth Innovation Promotion Association of the Chinese Academy of Sciences (2022293, D.Z.), and Photon Science Research Center for Carbon Dioxide. Y.S. and Q.L. acknowledge supports from Shanghai Sailing Program (No. 23YF1453500, Y.S., 23YF1453800, Q.L.). Y.S. acknowledge supports from China Postdoctoral Science Foundation (No. 2023M733617, Y.S.). The authors thank the staff from Shanghai Synchrotron Radiation Facility (SSRF) at BL02U2, BL17B1, BL16U1, BL13SSID, and User Experiment Assist System.
Author information
Authors and Affiliations
Contributions
D.Z. conceived the project. Y.S. and D.Z. developed the concept. Y.S. and H.S. conducted the experiments and data analysis. R.Q. performed the calculations. Q.L. and J.Z. analyzed the absorption spectrum data. W.Z., H.L., and M.L. performed the cycling tests. W.W. and Y.G. contributed to the calculations. Y.S., and D.Z. wrote the manuscript. X.L., C.Z., and D.Z. supervised the project. All authors discussed and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jiang Zhou, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sun, Y., Qi, R., Lei, Q. et al. Reversible multivalent carrier redox exceeding intercalation capacity boundary. Nat Commun 16, 343 (2025). https://doi.org/10.1038/s41467-024-55386-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55386-5
