
Abstract
Oxidative stress plays a critical role in postmenopausal osteoporosis, yet its impact on osteoblasts remains underexplored, limiting therapeutic advances. Our study identifies phospholipid peroxidation in osteoblasts as a key feature of postmenopausal osteoporosis. Estrogen regulates the transcription of glutathione peroxidase 4 (GPX4), an enzyme crucial for reducing phospholipid peroxides in osteoblasts. The deficiency of estrogen reduces GPX4 expression and increases phospholipid peroxidation in osteoblasts. Inhibition or knockout of GPX4 impairs osteoblastogenesis, while the elimination of phospholipid peroxides rescues bone formation and mitigates osteoporosis. Mechanistically, 4-hydroxynonenal, an end-product of phospholipid peroxidation, binds to integrin-linked kinase and triggers its protein degradation, disrupting RUNX2 signaling and inhibiting osteoblastogenesis. Importantly, we identified two natural allosteric activators of GPX4, 6- and 8-Gingerols, which promote osteoblastogenesis and demonstrate anti-osteoporotic effects. Our findings highlight the detrimental role of phospholipid peroxidation in osteoblastogenesis and underscore GPX4 as a promising therapeutic target for osteoporosis treatment.
Similar content being viewed by others

Introduction
Osteoblastic bone formation plays a pivotal role in maintaining skeletal homeostasis, and its disruption is a critical factor in the development of osteoporosis. Extensive researches have underscored the significant role of oxidative stress in both the onset and progression of osteoporosis1,2,3. However, the majority of studies have predominantly focused on the effects of common free radicals, such as superoxide anions, on osteoclast-mediated bone resorption4. Despite the availability of therapeutic strategies for osteoporosis, no treatments specifically target oxidative stress as part of the therapeutic approach5. The scarcity of effective strategies can be attributed to an incomplete understanding of the molecular mechanisms underlying the condition and the absence of clearly identified therapeutic targets. Although the decline in estrogen levels is widely recognized as the primary cause of postmenopausal osteoporosis6, the exact molecular mechanisms remain underexplored. Notably, the impact of oxidative metabolism, especially phospholipid peroxidation, on osteoblast-mediated bone formation and remodeling has been largely neglected. Furthermore, the intrinsic molecular factors driving oxidative stress under estrogen-deficient conditions have yet to be elucidated, representing a significant gap in the current understanding of osteoporosis pathogenesis.
Phospholipid peroxidation, a specific oxidative metabolic process mediated by lipoxygenases7, has garnered significant attention for its pivotal role in various pathological conditions8,9,10. Recent studies have indicated phospholipid peroxidation and ferroptosis, a regulated form of cell death driven by excessive phospholipid peroxidation, in the pathogenesis of osteoporosis11,12,13. Strategy targeting the inhibition of osteocyte ferroptosis has demonstrated potential in mitigating diabetic osteoporosis14. Additionally, the Nrf2 pathway has been identified as a crucial regulator of osteocyte ferroptosis-induced osteoclastogenesis12. However, the majority of these investigations have predominantly focused on osteoclasts, with relatively little attention directed toward the impact of phospholipid peroxidation on osteoblasts.
Glutathione peroxidase 4 (GPX4), a unique phospholipid hydroperoxidase, plays a critical role in catalyzing the reduction of phospholipid hydroperoxides, thereby safeguarding cells against oxidative stress15. This study highlights the essential role of GPX4 in reducing phospholipid peroxidation within osteoblasts and maintaining bone formation. Analysis of bone tissues from female osteoporotic patients and ovariectomized (OVX) mice reveals a correlation between impaired osteoblast differentiation and accumulation of phospholipid peroxides caused by GPX4 deficiency. This disturbance of the antioxidant defense system leads to excessive production of 4-hydroxynonenal (4-HNE), which forms adducts with integrin-linked kinase (ILK), leading to its ubiquitination-mediated degradation. The subsequent downregulation of ILK levels inhibits the transcription of Runt-related transcription factor 2 (RUNX2), thereby suppressing osteoblast activity and bone formation. Additionally, this study identifies 6-Gingerol and 8-Gingerol as novel allosteric activators of GPX4, demonstrating their anti-osteoporotic effects in both in vivo and in vitro models. These findings provide critical insights into the role of phospholipid peroxidation in osteogenesis dysfunction and emphasize the therapeutic potential of targeting GPX4 for the treatment of osteoporosis.
Results
The peroxidation of phospholipids is an essential metabolic feature in osteoporotic bone tissues in both human and mice
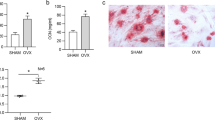
To elucidate the correlation between phospholipid peroxidation and osteoporosis, we initially quantified the levels of 4-HNE, a key end-product of phospholipid peroxidation, in bone tissues collected from aged patients undergoing femoral head replacement. Our results revealed a progressive elevation in 4-HNE content with age in femoral bone tissues (Fig. 1a). Similarly, elevated 4-HNE levels were observed in the femurs and tibias of both OVX mice (Fig. 1b, c) and aged mice (Fig. 1d, e), suggesting an association between phospholipid peroxidation and the progression of osteoporosis. Notably, 4-HNE accumulation was particularly prominent in the trabecular bone of OVX mice, particularly within osteoblasts (Fig. 1f). Additionally, 4-HNE staining colocalized with alkaline phosphatase (ALP), a well-established osteoblast marker, in femoral sections from OVX mice (Fig. 1g, h), indicating that osteoblasts in osteoporotic bone are adversely affected by phospholipid peroxidation.

a 4-HNE level in femurs of aged patients (n = 6 biologically independent samples). b 4-HNE level in long bone tissues of OVX and sham mice (n = 3 biologically independent samples). c 4-HNE content in bone tissues of OVX and sham mice detected by ELISA (n = 6 biologically independent samples). d 4-HNE level in femurs and tibias from both young (4 months) and aged (16 months) mice (n = 3 biologically independent samples). e 4-HNE content in bone tissues from both young (4 months) and aged (16 months) mice detected by ELISA (n = 6 biologically independent samples). f Representative immunohistochemical images of 4-HNE staining in femurs of mice. The red arrows indicated osteoblasts. Scale bar = 50 μm. g, h Representative immunofluorescence images and quantification of 4-HNE and ALP double staining in femurs of mice. 4-HNE (green), ALP (red) and DAPI (blue). The white arrow indicated double positive cells in trabecular bone. Scale bar = 20 μm. n = 5 (sham group), or 4 (OVX group) biologically independent samples. i The discriminative oxidized phospholipids are ranked in descending order by VIP scores. j Volcano plots of OVX-induced changes in oxPLs level log2 (fold change) versus significance (−log10 (P value)) by unpaired two-tailed t-test in bone tissues of sham and OVX mice (n = 6 biologically independent samples). oxCLs, oxidized cardiolipin. oxPCs, oxidized phosphatidylcholine. oxPEs, oxidized phosphatidylethanolamine. oxPGs, oxidized phosphatidylglycerol. oxPIs, oxidized phosphatidylinositol. oxPSs, oxidized phosphatidylserine. The significance in the volcano plot is defined by a fold change of more than 2 times and a P-value less than 0.05. k Representative micro-CT images showing the 3D microarchitecture of femurs from the indicated group. Scale bar = 500 μm. l Micro-CT measurements of BMD, BV/TV, Tb.N, Tb.Th in femurs, n = 5 independently biological samples. BMD, bone mineral density. BV/TV, ratio of bone volume to tissue volume. Tb.N, trabecular number. Tb.Th, trabecular thickness. Data are presented as means ± SD. Statistical analysis was performed with unpaired two-tailed t-test (b–e, h), one-way ANOVA with Tamheni test (a), Tukey’s post hoc test (l). Source data are provided as a Source Data file.
To further characterize the oxidized phospholipids (oxPLs) species present in osteoporotic bone, we performed relative content of phospholipids analysis using LC-MS. An Orthogonal Partial Least Squares Discriminant Analysis (OPLS-DA) score plot revealed a distinct separation between the OVX and sham-operated group (Supplementary Fig. 1a). Heatmaps highlighted significant differences in the oxPLs profiles, with enriched levels of oxidized phosphatidylethanolamine (oxPEs) and oxidized phosphatidylcholine (oxPCs) in OVX bone tissues (Supplementary Fig. 1b), as confirmed by the variable importance in projection analysis (VIP) (Fig. 1i). Similar oxPLs profiles were detected in aged mice (Supplementary Fig. 2a, b), where VIP analysis identified oxPCs as a key contributor (Supplementary Fig. 2c). Volcano plots further demonstrated a significant increase in oxPLs levels in both OVX (Fig. 1j) and aged mice (Supplementary Fig. 2d), consistent with prior study showing that neutralizing oxPLs alleviates senile osteoporosis11.
To validate the role of phospholipid peroxidation in osteoporosis, we administered vitamin E (α-tocopherol), a scavenger of lipid peroxyl radicals, intragastrically to OVX-operated mice daily, starting one week after OVX procedure. Micro-CT analysis demonstrated that inhibition of phospholipid peroxidation by vitamin E (100 mg/kg) significantly prevented OVX-induced bone loss (Fig. 1k) and markedly improved bone mineral density (BMD), bone volume to tissue volume ratio (BV/TV), trabecular number (Tb.N), and trabecular thickness (Tb.Th) in the femurs of OVX mice (Fig. 1l). These findings highlight the protective effect of vitamin E in OVX mice.
It is worth to mention that, normal mice were treated with vitamin E (100 mg/kg) via oral administration. Micro-CT analysis revealed a slightly but statistically significant reduction in BMD in normal mice (P < 0.05) (Supplementary Fig. 3), consistent with findings reported by Fujita et al16. These intriguing findings underscore the contrasting effects of vitamin E on bone metabolism under pathological versus physiological conditions. While vitamin E exhibited undesirable effects in healthy mice, its protective effects in mitigating OVX-induced bone loss underscores the critical involvement of phospholipid peroxidation in the pathogenesis and progression of osteoporosis.
GPX4 expression is downregulated in osteoblasts of OVX and aged mice
Phospholipid peroxidation is exquisitely regulated by key enzymes, including acyl-CoA synthetase long-chain family member 4 (ACSL4), lysophosphatidylcholine acyltransferase 3 (LPCAT3), and 15-lipoxygenase (ALOX15). Notably, GPX4 is the sole enzyme capable of neutralizing phospholipid peroxides17 (Fig. 2a). To elucidate the role of phospholipid peroxidation-related genes in osteoporosis, we performed qPCR assays, which revealed upregulated expressions of Alox15, Ptgs2, Lpcat3, Acsl4, and Tfrc, alongside a marked downregulation of Gpx4 in both OVX mice (Fig. 2b) and aged mice (Fig. 2g, Supplementary Fig. 2e). Similarly, GPX4 protein level was significantly reduced in bone tissues from aged patients (Fig. 2c), OVX mice (Fig. 2d), and aged mice (Fig. 2h), suggesting a potential link between GPX4 deficiency and phospholipid peroxide accumulation in osteoporotic conditions. Immunohistochemistry further demonstrated a substantial decrease in GPX4 level within osteoblasts of OVX mice (Fig. 2e). Furthermore, the colocalization of GPX4 with the osteoblast marker ALP was significantly diminished in the femurs of both OVX (Fig. 2f) and aged mice (Fig. 2i). In contrast, no difference was observed in the colocalization of GPX4 with the osteoclast marker CTSK between OVX and sham mice (Supplementary Fig. 2f). Collectively, these findings underscore the specific downregulation of GPX4 in osteoblasts within osteoporotic bone tissues.

a The schematic diagram of key enzymes involved in the production and reduction of lipid peroxidation. PUFA, polyunsaturated fatty acids. ACSL4, acyl-CoA synthetase long-chain family member 4. LPCAT3, lysophosphatidylcholine acyltransferase 3. ALOX15, 15-lipoxygenase. GPX4, glutathione peroxidase 4. PL, phospholipid. b Expression of lipid peroxidation-related genes in whole long bone from sham and OVX mice (n = 6 biologically independent samples). c Protein expression of GPX4 in femurs of female osteoporotic patients and its quantification (n = 6 biologically independent samples). d Protein level of GPX4 in whole long bone from OVX and sham mice and its quantification (n = 3 biologically independent samples). e Representative immunohistochemical images of GPX4 staining in femur from sham and OVX mice, scale bar = 100 μm. The dashed rectangles in red were enlarged and presented below, and the red arrow indicated osteoblasts. f Representative image of GPX4 and ALP double staining in femur from sham and OVX mice (left) and its quantification (right). GPX4 (green), ALP (red) and DAPI (blue), n = 3 biologically independent samples. Scale bar = 20 μm. The dashed rectangles in green were enlarged, and the white arrows indicated double positive cells in trabecular bone. g Relative mRNA expression of Gpx4 gene of aged (16 months) and young (4 months) mice (n = 6 biologically independent samples). h Protein expression of GPX4 in whole long bone from aged and young mice (left) and its quantification (right), n = 3 biologically independent samples. i Representative image of GPX4 and ALP double staining in femur from young (3 months) and aged (16 months) mice (left) and the quantification (right). GPX4 (green), ALP (red) and DAPI (blue), n = 4 biologically independent samples. Scale bar = 50 μm. The dashed rectangles in green were enlarged. OVX or sham surgery was performed on three-month-old mice. Tissue collection was conducted three months post-surgery. Data are presented as means ± SD. Statistical analysis was performed with unpaired two-tailed t-test (d, f–i) or one-way ANOVA with Tukey’s post hoc test (c). Source data are provided as a Source Data file.
Osteoblast differentiation is tightly regulated by key osteogenic transcription factors, including RUNX2, Osterix (OSX), and activating transcription factor 4 (ATF4), as well as key osteogenic genes such as ALP and collagen1α1 chain (COL1A1). Intriguingly, a significant downregulation of Runx2, Osx, Ocn, Alp, Atf4, and Col1α was observed in both OVX (Supplementary Fig. 4a) and aged mice (Supplementary Fig. 4b). These findings were corroborated by protein expression analyses, which demonstrated reduced levels of RUNX2 and ALP in aged mice (Supplementary Fig. 4c). Collectively, these results suggest a potential association between GPX4 deficiency and impaired osteoblast differentiation, contributing to diminished bone formation in osteoporotic condition.
Estrogen deficiency leads to inactivation of GPX4 transcription in osteoblasts
Given the observed downregulation of GPX4 expression in estrogen-deficient patients and mice, we further investigated the transactivation mechanism of GPX4 mediated by estrogen. Initially, we analyzed the mRNA expression of Gpx4 in MC3T3-E1 cells treated with estrogen (17β-estradiol) for different time periods. As expected, estrogen treatment upregulated Gpx4 gene expression in a time-dependent manner (Fig. 3a). Correspondingly, the protein expression of GPX4 also increased following estrogen exposure in MC3T3-E1 cells (Fig. 3b). To confirm the role of estrogen in GPX4 regulation, we employed fulvestrant, a widely used estrogen receptor antagonist. As shown in Fig. 3c, estrogen significantly elevated GPX4 expression, while fulvestrant effectively inhibited this effect. Estrogen primarily exerts its biological effects through estrogen receptors (ERs), ERα or ERβ, which form dimers, translocate to the nucleus, and bind to estrogen response elements (EREs) to modulate target gene transcription18. To identify the specific ER subtype responsible for GPX4 regulation, MC3T3-E1 cells were treated with Esr1 siRNA (encoding ERα) or Esr2 siRNA (encoding ERβ) for 24 hours, followed by estrogen treatment for 12 hours. The results revealed that knockdown of ERβ, but not ERα, abolished estrogen-induced GPX4 expression (Fig. 3d–g). Notably, when treatment with the protein synthesis inhibitor cycloheximide (CHX), estrogen could not alter the protein expression of GPX4 (Fig. 3h). These findings collectively support the hypothesis that estrogen regulates GPX4 transcription through ERβ.

a Relative mRNA expression of Gpx4 in MC3T3-E1 cells treated with estrogen (17β-estradiol, 1 μM) for different time periods (n = 3 independent cell culture preparations). b Protein level of GPX4 in MC3T3-E1 cells exposed to estrogen (17β-estradiol) at doses ranging from 10 nM to 1 μM for 24 hours (n = 3 independent cell culture preparations). c Effect of estrogen antagonist fulvestrant (1 μM) on GPX4 expression treated with estrogen (17β-estradiol, 1 μM), n = 3 independent cell culture preparations. d, e Validation of Esr2 (left) and Esr1 (right) siRNA sequences by western blot. f, g Relative mRNA expression of Gpx4 in MC3T3-E1 cells after knockdown of ERβ (left) or ERα (right), respectively (n = 3 independent cell culture preparations). h Protein expression of GPX4 in MC3T3-E1 cells treated with protein synthesis inhibitor (CHX, 100 μM) in the presence or absence of estrogen (17β-estradiol, 1 μM) (n = 3 independent cell culture preparations). Data are presented as means ± SEM. i Verification of ESR2-Flag plasmid by western blot. j Effect of ERβ on the activity of GPX4-luc reporter (n = 3 independent cell culture preparations). k Transcription factor ERβ binding sequence predicted in the JASPAR database. l Schematic diagram of GPX4 luciferase promoter. Luc, luciferase. m EMSA assays showing a direct interaction between ERβ protein and GPX4 E-box element. The experiment was repeated three times independently with similar results. n Workflow of ChIP experiment. o ChIP assays showing significant enrichment of ERβ at the E-box of GPX4 (n = 3 independent cell culture preparations). p Effect of ERβ on the activity of mutant GPX4-luc reporter (n = 8 independent cell culture preparations). q Schematic diagram illustrating the estrogen/ERβ-dependent transactivation of GPX4 expression. Data are presented as means ± SD unless specific description. Statistical analysis was performed with unpaired two-tailed t-test (a, f–g, j, o, p) or one-way ANOVA with Tukey’s post hoc test (b, c), or two-way ANOVA (h). Source data are provided as a Source Data file.
To further investigate the transcriptional regulation of GPX4 by ERβ, we conducted dual luciferase reporter assays and chromatin immunoprecipitation (ChIP) assays. The promoter region (~2 kb) of GPX4 was cloned into a luciferase reporter vector (pGL4.1 basic) to evaluate the direct impact of ERβ on GPX4 transcription (Fig. 3l). Overexpression of ERβ significantly enhanced GPX4 transcription in a dose-dependent manner (Fig. 3i, j). Bioinformatics analysis of the GPX4 promoter, utilizing the JASPAR database with the ERE consensus sequences (AGGTCA/TC/CCT) (Fig. 3k), predicted the presence of putative ERE regions within the GPX4 promoter (Fig. 3l). This interaction was further validated through electrophoretic mobility shift assays, which indicated direct binding of the ERβ protein to the E-box element of the GPX4 promoter (Fig. 3m). Additionally, ChIP analysis, following the manufacturer’s protocol (Fig. 3n), revealed significant enrichment of ERβ at the E-box of GPX4 promoter (Fig. 3o). To confirm the transactivation effect of ERβ on the GPX4 promotor, a mutant luciferase-GPX4 reporter was generated with a specific mutation on GPX4 E-box (Fig. 3l). This mutation abolished the transactivation activity of ERβ (Fig. 3p). Collectively, these findings suggest that estrogen activates GPX4 transcription by directly binding to the E-box element in its promoter region (Fig. 3q).
Osteoblast-specific knockout or inhibition of GPX4 damages bone formation
To elucidate the role of GPX4 in osteoblastogenesis and osteoporosis, we generated osteoblast-specific GPX4 knockout mice (Gpx4OBs−/−) and investigated their impact on osteoblast-mediated bone formation. Micro-CT analysis revealed that two-month-old Gpx4OBs−/− mice exhibited significantly reduced bone mass compared to their Gpx4flox/flox littermates (Fig. 4a). This reduction in bone mass was characterized by significant decreases in BV/TV, BMD and cortical bone thickness (Ct.Th) in the cortical bone of the femurs in Gpx4OBs−/− mice, alongside a reduction in Tb.Th and BV/TV in trabecular bone (Fig. 4b). Consistent with these findings, histological analysis using H&E staining (Fig. 4c) and von Kossa staining (Fig. 4d) revealed reduced trabecular bone and impaired bone mineralization in the femurs of Gpx4OBs−/− mice. Further analysis indicated a significant reduction in osteoblast numbers (Fig. 4e, f) and decreased expression of key bone-forming genes, including Runx2, Osx, Alp, Atf4, Col1α, Opn, and Ocn, in Gpx4OBs−/− bone tissues (Fig. 4g), suggesting impaired bone formation. Similarly, GPX4 knockdown in MC3T3-E1 cells resulted in diminished ALP activity and reduced mineral deposition (Fig. 4h), corroborating the in vivo findings.

a Representative micro-CT images of the three-dimensional long bone, trabecular bone, and cortical bone in femur of Gpx4OBs−/− mice and Gpx4flox/flox littermates. Scale bar = 1 mm. b Micro-CT measurements of Ct.BMD, Ct. BV/TV, Ct.Th, and BV/TV of trabecular bone, Tb.Th as well as Tb.N at the distal femur from Gpx4OBs−/− mice and Gpx4flox/flox mice (n = 6 independently biological samples). Ct.BMD, bone mass density of cortical bone. Ct. BV/TV, ratio of bone volume to total volume of cortical bone. Ct.Th, thickness of cortical bone. Trabecular bone BV/TV, ratio of bone volume to total volume of trabecular bone. Tb.Th, trabecular thickness. Tb.N, trabecular number. c Representative H&E staining images of femurs from Gpx4OBs−/− mice and Gpx4flox/flox mice. Scale bar = 100 μm. d Representative von Kossa staining images of femurs from Gpx4OBs−/− mice and Gpx4flox/flox mice. Scale bar = 1 mm. e–f Representative ALP immunohistochemical images (e) and quantification (f) of femur from Gpx4OBs−/− mice and Gpx4flox/flox mice. Scale bar = 20 μm. n = 4 (Gpx4flox/flox mice group) or 5 (Gpx4OBs−/− mice group) biologically independent samples. g qPCR analysis of genes expression related to bone formation (n = 5 independently biological samples). The tissues were harvested from Gpx4OBs−/− and Gpx4flox/flox mice at two months of age. h ALP staining in 1 week of osteogenic induction and alizarin red staining of mineralized matrix in MC3T3-E1 cells after 3 weeks of osteogenic induction post knockdown of GPX4. i ALP staining in osteoblasts after 1 week of osteogenic induction and alizarin red staining of mineralized matrix in MC3T3-E1 cells after 3 weeks of osteogenic induction treated with GPX4 siRNA and ferrostatin-1 (Fer-1, 5 μM). Data are presented as means ± SD. Statistical analysis was performed with unpaired two-tailed t-test. Source data are provided as a Source Data file.
Furthermore, we employed RSL3, a well-established GPX4 inhibitor19,20,21,22,23, to induce phospholipid peroxidation in the bone tissues of mice. Following a single intraperitoneal administration of RSL3 (20 mg/kg) in female mice, the compound was detected in bone tissues, exhibiting a half-life of 2.1 hours and reaching a maximum concentration (Cmax) of 481.40 ng/mg protein at 0.5 hour post-administration (Supplementary Fig. 6a). Remarkably, RSL3 remained detectable in bone tissues 24 hours post-administration, with a concentration of 0.68 ng/mg protein. To evaluate its long-term presence, we administered RSL3 at 20 mg/kg every other day for two months to nine-month-old female mice. Sustained drug exposure was confirmed, with RSL3 concentrations reaching 2.07 ng/mg protein in bone tissues 24 hours after the final treatment (Supplementary Fig. 6b). To verify the direct interaction of RSL3 with GPX4 in bone tissues, we utilized a RSL3 probe (RSL3-Alkyne) in a Click chemistry reaction-based pulldown assay (Supplementary Fig. 6c), which demonstrated dose-dependent formation of RSL3-Alkyne-GPX4 conjugates (Supplementary Fig. 6d). Furthermore, one- and two-month RSL3 treatments significantly reduced GPX4 levels (Supplementary Fig. 6e and Fig. 5g) and increased phospholipid peroxide accumulation (Supplementary Fig. 7a). These results demonstrate that RSL3 can be effectively distributed to bone tissues, inhibits GPX4, and induces phospholipid peroxidation.

a Representative micro-CT images of the three-dimensional microarchitecture of the long bone, trabecular bone and cortical bone of femurs from RSL3-treated mice and control mice (RSL3, 20 mg/kg, two months). Scale bar = 1 mm. b Micro-CT measurements of BMD, BV/TV, Tb.Th, Tb.N of femurs from RSL3-treated mice and control mice (n = 5 independently biological samples). BMD, bone mineral density. BV/TV, ratio of bone volume to tissue volume. Tb.Th, trabecular thickness. Tb.N, trabecular number. c Representative H&E staining images of femurs from both control and RSL3-treated mice (RSL3, 20 mg/kg, two months). Scale bar = 100 μm. d Representative von Kossa staining images of femurs from both control and RSL3-treated mice (RSL3, 20 mg/kg, two months). Scale bar = 1 mm. e Toluidine blue staining showing osteoblasts (red arrows indicated) (left) and the quantification (right) in the femurs of mice with RSL3 treatment for two months (n = 3 independently biological samples). Scale bar = 50 μm. f qPCR analysis of gene expression related to bone formation in the whole long bone tissues of mice with RSL3 treatment for two months (n = 6 independently biological samples). g Western blot analysis of GPX4, RUNX2, ALP, and OCN and the relative quantification in the whole long bone tissues of mice with RSL3 treatment for two months, n = 3 independently biological samples. RSL3 treatment was administered to nine-month-old mice, followed by tissue collection two months later. h Relative mRNA expression of Alp, Runx2, and Osx in MC3T3-E1 cells exposed to indicated concentration of RSL3 (n = 3 independently biological cell cultures). i. ALP staining in 1 week of osteogenic induction and alizarin red staining of mineralized matrix in mouse primary osteoblasts after 3 weeks osteogenic induction treated with indicated concentration of RSL3. Data were presented as means ± SD. Statistical analysis was performed with unpaired two-tailed t-test (b–e, g) or one-way ANOVA with Tamheni test (h). Source data are provided as a Source Data file.
Micro-CT analysis demonstrated that the inhibition of GPX4 with RSL3 caused significant bone loss, as indicated by a marked reduction in BMD, BV/TV and Tb. Th in femurs (Fig. 5a, b) and a significant decrease in BMD in vertebrae (Supplementary Fig. 6f, g). These observations were further supported by histological analysis, including H&E staining and von Kossa staining, which confirmed structural deterioration (Fig. 5c, d). Additionally, toluidine blue staining revealed a notable reduction in osteoblast numbers, as indicated by reduced Ob.S/BS (Fig. 5e). The inhibition of GPX4 by RSL3 also significantly suppressed the expression of bone formation-related genes (Runx2, Osx, Alp, Col1α, and Ocn) and proteins (RUNX2, ALP, and OCN) (Fig. 5f, g), indicating impaired osteogenic activity. Consistent with these in vivo findings, RSL3 treatment of osteoblast cells resulted in diminished expression of bone formation-related genes (Fig. 5h), impaired osteoblasts differentiation, and reduced mineralization capacity (Fig. 5i). Collectively, these results underscore the essential role of GPX4 in maintaining bone formation and highlight its involvement in osteoblast function and bone homeostasis.
Phospholipid peroxidation-mediated 4-HNE modification of ILK dampens RUNX2 signaling
Lipid peroxidation leads to the generation of 4-HNE, a highly reactive electrophilic compound that forms covalent adducts with lysine, histidine, and cysteine residues in proteins24,25. Consistent with the accumulation of 4-HNE-bound proteins observed in the bone tissues of aged patients and OVX mice (Fig. 1a, b, d), similar pattern was detected in RSL3-treated mice and MC3T3-E1 cells (Supplementary Fig. 7a, b), osteoblast-specific Gpx4 knockout mice (Supplementary Fig. 7c), and Gpx4 knockdown MC3T3-E1 cells (Supplementary Fig. 7d). Furthermore, exposure to 4-HNE reduced the expression of bone-forming genes (Runx2, Osx, Alp, and Col1α) (Supplementary Fig. 7e), and impaired osteoblastic differentiation and mineralization in MC3T3-E1 cells (Supplementary Fig. 7f). Notably, treatment with ferrostatin-1 (Fer-1), an inhibitor of phospholipid peroxidation, decreased the levels of 4-HNE-protein conjugates (Supplementary Fig. 7i) as well as restored the expression of bone-forming genes (Alp and Col1α) (Supplementary Fig. 7g) and proteins (RUNX2 and OSX) (Supplementary Fig. 7i) in the bones of OVX mice and MC3T3-E1 cells. Fer-1 also alleviated RSL3-induced suppression of osteoblast differentiation and mineralization (Fig. 4i, Supplementary Fig. 7h). Collectively, these findings highlight the inhibitory role of phospholipid peroxidation-mediated 4-HNE proteins modification in bone formation.
To elucidate the specific proteins involved in bone formation that are modified by 4-HNE, we performed LC-MS-based proteomics analysis (Fig. 6a). This approach identified several candidate proteins, including MAP kinase-activated protein kinase 3 (MAPK3), plastin-3 (PLS3), and integrin-linked protein kinase (ILK), all of which are implicated in bone formation or osteoblast differentiation (Fig. 6b). Notably, 4-HNE-modified ILK was significantly elevated in the GPX4-inhibited group (RSL3 treatment) compared to the control group (Fig. 6b). This observation was corroborated by co-immunoprecipitation (Co-IP) assays (Fig. 6c, d) and immunofluorescence staining (Fig. 6e), which revealed enhanced co-localization of 4-HNE and ILK in GPX4-inhibited MC3T3-E1 cells.

a Workflow for immunoprecipitation-MS-based proteomic. b Volcano plots of LC-MS identified proteins in RSL3-treated group and control group. (log2 (fold change)) versus significance (-log10 (P value)) by unpaired two-tailed t-test. The significance in the volcano plot is defined by a fold change of more than 4 times and a P-value less than 0.01. c, d Co-IP assay determined the level of ILK conjugated with 4-HNE in MC3T3-E1 cells treated with RSL3 (50 nM, 24 hours). e Representative confocal images showing the colocalization of 4-HNE (green) and ILK (red) in MC3T3-E1 cells treated with RSL3 (50 nM, 6 hours). Scale bars = 10 μm. f ILK expression in MC3T3-E1 cells treated with RSL3 for 24 hours (n = 3 independent cell culture). g Ilk mRNA expression in MC3T3-E1 cells treated with RSL3 for 24 hours (n = 3 independent cell culture). h Turnover of ILK by CHX chase assay in MC3T3-E1 cells treated with or without 4-HNE (n = 3 independent cell culture). CHX, 100 μM; 4-HNE, 10 μM. i The effect of MG132 (5 μM) or 3-MA (20 μM) on the RSL3 (50 nM)-induced ILK protein level (n = 3 independent cell culture). j Ubiquitination of ILK assessed by immunoblotting in MC3T3-E1 cells with RSL3 treatment (50 nM, 24 hours). The experiment was repeated three times independently with similar results. k ILK and RUNX2 expression in MC3T3-E1 cells after ILK knockdown (n = 3 independent cell culture). l Bone-forming genes expression in MC3T3-E1 cells after ILK knockdown (n = 3 independent cell culture preparations). m ALP staining in 1-week osteogenic-induced MC3T3-E1 cells after ILK knockdown. n The verification of ILK overexpression. o Bone-forming genes expression in MC3T3-E1 cells treated with RSL3 and ILK overexpression plasmid (RSL3, 50 nM, 24 hours) (n = 3 independent cell culture preparations). p Schematic diagram illustrating that phospholipid peroxidation-derived 4-HNE binds to ILK, resulting in ILK degradation, disturbing bone formation. Data are presented as means ± SD. Statistical analysis is performed with one-way ANOVA with Tukey’s post hoc test (f, g, i, k, o) or two-way ANOVA (h). Source data are provided as a Source Data file.
Protein modification by 4-HNE is known to either alter protein activity26 or promote degradation27. Our study demonstrated that GPX4 inhibition resulted in the downregulation of ILK protein (Fig. 6f) without affecting its mRNA levels (Fig. 6g), suggesting that 4-HNE-ILK adduct formation likely drives ILK degradation. As anticipated, ILK degradation occurred rapidly within 12 hours of CHX-induced inhibition of protein synthesis (Fig. 6h). Furthermore, the proteasome inhibitor MG132, instead of the autophagy inhibitor 3-MA, effectively reversed RSL3-induced ILK degradation (Fig. 6i). Additionally, GPX4 inhibition by RSL3 increased ILK ubiquitination levels in MC3T3-E1 cells (Fig. 6j).
Previous studies have reported the downregulation of ILK in osteoporotic patients28, and ILK deficiency in osteoblast has been shown to impair osteoblast differentiation and bone formation29, highlighting its critical role in osteogenesis. RUNX2 is a well-established transcription factor essential for osteoblast differentiation. Our findings demonstrated that ILK knockdown significantly decreased the protein and mRNA expression levels of RUNX2 (Fig. 6k, l), accompanied by reduced mRNA expression of downstream osteogenic markers, including Osx, Alp, and Col1α (Fig. 6l). This indicates that ILK positively regulates RUNX2 signaling. Moreover, ILK knockdown resulted in fewer ALP-positive staining cells (Fig. 6m), further supporting that ILK deficiency impairs RUNX2-mediated osteoblast differentiation. Conversely, ILK overexpression rescued the reduced expression of bone-forming genes induced by GPX4 inactivation (Fig. 6n, o). Collectively, these findings indicate that phospholipid peroxidation-derived 4-HNE modifies ILK, promoting its ubiquitination-dependent degradation. This degradation subsequently suppresses RUNX2 and OSX expression, ultimately disrupting bone formation (Fig. 6p).
Natural allosteric activators of GPX4 rescue osteogenesis and ameliorate osteoporosis
Recognizing the crucial role of GPX4 in osteoporosis, we aim to identify potential allosteric activators of GPX4 derived from natural products. As depicted in Fig. 7a, molecular docking was performed using a chemical library of natural products, targeting the reported allosteric site of GPX430. Candidate compounds were selected based on their docking scores, compliance with Lipinski’s Rule of Five criteria, and predicted binding mode with GPX4. Priority was given to compounds forming at least one intermolecular hydrogen bond with GPX4. Our initial screening identified the top 15 scoring compounds (Supplementary Table 2), among which three Gingerol derivatives, that is 6-, 8- and 10-Gingerol, exhibited significant inhibition of RSL3-induced cell death (Supplementary Fig. 9). Due to the relatively higher toxicity of 10-Gingerol (Supplementary Fig. 8), subsequent analyses focused on 6-Gingerol and 8-Gingerol. Computational docking predicted that 6-Gingerol forms intermolecular hydrogen bond with Lys31 and Phe100 of GPX4 thorough its phenolic hydroxyl group, and hydrophobic interactions with Asp21, Val27, and Phe103 (Fig. 7b). Similarly, 8-Gingerol forms a hydrogen bond with Lys31 of GPX4 via its carbonyl group, and engages Asp101 with its hydroxyl group, along with hydrophobic interactions with Asp23 (Fig. 7b). Notably, 6-Gingerol exhibited stronger binding to GPX4 than 8-Gingerol, potentially due to the longer carbon chain of 8-Gingerol, which may hinder optimal fitting within GPX4’s binding pocket.

a Workflow of GPX4 allosteric activator screening. b 6-, and 8-Gingerol predicted to bind with GPX4 protein by silico docking. The hydrogen bonds between the activator (green) and GPX4 (pale cyan, PDB entry 2OBI) are shown as orange lines. c Thermal stabilization of GPX4 in cell lysates incubated with DMSO or 6-, and 8-Gingerol (10 μM) determined by CETSA with heat treatment. Data were presented as the means ± SEM (n = 3 independent experiments). d The binding affinity between 6-Gingerol (Kd = 110.96 μM) or 8-Gingerol (Kd = 134.93 μM) and GPX4 examined by MST. e Effect of 6-, and 8-Gingerol on GPX4 enzyme activity (n = 3 independent samples). f Representative micro-CT images of the three-dimensional of trabecular bone and cortical bone in femur of mice treated with 6-, and 8-Gingerol. Scale bar = 1 mm. g Micro-CT measurements of BMD, BV/TV, Tb.N of distal femur of mice treated with 6-, and 8-Gingerol (n = 5 independently biological samples). BMD, bone mineral density. BV/TV, ratio of bone volume to tissue volume. Tb.N, trabecular number. h Expression of bone-forming genes in femurs of mice treated with 6-, and 8-Gingerol (n = 5 independently biological samples). i Protein expression of GPX4 and bone-forming proteins in femurs of mice treated with 6-, and 8-Gingerol (n = 5 independently biological samples). j 4-HNE level in the bone tissues of 6-, and 8-Gingerol-treated mice (n = 5 independently biological samples). Three-month-old mice received Gingerols one week after undergoing surgery. Bone tissues were collected after a 45-day treatment period. Data are presented as means ± SD unless specific description. Statistical analysis was performed with one-way ANOVA with Tukey’s post hoc test (e, j), LSD test (g, i), or two-way ANOVA (c). Source data are provided as a Source Data file.
To further validate the predicted interactions of 6-Gingerol and 8-Gingerol with GPX4, we performed a cellular thermal shift assay. The results demonstrated that both compounds stabilized GPX4 protein against temperature-induced denaturation (Fig. 7c). This interaction was further confirmed by microscale thermophoresis assay (Fig. 7d). Notably, 6- and 8-Gingerol significantly enhanced GPX4 activity both in the absence and presence of RSL3 (Fig. 7e, Supplementary Fig. 10a), supporting their role as potent allosteric GPX4 activators. Furthermore, 6- and 8-Gingerol were observed to increase GPX4 expression in both in vivo and in vitro models (Fig. 7i, Supplementary Fig. 10b–d). This dual mechanism, involving the enhancement of both GPX4 activity and expression, highlights the therapeutic potential of these compounds in restoring GPX4 function. Importantly, neither 6- nor 8-Gingerol prevented cell death in GPX4 knockout cells (Supplementary Fig. 10e), confirming that their protective effects are mediated specifically through GPX4. Additionally, the FENIX assay was employed to assess the lipid radicals trapping potential of these compounds. The results indicated minimal lipid radical scavenging capacity within the concentration range of 3.125 to 50 μM (Supplementary Fig. 10f). These findings suggest that 6- and 8-Gingerols exert their protective effects primarily through GPX4 activation, rather than direct lipid radicals scavenging.
Subsequently, treatment with 6- and 8-Gingerol (25 mg/kg) significantly increased BMD, trabecular bone BV/TV, and Tb.N in the femurs of OVX mice (Fig. 7f, g). These compounds also upregulated the expression of key osteogenic genes, including Runx2, Col1α, and Ocn (Fig. 7h), as well as the corresponding bone formation-related proteins RUNX2, OSX, and ALP (Fig. 7i). Additionally, treatment with 6- and 8-Gingerol reduced the level of 4-HNE in the bones of OVX mice (Fig. 7j). In vitro, 6- and 8-Gingerol restored ALP activity and calcium deposition suppressed by RSL3 treatment in MC3T3-E1 cells (Supplementary Fig. 10g). Collectively, these findings indicate a protective role of 6- and 8-Gingerol against OVX-induced osteoporosis. Furthermore, Micro-CT analysis revealed that 6- and 8-Gingerol at 25 mg/kg did not significantly affect bone parameters in normal mice, highlighting their selective efficacy in pathological bone loss conditions (Supplementary Fig. 3).
Furthermore, the potential effect of 8-Gingerol on bone parameters were evaluated in RSL3-treated mice. The results demonstrated that this GPX4 activator mitigated RSL3-induced bone mass loss (Supplementary Fig. 11a) and significantly improved trabecular bone parameters, including BMD, BV/TV, Tb.N, and Tb.Th (Supplementary Fig. 11b). Consistent with the observations in OVX mice, 8-Gingerol treatment also upregulated the expression of key osteogenic genes (Supplementary Fig. 11c, d) in RSL3-treated mice. Additionally, 8-Gingerol restored ALP activity suppressed by RSL3 (Supplementary Fig. 11e). Moreover, administration of 8-Gingerol significantly reduced the levels of phospholipid peroxidation-derived end products, including malondialdehyde and 4-HNE (Supplementary Fig. 11f, g).
Collectively, these in vivo and in vitro findings identify 6- and 8-Gingerol as natural allosteric activators of GPX4, and indicate that GPX4-targeted therapeutic strategies hold significant potential for the treatment of osteoporosis.
Discussion
Osteoporosis is a prevalent bone disorder affecting a significant global population and imposing considerable economic and health burdens31,32,33. Oxidative stress has been widely recognized as a critical factor in the development and progression of osteoporosis1,2. Although prior research has predominantly focused on the role of common free radicals, such as reactive oxygen species, in the pathogenesis of osteoporosis, no effective therapeutic agents specifically targeting these free radicals have been developed to date. Emerging evidence highlights phospholipid peroxidation and ferroptosis as potential mechanisms underlying osteoporosis12,13,14,34,35. Yang et al. demonstrated that targeting ferroptosis mitigates osteocyte glucolipotoxicity, thereby attenuating diabetic osteoporosis14. Additionally, recent study suggests that the Nrf2 signaling pathway plays a crucial role in osteocyte ferroptosis-mediated osteoclastogenesis12. Distinct from these findings, the present study underscores the essential role of estrogen-mediated GPX4 transcription in reducing phospholipid peroxidation within osteoblasts, thereby providing a novel mechanistic insight into osteoporosis pathogenesis.
GPX4 is a phospholipid-specific hydroperoxide enzyme that plays a crucial role in maintaining phospholipid redox homeostasis due to its unique ability to directly reduce phospholipid peroxides8,10. Using LC-MS-based phospholipids analysis, we characterized the profiles of oxPLs induced by GPX4 decline in osteoporotic tissues of mice. oxPLs are inherently unstable because the peroxided fatty acid chains of phospholipids can be rapidly hydrolyzed by phospholipase A2, producing peroxidized fatty acids36,37,38. These peroxidized fatty acids are further degraded into α, β-unsaturated aldehydes, with 4-HNE being among the most abundant. Compared to the lower electrophilic and instability of oxPLs, 4-HNE is a highly reactive electrophilic aldehyde that readily forms adducts with cellular macromolecules, particular proteins containing lysine, histidine, and cysteine residues24,25,39. Increasing evidences highlight the pivotal role of 4-HNE in post-translational modification of proteins21,27,40. Our previous research demonstrated that 4-HNE forms adducts with the critical chondrogenesis transcription factor SOX9, disturbing bone development27. In this study, western blot analysis revealed elevated levels of 4-HNE-bound proteins in osteoporotic bone tissues, with a prominent band around 45-60 kDa. This observation was consistent across aged humans, mice, and OVX mice. Subsequent LC-MS-based proteomics analysis identified ILK (50 kDa) as a specific bone formation protein modified by 4-HNE.
Intriguingly, apart from the disruption of osteoblastogenesis and the loss of bone mass observed in osteoblast-specific Gpx4 knockout adult mice, other phenotypic features, including reduced body weight, and limb shortening were also noted in these mice, which bear resemblance to certain characteristics of Sedaghatian-type spondylometaphyseal dysplasia (SSMD). SSMD is a very rare genetic disorder characterized by severe metaphyseal chondrodysplasia, mild limb shortening, platyspondyly, cardiac conduction defects, and central nervous system abnormalities. Duarte et al. investigated two unrelated families and identified truncating mutations in GPX4, marking the first report linking such mutations to SSMD41. Specifically, a heterozygous stop mutation in exon 3 of GPX4 was detected in the parents of a child diagnosed with SSMD, highlighting the pathogenic role of GPX4 mutation in this disease. Notably, the catalytic site of GPX4 responsible for reducing phospholipid peroxidation is located within exon 315. Based on this, it is plausible to hypothesize that the loss of GPX4 in osteoblasts during skeletal development may lead to dysplasia resembling SSMD, while its loss in adult bone tissues might result in osteoporosis. Our investigation into the role of GPX4 in reducing phospholipid peroxidation of osteoblasts not only elucidates the mechanisms underlying osteoporosis, but also provides a potential perspective on the pathogenesis of SSMD.
To confirm the involvement of phospholipid peroxidation in osteoblastogenesis dysfunction, we established a bone-specific GPX4 inactivation model in female mice using the tool compound RSL3. Despite its limitations for in vivo applications, including instability, toxicity, and non-specificity, RSL3 remains a widely used agent in phospholipid peroxidation and ferroptosis research19,20,21,22,23. Using LC-MS analysis and a RSL3-Alkyne mediated GPX4 pulldown assays, we demonstrated the accessibility of RSL3 to bone tissue and its binding to GPX4 in murine bone. Moreover, RSL3 administration significantly reduced GPX4 protein levels and led to the accumulation of phospholipid peroxides in bone tissue, validating its efficacy as a GPX4 inhibitor in this model. Colllectively, the RSL3-established GPX4 inhibition model complements our findings from clinical bone samples, OVX mice, aged mice, and osteoblast-specific Gpx4 knockout mice. This integrative approach enhances the robustness of our conclusion and reinforces the critical role of GPX4 in maintaining bone homeostasis.
The essential role of GPX4 in bone formation highlights its potential as novel therapeutic target for osteoporosis. Notably, two natural GPX4 activators 6-Gingerol and 8-Gingerol, have been identified and validated in our study. Interestingly, both compounds were shown to upregulate GPX4 expression, suggesting a potential feedback mechanism mediated by GPX4 activation or other yet-to-be-elucidated mechanisms. In OVX mice, 6-Gingerol and 8-Gingerol exhibited significant bone-protective effects by enhancing bone mineral density and reducing phospholipid peroxidation, underscoring their therapeutic potential in osteoporosis management. Importantly, these compounds are derived from ginger, a traditional Chinese medicine known for its dual role as both food and medicine. Recent clinical trial has further supported ginger’s potential anti-osteoporosis activity42. Additionally, prior study has documented the anti-ferroptotic effect of 6-Gingerol in a diabetic cardiomyopathy model43. Although the underlying mechanism in this study are far than well-grounded, their results substantiate our discovery on the attenuating effect of Gingerols on phospholipid peroxidation. Collectively, our findings on 6- and 8-Gingerols not only highlight their promise as new anti-osteoporosis agents, but also provides a scientific foundation for the traditional use of ginger in addressing bone-related disorders.
Another significant finding in this study pertains to the differential effects of vitamin E on bone mass in normal mice versus OVX mice. Initially, we investigated vitamin E as a pharmacological inhibitor of phospholipid peroxidation, inspired by previous studies44,45,46,47,48. As anticipated, the inhibition of phospholipid peroxidation by vitamin E effectively mitigated OVX-induced bone loss. Previous researches have consistently demonstrated the beneficial effects of vitamin E in osteoporotic animal models49,50 and epidemiological studies51,52,53,54,55, primarily attributing its anti-osteoporotic properties to its antioxidative function. In contrast to OVX mice, the same dosage of vitamin E slightly reduced BMD in normal mice, with statistically significant differences observed. Our findings are consistent with those of Fujita et al., who reported notable effects of vitamin E in normal animals16. Their study indicated that vitamin E promotes osteoclast fusion through the p38α/Mitf/Tm7sf4 pathway rather than through its antioxidative activity. The divergent effect of vitamin E on bone mass in normal and osteoporotic conditions may be attributed to its distinct performances on osteoclasts and osteoblasts, which operate differently under physiological and pathological conditions56,57. While originally intended as a tool to explore phospholipid peroxidation in bone formation dysfunction, our unexpected findings underscore the dualistic effect of vitamin E on bone metabolism. These findings highlight the therapeutic potential of vitamin E in osteoporosis, but cautioning against indiscriminate use in healthy individuals.
In conclusion, this study provides robust evidence that GPX4 deficiency exacerbates bone loss and disrupts bone formation, ultimately contributing to the pathogenesis of osteoporosis, as summarized in Fig. 8. Furthermore, the findings highlight the critical role of phospholipid peroxidation in the underlying mechanisms of osteoporosis. Consequently, therapeutic strategies targeting the modulation of GPX4 activity and the prevention of phospholipid peroxidation hold significant potential for the treatment of osteoporosis.

Phospholipid peroxidation in osteoblasts plays a pivotal role in postmenopausal osteoporosis, driven by diminished estrogen-regulated GPX4 expression. The lipid peroxidation byproduct 4-HNE disrupts RUNX2 signaling through the degradation of ILK. While the natural GPX4 activators 6- and 8-Gingerols promote osteoblastogenesis and demonstrate significant anti-osteoporotic effects.
Methods
Ethics statement
This study was approved by the Committees of Clinical Ethics in The First Affiliated Hospital of Jinan University. All research was conducted in accordance with relevant guidelines and regulations, and prior informed consent was obtained from all participants. The animal experimental procedures were approved by the Committees of Animal Ethics and Experimental Safety of Jinan University.
Animals
Gpx4-floxed (Gpx4flox/flox) mice were obtained from the Jackson laboratory (#027964, Bar Harbor, Maine, USA). Sp7-Cre mice were generated by the Beijing Biocytogen Co., Ltd (#110131, Beijing, China). Wild-type C57BL/6J female mice were purchased from Guangdong Medical Laboratory Animal Center (Guangzhou, China). The mice were housed in a room at a mean constant temperature (24 ± 2 °C), a relative humidity of 50–60%, and provided free access to standard pellet chow and water under a 12-hour light-dark cycle. The experimental procedures were approved by the Committees of Animal Ethics and Experimental Safety of Jinan University.
Generation of osteoblast-specific Gpx4 knockout mice
To establish the osteoblast-specific Gpx4 knockout mice, the Sp7-Cre mice was firstly constructed (Supplementary Fig. 5a). Specifically, Cre was introduced between the coding sequence of exon2 and 3’UTR of Osx gene. Self-cleaving 2 A peptide was used to let Sp7 and Cre expressed at the same time and at the same level. To avoid disrupting poly A signal of Sp7 and promoter region of downstream gene Aaas, Neo cassette flanked by Frt sites was inserted within the non-conserved region of intron 1. This Sp7-Cre building technique dose not impact the cortical or trabecular bone structures of the mice (Supplementary Fig. 5f, g) and has been extensively utilized by several research groups58,59,60,61. Based on the principle of Cre-LoxP system, Gpx4flox/flox mice were mated with Sp7-Cre mice to obtain the Gpx4flox/-Cre mouse generation. These heterozygotes were further crossed with Gpx4flox/flox mice to produce osteoblast-specific Gpx4 knockout mice Gpx4flox/flox Sp7-Cre (Gpx4OBs−/−). The genotype of mice was confirmed by tail identification test, western blot and qPCR. GPX4 expression was reduced in bone tissues but not in other major tissues of Gpx4OBs−/− mice (Supplementary Fig. 5b–d), suggesting the successful generation of Gpx4OBs−/− transgenic mouse model. The body weight of Gpx4OBs−/− mice was monitored as well (Supplementary Fig. 5e).
OVX osteoporotic mouse model and drug treatments
Ovariectomy surgery was employed to establish osteoporotic model in three-month mice. One week after ovariectomy surgery, mice were daily orally received the administration of 6-Gingerol (25 mg/kg), 8-Gingerol (25 mg/kg), or Vitamin E (α-tocopherol, 100 mg/kg, #S0079, Beyotime, China) for 45 days. For ferrostatin-1 (Fer-1) treatment, the drug was intraperitoneally injected to OVX-operated mice (10 mg/kg) for one month. At the end of all treatments, bilateral femurs and tibias were harvested for subsequent analysis.
RSL3-established lipid peroxidation in murine bones and 8-Gingerol treatment
To investigate the role of GPX4 and phospholipid peroxidation in osteoporosis pathogenesis, a tool chemical RSL3 (20 mg/kg, i.p.) (#S8155, Selleck, USA) was injected into the nine-months old mice every other day for two months, followed by collection of long bone tissues for further measurement. Another bath of animals was utilized to assess the effect of 8-Gingeorl on RSL3-induced bone loss. Mice were treated with RSL3 (20 mg/kg, i.p.) 8-Gingerol (25 mg/kg or 50 mg/kg, i.g.) (#PCS0532, Phytochemical pure biotechnology, China) every other day for one month. 8-Gingerol was pretreated one hour prior to RSL3 administration.
Collection of human specimens
We obtained bone specimens from female patients with fracture aged between 60 and 90 years old from The First Affiliated Hospital of Jinan University. We included patients who had fracture due to falling without obvious violence (inclusion criteria), while excluded patients who had malignancy, diabetes or other severe diseases in the previous five years (exclusion criteria). Totally, we collected 18 clinical bone samples. The study protocol was approved by the Committees of Clinical Ethics in The First Affiliated Hospital of Jinan University.
Primary osteoblasts isolation and in vitro osteoblastic differentiation
Primary osteoblasts were isolated from calvariae of three-day old C57BL/6J mice according to the previous report62. Briefly, calvariae were washed twice with PBS and then subjected to four cycles of digestion with 4 mL pancreatin and collagenase mixture at 37 °C. The supernatants were collected and centrifuged at 300 × g for 5 minutes, and osteoblasts were re-suspended in α-MEM with 10% FBS. The medium was changed one day after osteoblast isolation. To induce osteoblastic differentiation, MC3T3-E1 cells or mouse primary osteoblast cells were cultured in 6-cm cell dishes or 12-well plates with medium containing 50 μg/ml of ascorbic acid and 10 mM β-glycerophosphate. The medium was supplemented with RSL3 and changed every three days. The cells were harvested for qPCR, Western blot analysis or ALP staining after seven-day culture.
ALP and ARS staining
A BCIP/NBT Alkaline Phosphatase Color Development Kit (#C3206, Beyotime, China) was used for ALP staining according to the manufacturer’s instruction. In brief, cells in plates were washed with PBS three times for 5 minutes each and then fixed with 4% paraformaldehyde for 15 minutes. At the end of fixing, cells were rinsed with deionized water for 3–5 times for 5 minutes each, followed by incubation of BCIP/NBT solution for 24 hours in the dark. BCIP/NBT solution was discarded and cells were rinsed with deionized water once or twice.
To perform ARS staining, MC3T3-E1 cells or mouse primary osteoblast cells were fixed in 70% ice-cold ethanol for one hour, and subsequently rinsed with deionized water and stained with 40 mM Alizarin red S (#A5533, Sigma) at pH 4.0 with gentle agitation. Cells were rinsed five times with deionized water and once with PBS for 15 minutes while gently shaking.
qPCR analysis
Total RNA from bone tissue or cells was extracted with TRIzol reagent (#DP424, Tiangen biotech) and complementary DNAs (cDNAs) were synthesized using One-Step gDNA Removal and cDNA Synthesis SuperMix kit (#AT311, Transgen biotech) according to the manufacturer’s instruction. qPCR reactions were performed on an Applied Biosystems using Top Green qPCR SuperMix kit (#AQ131, Transgen biotech) to measure expression levels. The following primers were used as shown in the Supplementary Table 1, and Gapdh was used for internal control.
Western blot and Co-IP assay
Bone tissue and cells were lysed in lysis buffer (#P0013, Beyotime) on ice for 30 minutes. Protein fraction was obtained by centrifugation at 12,000 × g at 4 °C for 10 minutes, and the protein concentration was monitored by BCA protein assay (#23225, Thermofisher scientific). The protein was denatured with loading buffer for 10 minutes and separated by SDS-PAGE gel and transferred to PVDF membranes. The membranes were incubated with primary antibodies overnight at 4 °C, followed by incubation with anti-mouse or anti-rabbit secondary antibodies for 2 hours at room temperature, and visualized using Multi-Imaging System (#Tanon 5200, Tanon Science & technology). Primary antibodies used were: 4-HNE (#ab46545, Abcam, 1:1000), GPX4 (#ab125066, Abcam, 1:1000), β-actin (#FD0060, Fude biological technology, 1:5000), GAPDH (#FD0063, Fude biological technology, 1:5000), RUNX2 (#12556, Cell Signaling Technology, 1:1000), ALP (#sc-365765, Santa Cruz; #PA5-106391, Thermofisher Scientific, 1:1000), OCN (#AB10911, Sigma-Aldrich, 1:1000), ILK (#3856, Cell Signaling Technology, 1:1000), Ubiquitin (#sc-8017, Santa Cruz, 1:1000), Flag (#14793, Cell Signaling Technology, 1:1000), ERβ (#sc-390243, Santa Cruz, 1:500), and ERα (#sc-787, Santa Cruz, 1:500).
To confirm the protein-protein interaction, Co-IP assay was performed. MC3T3-E1 cells were treated with RSL3 for 24 hours and protein fraction was prewashed using protein A/G beads for 4 hours at 4 °C. Subsequently, primary antibodies (anti-4-HNE or anti-ILK), or normal mouse IgG (#sc-2025, Santa Cruz), normal rabbit IgG (#2729S, Cell Signaling Technology) were incubated for overnight and then added protein A/G beads for another 4 hours on the next day. After centrifugation at 12,000 × g for 10 minutes, the immune complexes were washed 3 times with the cold lysis buffer and eluted with SDS loading buffer. Finally, the complexes were denatured at 100 °C for 10 minutes and loaded onto gel for western blot analysis.
Measurement of malonaldehyde level
Bone tissues were subjected to lysis in a buffer solution on ice for 30 minutes, followed by centrifugation at 12,000 × g and for 10 minutes. The protein concentration of the supernatant was quantified using BCA protein assay. The malonaldehyde content was measured by a commercial assay kit (#S0131, Beyotime) according to the manufacturer’s protocol.
ALP activity measurement
Bone tissues were lysed with lysis buffer on ice for 30 minutes, and centrifuged at 12,000 × g at 4 °C for 10 minutes to collect the supernatant. ALP activity was determined by a commercial assay kit (#P0321, Beyotime) following the manufacturer’s protocol.
LC-MS/MS-based phospholipidomics analysis of phospholipids
The extraction of lipids and phosphorus quantification for bone tissue was based on the Folch procedure63. Deuterated PE 16:0-d31/18:1, deuterated PC 16:0-d31/18:1, and deuterated PI 16:0-d31/18:1 were used as internal standard substance. The phospholipids were separated by a Dionex UltiMate 3000 DGLC standard system at a flow rate of 0.2 mL/min on normal-phase column (Luna 3 μm, Silica, 150 × 2.0 mm, Phenomenex) followed by MS analysis with Q-Exactive Hybrid Quadrupole-Orbitrap mass spectrometer (Thermofisher Scientific). The column temperature was maintained at 40 °C, and the analysis was conducted using gradient solvents (A and B) consisting of 10 mM ammonium formate. Solvent A was composed of isopropanol/hexane/water (285:215:5, v/v/v), and solvent B was contained with isopropanol/hexane/water (285:215:40, v/v/v). All solvents utilized were LC-MS grade. The gradient elution procedure was executed in accordance with the following program: 0 minute, 10% B; 20 minute, 32% B; 30 minute, 70% B; 32 minute, 100% B; 58 minute, 100% B; 60 minute, 10% B; 75 minute, 10% B. Negative ion mode was selected for the analysis, with a resolution of 70,000 for full MS and 17,500 for MS2 in data-dependent scan mode. The scan range for MS analysis was set at m/z400-1800, with a maximum injection time of 600 ms using one microscan. The capillary spray voltage was set at −3.0 kV, and the capillary temperature was fixed at 320 °C. The S-lens Rf level was set at 60, and a maximum injection time of 200 ms was employed for MS2 (high-energy collisional dissociation) analysis, with normalized collision energy and isolation window set at 24, 1.0 Da. MS data acquired were analyzed with Xcalibur (4.2 SP1, Thermo Fisher Scientific) software to determine relative content of phospholipids.
Molecular docking
Molecular docking was conducted using Accelrys Discovery Studio (version 3.5, Accelrys, San Diego, CA, USA) to elucidate the binding modes of compounds with the GPX4 protein. The X-ray crystal structure of GPX4 (PDB ID: 2OBI) was retrieved from the Protein Data Bank and used as the template. Prior to docking, the protein was prepared by removing water molecules and adding hydrogen atoms, followed by the application of the CHARMM force field. The reported allosteric site of GPX430 was identified using Accelrys Discovery Studio. The compounds from the ChemDiv compound library, provided in SDF format, underwent 3D conformer generation and energy minimization using the CHARMM force field. Subsequently, the compounds were docked to GPX4 using the LibDock module with default parameters.
Micro-CT analysis
The micro-computed tomography (micro-CT) system (Skyscan 1176 scanner Aartselaar, Belgium) was used to scan the distal femurs and vertebrae of mice from each group ex vivo. The samples were scanned at a normal pixel of 8 μm at 70 kV and 100 mA x-ray source using Skyscanner software. The images were reconstructed by NRecon software, and the trabecular parameters of femurs were analyzed by CT analyzer software. The following parameters were calculated by segmenting all trabecular bone from each slice for 3D reconstruction.
Hematoxylin and eosin (H&E) staining
The distal femurs of mice from each group were fixed in phosphate-buffered 4% paraformaldehyde (PFA) solution and then decalcified in 10% ethylenediaminetetraacetic acid for 21 days. The tissues were embedded in paraffin and sectioned into 4.5-μm slices. H&E staining following the manufacturer’s protocol. After deparaffinization and rehydration, the slices were stained by hematoxylin and eosin and the morphological study of samples was conducted with an automatic scanning microscope (Precipoint M8, Freising).
Immunohistochemistry
The bone tissue slices were deparaffinized and rehydrated, and then permeabilized with 0.2% triton-X for 15 minutes. The slices were blocked with 5% BSA for one hour and incubated with primary antibodies against GPX4 (#ab125066, Abcam, 1:200), ALP (#sc-365765, Santa Cruz, 1:200), CTSK (#sc-48353, Santa Cruz, 1:200), or 4-HNE (#MA5-27570, Thermofisher, 1:200) at 4 °C overnight. The slices were incubated with either Alexa-Flour 555 and 488 secondary antibodies or HRP-conjugated secondary antibodies on the next day. Images were obtained by an automatic scanning microscope (Precipoint M8, Freising) or the Zeiss Laser Scanning Microscope.
siRNA and plasmid transfection
Cells were cultured in 6-cm dishes till the cell density around 60%. Subsequently, cells were transfected with control siRNA (no silencing) and Gpx4 siRNA or Ilk siRNA using lipofectamine 2000 following the manufacture’s instruction. The sequences of Gpx4 siRNA in the experiment were shown below: AGTTTGACATGTACAGCAA (Gpx4 siRNA-1), GGAAATGGATGAAAGTCCA (Gpx4, siRNA-2); GCACCAATTTCGTTGTGGA (Ilk siRNA-1), GTTCGAGACTGGAGTACAA (Ilk siRNA-2).
Cells were seeded in 6-wells plate and grew until they reached approximately 60% confluence. Next, they were transfected with ERβ or ILK-Flag plasmid using NeofectTM DNA transfection reagent. After 24-hour transfection, cells were harvested for further detection.
Cell viability assay
Cells (8000 cells per well) were seeded in 96-well plates and exposed to RSL3 for 24 hours. Cell viability was then assessed by MTT assay. Briefly, 5 mM MTT solution was added to each well and incubated at 37 °C for 4 hours. The supernatant was removed and 200 μl of DMSO was added to dissolve the formazan crystals. The absorbance was measured at 570 nm by using spectrophotometer.
Paf1 cells were treated with tamoxifen (10 μM) for 48 hours to induce GPX4 knockout. These cells were administrated with or without 6- and 8-Gingerols at dose ranging from 3.125 to 50 μM. After 48 hours treatment, cell viability was assessed by MTT assay.
Estrogen treatment
MC3T3-E1 cells were seeded in 6-well plates and cultured for 24 hours. To evaluate the effect of estrogen (17β-estradiol, #S1709, Selleck) on GPX4 expression in a time-dependent manner, the cells were treated with or without 1 μM 17β-estradiol for 3, 6, 12, or 24 hours. At the indicated time points, the cells were harvested for qPCR analysis. To assess the effect of estrogen on GPX4 expression in a dose-dependent manner, the cells were treated with various concentrations of 17β-estradiol (0.01, 0.1, and 1 μM) for 24 hours. After treatment, the cells were collected for western blot analysis. To confirm the role of estrogen in GPX4 expression, the cells were treated with 1 μM 17β-estradiol with or without 1 μM fulvestrant (#HY-13636, Med Chem Express), an estrogen antagonist, for 24 hours. Subsequently, the cells were harvested for western blot analysis.
Toluidine blue staining
The femurs were harvested and fixed in a 4% PFA solution for 24 hours, followed by dehydration in an ascending alcohol series. Subsequently, they were embedded in glycol methacrylate resin and polymerized. The tissues were sectioned at a thickness of approximately 15 μm. The sections were incubated in a 0.1% toluidine blue solution prepared in a 0.1 M sodium acetate buffer for 3 to 10 minutes. The ratio of osteoblast surface to bone surface (Ob.S/BS) was evaluated using ImageJ.
Von Kossa staining
The femurs were harvested and fixed into 4% PFA solution for 24 hours. Von kossa staining was performed according to a previously publication59. Briefly, tissues were embedded in light-cured resin and sectioned at a thickness of approximately 15 μm. The sections were treated with 100 μl of 2% silver nitrate solution and exposed to strong light until the mineralized bone turned black. The sections were then rinsed with distilled water and briefly immersed in 5% sodium thiosulfate before staining with van Gieson working solution for 5 minutes. The sections were mounted with permount medium and examined under a light microscope.
Luciferase reporter experiment
To investigate the regulation of ERβ on GPX4 transcription, Hek293 cells were co-transfected with GPX4 luciferase reporter (GPX4-luc, 100 ng), pRL-nTK (a renilla luciferase reporter, 10 ng) and ESR2-Flag plasmid using NeofectTM DNA transfection reagent for 24 hours. Cells lysates were prepared in passive lysis buffer and luciferase activities were determined by Dual-Luciferase Reporter Assay system (Promega, Madison). Firefly luciferase activity was normalized to renilla luciferase activity.
To explore the role of 6- and 8-Gingerols on GPX4 expression, Hek293 cells were transfected with GPX4 luciferase reporter (GPX4-Luc, 100 ng) and pRL-nTK (a renilla luciferase reporter, 10 ng) using NeofectTM DNA transfection reagent for 24 hours. These cells were treated with Gingerols (25 μM) for 24 hours. Cells lysates were prepared in passive lysis buffer and luciferase activities were determined by Dual-Luciferase Reporter Assay system (Promega, Madison). Firefly luciferase activity was normalized to renilla luciferase activity.
Electrophoretic mobility shift assay
Electrophoretic mobility shift assay (EMSA) was conducted using a chemiluminescent EMSA kit (#GS009, Beyotime) following its protocol. Nuclear protein was incubated with biotin-labeled probe in EMSA binding buffer (unlabeled probe or unlabeled mutated probe was used for competitive experiments). The mixture was subjected to nondenaturing polyacrylamide gel electrophoresis, and transferred to a Hybond-N+ membrane. After cross-linking, blocking, washing, and balancing, the membrane was incubated with enhanced chemiluminescent and visualized by Multi-Imaging System (#Tanon 5200, Tanon Science & technology). The oligonucleotide sequences are shown below (labeled or unlabeled probe sequence, GGCAGGCTCATAGCACCCCCCAAGTCAACCCGGCCCTGA; unlabeled mutated probe sequence, GGCAAACTCGCAGCATTTCCCAAATCAGCCCAACCCTGA).
ChIP analysis
ChIP assay was performed using a SimpleChIp plus Enzymatic Chromatin IP kit according to the manufacturer’s instructions (#9004, Cell Signaling Technology). Briefly, Hek293 cells were transfected with ESR2-Flag plasmid or related vector for 24 hours, followed by crosslinked with 1% formaldehyde for 15 minutes at room temperature. The crosslinked chromatin was digested with micrococcal nuclease and immunoprecipitated (IP) with anti-ERβ antibody or normal IgG (a negative control) overnight at 4 °C. The IP complex was de-crosslinked at 65 °C for 4 hours. The DNA was purified and analyzed by qPCR with specific primers. The primers of ESR2 were: forward AGAAACCAAACCCCCTCGAC; reverse GGACTTGTTGTTGGCGACTG. The results were normalized to the input value.
Bottom-up proteomics for 4-HNE adducts analysis
To examine the protein modification by 4-HNE, MC3T3-E1 cells were exposed to RSL3 for 24 hours and then lysed to obtain the protein fraction. The protein fractions were prewashed by the protein A/G beads at 4 °C for 2 hours to remove nonspecific binding proteins and then incubated with anti-4-HNE primary antibody or normal mouse IgG antibody at 4 °C overnight on a shaker. The protein bound to 4-HNE were eluted with a buffer containing 6 M urea, 200 mM DTT, and 500 mM IAA in the dark at room temperature for 30 minutes. The eluted proteins were digested with trypsin (1 μg/μL) in the presence of 2 M urea, 1 mM CaCl2 at 37 °C for 16 hours. The resulting peptides were purified by ODS C18 SPE column (Agilent) and analyzed by an EASY-nLC 1200 chromatography system (Thermo Scientific, Waltham, MA) coupled to an Fusion Lumos instrument (Thermofisher Scientific, San Jose, CA). Peptides were separated by reversed-phase chromatography using a nano-LC column (PepMap RSLC C18, 150 μm × 25 cm, 1.9 μm particles, Thermo Scientific) at a flow rate of 600 nL/min using linear gradients of solvent A (0.1% formic acid in H2O) and solvent B (0.1% formic acid, 80% ACN in H2O). The gradient elution procedure was executed in accordance with the following program: 0 minute, 5% B; 11 minute, 13%B; 32 minute, 32% B; 46 minute, 48 % B; 47 minute, 95% B; 60 minute, 95% B. The data dependent mass spectrometer acquisition was operated in top-speed mode with the following settings: MS1 scan was acquired from 350 to 1500 m/z with a resolution of 120 k, the auto gain control (AGC) was set to 8e5, and the maximum injection time was set to 50 ms. MS2 scans were performed at a resolution of 15 k with an isolation window of 1.6 m/z, the higher collision energy dissociation (HCD) collision energy was set to 31%, the AGC target was set to 5e4, and the maximal injection time was set to 100 ms.
Raw data were automatically processed by the Sequest HT engine of the Proteome Discoverer v2.4 (Thermofisher Scientific) against a database of Uniport-Mus musculus with the following parameters: 1) enzyme: trypsin; 2) fixed modification: carbamidomethylation; 3) variable modifications: 4-Hydroxynonenal, 156.1 Da; 4) peptide mass tolerance: 10 ppm; 5) MS/MS mass tolerance: 0.02 Da; 6) peptide charges: 1 + , 2 + , and 3 + ; 7) instrument: ESI FT; and 8) allow up to two missed cleavage. The occurrence of false positives was estimated by running searches using the same parameters against decoy databases. The false discovery rate for proteins and peptides determination was set to 0.01. At least two unique peptide identifications were required for each protein.
Cellular thermal shift assay
To assess the effect of 6-, 8-Gingerol on GPX4 binding, cells were lysed by three cycles of freeze-thawing in liquid nitrogen and then divided into two equal aliquots. One aliquot was incubated with 6-, 8-Gingerol (10 μM) and the other aliquot with DMSO as a control for 1 hour at 37 °C. The lysates were then subjected to different heat shock temperatures (40-70 °C) for 4 minutes each, followed by 4 minutes of cooling at room temperature. The samples were centrifuged at 12, 000 × g for 10 minutes at 4 °C to separate the soluble and insoluble fraction. The soluble fractions were collected and analyzed by western blot.
Microscale thermophoresis assay
To measure the binding affinity of 6- or 8-Gingerol to GPX4, the compounds were dissolved in microscale thermophoresis assay buffer (50 mM HEPES, 150 mM NaCl, 10 mM MgCl2, 0.05 %Tween-20, pH 7.0). The compound and GPX4 enzyme mixtures were incubated at room temperature for 30 minutes and then subjected to microscale thermophoresis (NanoTemper Monolith Instrument NT.115). The data were analyzed by MO Affinity Analysis v2.3.
Immunofluorescence staining
To detect the localization of 4-HNE and ILK in cells, immunofluorescence staining was performed as follows. Cells were fixed with 4% PFA solution for 15 minutes at room temperature and then permeabilized with 0.2% triton-X for 15 minutes. After blocking with 5% BSA for 1 hours, cells were incubated with anti-4-HNE and anti-ILK primary antibodies (1:100) at 4 °C overnight. The next day, cells were washed and incubated with Alexa-Flour 555 and 488 secondary antibodies (1:300) at room temperature for 2 hours. The nuclei were stained with DAPI (1:1000) at room temperature for 10 minutes. The samples were imaged by a confocal laser scanning microscope (LSM 880 with AiryScan, Carl Zeiss).
Assessment of GPX4 enzyme activity
To evaluate the effect of 6-Gingerol or 8-Gingerol on GPX4 activity, the following procedure was performed. Hek293 cells were transfected with the GPX4-overexpression plasmid for 24 hours and then lysed to obtain crude GPX4 enzyme. 6- or 8-Gingerol was dissolved in the reaction buffer (100 mM Tris-HCl, 5 mM NEDTA, 3 mM GSH, 1.5 mM NADPH, 2 μM PEOOH, 0.1%Triton X-100, pH 7.4) to different concentrations (100 μM, 200 μM or 400 μM). The compound and enzyme mixtures were incubated at room temperature for 30 minutes in the presence or absence of RSL3 (5 μM) and then quenched with chloroform: methanol (2:1, v/v) solution. The lipids were extracted by the Folch method and separated by a Dionex UltiMate 3000 DGLC standard system at a flow rate of 0.35 mL/min on normal-phase column (Luna 3 μm, Silica, 150 × 2.0 mm, Phenomenex) followed by MS analysis with Q-Exactive Hybrid Quadrupole-Orbitrap mass spectrometer (Thermofisher Scientific). The column temperature was maintained at 40 °C, and the analysis was conducted using gradient solvents (A and B) consisting of 10 mM ammonium formate. Solvent A was composed of isopropanol/hexane/water (285:215:5, v/v/v), and solvent B was contained with isopropanol/hexane/water (285:215:40, v/v/v). All solvents utilized were LC-MS grade. The gradient elution procedure was executed in accordance with the following program: 0 minute, 10% B; 5 minute, 45% B; 6 minute, 100% B; 13 minute, 100% B; 13.5 minute, 10% B; 18 minute, 10% B. Negative ion mode was selected for the analysis, with a resolution of 70,000 for full MS scan. The scan range for MS analysis was set at m/z 400–1800, with a maximum injection time of 600 ms using one microscan. The capillary spray voltage was set at −3.0 kV, and the capillary temperature was fixed at 320 °C. The S-lens Rf level was set at 60.
FENIX assay
Liposomes were prepared from egg phosphatidylcholine (egg PC, #840051 P, Sigma-Aldrich) in phosphate-buffered saline (PBS, 10 mM, Chelex-100 treated, pH 7.4) and extruded to 100 nm using an Avestin LiposoFastmini extruder equipped with a 100 nm polycarbonate membrane, based on previous reports64,65,66. Subsequently, 2 mM liposomes (from the aforementioned suspension) and 1 μM STY-BODIPY (#27089, Cayman) were added to a black 96-well plate, and the final volume was adjusted to 294 μL using PBS. The plate was incubated at 37 °C with shaking for 3 minutes. Then, 4 μL of the initiator (DTUN, 0.2 mM in EtOH, #GC48650, GLPBIO) and 2 μL of the stock solutions of PMC, 6-Gingerol, or 8-Gingerol were added to the wells of the 96-well plate. The plate was incubated for an additional 3 minutes at 37 °C in a BioTek Synergy H1 plate reader. Reaction progress was monitored by fluorescence (λex = 488 nm, λem = 518 nm).
4-HNE level detected by ELISA
Bone tissues were homogenized in cold PBS and centrifuged at 12, 000 × g at 4 °C for 10 minutes to collect the supernatant. The protein concentration was monitored by BCA protein assay (#23225, Thermofisher scientific), and the content of 4-HNE was quantified using a mouse 4-HNE ELISA Kit (FineTest Biotech, EM1583). All samples were normalized to protein content.
LC-MS detection of RSL3 level in bone tissues
To investigate RSL3 metabolism in bone tissues, two-month-old mice were intraperitoneally administered RSL3 at a dose of 20 mg/kg. Bone tissues were collected at specific time points, including 0.5, 1, 2, 4, 8, 12, and 24 hours post-administration. Prior to homogenization, the bone tissues were thoroughly washed with cold PBS to remove the majority of bone marrow cells. The tissues were then homogenized in saline, and protein concentration was determined using a BCA assay. For LC-MS sample preparation, the homogenate was mixed with 1000 μL of acetonitrile and vortexed thoroughly. The mixture was centrifuged at 12,000 × g for 20 minutes at 4 °C, and the supernatant was collected and dried under vacuum. The residue was reconstituted in 50 μL of methanol, and after a second centrifugation at 12,000 × g for 20 minutes at 4 °C, the supernatant was subjected to LC-MS analysis. RSL3 detection was carried out using a Waters I-Class system coupled with an AB SCIEX 7500 Q-Trap mass spectrometer. Chromatographic separation was achieved using reversed-phase column (1.8 μm, Premier HSS T3, 100 × 2.1 mm, Waters Acquity). The mobile phases were consisted of water (A) and acetonitrile (B), with the following elution gradient: 0 min, 5% B; 4.0-5.5 min, 95% B; 6.0–8.5 min, 5% B. The column temperature was maintained at 40 °C, with an injection volume of 2 μL. Mass spectrometry was performed in negative ion mode with multiple reaction monitoring. The ion source gas 1 and gas 2 were set at 35 and 70, respectively, with a curtain gas of 40, an ion source temperature of 400 °C, and a spray voltage of -3.0 kV. RSL3 content was expressed as ng/mg of protein.
The RSL3 content in the bone tissues of mice that underwent a two-month administration of RSL3 (20 mg/kg) was also measured using the aforementioned method.
Detection of RSL3-bound GPX4 in bone tissues
Two-month-old mice were administered RSL3 or RSL3-Alkyne intraperitoneally at doses of 20 mg/kg or 50 mg/kg for 1 hour. Following treatment, bone tissues were harvested and rinsed thoroughly with cold PBS to remove most of the bone marrow cells. The tissues were then homogenized in lysis buffer, and protein concentrations were standardized to 1 mg. A Click chemistry reaction was subsequently conducted using a reaction mixture containing 100 mM Tris((1-benzyl-4-triazolyl)methyl)amine, 1 M Tris(2-carboxyethyl)phosphine hydrochloride, 1 M CuSO4, and 100 mM Bio-N3. The reaction was carried out at room temperature for 2 hours. Post-reaction, proteins were precipitated using ice-cold acetone. The precipitate was dissolved in 1% SDS-PBS solution and incubated with beads (#29202, Thermofisher) at 4 °C overnight to pull down proteins in the bone tissues. After incubation of beads, the precipitate was sequentially washed with 1 mL of 1% SDS-PBS solution, 0.1% SDS-PBS solution, and PBS twice each on the second day. The precipitate was collected following centrifugation and incubated with 2 × loading buffer at 100 °C for 10 minutes. After a second centrifugation, the supernatant was subjected to western blot analysis to assess GPX4 expression levels in the bone tissues. The input control was the part of supernatant before Click chemistry reaction-pulldown assay. The western blot assay was consistent with the regular protocol.
Statistical analysis
Statistical analysis was performed using IBM SPSS Statistics 25.0 (SPSS Inc. USA). The Levene test was applied to check the assumptions of homogeneity of variance and normality of distribution. Data were presented as means ± SD or means ± SEM of unpaired two-tailed t-test, one-way ANOVA or two-way ANOVA. P < 0.05 is considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available within the main text and its Source Data file. The relevant proteomics data have been deposited to the ProteomeXchange Consortium via the MassIVE (https://proteomecentral.proteomexchange.org/ui) partner repository with the data set identifier PXD058306. LC-MS data of phospholipids analysis have been deposited to the MassIVE (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) with the data set identifier MSV000096539. Source data are provided with this paper.
References
Manolagas, S. C. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev 31, 266–300 (2010).
Hendrickx, G., Boudin, E. & Van Hul, W. A look behind the scenes: the risk and pathogenesis of primary osteoporosis. Nat Rev Rheumatol 11, 462–474 (2015).
Riegger, J., Schoppa, A., Ruths, L., Haffner-Luntzer, M. & Ignatius, A. Oxidative stress as a key modulator of cell fate decision in osteoarthritis and osteoporosis: a narrative review. Cell Mol Biol Lett 28, 76 (2023).
Agidigbi, T. S. & Kim, C. Reactive Oxygen Species in Osteoclast Differentiation and Possible Pharmaceutical Targets of ROS-Mediated Osteoclast Diseases. Int J Mol Sci 20 (2019).
Reid, I. R. & Billington, E. O. Drug therapy for osteoporosis in older adults. Lancet 399, 1080–1092 (2022).
Almeida, M. et al. Estrogens and Androgens in Skeletal Physiology and Pathophysiology. Physiol Rev 97, 135–187 (2017).
Jiang, X., Stockwell, B. R. & Conrad, M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol 22, 266–282 (2021).
Sun, J. et al. Midbrain dopamine oxidation links ubiquitination of glutathione peroxidase 4 to ferroptosis of dopaminergic neurons. J. Clin. Investig. 133 (2023).
Ma, X. H. et al. ALOX15-launched PUFA-phospholipids peroxidation increases the susceptibility of ferroptosis in ischemia-induced myocardial damage. Signal Transduct Target Ther 7, 288 (2022).
Tu, L. F. et al. GPX4 deficiency-dependent phospholipid peroxidation drives motor deficits of ALS. J Adv Res 43, 205–218 (2023).
Palmieri, M. et al. Neutralization of oxidized phospholipids attenuates age-associated bone loss in mice. Aging Cell 20, e13442 (2021).
Jiang, Z. et al. Ferroptosis in Osteocytes as a Target for Protection Against Postmenopausal Osteoporosis. Adv Sci (Weinh) 11, e2307388 (2024).
Klein, R. F. et al. Regulation of bone mass in mice by the lipoxygenase gene Alox15. Science 303, 229–232 (2004).
Yang, Y. et al. Targeting ferroptosis suppresses osteocyte glucolipotoxicity and alleviates diabetic osteoporosis. Bone Res 10, 26 (2022).
Ingold, I. et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 172, 409–422.e421 (2018).
Fujita, K. et al. Vitamin E decreases bone mass by stimulating osteoclast fusion. Nat Med 18, 589–594 (2012).
Yant, L. J. et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med 34, 496–502 (2003).
Deroo, B. J. & Korach, K. S. Estrogen receptors and human disease. J Clin Invest 116, 561–570 (2006).
Zhang, L. et al. HDAC3 aberration-incurred GPX4 suppression drives renal ferroptosis and AKI-CKD progression. Redox Biol 68, 102939 (2023).
Yang, J. et al. Cetuximab promotes RSL3-induced ferroptosis by suppressing the Nrf2/HO-1 signalling pathway in KRAS mutant colorectal cancer. Cell Death Dis 12, 1079 (2021).
Jia, M. et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat Immunol 21, 727–735 (2020).
Cui, Y. et al. Microglia and macrophage exhibit attenuated inflammatory response and ferroptosis resistance after RSL3 stimulation via increasing Nrf2 expression. J Neuroinflammation 18, 249 (2021).
Chen, H. et al. Aspirin promotes RSL3-induced ferroptosis by suppressing mTOR/SREBP-1/SCD1-mediated lipogenesis in PIK3CA-mutant colorectal cancer. Redox Biol 55, 102426 (2022).
Wronski, A., Gegotek, A. & Skrzydlewska, E. Protein adducts with lipid peroxidation products in patients with psoriasis. Redox Biol 63, 102729 (2023).
Andringa, K. K., Udoh, U. S., Landar, A. & Bailey, S. M. Proteomic analysis of 4-hydroxynonenal (4-HNE) modified proteins in liver mitochondria from chronic ethanol-fed rats. Redox Biol 2, 1038–1047 (2014).
Zhai, X. et al. Carbonylation of Runx2 at K176 by 4-Hydroxynonenal Accelerates Vascular Calcification. Circulation 149, 1752–1769 (2024).
Niu, J. et al. Phospholipid peroxidation-driven modification of chondrogenic transcription factor mediates alkoxyl radicals-induced impairment of embryonic bone development. Redox Biol 56, 102437 (2022).
Zhou, Q. et al. Differentially expressed proteins identified by TMT proteomics analysis in bone marrow microenvironment of osteoporotic patients. Osteoporos Int 30, 1089–1098 (2019).
Dejaeger, M. et al. Integrin-Linked Kinase Regulates Bone Formation by Controlling Cytoskeletal Organization and Modulating BMP and Wnt Signaling in Osteoprogenitors. J Bone Miner Res 32, 2087–2102 (2017).
Li, C. et al. Novel Allosteric Activators for Ferroptosis Regulator Glutathione Peroxidase 4. J Med Chem 62, 266–275 (2019).
Cui, Z. et al. Estimation and projection about the standardized prevalence of osteoporosis in mainland China. Arch Osteoporos 15, 2 (2019).
Collaborators, G. B. D. F. Global, regional, and national burden of bone fractures in 204 countries and territories, 1990-2019: a systematic analysis from the Global Burden of Disease Study 2019. Lancet Healthy Longev 2, e580–e592 (2021).
Clynes, M. A. et al. The epidemiology of osteoporosis. Br Med Bull 133, 105–117 (2020).
Ru, Q. et al. Fighting age-related orthopedic diseases: focusing on ferroptosis. Bone Res 11, 12 (2023).
Gao, Z., Chen, Z., Xiong, Z. & Liu, X. Ferroptosis - A new target of osteoporosis. Exp Gerontol 165, 111836 (2022).
Zhong, H. & Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: focusing on mitochondria. Redox Biol 4, 193–199 (2015).
Czyzowska, A. et al. Elevated phospholipid hydroperoxide glutathione peroxidase (GPX4) expression modulates oxylipin formation and inhibits age-related skeletal muscle atrophy and weakness. Redox Biol 64, 102761 (2023).
Spickett, C. M. The lipid peroxidation product 4-hydroxy-2-nonenal: Advances in chemistry and analysis. Redox Biol 1, 145–152 (2013).
Uchida, K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res 42, 318–343 (2003).
Liu, L. et al. Deubiquitinase OTUD5 as a Novel Protector against 4-HNE-Triggered Ferroptosis in Myocardial Ischemia/Reperfusion Injury. Adv Sci (Weinh) 10, e2301852 (2023).
Smith, A. C. et al. Mutations in the enzyme glutathione peroxidase 4 cause Sedaghatian-type spondylometaphyseal dysplasia. J Med Genet 51, 470–474 (2014).
Salekzamani, Y., Shakouri, S. K., Dolatkhah, N., Saleh, P. & Hashemian, M. The effect of ginger and curcumin co-supplementation in postmenopausal women with osteoporosis: a randomised, triple-blind, placebo-controlled clinical trial. J Herb Med 42 (2023).
Wu, S. et al. 6-Gingerol Alleviates Ferroptosis and Inflammation of Diabetic Cardiomyopathy via the Nrf2/HO-1 Pathway. Oxid Med Cell Longev 2022, 3027514 (2022).
Matsushita, M. et al. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med 212, 555–568 (2015).
Kang, R. et al. Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe 24, 97–108.e104 (2018).
Kagan, V. E. et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13, 81–90 (2017).
Hu, Q. et al. GPX4 and vitamin E cooperatively protect hematopoietic stem and progenitor cells from lipid peroxidation and ferroptosis. Cell Death Dis 12, 706 (2021).
Carlson, B. A. et al. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol 9, 22–31 (2016).
Ibrahim, N. et al. Osteoprotective Effects in Postmenopausal Osteoporosis Rat Model: Oral Tocotrienol vs. Intraosseous Injection of Tocotrienol-Poly Lactic-Co-Glycolic Acid Combination. Front Pharmacol 12, 706747 (2021).
Feresin, R. G. et al. Effects of vitamin e on bone biomechanical and histomorphometric parameters in ovariectomized rats. J Osteoporos 2013, 825985 (2013).
Vallibhakara, S. A., Nakpalat, K., Sophonsritsuk, A., Tantitham, C. & Vallibhakara, O. Effect of Vitamin E Supplement on Bone Turnover Markers in Postmenopausal Osteopenic Women: A Double-Blind, Randomized, Placebo-Controlled Trial. Nutrients 13 (2021).
Shi, W. Q. et al. Association of dietary and serum vitamin E with bone mineral density in middle-aged and elderly Chinese adults: a cross-sectional study. Br J Nutr 115, 113–120 (2016).
Shen, C. L. et al. Tocotrienol supplementation suppressed bone resorption and oxidative stress in postmenopausal osteopenic women: a 12-week randomized double-blinded placebo-controlled trial. Osteoporos Int 29, 881–891 (2018).
Michaelsson, K., Wolk, A., Byberg, L., Arnlov, J. & Melhus, H. Intake and serum concentrations of alpha-tocopherol in relation to fractures in elderly women and men: 2 cohort studies. Am J Clin Nutr 99, 107–114 (2014).
Holvik, K. et al. Low serum concentrations of alpha-tocopherol are associated with increased risk of hip fracture. A NOREPOS study. Osteoporos Int 25, 2545–2554 (2014).
Neve, A., Corrado, A. & Cantatore, F. P. Osteoblast physiology in normal and pathological conditions. Cell Tissue Res 343, 289–302 (2011).
Henriksen, K., Bollerslev, J., Everts, V. & Karsdal, M. A. Osteoclast activity and subtypes as a function of physiology and pathology-implications for future treatments of osteoporosis. Endocr Rev 32, 31–63 (2011).
Wang, Y. et al. Inactivation of Ihh in Sp7-Expressing Cells Inhibits Osteoblast Proliferation, Differentiation, and Bone Formation, Resulting in a Dwarfism Phenotype with Severe Skeletal Dysplasia in Mice. Calcif Tissue Int 111, 519–534 (2022).
Sun, Y. et al. The long noncoding RNA lnc-ob1 facilitates bone formation by upregulating Osterix in osteoblasts. Nat Metab 1, 485–496 (2019).
Liu, X. et al. Fam20c regulates the calpain proteolysis system through phosphorylating Calpasatatin to maintain cell homeostasis. J Transl Med 21, 417 (2023).
Liu, J. et al. Increased PLEKHO1 within osteoblasts suppresses Smad-dependent BMP signaling to inhibit bone formation during aging. Aging Cell 16, 360–376 (2017).
Cappariello, A. et al. Osteoblast-Derived Extracellular Vesicles Are Biological Tools for the Delivery of Active Molecules to Bone. J Bone Miner Res 33, 517–533 (2018).
Luo, X. et al. Oxygenated phosphatidylethanolamine navigates phagocytosis of ferroptotic cells by interacting with TLR2. Cell Death Differ 28, 1971–1989 (2021).
Shah, R., Farmer, L. A., Zilka, O., Van Kessel, A. T. M. & Pratt, D. A. Beyond DPPH: Use of Fluorescence-Enabled Inhibited Autoxidation to Predict Oxidative Cell Death Rescue. Cell Chem Biol 26, 1594–1607.e1597 (2019).
Mishima, E. et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature 608, 778–783 (2022).
Wu, Z. et al. Hydropersulfides Inhibit Lipid Peroxidation and Protect Cells from Ferroptosis. J Am Chem Soc 144, 15825–15837 (2022).
Acknowledgements
This study was partly supported by the National Natural Science Foundation of China (T2341004, 82125038 to R.R.H., 82174054, 82321004 to Y.F.L., 82274123 to W.J.D., and 82350003 to X.G.W.), the Natural Science Foundation of Guangdong (2023B1515040016, 2023B0303000026 to Y.F.L., 2021B1515120023 to R.R.H. and 2020A1515110596 to S.H.O.Y.), the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01Y036 to R.R.H.), Guangdong-Hong Kong-Macao Universities Joint Laboratory for the Internationalization of Traditional Chinese Medicine (2023LSYS002), and Guangzhou Key Laboratory of Traditional Chinese Medicine & Disease Susceptibility (2024A03J090) to R.R.H., Science and Technology Program of Guangzhou (202102010116) to Y.F.L., and the Research Fund Program of Guangdong Provincial Key Laboratory of Speed Capability Research. The authors (R.R.H. and Y.F.L.) also gratefully acknowledge the support of the K.C. Wong Education Foundation. We also thank Prof. Zheng-Qiu Li (Jinan University, Guangzhou, China) for generously providing RSL3-Alkyne probe.
Author information
Authors and Affiliations
Contributions
Q.Y.Z. conducted the most of the experiments, acquired and analyzed data, and prepared the figures, tables, and manuscript. H.B.G. performed a part of LC-MS/MS-based experiments, acquired and analyzed data. M.Y.J. was responsible for part of the in vivo experiments, including the evaluation of 6-/8-Gingerol. F.J.J. isolated primary osteoblasts, and performed osteoblast staining, toluidine blue staining, and part of micro-CT analysis. G.W. performed the molecular docking analysis. C.Y.Y. assessed the toxicity and anti-ferroptosis effects of 6-/8-Gingerol in vitro. X.L. contributed to a portion of the LC-MS/MS-based experiments. W.Y.S. assisted in conducting the LC-MS/MS-based experiments and analyzing data. Y.F.C. supported the LC-MS platform and consulted regarding the data analysis. M.J.L. conducted the micro-CT analysis of Osx-cre mice, analyzed the associated data. G.L.J. and H.J.W. collected the clinical samples. S.H.O.Y., Y.P.W., W.J.D., L.L., X.X.S. and H.K. partly advised the research. X.G.W. supervised and advised the project. R.R.H. and Y.F.L. conceived and designed the research, revised the manuscript. All of the authors have approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Elena Ambrogini, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, QY., Gong, HB., Jiang, MY. et al. Regulation of enzymatic lipid peroxidation in osteoblasts protects against postmenopausal osteoporosis. Nat Commun 16, 758 (2025). https://doi.org/10.1038/s41467-025-55929-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-55929-4
This article is cited by
-
ACT001 improves OVX-induced osteoporosis by suppressing the NF-κB/NLRP3 signaling pathway
Molecular Medicine (2025)
