
Abstract
Electrochemiluminescence (ECL) is a light-emitting process in which the stability of electrochemically generated radicals has a crucial impact on the efficiency and durability of excited state generation. Therefore, deciphering a relationship between radical stability and ECL performance is highly appealing. In this work, three sp2 carbon-conjugated covalent organic framework (COF) reticular nanoemitters compositing of same pyrene luminophores but different acrylonitrile linkers are designed with progressive electron affinities, named as CN-COF-1, 2, and 3. By precisely modulating the electron affinity of CN-COFs, a volcano relationship between ECL and radical stability is discovered with 78 folds enhancement in ECL intensity. Density functional theoretical calculations indicate that CN-COF-2 exhibits moderate radical stabilization capacity as well as efficient electron transport between the pyrene cores, facilitating ECL generation. Significantly, the appropriate radical stability of CN-COF-2 not only achieves the self-enhanced cathodic ECL but also promotes durability of the ECL intensity. The rational regulation of radical stability paves the way for developing efficient reticular nanoemitters and decoding the ECL fundamentals.
Similar content being viewed by others
Introduction
Electrochemiluminescence (ECL), as an analytical method, exhibits great potential to be widely applied in clinical and biological research due to high sensitivity, low background, and good reliability1,2,3,4. During the light-emitting process, the excited states are produced through electron transfer among the electrochemically generated radicals in the vicinity of working electrodes5,6. However, the possible decomposition of radicals may cause the chemical irreversibility of redox processes during the ECL generation, leading to low ECL efficiency7. Thus, several strategies have been employed to enhance the stability of anion/cation radicals. For example, accumulating radicals by pre-reduction or -oxidation electrolysis8,9, improving the emitter radical stability by introducing donor or acceptor groups10, and regulating hydrophobic microenvironment for coreactant radicals11. According to our previous work12, the excessively stable radical could impede the generation of excited states due to the completely separated charges. To mitigate the negative effects caused by the vulnerability or overstability of reactive radicals, it’s highly appealing to establish a relationship between radical stability and ECL generation.
Covalent organic frameworks (COFs) are an emerging class of metal-free crystalline nanoemitters linked by covalent bonds, integrating the advantages of large porosity, regular channels, and conductive reticular structures, which facilitate charge and mass transfer among the skeleton13,14,15. Especially, due to the predesignable topologies and functionalities of COF materials, several strategies have been conducted to boost ECL performance, such as construction of donor-acceptor pairs to stabilize radicals16,17, integration of luminophore and co-reactant units to shorten the distance between anion/cation radicals18,19, and introduction of conjugated linkages for extended π-conjugation to improve charge transfer efficiency among reactive radicals20. Among the COF emitters, sp2c-COFs with acrylonitrile linkers hold an opportunity to investigate the dependence of ECL on radical stability21,22,23. On the one hand, charge transfer between radicals could be accelerated by the full-conjugated skeletons offered by C=C bonds, excluding the inefficiency of charge transfer24. On the other hand, the anion radicals generated during ECL could be stabilized by the electron-withdrawing acrylonitrile linkers, providing a radical stability-dependent ECL generation25. To our best knowledge, there is no relevant report on the relationship between the ECL performance and the electron affinity of sp2c-COFs, attributed to the weak reversibility of the Knoevenagel condensation reaction.
In this work, we designed and synthesized three acrylonitrile-linked covalent organic frameworks (CN-COFs) by cyanomethyl-contained monomers via Knoevenagel condensation (Fig. 1a and Supplementary Figs. 1–3), and investigated their relationship between radical stabilities and ECL behaviors. By means of regulating the electron affinity of acrylonitrile linkers, the radical stability of COF nanoemitters could be finely controlled. As shown in Fig. 1b, the electrostatic potential (ESP) of three COFs illustrated electron-withdrawal capacity of nitrile groups in acrylonitrile linkers, indicating progressive electron affinity of CN-COFs. Compared to CN-COF-1, CN-COF-2 with fewer phenyl units demonstrated a stronger electron affinity to stabilize and accumulate anion radicals, resulting in 6 folds enhancement of ECL intensity. In contrast, CN-COF-3 with double nitrile moieties as electron acceptors, exhibited a 78-fold decrease of ECL signal than CN-COF-2 due to the excessive electron affinity to trap anion radicals, verified by the electron paramagnetic resonance (EPR) spectra and electron doping simulation. Thus, a volcano-shaped relationship between ECL and electron affinity of acrylonitrile linkage was established. Theoretically, CN-COF-2 with one nitrile group exhibited appropriate radical stabilization ability, resulting in efficient intrareticular charge transfer (IRCT) between pyrene moieties. Furthermore, CN-COF-2 achieved durable ECL performance for 600 s as a cathode material. These findings not only demonstrate the potential of CN-COF-2 as a promising metal-free ECL emitter but also provide valuable insights into the strategy of boosting ECL performance through controlling radical stability associated with electron affinity in reticular nanoemitters.

a Schematic illustration of synthesis of CN-COFs composing of the pyrene luminophore and different electron-withdraw acrylonitrile linkers. b ESP distributions of CN-COF-1, CN-COF-2, and CN-COF-3.
Results
CN-COF synthesis and characterization
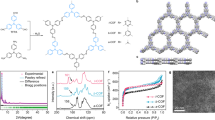
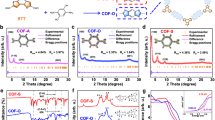
The structural difference of CN-COF-1, 2, and 3 lies in the acrylonitrile linker, in which the CN-COF-1, CN-COF-2, and CN-COF-3 own one nitrile group and one benzene unit, one nitrile group, and two nitrile groups and one benzene unit, respectively. The crystalline structures of three pyrene-based sp2c-COFs were examined via powder X-ray diffraction (PXRD) analysis. As illustrated in Fig. 2a–c, distinct diffraction peaks at 4.1°, 5.0°, and 3.6° were observed, corresponding to the (110) facets of CN-COF-1, CN-COF-2, and CN-COF-3, respectively. Also, diffraction peaks at 8.1°, 10.3°, and 7.3° were observed, assigned to the (220) facets, indicating the well-defined crystalline structures. Meanwhile, Pawley refinements yielded good fits with the experimental PXRD data, where low residual values and acceptable profile differences underscored the high crystallinity of COFs. Furthermore, experimental PXRD patterns of all three COFs matched well with eclipsed AA stacking arrangement. The optimized lattice parameters were a = 36.56 Å, b = 29.34 Å, c = 3.44 Å for CN-COF-1, a = 30.13 Å, b = 23.40 Å, c = 3.43 Å for CN-COF-2, and a = 34.78 Å, b = 35.08 Å, c = 3.50 Å for CN-COF-3. The regular structures brought by high crystallinity and AA stacking arrangement promoted facile charge transfer within the frameworks, thereby augmenting the ECL process26.

The PXRD patterns, Pawley refinements, AA stacking simulation, difference, and Bragg position of (a) CN-COF-1 (Rwp = 10.71%; Rp = 7.49%), b CN-COF-2 (Rwp = 4.22%; Rp = 3.13%), and c CN-COF-3 (Rwp = 5.67%; Rp = 4.57%). d Solid-state 13C CP-MAS spectra of CN-COFs. e N2 sorption isotherms and pore size distribution (inset) of CN-COFs. f HR-TEM image of CN-COF-2.
Subsequently, Fourier transform-infrared (FT-IR) spectra were performed to further verify the successful synthesis of three COFs, where the peak at 1700 cm−1 for aldehyde groups exhibited a notable reduction while the peak at 2248 cm−1 for nitrile groups preserved well as illustrated in Supplementary Figs. 4–6, resulting from a highly efficient polymerization process. Besides, the 13C cross-polarization magic-angle-spinning (CP-MAS) nuclear magnetic resonance (NMR) spectra of three CN-COFs showed clear patterns of nitrile group C atoms at 117 ppm, C atoms attached to nitrile group at 110 ppm as well as the other C atoms of C=C bond at 141 ppm (Fig. 2d)22,27. Next, the chemical stability of CN-COFs was evaluated by comparing the crystal structure change before and after immersion in strong acid (6 M HCl) or base (6 M NaOH) (Supplementary Fig. 7). Based on the PXRD patterns, crystal structures of CN-COFs with robust C=C bonds remained basically intact, verifying the excellent stability of the olefin linkage towards acidic or alkaline environment. The thermogravimetric assessment of three COFs illustrated that all COFs maintained stable up to 350 °C (Supplementary Fig. 8). These findings collectively verified the robustness and thermal resilience of the CN-COFs. Furthermore, the nitrogen adsorption isotherms at 77 K were measured to identify the porous structure of three COFs (Fig. 2e and Supplementary Figs. 9–11). The specific Brunauer-Emmett-Teller (BET) surface areas of three COFs were found to be 727 m2 g−1, 806 m2 g−1, and 638 m2 g−1, where high specific surface areas promoted the accessibility of coreactants. Subsequently, the pore size distribution of three COFs was calculated by NLDFT model, with a similar diameter of 1.6 nm, 1.5 nm, and 1.7 nm, respectively. Obviously, CN-COF-2 and CN-COF-3 exhibited fibrous morphology while CN-COF-1 exhibited granular morphology according to scanning electron microscopy (SEM) imaging (Supplementary Fig. 12). In addition, the layered morphology could be clearly identified by high-resolution transmission electron microscopy (HR-TEM), thus providing further proof of the crystal structures of three COFs (Fig. 2f and Supplementary Figs. 13–15). The above results demonstrated the successful synthesis of three acrylonitrile-linked CN-COFs with high crystalline structures.
Meanwhile, UV-vis diffuse reflection spectra (DRS) were performed, compared to CN-COF-1 and CN-COF-2, CN-COF-3 with double nitrile linkage of highest electron affinity showed apparent redshift of absorbance, indicating its largest donor-acceptor (D-A) contrast versus pyrene unit (Fig. 3a)17,28. The increased electron affinity of acrylonitrile linker in CN-COF-2 was accomplished by reduction of the phenyl units compared with CN-COF-1 while the double amount of embedded nitrile groups in acrylonitrile linker of CN-COF-3 exhibited considerably higher electron affiliation. And the band gaps of CN-COF-1, CN-COF-2, and CN-COF-3 were ascertained in a decreasing order: 2.36 eV, 2.22 eV, and 2.14 eV (Supplementary Fig. 16). Also, the valence band X-ray photoelectron spectroscopy measurement was conducted to determine the position of valence bands for CN-COFs, which were 0.74 V, 0.93 V, and 1.47 V (vs. NHE) (Supplementary Fig. 17), and thus their conduction band maximums (CBMs) could be calculated to be −1.62 V, −1.29 V, and −0.67 V (vs. NHE). The band alignments of CN-COFs were illustrated in Fig. 3b, which were consistent with theoretical results (Supplementary Table 1). The lowest VBM and CBM energy levels of CN-COF-3 indicated its highest electron-accepting ability. Moreover, steady-state fluorescence spectroscopy showed that the emission peak wavelengths of three CN-COFs were 533 nm, 558 nm, and 613 nm (Supplementary Fig. 18), revealing an order consistent with the UV absorption spectra. These complementary analyses identified the successful synthesis of acrylonitrile-linked COFs with progressive electron affinity, providing an emissive platform for investigating the relationship between ECL and radical stability.

a UV-vis DRS spectra and (b) schematic band alignments of CN-COFs. c ECL curves of CN-COFs modified GCEs in 0.10 M PBS (pH = 7.4) containing 20 mM K2S2O8 (PMT = 400 V). Scan rate: 0.10 V s−1. Inset: Magnified ECL curves of three COFs. HOMO-1, HOMO, LUMO, and LUMO + 1 of (d) CN-COF-1, e CN-COF-2, and f CN-COF-3. Blue and Red dotted boxes represent the HOMO and LUMO distribution, respectively.
ECL properties of CN-COFs
The ECL behavior of three CN-COFs was evaluated in presence of potassium persulfate (K2S2O8) as coreactant for cathode ECL measurement (Supplementary Fig. 19)29,30, and the coreactant concentrations and scan rates were optimized (Supplementary Figs. 20, 21). Compared with CN-COF-1, CN-COF-2 exhibited a 6-fold enhancement of ECL intensity (Fig. 3c) due to anion radicals stabilization originated from electron-withdrawing acrylonitrile linker in the COF skeleton, thereby enhancing the ECL efficiency. In contrast, CN-COF-3 exhibited a 78-fold reduction in ECL intensity than CN-COF-2 since the strong electron affiliation of CN-COF-3, thus overly stabilizing anion radicals and impeding the radical annihilation. Therefore, the moderate electron affinity of CN-COF-2 resulted in rational radical stability and hence boosted ECL intensity, indicating its great potential as an ECL emitter in an aqueous solution. In order to investigate the influence of crystallinity, we also synthesized CN-COFs-w with relatively lower crystallinity, whose ECL intensities showcased a certain degree of reduction but still was in consistent with the relationship we previously observed (Supplementary Fig. 22).
According to the CV curve of CN-COF-2 (Supplementary Fig. 23), the anode peak was at +1.26 V, signifying the oxidation of pyrene unit, while the cathode onset potential was at −1.85 V, corresponding to the reduction of pyrene units. ECL transients of CN-COF-2 obtained by stepping pulse were conducted to further investigate stability of anion radicals and cation radicals during the ECL generation process. ECL emissions were generated when pulsed between +1.40 V and −2.20 V, where both anodic and cathodic steps demonstrated efficient ECL emissions, proving the efficient pathway of ECL generation via IRCT between the anion and cation radicals at pyrene units of CN-COF-2 (Supplementary Fig. 24)31.
Progressive radical stability of CN-COFs
Next, the comprehensive investigation of the molecular orbital (MO) distributions of CN-COFs offered invaluable insights into their electrochemical behavior as well as charge transfer characteristics (Fig. 3d–f)32. Specifically, a detailed analysis of their highest occupied molecular orbitals (HOMO), HOMO-1, lowest unoccupied molecular orbitals (LUMO), and LUMO + 1 has been conducted. On the one hand, HOMO and HOMO-1 orbitals of CN-COFs exhibited significant spatial distribution primarily concentrated within the pyrene units. This observed localization could be attributed to the electron-donating propensity inherent to pyrene moieties, thereby hinting at the propensity for cation radical formation at these sites27,33. Conversely, the LUMO of the CN-COFs predominantly localized at the acrylonitrile linkers, due to their electron-accepting nature. Since the comparatively modest electron affinity of CN-COF-1 and CN-COF-2, the LUMO of these frameworks also exhibited discernible localization within the pyrene units. Meanwhile, it could be observed that there existed almost no LUMO distribution at pyrene units in CN-COF-3 owing to its considerably strong electron affinity. Furthermore, the distinct distribution patterns for the LUMO + 1 orbital of CN-COF-3, still predominantly distributed on the acrylonitrile linker, in contrast to those of CN-COF-1 and CN-COF-2, which majorly localized on the pyrene units (Supplementary Fig. 25), indicating the electron-accepting ability of pyrene units during the ECL transient experiments. Overall, the distinct localization of molecular orbitals in CN-COFs verified the distinct electron-withdrawing capacity of the acrylonitrile linker in COF skeleton.
To further elucidate the electron affinity capacity of CN-COFs, the charge density of CN-COFs fragment upon the addition of an electron was illustrated in Fig. 4a–c. The enhancement of the electron affinity of CN-COFs was visualized by the increased proportion of charge distribution at the acrylonitrile moiety. Notably, in the case of CN-COF-3, negligible charge distribution was observed at the pyrene units, suggesting a strong electron-withdrawing capacity inherent to CN-COF-3, enabling it to effectively stabilize anion radicals at the acrylonitrile linkers. Time-correlated single-photon counting traces (TCSPCs) were further performed to investigate the electron transfer among the framework where the average lifetimes of CN-COF-1, CN-COF-2, and CN-COF-3 were 3.32 ns, 2.73 ns, and 4.18 ns (Fig. 4d), respectively. The shortest PL lifetime of CN-COF-2 indicated the rapid charge transfer among the skeleton while the excessive stability of radials in CN-COF-3 oppositely slowed down the charge transport34,35. Furthermore, according to electronic impedance spectroscopy (EIS) analysis (Fig. 4e), a smaller semicircle radius clearly reflected that CN-COF-2 exhibited a lower charge transfer resistance than other two COFs36, which favored the electron-transfer reaction between reactive radicals, in accordance with the TCSPC lifetimes. The ideal radical stability endowed CN-COF-2 with efficient electron transport during the ECL generation process.

Charge distribution of (a) CN-COF-1, b CN-COF-2, and c CN-COF-3 fragment by adding one electron. Ellipse shadows represent the charge distribution of CN-COFs. d TCSPC traces and e EIS Nyquist plots of CN-COFs.
As shown in Fig. 5a, for CN-COF-2, the correspondence between the wavelengths of the ECL spectrum and the PL spectrum indicated the band gap ECL mode29. Meanwhile, the ECL spectra of CN-COF-1 and CN-COF-3 showed a redshift relative to their photoluminescence spectra, probably attributing to the surface state transition mode (Supplementary Figs. 26, 27)37. Next, continuous cyclic voltammetry experiments were conducted to examine the durability of ECL signals of three CN-COFs. Due to the unsteady anion radicals in CN-COF-1, the ECL signal could be kept for around 200 s and declined gradually later (Supplementary Fig. 28). After improving the stability of anion radicals associating with the electron affinity in CN-COF-2, the strong and stable ECL signal exhibited repeatability for around 600 s (Fig. 5b). In contrast, since the difficult charge movement associated with overstable anion radicals, the ECL signal of CN-COF-3 dropped from the second loop and kept weak (Supplementary Fig. 29). Furthermore, the relative radical stability of CN-COFs could be evaluated by the electron paramagnetic resonance (EPR) analysis38,39. As shown in Fig. 5c, upon photoirradiation, CN-COF-3 exhibited highest growth ratio of EPR intensity while CN-COF-1 exhibited the lowest growth ratio. Afterwards, EPR intensity gradually decreased upon light-off, indicating the reversible change of radical concentrations (Supplementary Fig. 30). In addition, the largest g-value shift of CN-COF-3 was observed (gCN-COF-1 = 2.0032, gCN-COF-2 = 2.0031, and gCN-COF-3 = 2.0077) under irradiation (Supplementary Fig. 31), suggesting the highest localization of electron at the acrylonitrile linker of CN-COF-340.

a ECL and PL spectra of CN-COF-2. b Continuous ECL signals of CN-COF-2 modified GCE in 0.10 M PBS containing 20 mM K2S2O8 (PMT voltage = 400 V). c EPR spectra of CN-COFs solid under white irradiation and in dark. d Differential charge density between 1st excited state and ground state of CN-COFs. Electron density gain and loss are donated as green and blue, respectively. e Dependence of volcano-shaped ECL on radical stability of CN-COFs via IRCT between pyrene units.
Volcano-shaped reticular ECL mechanism
Furthermore, to illustrate the electron movement between reactive radicals under electrochemical excitation, the differential charge density between 1st excited state and ground state was calculated (Fig. 5d)41,42. For CN-COF-1 and CN-COF-2, the differential charge density distribution demonstrated electron density loss on one pyrene units and electron density gain of the other pyrene units and acrylonitrile linkers, suggesting the existence of IRCT process between two pyrene units. Meanwhile, the region of increased charge density in CN-COF-3 only located at acrylonitrile linkers, indicating that almost no IRCT occurred between pyrene units. Besides, the charge transfer numbers between the two pyrene units of CN-COF-1 and CN-COF-2 were calculated to be 0.01409 e and 0.05036 e, respectively, much larger than that of CN-COF-3. The increased amount of charge transfer number for CN-COF-2 was attributed to the ideal electron affinity and resulting stabilized anion radicals. Meanwhile, the impeded electron transport between anion and cation radicals resulted in weak and unstable ECL signal for CN-COF-3.
Finally, a volcano relationship between ECL intensity and radical stability associated with electron affinity was established in Fig. 5e. With the presence of acrylonitrile linker, electron affinity of COFs could be enhanced to stabilize anion radicals. CN-COFs were electrochemically reduced to anion radical (COF-•) by injecting electron into the LUMOs. Meanwhile, COF+• was obtained by oxidation of SO4-•, and ECL is generated by the annihilation between COF−• and COF+• radicals via IRCT (Supplementary Fig. 32)30. Due to the weak electron affinity of CN-COF-1, anion radicals were in an ‘understability’ state, leading to relatively low and unstable ECL emission. Subsequently, the ECL intensity and durability could be improved via increasing the stability of radicals, boosting IRCT between COF-• and COF+•. However, further enhancing the electron affinity of CN-COF-3, ‘overstable’ COF-• could impede the charge transfer towards COF+•, leading to the fragility of the resultant ECL emission. Furthermore, CN-COF-4 with decreased electron affinity of acrylonitrile linkers was designed and synthesized, which exhibited a 2.4-fold enhancement in ECL intensity than that of CN-COF-3, ascertaining the proposed relationship between radical stability and ECL performance (Supplementary Fig. 33). Overall, the rational modulating electron affinity of COFs paved the way for developing efficient reticular ECL nanoemitters.
Discussion
In summary, we successfully synthesized three fully conjugated COFs containing pyrene luminophores and tailored acrylonitrile linkers for deciphering ECL mechanism. By precisely modulating the electron affinity of COFs through the number of cyano and benzene rings in acrylonitrile linkers, we achieved control over both anion radical stability and charge transfer process, which has been further proven by theoretical calculations. With the increase of electron affinity of CN-COFs, a volcano plot of ECL and radical stability associated with electron affinity is achieved with 78 folds enhancement in ECL intensity. Also, due to appropriate anion radical stabilization, efficient ECL signal of CN-COF-2 exhibited remarkable durability for around 600 s as cathode material. The manipulating electron affinity of COF not only presents an innovative approach for the development of highly efficient and durable ECL nanoemitters, but also expands the applications of COFs in advanced electronic devices.
Methods
Reagents
4,4′,4′′,4′′′-(Ppyrene-1,3,6,8-tetrayl) tetrabenzaldehyde (TFPPy, 95%), 4′,4′′′,4′′′′′,4′′′′′′′-(pyrene-1,3,6,8-tetrayl)tetrakis(([1,1’-biphenyl]-4-carbaldehyde)) (TFBPy, 95%), 2,2′,2′′,2′′′-(pyrene-1,3,6,8-tetrayltetrakis(benzene-4,1-diyl))tetraacetonitrile (TCPPy, 95%), and 2,2′-(1,4-phenylene)diacetonitrile (PDAN, 95%) were purchased from Jilin Chinese Academy of Sciences-Yanshen Technology Co., Ltd. Cesium carbonate (Cs2CO3, 99%) was bought from Shanghai Macklin Biochemical Technology Co., Ltd. 1,4-Dioxane (AR) and tetrahydrofuran (THF, AR) were received from Nanjing Chemical Reagent Co., Ltd. 1,2-dichlorobenzene (AR) and N,N-dimethylformamide (DMF, AR) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. Potassium phosphate dibasic trihydrate (99.0%) was obtained from Shanghai Titan Scientific Co., Ltd. Phosphate buffer solutions (PBS, 0.10 M, pH = 6.0–8.0) were prepared by mixing stock solutions of KH2PO4 and K2HPO4. All chemicals were used without further purification unless otherwise noted.
Synthesis of CN-COFs
Typically, for CN-COF-1, 46.2 mg (0.05 mmol) of TFBPy, 33.1 mg (0.05 mmol) of TCPPy, 60 mg of Cesium carbonate and solution of o-dichlorobenzene/N,N-dimethylformamide (1/1: v/v, 2.0 mL) were charged into a 5 mL Pyrex tube. The mixture was degassed by three freeze-pump-thaw cycles. The Pyrex tube was flame-sealed under vacuum and then heated at 120 °C for 3 days. After cooling, the mixture was filtered and washed with THF several times to remove unreacted monomers, the catalyst, and the solvent. The solid was further purified by Soxhlet extraction using THF for 1 day and subjected to supercritical CO2 drying to obtain yellow powders. The synthesis of CN-COF-2 and CN-COF-3 followed the similar procedure with the replacement of reagents and solvent, 30.9 mg (0.05 mmol) of TFPPy, 33.1 mg (0.05 mmol) of TCPPy, and 2.0 mL N,N-dimethylformamide for CN-COF-227 while 30.9 mg (0.05 mmol) of TFPPy, 15.6 mg (0.10 mmol) of PDAN, and solution of o-dichlorobenzene/n-butanol (1/1: v/v, 2.0 mL) for CN-COF-322.
General characterization
Powder X-ray diffraction (PXRD) analysis was performed by a D8 ADVANCE (Bruker) employing Cu Kα line focused radiation at 40 kV, 40 mA powder. Samples were placed on a silicon zero background sample holder, and then the sample surface was leveled with a clean microscope slide. Samples were rotated as diffraction data were collected using a continuous 2θ scan from 1–30°. Nitrogen sorption isotherms were measured at 77 K with a JW-BK200B (JWGB SCI. & TECH. Co., Ltd.) or a Micrometrics ASAP 3020 volumetric gas sorption instrument. All samples were degassed under vacuum at 120 °C for 2 h before measurement. Surface parameters were determined using BET adsorption models, and pore size distributions were determined using the NLDFT model equipped in the instruments. Scanning electron microscopy (SEM) images were obtained using a Hitachi S-4800. Accelerating the voltage to 5 kV with Au coating of the sample was used to image the morphology. Transmission electron microscopy (TEM) images were collected using a JEOL JEM-2800 transmission electron microscopy, operated at an acceleration voltage of 200 kV. Fourier transform-infrared spectroscopy (FT-IR) data of samples pelletized with KBr powder in the range of 4000 − 400 cm−1 were recorded with a Bruker Tenson-27 FT-IR spectrometer. Solid-state 13C cross-polarization magic angle spinning (CP/MAS) NMR spectra were collected by an Agilent-NMR-vnmrs600. Thermogravimetric analysis (TGA) was carried out on a NETZSCH TG-DSC_STA449F3 with a heating rate of 10.0 K min−1 under nitrogen flow (50 mL min−1). Valence band X-ray photoelectron spectroscopy (VBXPS) was carried out using a Nexsa system (Thermo Fisher). Photoluminescence (PL) spectra were collected on Edinburgh FLS980 using a quartz cell. Solid-state Ultraviolet-visible (UV-Vis) spectra of COF samples were recorded on Shimadzu UV-3600 in a S3 diffuse reflection mode.
CV and ECL measurement
Cyclic voltammetry (CV) curves were collated using CHI-660D electrochemical workstation (CHI instruments Inc., China). Step pulse (SP) and electrochemiluminescence (ECL) experiments were carried out in a self-made cell on MPIA multifunctional electrochemical and chemiluminescent analytical system (Xi’an Remex Analytical Instrument Co., Ltd. China) using a three-electrode system including a glassy carbon electrode (GCE) working electrode, a Pt counter electrode, and an Ag/AgCl (saturated) reference electrode. COF powders were dispersed in 1,4-dioxane and sonicated for 1 h to form 1.0 mg mL−1 COF/1,4-dioxane suspensions. ECL spectra were recorded on a homemade ECL spectrum analyzer consisting of a Princeton Acton P-2300 monochromator equipped with a grating (grating density: 50 g mm−1; blazed wavelength: 600 nm), a liquid N2 cooled Princeton PyLoN digital charge-coupled device (CCD) detector, and a CHI-660D electrochemical workstation17.
Calculation parameters
The first-principles calculations of COF units were carried out within the Gaussian 16 package43. The hybrid functional B3LYP with 6-31 G(d, p) basis set was used for geometry optimization. Based on the optimized structures, the electronic characters including the highest occupied, lowest unoccupied molecular orbitals, and the neighboring molecular orbitals are obtained. The corresponding spatial distribution of the electron density is illustrated. The electron difference between the one electron doped COF units and the pristine one is calculated by using the formula: Δρ = ρ_1e – ρ, in which ρ_1e and ρ denote the total electron density of one electron doped and pristine COF units. The Time dependent DFT (TDDFT) with the same basis set and functional was used to calculate and analyze the excited states. The optimized ground structures were used in TDDFT calculations for the vertical excitation energies of the COF units. In addition, orbital composition analysis, hole-electron analysis and the charge transfer between ligands of the excited states were also performed using the Multiwfn44. For the periodic COF systems, the calculations were performed based on density functional (DFT) framework implemented with the VASP package45,46, where the generalized gradient approximation (GGA) method in Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional was adopted to describe the exchange-correlation potential47, and Grimme’s D3 dispersion corrections was also utilized to describe the interlayer van der Waals (vdW) interactions48. The periodic boundary condition (PBC) was applied and the input geometry was built from crystal structure. Electronic and force convergence criteria were set to be 10−5 eV and 0.02 eV/Å. The plane-wave cutoff energy was 520 eV and the reduced Brillouin zone sampled with a gamma-centered 1 × 1 × 1. The c-axis in our model represents a vacuum layer with a thickness exceeding 20 Å, effectively isolating the monolayer and preventing interlayer interactions. As known, the PBE functional tends to underestimate the bandgaps of the semiconductor, and a more accurate hybrid functional HSE06 was adopted in the electronic structure calculations49. The energy levels versus to the vacuum of the 2D COFs are deduced by analyzing the potential perpendicular to the layer. The vaspkit package was used in data processing and analysis50.
Data availability
The data that support the findings of this study are available within the article and supplementary information files, or from the corresponding author upon request.
References
Wang, Y. et al. Self-luminescent lanthanide metal–organic frameworks as signal probes in electrochemiluminescence immunoassay. J. Am. Chem. Soc. 143, 504–512 (2020).
Zanut, A. et al. Dye‐doped silica nanoparticles for enhanced ECL‐based immunoassay analytical performance. Angew. Chem. Int. Ed. 59, 21858–21863 (2020).
Descamps, J., Colin, C., Tessier, G., Arbault, S. & Sojic, N. Ultrasensitive imaging of cells and sub‐cellular entities by electrochemiluminescence. Angew. Chem. Int. Ed. 62, e202218574 (2023).
Hu, L. & Xu, G. Applications and trends in electrochemiluminescence. Chem. Soc. Rev. 39, 3275–3304 (2010).
Luo, R., Zhu, D., Ju, H. & Lei, J. Reticular electrochemiluminescence nanoemitters: structural design and enhancement mechanism. Acc. Chem. Res. 56, 1920–1930 (2023).
Dong, J. et al. Direct imaging of single-molecule electrochemical reactions in solution. Nature 596, 244–249 (2021).
Rashidnadimi, S., Hung, T. H., Wong, K.-T. & Bard, A. J. Electrochemistry and electrogenerated chemiluminescence of 3,6-di(spirobifluorene)-N-phenylcarbazole. J. Am. Chem. Soc. 130, 634–639 (2008).
Jin, Z. et al. Electroactive metal-organic frameworks as emitters for self-enhanced electrochemiluminescence in aqueous medium. Angew. Chem. Int. Ed. 59, 10446–10450 (2020).
Peng, H. et al. Pre‐oxidation of gold nanoclusters results in a 66% anodic electrochemiluminescence yield and drives mechanistic insights. Angew. Chem. Int. Ed. 58, 11691–11694 (2019).
Oh, J. W. et al. Enhancement of electrogenerated chemiluminescence and radical stability by peripheral multidonors on alkynylpyrene derivatives. Angew. Chem. Int. Ed. 48, 2522–2524 (2009).
Nie, Y., Zhang, Y., Cao, W., Chai, Y.-q & Yuan, R. Ligand-based shielding effect induced efficient near-infrared electrochemiluminescence of gold nanoclusters and its sensing application. Anal. Chem. 95, 6785–6790 (2023).
Luo, R. et al. Reticular ratchets for directing electrochemiluminescence. J. Am. Chem. Soc. 146, 16681–16688 (2024).
Lyle, S. J., Waller, P. J. & Yaghi, O. M. Covalent organic frameworks: organic chemistry extended into two and three dimensions. Trends Chem. 1, 172–184 (2019).
Rodriguez-San-Miguel, D. & Zamora, F. Processing of covalent organic frameworks: an ingredient for a material to succeed. Chem. Soc. Rev. 48, 4375–4386 (2019).
Ding, S. Y. & Wang, W. Covalent organic frameworks (COFs): from design to applications. Chem. Soc. Rev. 42, 548–568 (2013).
Li, Y.-J. et al. A general design approach toward covalent organic frameworks for highly efficient electrochemiluminescence. Nat. Commun. 12, 4735 (2021).
Luo, R. et al. Intrareticular charge transfer regulated electrochemiluminescence of donor–acceptor covalent organic frameworks. Nat. Commun. 12, 6808 (2021).
Meng, X. et al. Bimodal oxidation electrochemiluminescence mechanism of coreactant-embedded covalent organic frameworks via postsynthetic modification. Angew. Chem. Int. Ed. 63, e202402373 (2024).
Zhu, D. et al. Dual intrareticular oxidation of mixed-ligand metal-organic frameworks for stepwise electrochemiluminescence. J. Am. Chem. Soc. 143, 3049–3053 (2021).
Zhang, J. L. et al. Conductive covalent organic frameworks with conductivity- and pre-reduction-enhanced electrochemiluminescence for ultrasensitive biosensor construction. Anal. Chem. 94, 3685–3692 (2022).
Luo, Q.-X. et al. Construction of sp2 carbon-conjugated covalent organic frameworks for framework-induced electrochemiluminescence. ACS Appl. Electron. Mater. 3, 4490–4497 (2021).
Jin, E. Q. et al. Two-dimensional sp2 carbon-conjugated covalent organic frameworks. Science 357, 673–676 (2017).
He, T., Geng, K. & Jiang, D. All sp2 carbon covalent organic frameworks. Trends Chem. 3, 431–444 (2021).
Qi, H., Chen, Y.-H., Cheng, C.-H. & Bard, A. J. Electrochemistry and electrogenerated chemiluminescence of three phenanthrene derivatives, enhancement of radical stability, and electrogenerated chemiluminescence efficiency by substituent groups. J. Am. Chem. Soc. 135, 9041–9049 (2013).
Zhang, A., Jiang, W. & Wang, Z. Fulvalene‐embedded perylene diimide and its stable radical anion. Angew. Chem. Int. Ed. 59, 752–757 (2019).
Chen, R. et al. Rational design of isostructural 2D porphyrin-based covalent organic frameworks for tunable photocatalytic hydrogen evolution. Nat. Commun. 12, 1354 (2021).
Wang, Y. et al. Linkages make a difference in the photoluminescence of covalent organic frameworks. Angew. Chem. Int. Ed. 62, e202310794 (2023).
Bai, Y. et al. Accelerated discovery of organic polymer photocatalysts for hydrogen evolution from water through the integration of experiment and theory. J. Am. Chem. Soc. 141, 9063–9071 (2019).
Zhao, T. et al. Ultrafast condensation of carbon nitride on electrodes with exceptional boosted photocurrent and electrochemiluminescence. Angew. Chem. Int. Ed. 59, 1139–1143 (2019).
Ding, Z. et al. Electrochemistry and electrogenerated chemiluminescence from silicon nanocrystal quantum dots. Science 296, 1293–1297 (2002).
Shen, M. et al. Electrochemistry and electrogenerated chemiluminescence of dithienylbenzothiadiazole derivative. differential reactivity of donor and acceptor groups and simulations of radical cation−anion and dication−radical anion annihilations. J. Am. Chem. Soc. 132, 13453–13461 (2010).
Qian, Y. et al. Computation-based regulation of excitonic effects in donor-acceptor covalent organic frameworks for enhanced photocatalysis. Nat. Commun. 14, 3083 (2023).
Zhang, J.-L. et al. Highly stable covalent organic framework nanosheets as a new generation of electrochemiluminescence emitters for ultrasensitive microrna detection. Anal. Chem. 93, 3258–3265 (2021).
Hou, Y. et al. Efficient photosynthesis of hydrogen peroxide by cyano-containing covalent organic frameworks from water, air and sunlight. Angew. Chem. Int. Ed. 63, e202318562 (2024).
Kou, M. et al. Molecularly engineered covalent organic frameworks for hydrogen peroxide photosynthesis. Angew. Chem. Int. Ed. 61, e202200413 (2022).
Ghosh, S. et al. Identification of prime factors to maximize the photocatalytic hydrogen evolution of covalent organic frameworks. J. Am. Chem. Soc. 142, 9752–9762 (2020).
Liu, S. et al. Electrochemiluminescence tuned by electron-hole recombination from symmetry-breaking in wurtzite ZnSe. J. Am. Chem. Soc. 138, 1154–1157 (2016).
Lü, B. et al. Stable radical anions generated from a porous perylenediimide metal-organic framework for boosting near-infrared photothermal conversion. Nat. Commun. 10, 767 (2019).
Ye, X. et al. Organic radicals stabilization above 300 °C in Eu-based coordination polymers for solar steam generation. Nat. Commun. 13, 6116 (2022).
Aljarb, A. et al. Interfacial reconstructed layer controls the orientation of monolayer transition-metal dichalcogenides. ACS Nano 17, 10010–10018 (2023).
Le Bahers, T., Adamo, C. & Ciofini, I. A qualitative index of spatial extent in charge-transfer excitations. J. Chem. l Theory Comput. 7, 2498–2506 (2011).
Yuan, J., Yuan, Y., Tian, X., Liu, Y. & Sun, J. Insights into the photobehavior of fluorescent oxazinone, quinazoline, and difluoroboron derivatives: molecular design based on the structure–property relationships. J. Phy. Chem. C. 121, 8091–8108 (2017).
Frisch, M. J. et al. Gaussian 16 Rev. C.01; Wallingford, CT, 2016.
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phy. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phy. Rev. Lett. 77, 3865–3868 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phy. 132, 154104 (2010).
Heyd, J., Scuseria, G. E. & Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phy. 118, 8207–8215 (2003).
Wang, V., Xu, N., Liu, J.-C., Tang, G. & Geng, W.-T. VASPKIT: a user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phy. Commun. 267, 108033 (2021).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22274071, 22073087, 22225301, 22303092, 22301181), the CAS Project for Young Scientists in Basic Research (YSBR-004), the Strategic Priority Research Program of the CAS (XDB0450101), the Fundamental Research Funds for the Central Universities (020514380274, WK2060000038, 20720220009), Innovation Program for Quantum Science and Technology (2021ZD0303302), the Super Computer Centre of USTCSCC and SCCAS, the Postdoctoral Fellowship Program of CPSF (GZB20240303), the China Postdoctoral Science Foundation (2024M751374), the Jiangsu Funding Program for Excellent Postdoctoral Talent (2024ZB547), and Shanghai Sailing Program (23YF1429000).
Author information
Authors and Affiliations
Contributions
J.P. Lei and K. Xi conceived the idea. H.C. Xu and R.G. Luo designed the experiments and collected the data. H.F. Lv accomplished the theoretical calculation. T.R. Liu, Q.B. Liao, Y.D. Wang, and Z.Y. Zhong assisted with the experiments and characterizations. H.C. Xu and R.G. Luo wrote the manuscript. X.J. Wu, J.P. Lei, and K. Xi supervised and coordinated all investigators for this project. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Qianrong Fang, Ying Zhuo and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, H., Luo, R., Lv, H. et al. Deciphering a volcano-shaped relationship between radical stability and reticular electrochemiluminescence. Nat Commun 16, 1924 (2025). https://doi.org/10.1038/s41467-025-56009-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56009-3
