
Abstract
Compared to the sluggish kinetics observed in methanol-mediated side-chain alkylation of methyl groups with sp3 C–H bonds, CO2 hydrogenation emerges as a sustainable alternative strategy, yet it remains a challenge. Here, as far as we know, it is first reported that using CO2 hydrogenation replacing methanol can conduct the side-chain alkylation of 4-methylpyridine (MEPY) over a binary metal oxide-zeolite Zn40Zr60O/CsX tandem catalyst (ZZO/CsX). This ZZO/CsX catalyst can achieve 19.6% MEPY single-pass conversion and 82% 4-ethylpyridine (ETPY) selectivity by using CO2 hydrogenation, which is 6.5 times more active than methanol as an alkylation agent. The excellent catalytic performance is realized on the basis of the dual functions of the tandem catalyst: hydrogenation of CO2 on the ZZO and activation of sp3 C–H bond and C–C bond coupling on the CsX zeolite. The thermodynamic and kinetic coupling between the tandem reactions enables the highly efficient CO2 hydrogenation and C–C bond coupling. In-situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and density functional theory (DFT) calculations suggest that the CHxO* (CH2O*) species, rather than methanol produced from CO2 hydrogenation, is the key intermediate to achieve the C–C bond coupling.
Similar content being viewed by others

Introduction
CO2 has become a strategic carbon resource for the fabrication of high-value-added chemicals, not just as a greenhouse gas1,2. The proposal on carbon capture and utilization (CCU) has received increasing attention worldwide3,4,5,6,7. Utilization of CO2 as a feedstock to synthesize various valuable products, such as methanol8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31, higher acohols32,33,34,35,36, lower olefins37,38,39,40,41,42,43, and aromatics44,45,46,47,48, is emerging as a complementary alternative to fossil-derived chemical production. However, CO2 is located at the bottom of the energetics ladder in thermodynamics, which requires considerable Gibbs energy (CO2 ∆fGo = -393.5 kJ/mol) input to convert it. The employment of H2 derived from renewable energy can make the conversion of CO2 thermodynamically possible and practically significant49,50,51,52,53,54,55,56,57,58,59,60.
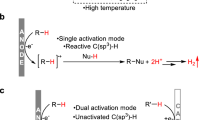
Methanol, a critical platform chemical synthesized by CO2 hydrogenation, can be used as an alkylation agent for methylation of toluene to paraxylene61,62,63 and side-chain alkylation of 4-methylpyridine (MEPY) to 4-ethylpyridine (ETPY). However, compared with the methylation of toluene, the side-alkylation of MEPY is more difficult because of the inert sp3 C–H bond of MEPY. More importantly, side-chain alkylation of MEPY using methanol requires the first dehydrogenation of methanol to formaldehyde, which is thermodynamically and kinetically unfavorable64,65,66,67. Although formaldehyde was also considered as an alkylation agent68, its toxicity limited its application. ETPY and its derivatives are important intermediates of agricultural chemicals and medicines, such as antihistamine69,70. The current synthesis procedure involves the reaction of pyridine and acetic anhydride with zinc, producing an amount of waste and thus is not environment-friendly71. Therefore, it is crucial to develop new strategies to replace methanol and formaldehyde to achieve efficient side-chain alkylation.
It has been reported that toluene can be methylated by CO2 and H2 over dual-functional catalyst ZnZrO/ZSM-572. The results demonstrate that the reactive methylation species (H3CO*) are generated more easily by CO2 hydrogenation than by methanol dehydrogenation. Our group also reported that the hydrogenation of CO2 can synthesize the lower olefins and aromatics over tandem catalysts, with CHxO species as a key intermediate governing the thermodynamics and kinetics of C–C bond coupling39,45. Considering the key to overcome the disadvantages of using methanol or formaldehyde as agents, CHxO species formed by CO2 hydrogenation should be adopted to carry out the side-chain alkylation of MEPY.
This work highlights the potential for utilization of the greenhouse gas CO2 as alternative C1 resource. The critical intermediates through hydrogenation of CO2 enables the side-chain alkylation of MEPY to ETPY over tandem catalysts. The optimized ZZO/CsX catalyst shows the MEPY conversion of 19.6% and ETPY selectivity of 82% through CO2 hydrogenation, which is far higher than by using methanol as an alkylation agent (Conversion of 3.0% and ETPY selectivity of 61% by using CsX catalyst). DFT calculation further confirms that the formation of CHxO* (CH2O*) intermediate species through CO2 hydrogenation boosts the side-chain alkylation of MEPY.
Results and discussion
A series of χ%ZnO-ZrO2 binary oxides (χ% represent molar percentage of Zn, metal base; and ZnO-ZrO2 denoted as the ZnZrO) were prepared by the sol-gel method73, the CsX zeolite was prepared by ion-exchange of X zeolite with CsOH74 (the detailed preparation was shown in Methods). Tandem catalysts were obtained through physical mixing of ZnZrO and CsX39,45. In a typical tandem catalysis process, both CO2 hydrogenation and 4-methylpyridine (MEPY) methylation reaction were carried out in a fixed-bed reactor. The catalytic performances of series tandem catalysts were evaluated, as shown in Fig. 1. The performance of ZnZrO/CsX tandem catalysts increases firstly and then decreases with increasing the Zn/(Zn+Zr) molar ratio (Fig. 1a). The catalytic activity achieves the optimized results with the MEPY conversion of 12.8% and ETPY selectivity of 86% when the Zn/(Zn+Zr) molar ratio is close to 40%. Hereafter, the Zn/(Zn+Zr) molar ratio of 40% represents the optimized catalyst (Zn40Zr60O/CsX denoted as the ZZO/CsX) for ZnZrO/CsX tandem catalysts. With increasing the reaction temperature, the conversion of MEPY varies greatly and the selectivity of ETPY changes slightly. The highest conversion of MEPY is reached at 380 °C (Fig. 1b). The varying of the mass ratio of ZZO and CsX also affects the catalytic performance of the ZZO/CsX tandem catalysts slightly, and the optimized mass ratio of ZZO/CsX is 1/6, while the mass content of CsX is far higher than ZZO (Fig. 1c), and the gas hourly space velocity (GHSV) and pressure were also evaluated (Supplementary Fig. 1a, Supplementary Fig. 1b, Supplementary Information). Figure 1d (left panel) shows a representative result that the side-chain alkylation of MEPY gives 82% selectivity for ETPY at the MEPY single-pass conversion of 19.6% under reaction conditions of 2 MPa, 0.6 h−1, and 380 °C. This reaction over the ZZO catalyst only produces trace ETPY and side-product DMPY, while no ETPY is detected for CsX alone, indicating the tandem catalyst can efficiently convert MEPY with CO2 hydrogenation to ETPY. Figure 1d (right panel) shows that the methanol and CO are produced through CO2 hydrogenation on ZZO catalyst, while only large amounts of CO and trace methane are detected over CsX. These results lead us to conclude that tandem reactions take place on the ZZO/CsX tandem catalyst: ZZO generates methanol from CO2 hydrogenation, and CsX is responsible for the MEPY conversion and C–C bond coupling. Though this tandem catalyst shows the deactivation behavior due to coke formation67 (Supplementary Fig. 2a, Supplementary Fig. 3), this coke formation may arise from the conversion of intermediates (CH₂O* species) during the CO₂ hydrogenation process on the zeolite, as well as from the coupling of the pyridine ring through intermediates (CH2O*) in CO₂ hydrogenation, ultimately leading to deactivation. The catalytic activity can be regenerated through the thermal treatment of catalysts with air (Supplementary Fig. 2b).

a Catalytic activity on χ%ZnZrO/CsX with different Zn/(Zn+Zr) molar ratios at 350 °C; m (ZnZrO): m (CsX) = 1: 9. b The effect of temperature on the catalytical performance; m (ZZO): m (CsX) = 1: 6. c Catalytic activity of ZZO/CsX with different m (ZZO): m (CsX) mass ratios at 350 °C. d Side-alkylation of MEPY with CO2 hydrogenation over ZZO/CsX (m (ZZO): m (CsX) = 1: 6), ZZO, CsX. CO2 hydrogenation at 380 °C on ZZO and CsX, respectively, GHSV = 16,000 mL g−1 h−1; Standard reaction conditions: VHSV = 0.6 mL g−1 h−1, P = 2.0 MPa, H2/CO2/Ar = 72/24/4 and GHSV = 16000 mL g−1 h−1. The color shadings represent the selectivity, and the red square represents the conversion.
Powder X-ray diffraction (XRD) patterns (Fig. 1a) of χ%ZnZrO (with ZnO molar content of 5%) show the same phase structure of tetragonal ZrO2 (t-ZrO2)8,75 and the XRD from the (101) spacing of ZrO2 slightly shifts to the higher angle in comparison with t-ZrO2 (Supplementary Fig. 4a), indicating that it is in solid solution structure8,76. Raman spectra8,75,77 also confirm the presence of tetragonal phase ZrO2 (Supplementary Fig. 4b). With increasing the content of ZnO (>5%) in χ%ZnZrO, a new phase that assigned to ZnO was observed and gradually dominated (Fig. 1a, Supplementary Fig. 4a, Supplementary Fig. 4b), showing the co-existence of ZnO and ZnZrO solid solution phase, and the X-ray photoelectron spectroscopy (XPS) results of ZnZrO indicate the presence of oxidation state of Zn2+ and Zr4+ species (Supplementary Fig. 5). The ion-exchanged NaX after CsOH treatment (CsX) shows the same FAU framework structure of pristine NaX with high crystallinity74 (Fig. 2b). In addition, a new peak centered at 25.6° was observed, which can be attributed to the Cs2O phase (PDF number 09-0104), the XRD of mixed ZZO/CsX sample showed the almost similar signal to the CsX, and the three new peaks were mainly attributed to the ZZO (Supplementary Fig. 6). The XPS results of CsX show the two different Cs species on CsX74,78,79,80, the peak centered at 724.34 eV can be attributed to the cesium ions (Cs+) that balances the negative charge of the zeolite framework through replacing the Na+, while the peak centered at 725.15 eV can be assigned to Cs2O that deposited on CsX (Fig. 2c, Supplementary Fig. 7). X-ray absorption near-edge structure (XANES) measurement was further performed to characterize the oxidation state of Cs81,82. As shown in Fig. 2d, the Cs K-edge for both CsX-L (CsX-L was exchanged with low concentration 0.04 mol/L CsOH) and CsX (CsX was exchanged with concentration 0.4 mol/L CsOH) catalysts are located at the low-energy positions as compared with that of the Cs2CO3 standard sample, indicating the lower oxidation states of the CsX catalyst, which is consistent with the XPS result. The particle size of ZZO is in the range of 10–30 nm (Fig. 2e), and HRSTEM shows (101) facet is the dominant surface exposed in t-ZrO2, the HAADF-STEM displays that the Zn and Zr elements are relatively uniformly distributed in ZZO particles (Fig. 2i, j) with the presence of trace ZnO particles. The prepared CsX shows the nanosphere morphology ranging from 2 to 5 μm (Fig. 2k, l). The two components of tandem catalysts fabricated by physical mixing of ZZO and CsX keep their individual structures. Due to the smaller nanoparticles of ZZO compared to CsX, the ZZO particles are highly dispersed on the outer surface of CsX in ZZO/CsX tandem catalysts (Fig. 2m, n).

a XRD patterns of individual χ%ZnZrO; PDF number: t-ZrO2, 49-1642; ZnO, 36–1451. b XRD patterns of CsX and NaX, respectively. c XPS spectra of Cs species for individual CsX zeolite. d The Cs K-edge for individual CsX-L and CsX catalysts. e The TEM (f, g) high-resolution STEM of individual ZZO catalyst and (h–j) aberration-corrected scanning TEM–high-angle annular dark-field images and element distribution of individual ZZO catalyst; Scanning electron microscopy images of individual CsX (k, l). m, n Scanning electron microscopy images of mixed ZZO/CsX (1:6).
To investigate the function of CsX in ZZO/CsX tandem catalysts, the NaX exchanged with different metal cations were employed to construct tandem catalysts. The ZZO/CsX tandem catalysts give the maximum conversion of MEPY and STY of ETPY, indicating that CsX shows the highest activity for the activation of sp3 C–H bond and C–C bond coupling (Fig. 3a). This performance was associated with its strong basic properties among these modified X zeolites. To understand the critical effect of Cs species of CsX for the C–C bond coupling, the Cs2O-SiO2 catalyst was prepared by depositing Cs2O on the surface of SiO2. Unfortunately, the ZZO/Cs2O-SiO2 tandem catalysts show no activity for side-chain alkylation of MEPY through CO2 hydrogenation (Fig. 3b). On the contrary, the CsX-L catalyst with a lower content of Cs was controllably prepared, and the Cs species are mainly Cs+. Compared with ZZO/Cs2O-SiO2, ZZO/CsX-L tandem catalysts show an MEPY conversion of 16%, which is slightly lower than the ZZO/CsX (Fig. 3b). These results suggest that the Cs+ balanced the negative charge of zeolite framework, rather than the Cs2O species deposited on CsX, is responsible for MEPY activation and C–C bond coupling. The base and acid properties of ZnZrO and CsX were conducted by using CO2-TPD and NH3-TPD (Supplementary Fig. 8, Supplementary Fig. 9), the Fourier transform infrared (FTIR) spectroscopy of adsorbed pyridine75 was further performed to distinguish the strength of acid sites for NaX and CsX. Figure 3c, d shows the bands at 1592 and 1442 cm−1 for NaX (Fig. 3c) and at 1579 and 1438 cm−1 for CsX (Fig. 3d), indicating the existence of the Lewis acid sites on both NaX and CsX. With increasing the desorption temperature of pyridine from 50 to 100 and 200 °C, the intensity of peaks for all acid sites decreases. Further increasing the desorption temperature from 200 to 300 and 400 °C, obvious signals of pyridine adsorbed on NaX can still be observed, however, pyridine has completely desorbed on the CsX, indicating the weaker Lewis acidity of CsX than NaX. It is known that these peaks of Lewis acid sites can be assigned to the adsorption of pyridine on the Na+ and Cs+ sites in NaX and CsX through the N atom of pyridine. Thus, it is deduced that, compared with NaX, the weaker adsorption of the pyridine moiety of MEPY on CsX facilitates the sp3 C–H bond activation and C–C bond formation over Cs+ sites under the reaction temperature of 380 °C.

a Side-alkylation of MEPY on tandem catalysts with different cation-exchanged X zeolite, m (ZZO): m (LiX, NaX, KX, CsX, MgX or CaX) = 1: 9. b Side-alkylation of MEPY on ZZO/CsX (1: 6), ZZO/Cs2O-SiO2 (1: 6), ZZO/CsX-L (1: 6), T = 380 °C. Standard reaction conditions: VHSV = 0.6 mL g−1 h−1, P = 2.0 MPa, H2/CO2/Ar = 72/24/4 and GHSV = 16,000 mL g−1 h−1. c, d FT-IR spectra of adsorbed pyridine at different temperatures for individual X and CsX zeolites; Lewis Acid (LA); The color shadings represent the selectivity, and the red square represents the conversion.
In order to understand the working mechanism of the tandem catalyst39,45, the side-chain alkylation of MEPY with CO2 hydrogenation on ZZO/CsX and with methanol on CsX was compared at the same reaction temperature of 380 °C (Fig. 4a). Surprisingly, we found that the conversion of MEPY with CO2 hydrogenation on ZZO/CsX is 6.5 times of the side-chain alkylation with methanol on CsX, indicating that the side-chain alkylation of MEPY with CO2 hydrogenation does not undergo the same reaction pathway with methanol as an intermediate. To further clarify the tandem catalysis, the catalytic activity of the tandem catalyst was compared with the different proximity of the two components in a tubular fixed-bed reactor (Fig. 4b). When ZZO and CsX were mixed in a granule (200−450 μm), the conversion of MEPY decreases sharply from 19.6 to 12% (Fig. 4b, I and II). The conversion of MEPY decreases continuously when the two components of ZZO and CsX are placed as dual bed layers (Fig. 4b, III). When ZZO and CsX particulates are separated in space with a quartz sand layer (Fig. 4b, IV), the conversion of MEPY severely deteriorates. The conversion of MEPY drops dramatically with decreasing the contact area of the two components, suggesting that the excellent conversion of MEPY for tandem catalyst is due to the effective synergy between ZZO and CsX, which further implies that the gaseous methanol is not the main intermediate for side-chain alkylation of MEPY.

a Side-alkylation of MEPY with CO2 hydrogenation on ZZO/CsX and with methanol on CsX (molar ratio of methanol to MEPY is 1:1, VHSV was calculated as 0.6 mL g−1 h−1 based on MEPY). b Side-alkylation of MEPY with CO2 hydrogenation over ZZO/CsX, integrated catalyst components ZZO and CsX with different proximity. Standard reaction conditions: VHSV = 0.6 mL g−1 h−1, P = 2.0 MPa, T = 380 °C, H2/CO2/Ar = 72/24/4 and GHSV = 16000 mL g−1 h−1, m (ZZO): m (CsX) = 1: 6; (ZZO/CsX denoted as the ZZO/CsX). The color shadings represent the selectivity, and the red square represents the conversion; (I): ZZO and CsX were mixed in powder; (II): ZZO and CsX were mixed in a granule; (III): ZZO and CsX are placed as dual bed layers; (IV): ZZO and CsX particulates are separated in space with a quartz sand layer.
To understand the coupling mechanism in reaction kinetics, the surface species formed on ZZO were detected by an in-situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS)8,83 (Fig. 5). The HCOO* and CH3O* species are distinctly observed for ZZO (Fig. 5a, b). The IR peaks at 1599 and 1368 cm−1 are assigned to the asymmetric and symmetric OCO stretching vibrations of adsorbed bidentate HCOO* species, respectively. The peaks at 2884 and 1387 cm−1 are assigned to the stretching vibration ν(CH) and bending vibration δ(CH), respectively. The peaks at 2938 and 2828 cm−1 are attributed to CH3O* species. The peaks at 2884 and 2828 cm−1 were used to determining the tendency of changes of HCOO* and H3CO* species, meanwhile, the products were also detected by mass spectrometry (MS)8,83. As seen in Fig. 5c, when CO2 + H2 was switched to CO2 + D2, the HCOO* and CH3O* species decrease, while the DCOO* and CD3O* species increase, and the D-substituted products were also detected by MS. When CO2 + H2 was switched to D2, the HCOO* and H3CO* species gradually decrease (Fig. 5d). Correspondingly, two new peaks at 2165 and 2052 cm−1 for DCOO* and CD3O* species appear, and then decrease slowly (Fig. 5e, f). Also, the D-substituted products show the same tendency of DCOO* and CD3O* species (Fig. 5f). According to these evidences, the CO2 hydrogenation mainly undergoes the formate reaction pathway, and the CHxO* (CH2O*) species is the key intermediate for the hydrogenation of HCOO* to CH3O*, the synergetic effect between Zn and Zr sites play a key role for generating the key intermediates CHxO8,39,45,83.

a, b In-situ DRIFT spectra of surface species formed from the CO2 + H2 reaction on individual ZZO catalyst. (c) DRIFT-MS of CO2 + H2 and subsequently switched to CO2 + D2 reactions on individual ZZO catalyst. d, e In situ DRIFT spectra of surface species from CO2 + H2 and subsequently switched to D2 on individual ZZO catalyst. f DRIFT-MS of CO2 + H2 and subsequently switched to D2 on individual ZZO catalyst. Reaction conditions: ZZO catalyst, 300 °C, 20 ml/min Ar + 10 ml/min CO2 + 30 ml/min H2 (D2).
According to the above results, it is suggested that the CHxO* (CH2O*) as key intermediates in CO2 hydrogenation conduct the coupling of kinetics between the two components of ZZO and CsX of tandem catalysts. To further confirm this speculation, Density functional theory (DFT) calculations were performed to understand the reaction mechanism84,85,86,87,88 (details in the Supplementary Information). The two major intermediates, i.e., CH2O* and methanol, which can transfer from ZZO into the micropore of CsX, were evaluated. Using methanol as the intermediate involves the dehydrogenation of methanol to formaldehyde species (CH2O*) in CsX. With the adsorption of the oxygen atom of methanol on Cs+ of CsX, the C–H and O–H bonds were activated, which resulted in the dehydrogenation of CH3OH* and the formation of CH2O* and two H* (Fig. 6 a2). It is found that the dehydrogenation of CH3OH* to CH2O* and H* needs to overcome an extremely high activation barrier of 437 kJ/mol (Fig. 6b). Although coupling of H* species to molecular H2 can stabilize the system (Fig. 6 a3), the formation of CH2O* and H2 from CH3OH is still an endothermic reaction process on Cs+. These results strongly suggest that the dehydrogenation of methanol to CH2O* is thermodynamically and kinetically unfavorable over the CsX catalytic component.

a Optimized geometries of all reaction intermediates and transition states. Reaction Gibbs free energy diagram (T = 380 °C, P = 2.0 Mpa) of (b): methanol dehydrogenation process over CsX zeolite. c CH2O reacting with MEPY on CsX zeolite.
In principle, according to our previous works39,45 and above experimental results, it is proposed that the CH2O* can be as the intermediate to participate the C–C bond coupling over zeolite. Compared with methanol, the CH2O* as the intermediate can avoid the thermodynamically unfavorable dehydrogenation of methanol. As shown in Fig. 6, firstly, the CH2O* adsorbs on Cs+ of CsX channel by forming the O-Cs bond, and MEPY* interacts with CsX through binding of N with Cs+, as well as the H (CH3) activated by the O (Si–O–Al) of CsX zeolite (Fig. 6c, b1). With the attacking of the C of -CH3 moiety to the C site of CH2O*, the H (CH3) also binds with the O (CH2O*) and forms a four-numbered ring (H–C–C–O), which is the transition state for the C–C bond coupling (Fig. 6c, bTS1). The formation of HOCH2-CH2py* needs to overcome an activation barrier of 270 kJ/mol with the cleavage of C–H bond (CH3) and the formation of O (CH2O*)-H (CH3) bond (Fig. 6c, b2), which is energetically the most difficult reaction step. The dehydration of HOCH2-CH2py* occurs smoothly with protonating the OH group (Fig. 6c, bTS2), and the VYPY* is formed with the cleavage of the C-O bond (H2O* formation) (Fig. 6c, b3). The ETPY* was produced with the desorption of H2O* (Fig. 6c, b4) and the hydrogenation of VYPY* (Fig. 6c, b5). Finally, the product ETPY* is formed through hydrogenation (Fig. 6c, b6), and the formaldehyde route experiments also verified this mechanism (Supplementary Fig. 10). In addition, the pathways for by-product formation are also proposed and shown in Supplementary Fig. 11.
DFT results suggest that the synthesis of ETPY through CH2O* intermediate only needs to overcome the highest activation energy barrier of 270 kJ/mol, which is far lower than that of methanol dehydrogenation to CH2O (437 kJ/mol) (Fig. 6b). Therefore, it is concluded that the key intermediate for realizing the kinetics coupling of ZZO/CsX tandem catalysts is CH2O* but not methanol. Based on the surface reaction kinetics and DFT calculations, it is proposed that the tandem reaction proceeds as follows in Fig. 7: Firstly, the generation of key intermediates CHxO species via CO2 hydrogenation mainly takes place on the ZnZrO catalyst, and then migrate/transfer on to the CsX catalyst; Secondly, the CsX catalyst activates of sp3 C–H bond of the 4-methylpyridine (MEPY) to form the carbanion; Finally, the coupling of C–C bond occurs between the migrated/transferred CHxO species and the carbanion on the CsX catalyst to produce the 4-ethylpyridine (ETPY).

The generation of key intermediates CHxO species via CO2 hydrogenation take place on ZnZrO catalyst; The CsX catalyst activates of sp3 C–H bond of the 4-methylpyridine (MEPY) form the carbanion; The coupling of C–C bond occurs between the migrated/transferred CHxO species and the carbanion on CsX catalyst to produce the 4-ethylpyridine (ETPY).
In summary, the direct synthesis of ETPY through CO2 hydrogenation and sp3 C–H bond activation of MEPY was realized by a tandem catalyst, Zn40Zr60O/CsX. CO2 and H2 were activated on Zn40Zr60O and the activation of the sp3 C–H bond and coupling of C–C bond were conducted on CsX. The selectivity of ETPY can reach 82 % with the MEPY conversion of 19.6%. Compared with the direct methanol route, the synthesis of ETPY through CO2 hydrogenation shows a 6.5-fold enhancement of MEPY conversion. The migrating of the key CHxO* (CH2O*) intermediate modulate the coupling of thermodynamics and kinetics of tandem catalysis, significantly facilitating the activation of sp3 C–H bond and coupling of C–C bond. This work highlights the potential of key intermediate CHxO* (CH2O*) generated in situ through CO2 hydrogenation. It enables the C–C bond coupling by activating the sp3 C–H bond, opening a new avenue for synthesizing high-value-added chemicals through CO2 hydrogenation.
Methods
Chemicals
Zn(NO3)2·6H2O, Ca(NO3)2·4H2O, Mg(NO3)2·4H2O, were purchased Sinopharm (China), Zr(NO3)4·5H2O, citric acid, 4-methylpyridine, diphenyl, cesium hydroxide, LiOH, KOH, were obtained from Macklin Biochemical, PVP (polyvinyl pyrrolidone) K30, average molecular weight 5800 were obtained from Sigma-Aldrich, CO2/H2/Ar [24/72/4(v/v/v)] were obtained from Lanzhou Yulong Gas Co., Ltd.
Preparation of catalysts
A series of χ%ZnO-ZrO2 catalysts (χ% represent molar percentage of Zn, metal base) were synthesized by using the sol-gel method73. Typically, 11.9 g Zn(NO3)2·6H2O, 25.8 g Zr(NO3)4·5H2O, 38.4 g citric acid and 10.0 g PVP were dissolved in 100 mL deionized water at 50 °C (Zn2++Zr4+=1 mol/L) under the vigorously stir. After Zn(NO3)2·6H2O, Zr(NO3)4·5H2O, citric acid and PVP completely dissolved, the mixture was mildly evaporated at 90 °C until a viscous gel was obtained. Then, the sample was dried at 200 °C for 5 h, grind, and calcined at 600 °C at a rate of 5 °C/min for 10 h.
CsX catalysts were prepared by the ion exchange of NaX (from Nankai Catalysts;13X) with two times. The detailed preparation procedures are as follows: 30 g NaX was treated in a 250 mL eggplant shaped flask at 80 °C for 2.5 h with a 0.4 mol/L solution of cesium hydroxide (liquid/solid ratio: 5 mL/g). The slurries were centrifuged and washed with deionized for three times. Then, the samples obtained dried at 80 °C overnight and calcined in air at 540 °C for 3 h. The second ion exchange procedure is same as the above-mentioned process. LiX obtained with a 0.4 mol/L solution of LiOH; KX obtained with a 0.4 mol/L solution of KOH; CaX obtained with a 0.4 mol/L solution of Ca(NO3)2·4H2O; MgX obtained with a 0.4 mol/L solution of Mg(NO3)2·4H2O, the other conditions are same as the procedure of CsX; CsX-L was exchanged only one times with 0.04 mol/L CsOH, and the other conditions are same as the procedure of CsX.
Finally, the ZnZrO catalyst and the CsX catalyst were ground in an agate mortar for about 0.5 h to fully mix to obtain the physical mixed ZnZrO/CsX catalyst.
Catalyst Evaluation
Catalytic performance was evaluated on a high-pressure fixed-bed reactor equipped with a quartz sample tube (The diameter:10 mm). Typically, 1.0 g catalyst (40-80 mesh) were loaded into the quartz sample tube without mixed quartz sand. Before evaluating the catalytical performances, raised to the specified temperature under the protection of argon. For the investigation of 4-methylpyridine side chain alkylation with CO2 and H2, a mixture gas of CO2/H2/Ar [24/72/4(v/v/v)] was induced into a quartz sample tube, and then 4-methylpyridine was carried into reaction tube (393 K) at a constant flow rate by using a piston pump. Typically, the reaction was conducted at 653 K and 2 MPa, with a GHSV of 16000 mL·g-1·hour-1 and 4-methylpyridine volume hourly space velocity (VHSV) of 0.6 mL g-1·hour-1. Before collecting the samples, the reaction is firstly running for 2.5 hours to reach a steady state, and then collected the sample for the next 8 h. The sample was condensed at −20 °C by using a cooling condenser. In the qualitative and quantitative analysis, we use diphenyl as an internal standard to determine the N balance, and the N balance was above 96%. The liquid products were analyzed by using the gas chromatography, the conversion of 4-methylpyridine was calculated as follows:
where f1–f7 refer to the 4-methylpyridine, 1,4-dimethylpiperidine, 4-ethylpyridine, 4-isopropylpyridine, 4-vinylpyridine, 4-(tert-butyl) pyridine and 2,4-dimethylpyridine molar correction factor respectively. Ai refers to the peak area in FID of product-i.
The selectivity calculation method is as follows:
where f1–f6 refer to the, 4-dimethylpiperidine, 4-ethylpyridine, 4-isopropylpyridine, 4-vinylpyridine, 4-(tert-butyl) pyridine and 2,4-dimethylpyridine molar correction factor respectively. Ai refers to the peak area in FID of product-i.
The space-time yield (STY) defined as quantity of product per unit mass per unit time (mg.g−1·h−1), the STY of 4-ethylpyridine (ETPY) was denoted as STY(ETPY), and the highest space-time yield (STY) of ETPY is 106 mg.g−1·h−1 for ZZO/CsX(1:6) tandem catalyst at 380 °C in Fig. 1b.
The catalysts (ZnZrO, CsX or ZnZrO/CsX) used in the catalyst evaluation were provided in Figs. 1–5 in the main text and Supplementary Information.
Powder x-ray diffraction (XRD)
The XRD results were collected on a Philips PW1050/81 diffractometer operating in Bragg-Brentano focusing geometry and using CuKα radiation (λ = 1.5418 Å) from a generator operating at 40 kV and 30 mA.
Aberration-corrected high-angle annular dark-field (HAADF)-scanning
Aberration-corrected HAADF-STEM images and STEM-EDX mapping images were captured using a probe aberration-corrected STEM (Cubed Titan G2 60-300, FEI, USA) operated at 300 kV.
The morphology of the catalyst was observed by SEM. Manufacturer: Thermo Fisher Scientific, Instrument Model: Apreo S, Electron Gun: Field Emission FEG, Accelerating Voltage 200–30 kV.
Brunauer–Emmett–Teller (BET)
The specific surface area and pore size distribution of the samples were determined by N2 physisorption instrument (MicrotracBEL, BELSORP-mini II). Before the N2 physisorption, the catalysts (~0.2 g) were degassed at 300 °C for 2 h. The specific surface area and the pore size distribution were calculated by the Brunauer- Emmett-Teller (BET) and the DFT methods, respectively.
Temperature programmed desorption (TPD)
CO2 and NH3 Temperature-programed desorption (TPD) were performed on a Micrometrics ASAP 2920 unit. Firstly, 100 mg catalyst was placed in a quartz reactor and pretreated at 400 °C for 3 h under argon, and then cool down to 50 °C; Secondly, the argon was switched to CO2/NH3 (10 vol % in He) for 1 h at 50 °C with a 50 mL/min; Then, CO2/NH3 was switched to He and kept at 50 °C for 1 h to remove the physisorbed CO2/NH3, and when the baseline was stable, the temperature was heated to 800 °C at a heating rate of 10 °C min−1 under He, and the signal of desorption for CO2/NH3 was detected by a thermal conductivity detector (TCD).
X-ray photoelectron spectroscopy (XPS)
XPS was performed using a Thermo ESCALAB 250Xi with Al K radiation (15 kV, 10.8 mA, hν = 1486.6 eV) under an ultrahigh vacuum (5 × 10–7 Pa), calibrated internally by the carbon deposit C(1 s) (Eb = 284.8 eV).
The catalysts (ZnZrO, CsX or ZnZrO/CsX) used in the catalyst characterization were provided in Fig. 1-5 in the main text and Supplementary Information.
Fourier transform infrared (FTIR) spectra
FTIR spectra of pyridine was performed for both NaX and CsX zeolites. The sample (about 30 mg) was pressed into a wafer, and then treated under vacuum at 400 °C for 1 h. After cooling to 50 °C, pyridine was introduced to the transmission cell until saturation. Then, the signal vs. the reference signal was recorded at different temperature (100 °C, 200 °C, 300 °C and 400 °C) until the signal unchanged.
Data availability
All data needed to support the findings of this study are included in the main text or in the Supplementary Information. Any additional information may be available from the corresponding authors upon request.
References
Jenkinson, D. S., Adams, D. E. & Wild, A. Model estimates of CO2 emissions from soil in response to global warming. Nature 351, 304–306 (1991).
Falkowski, P. et al. The global carbon cycle: a test of our knowledge of earth as a system. Science 290, 291–296 (2000).
Gao, W. L. et al. Industrial carbon dioxide capture and utilization: state of the art and future challenges. Chem. Soc. Rev. 49, 8584–8686 (2020).
He, M. Y., Sun, Y. H. & Han, B. X. Green carbon science: efficient carbon resource processing, utilization, and recycling towards carbon neutrality. Angew. Chem. Int. Ed. 61, e202112835 (2022).
Hepburn, C. et al. The technological and economic prospects for CO2 utilization and removal. Nature 575, 87–97 (2019).
Markewitz, P. et al. Worldwide innovations in the development of carbon capture technologies and the utilization of CO2. Energy Environ. Sci. 5, 7281–7305 (2012).
Osman, A. I., Hefny, M., Abdel Maksoud, M. I. A., Elgarahy, A. M. & Rooney, D. W. Recent advances in carbon capture storage and utilisation technologies: a review. Environ. Chem. Lett. 19, 797–849 (2020).
Wang, J. J. et al. A highly selective and stable ZnO-ZrO2 solid solution catalyst for CO2 hydrogenation to methanol. Sci. Adv. 3, e1701290 (2017).
Meng, C. et al. Oxygen-deficient metal oxides supported nano-intermetallic InNi3C0.5 toward efficient CO2 hydrogenation to methanol. Sci. Adv. 7, eabi6012 (2021).
Graciani, J. et al. Highly active copper-ceria andcopper-ceria-titania catalysts for methanol synthesis from CO2. Science 345, 546–550 (2014).
Dang, S. S. et al. Rationally designed indium oxide catalysts for CO2 hydrogenation to methanol with high activity and selectivity. Sci. Adv. 6, eaaz2060 (2020).
Kattel, S., Ramirez, P. J., Chen, J. G., Rodriguez, J. A. & Liu, P. Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts. Science 355, 1296–1299 (2017).
An, B. et al. Confinement of ultrasmall Cu/ZnOx nanoparticles in metal-organic frameworks for selective methanol synthesis from catalytic hydrogenation of CO2. J. Am. Chem. Soc. 139, 3834–3840 (2017).
Bahruji, H. et al. Pd/ZnO catalysts for direct CO2 hydrogenation to methanol. J. Catal. 343, 133–146 (2016).
Chen, T. Y. et al. Unraveling highly tunable selectivity in CO2 hydrogenation over bimetallic In-Zr oxide catalysts. ACS Catal. 9, 8785–8797 (2019).
González-Garay, A. et al. Plant-to-planet analysis of CO2-based methanol processes. Energy Environ. Sci. 12, 3425–3436 (2019).
Han, Z. et al. CO2 hydrogenation to methanol on ZnO-ZrO2 solid solution catalysts with ordered mesoporous structure. J. Catal. 396, 242–250 (2021).
Hu, J. T. et al. Sulfur vacancy-rich MoS2 as a catalyst for the hydrogenation of CO2 to methanol. Nat. Catal. 4, 242–250 (2021).
Jiang, X., Nie, X. W., Guo, X. W., Song, C. S. & Chen, J. G. Recent advances in carbon dioxide hydrogenation to methanol via heterogeneous catalysis. Chem. Rev. 120, 7984–8034 (2020).
Kar, S., Sen, R., Goeppert, A. & Prakash, G. K. S. Integrative CO2 capture and hydrogenation to methanol with reusable catalyst and amine: toward a carbon neutral methanol economy. J. Am. Chem. Soc. 140, 1580–1583 (2018).
Larmier, K. et al. CO2 -to-methanol hydrogenation on zirconia-supported copper nanoparticles: reaction intermediates and the role of the metal-support interface. Angew. Chem. Int. Ed. 56, 2318–2323 (2017).
Lee, K. et al. Atomic Pd-promoted ZnZrO solid solution catalyst for CO2 hydrogenation to methanol. Appl Catal. B Environ. 304, 120994 (2022).
Li, W. H. et al. CO2 hydrogenation on unpromoted and M-promoted Co/TiO2 catalysts (M = Zr, K, Cs): effects of crystal phase of supports and metal–support interaction on tuning product distribution. ACS Catal. 9, 2739–2751 (2019).
Martin, O. et al. Indium oxide as a superior catalyst for methanol synthesis by CO2 hydrogenation. Angew. Chem. Int. Ed. 55, 6261–6265 (2016).
Men, Y. L. et al. Highly dispersed Pt-based catalysts for selective CO2 hydrogenation to methanol at atmospheric pressure. Chem. Eng. Sci. 200, 167–175 (2019).
Studt, F. et al. Discovery of a Ni-Ga catalyst for carbon dioxide reduction to methanol. Nat. Chem. 6, 320–324 (2014).
Wang, J. Y. et al. CO2 hydrogenation to methanol over In2O3-based catalysts: from mechanism to catalyst development. ACS Catal. 11, 1406–1423 (2021).
Wang, Z. Q. et al. High-performance and long-lived Cu/SiO2 nanocatalyst for CO2 hydrogenation. ACS Catal. 5, 4255–4259 (2015).
Yang, C. S. et al. Strong electronic oxide-support interaction over In2O3/ZrO2 for highly selective CO2 hydrogenation to methanol. J. Am. Chem. Soc. 142, 19523–19531 (2020).
Zhang, J. Z. et al. Neighboring Zn-Zr sites in a metal-organic framework for CO2 hydrogenation. J. Am. Chem. Soc. 143, 8829–8837 (2021).
Wu, C. et al. Inverse ZrO2/Cu as a highly efficient methanol synthesis catalyst from CO2 hydrogenation. Nat. Commun. 11, 5767 (2020).
Bai, S. X. et al. Highly active and selective hydrogenation of CO2 to ethanol by ordered Pd-Cu nanoparticles. J. Am. Chem. Soc. 139, 6827–6830 (2017).
Ding, L. P. et al. CO2 hydrogenation to ethanol over Cu@Na-Beta. Chem 6, 2673–2689 (2020).
He, Z. H. et al. Water-enhanced synthesis of higher alcohols from CO2 hydrogenation over a Pt/Co3O4 catalyst under milder conditions. Angew. Chem. Int Ed. 55, 737–741 (2016).
Liu, S. H. et al. Moderate surface segregation promotes selective ethanol production in CO2 hydrogenation reaction over CoCu catalysts. Angew. Chem. Int. Ed. 61, e202109027 (2022).
Wang, L. X. et al. Selective hydrogenation of CO2 to ethanol over cobalt catalysts. Angew. Chem. Int. Ed. 57, 6104–6108 (2018).
Dang, S. S. et al. Selective transformation of CO2 and H2 into lower olefins over In2O3-ZnZrOx /SAPO-34 bifunctional catalysts. ChemSusChem 12, 3582–3591 (2019).
Gao, P. et al. Direct production of lower olefins from CO2 conversion via bifunctional catalysis. ACS Catal. 8, 571–578 (2017).
Li, Z. L. et al. Highly selective conversion of carbon dioxide to lower olefins. ACS Catal. 7, 8544–8548 (2017).
Ma, Z. Q. & Porosoff, M. D. Development of tandem catalysts for CO2 Hydrogenation to olefins. ACS Catal. 9, 2639–2656 (2019).
Orege, J. I. et al. Highly stable Sr and Na co-decorated Fe catalyst for high-valued olefin synthesis from CO2 hydrogenation. Appl Catal. B: Environ. 316, 121640 (2022).
Wang, S. et al. Selective conversion of CO2 into propene and butene. Chem 6, 3344–3363 (2020).
Zhang, Z. Q. et al. Selective hydrogenation of CO2 and CO into olefins over sodium- and zinc-promoted iron carbide catalysts. J. Catal. 395, 350–361 (2021).
Cui, X. et al. Selective production of aromatics directly from carbon dioxide hydrogenation. ACS Catal. 9, 3866–3876 (2019).
Li, Z. L. et al. Highly selective conversion of carbon dioxide to aromatics over tandem catalysts. Joule 3, 570–583 (2019).
Ni, Y. M. et al. Selective conversion of CO2 and H2 into aromatics. Nat. Commun. 9, 3457 (2018).
Wang, Y. et al. Rationally designing bifunctional catalysts as an efficient strategy to boost CO2 hydrogenation producing value-added aromatics. ACS Catal. 9, 895–901 (2018).
Zhou, C. et al. Highly active ZnO-ZrO2 aerogels integrated with H-ZSM-5 for aromatics synthesis from carbon dioxide. ACS Catal. 10, 302–310 (2019).
Alvarez, A. et al. Challenges in the greener production of formates/formic acid, methanol, and DME by heterogeneously catalyzed CO2 hydrogenation processes. Chem. Rev. 117, 9804–9838 (2017).
Centi, G. & Perathoner, S. Opportunities and prospects in the chemical recycling of carbon dioxide to fuels. Catal. Today 148, 191–205 (2009).
Dorner, R. W., Hardy, D. R., Williams, F. W. & Willauer, H. D. Heterogeneous catalytic CO2 conversion to value-added hydrocarbons. Energy Environ. Sci. 3, 884–890 (2010).
Gao, P., Zhang, L. N., Li, S. G., Zhou, Z. X. & Sun, Y. H. Novel heterogeneous catalysts for CO2 hydrogenation to liquid fuels. ACS Cent. Sci. 6, 1657–1670 (2020).
Goeppert, A., Czaun, M., Jones, J. P., Surya Prakash, G. K. & Olah, G. A. Recycling of carbon dioxide to methanol and derived products - closing the loop. Chem. Soc. Rev. 43, 7995–8048 (2014).
Liu, C. et al. Gallium nitride catalyzed the direct hydrogenation of carbon dioxide to dimethyl ether as primary product. Nat. Commun. 12, 2305 (2021).
Olah, G. A. Beyond oil and gas: the methanol economy. Angew. Chem. Int. Ed. 44, 2636–2639 (2005).
Otto, A., Grube, T., Schiebahn, S. & Stolten, D. Closing the loop: captured CO2 as a feedstock in the chemical industry. Energy Environ. Sci. 8, 3283–3297 (2015).
Porosoff, M. D., Yan, B. H. & Chen, J. G. Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: challenges and opportunities. Energy Environ. Sci. 9, 62–73 (2016).
Wang, W., Wang, S. P., Ma, X. B. & Gong, J. L. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 40, 3703–3727 (2011).
Wei, J. et al. Directly converting CO2 into a gasoline fuel. Nat. Commun. 8, 15174 (2017).
Wei, J., Yao, R. W., Han, Y., Ge, Q. J. & Sun, J. Towards the development of the emerging process of CO2 heterogenous hydrogenation into high-value unsaturated heavy hydrocarbons. Chem. Soc. Rev. 50, 10764–10805 (2021).
Zhu, Z., Chen, Q., Xie, Z., Yang, W. & Li, C. The roles of acidity and structure of zeolite for catalyzing toluene alkylation with methanol to xylene. Micropor. Mesopor. Mat. 88, 16–21 (2006).
Alabi, W., Atanda, L., Jermy, R. & Al-Khattaf, S. Kinetics of toluene alkylation with methanol catalyzed by pure and hybridized HZSM-5 catalysts. Chem. Eng. J. 195-196, 276–288 (2012).
Huang, X. et al. Catalyst design strategies towards highly shape-selective HZSM-5 for para-xylene through toluene alkylation. Green. Energy Environ. 5, 385–393 (2020).
Shreiber, E. H., Rhodes, M. D. & Roberts, G. W. Methanol dehydrogenation with Raney copper in a slurry reactor. Appl Catal. B: Environ. 23, 9–24 (1999).
Merko, M., Busser, G. W. & Muhler, M. Non‐oxidative dehydrogenation of methanol to formaldehyde over bulk β‐Ga2O3. ChemCatChem 14, e202200258 (2022).
Palomares, A. E., Eder-Mirth, G., Rep, M. & Lercher, J. A. Alkylation of toluene over basic catalysts - key requirements for side chain alkylation. J. Catal. 180, 56–65 (1998).
Hong, Z., Xiong, C. F., Zhao, G. Q. & Zhu, Z. R. Side-chain alkylation of toluene with methanol to produce styrene: an overview. Catal. Sci. Technol. 9, 6828–6840 (2019).
Madhavi, G., Kulkarni, S. J., Murthy, K. V. V. S. B. S. R., Viswanathan, V. & Raghavan, K. V. Side-chain alkylation of 4-picoline with formaldehyde over alkali-modified zeolites. Appl Catal. A: Gen. 246, 265–282 (2003).
Chen, F. et al. Hydrogenation of pyridines using a nitrogen-modified titania-supported cobalt catalyst. Angew. Chem. Int. Ed. 57, 14488–14492 (2018).
Wang, H., Liu, J., Qu, J. P. & Kang, Y. B. Overcoming electron-withdrawing and product-inhibition effects by organocatalytic aerobic oxidation of alkylpyridines and related alkylheteroarenes to ketones. J. Org. Chem. 85, 3942–3948 (2020).
Wilbert, G., Reich, L. & Tenenbaum, L. Improved synthesis of 4-ethylpyridine. J. Org. Chem. 22, 694–695 (1957).
Zuo, J. et al. Selective methylation of toluene using CO2 and H2 to para-xylene. Sci. Adv. 6, eaba5433 (2020).
Zhang, W. et al. Effective conversion of CO2 into light olefins over a bifunctional catalyst consisting of La-modified ZnZrOx oxide and acidic zeolite. Catal. Sci. Technol. 12, 2566–2577 (2022).
Yu, Q. et al. Role of ball milling during Cs/X catalyst preparation and effects on catalytic performance in side-chain alkylation of toluene with methanol. Chin. J. Catal. 41, 1268–1278 (2020).
Cheng, J. et al. Selective upcycling of polyethylene terephthalate towards high-valued oxygenated chemical methyl p-methyl benzoate using a Cu/ZrO2 catalyst. Angew. Chem. Int. Ed. 63, e202319896 (2024).
Tada, S. et al. Active sites on ZnxZr1–xO2–x solid solution catalysts for CO2-to-methanol hydrogenation. ACS Catal. 12, 7748–7759 (2022).
Gebicki, W., Osuch, K., Jastrzebski, C., Golacki, Z. & Godlewski, M. Raman scattering study of ZnO:Ti and ZnO:Mn bulk crystals. Superlattice Microst. 38, 428–438 (2005).
Hong, Z. et al. Ammonia pools effect in Cs modified X zeolites for side-chain alkylation of toluene with methanol. Chem. Eng. J. 474, 145650 (2023).
Hong, Z., Zhao, G., Huang, F., Wang, X. & Zhu, Z. Enhancing the side-chain alkylation of toluene with methanol to styrene over the Cs-modified X zeolite by the assistance of basic picoline as a co-catalyst. Green. Energy Environ. 7, 1241–1252 (2022).
Han, H. et al. Effects of cesium ions and cesium oxide in side-chain alkylation of toluene with methanol over cesium-modified zeolite X. Ind. Eng. Chem. Res 55, 1849–1858 (2016).
Park, M., Kim, S., Takahashi, Y. & Jeong, H. Y. Thermal stabilization of extraframework Cs+ in zeolite 13X. J. Nucl. Mater. 572, 154078 (2022).
Doskocil, E. J. & Davis, R. J. Spectroscopic characterization and catalytic activity of zeolite x containing occluded alkali species. J. Catal. 188, 353–364 (1999).
Feng, Z. et al. Asymmetric sites on the ZnZrOx catalyst for promoting formate formation and transformation in CO2 hydrogenation. J. Am. Chem. Soc. 145, 12663–12672 (2023).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Liu, C., Li, G., Hensen, E. J. M. & Pidko, E. A. Nature and catalytic role of extraframework aluminum in faujasite zeolite: a theoretical perspective. ACS Catal. 5, 7024–7033 (2015).
Liu, C., Li, G., Hensen, E. J. M. & Pidko, E. A. Relationship between acidity and catalytic reactivity of faujasite zeolite: a periodic DFT study. J. Catal. 344, 570–577 (2016).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22072061), and the National Key R&D Program of China (2023YFB4104501). The authors thank the staff of beamline BL13SSW at Shanghai Synchrotron Radiation Facility for experiment supports.
Author information
Authors and Affiliations
Contributions
Q.M. and J.C. contributed equally. Q.M. and J.C. conceived and conducted the experiments, and analyzed data; X.W., J.X., R.Z., Z.M., and H.Y. offered the help in the experiments and data analysis; W.F. and J.Z. conducted the EXAFS experiments, and analyzed data; G.L. conducted the DFT calculations, and analyzed data; J.B. offered the help in the DFT calculations. The manuscript was written by Z.L.; Z.L. and C.L. proposed the project, analyzed data and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Lei Li who co-reviewed with Zhe Hong; and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, Q., Cheng, J., Wu, X. et al. C–C bond coupling with sp3 C–H bond via active intermediates from CO2 hydrogenation. Nat Commun 16, 140 (2025). https://doi.org/10.1038/s41467-024-55640-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55640-w
