
Abstract
Steroid hormones are powerful endocrine regulators, but little is known about how environmental conditions modulate steroidogenesis to reprogram developmental fates. Here, we use the Drosophila prothoracic gland (PG) to investigate how a nutrient restriction checkpoint (NRC) ensures or blocks developmental progression and sexual maturation via regulating steroidogenesis. Extensive transcriptome analysis of the PG reveals that pre-NRC starvation significantly downregulates mitochondria-associated genes. Pre-NRC starvation reduces prothoracicotropic neuropeptide hormone signaling, insulin signaling, and TORC1 activity in PG cells, which prevent mitochondrial fragmentation and import of Disembodied, a key steroidogenic enzyme. Ultimately, pre-NRC starvation causes severe mitophagy and proteasome dysfunction, blocking steroidogenesis and metamorphosis. By contrast, post-NRC starvation does not impair mitochondrial homeostasis in PG cells but reduces sit expression and induces moderate autophagy to promote steroidogenesis, leading to precocious metamorphosis. This study constitutes a paradigm for exploring how steroid hormone levels are controlled in response to environmental stress during developmental checkpoints.
Similar content being viewed by others

Introduction
In animals, juvenile growth is typically associated with developmental checkpoints that control sexual maturation, which ensures completion of development into an adult, and a species-dependent final body size that maximizes fitness and survival in a range of environments1. Malnutrition or starvation can delay the onset of puberty in mammals, which involves a cascade of neuropeptide signals to stimulate sex steroid hormone production in gonads to initiate maturation2,3. Insects have two nutrient restriction checkpoints (NRC), known as critical weight and minimal viable weight. Critical weight is reached when subsequent starvation no longer delays the onset of puparium formation, whereas minimal viable weight is the lowest larval weight needed to initiate and complete metamorphosis. The NRC allows animals to adapt to environmental stress, especially nutritional stress like starvation. The attainment of critical weight commits larvae to rapidly produce sufficient amounts of 20-hydroxyecdysone (20E, the main active form of insect steroid hormones) to trigger metamorphosis, the juvenile-to-adult transition in insects4. Both mammalian puberty and insect metamorphosis are controlled by steroid hormones, suggesting a conserved system for triggering maturation. Steroid hormone synthesis (hereafter, steroidogenesis) is regulated by a variety of intrinsic signals in response to external environments5,6,7. However, much remains unknown about the cellular and molecular mechanisms by which steroid-producing endocrine organs effectively integrate environmental cues with internal signals to coordinate the transition from juvenile-to-adult stages in animals.
In insects, control over steroidogenesis is multi-layered and complex. In the fruit fly, Drosophila melanogaster, the larval ring gland (RG) is composed of the prothoracic gland (PG), which produces ecdysone (the immediate precursor of 20E), the corpora allata, and the corpora cardiaca. In PG cells, dietary cholesterol is converted to ecdysone through a series of proteins referred to as the Halloween enzymes, including the final three cytochrome P450 enzymes, Phantom (Phm), Disembodied (Dib), and Shadow (Sad)5,7. Upon ingestion of dietary sterols, cholesterol or other suitable sterols are transported into PG cells through receptor-mediated endocytosis. Within PG cells, steroidogenesis requires sterol/steroid trafficking, which in turn relies on the Niemann-Pick complex 1 (Npc1) and the fatty acid elongase stuck in traffic (Sit), as well as molecular components involved in autophagy. Newly synthesized ecdysone exits PG cells via vesicle-mediated secretion8,9,10,11,12. Halloween gene expression is regulated by a complex network of transcription factors, some of which are activated by extracellular signals. For example, environmental cues impinge on the neurons producing prothoracicotropic hormone (PTTH) and insulin/insulin-like peptides (ILPs) to adjust steroidogenic activity in the PG. PTTH binds to its receptor Torso and activates the Ras/Raf/Erk pathway and transcription factors such as nuclear receptor Hr4. ILPs bind to InR to activate the PI3K/Akt pathway and target rapamycin complex 1 (TORC1). TORC1 stimulates endocycles in PG cells through the transcription factor Snail13,14,15,16,17,18,19,20. Steroidogenesis is autoregulated by a number of transcription factors in the 20E-triggered transcription cascade, while it is inhibited by juvenile hormone signaling that antagonizes 20E action in the PG21,22,23. Once ecdysone is released from the PG into the hemolymph, it is converted to 20E in target tissues by Shade (Shd), also a cytochrome P450 enzyme, where 20E executes its physiological and developmental roles5.
In Drosophila, critical weight and minimal viable weight largely overlap, and together referred to as the NRC9,24. Under normal conditions, i.e., a non-restricting diet, NRC is passed in the early third instar (~78 hours after egg laying = 78 h AEL). Pre-NRC starvation blocks steroidogenesis and leads to developmental arrest, whereas post-NRC starvation accelerates steroidogenesis and irreversibly induces precocious metamorphosis4,19. In post-NRC PG cells, starvation modulates cholesterol storage by reducing TORC1-induced expression of Npc1 and sit and stimulates cholesterol mobilization via the activation of TORC1-inhibited autophagy, causing precocious steroidogenesis and metamorphosis. In addition, upon starvation, the corpora cardiaca releases stored ILPs to maintain steroidogenesis in the PG8,9,10,24. In pre-NRC PG cells, starvation decreases TORC1-mediated expression of snail and the number of endocycles, however, it is unclear how endocycling is mechanistically linked to steroidogenesis. Starvation also induces extensive autophagy in PG cells, which alters cholesterol trafficking and decreases steroidogenesis. Further, starvation induces the expression and secretion of Hedgehog in the midgut, which remotely suppresses steroidogenesis in the PG9,19,20,25. In contrast to the well-studied post-NRC responses to nutrient restriction, there is little clarity on the cellular and molecular mechanisms by which pre-NRC starvation blocks steroidogenesis in the PG.
To examine the consequences of pre-NRC starvation, we reared Drosophila larvae under three different regimens. Control larvae were continuously fed until 108 h AEL, pre-NRC starvation larvae were initially fed for 72 hours, but then starved from 72 h to 108 h AEL, whereas post-NRC starvation larvae were fed the first 84 hours, followed by starvation from 84 h to 108 h AEL. We then isolated >10,000 RGs, which are very small and comprise ~100 cells, by dissection at 108 h AEL, and prepared RG samples for comparative transcriptome analysis. The RNA-Seq data showed strong enrichment of genes with roles in the mitochondria (mitochondria-associated genes) (81/554≅14.6%). Based on the data from a previous PG-specific RNAi screen8, we found that 82 of the 554 genes were indispensable for larval development, including seven mitochondria-associated genes (7/82≅8.5%). Thus, focusing on mitochondria function and regulation should provide a new angle in understanding how steroid hormone dynamically switches developmental fates. By utilizing cellular, biochemical, molecular, and genetic approaches, we then showed that pre-NRC starvation disrupted normal mitochondrial homeostasis in PG cells, which in turn blocked steroidogenesis. This mechanism was not observed in response to post-NRC starvation. Our comprehensive study revealed that nutrient status alters developmental fates via a switch in mitochondrial homeodynamics in Drosophila PG cells, shedding light on the general mechanisms by which environmental conditions impact endocrine control during developmental checkpoints in animals.
Results
Pre-NRC starvation downregulates mitochondria-associated genes in the PG
Our three different rearing strategies, i.e., controls fed with a standard diet, pre-NRC starvation, and post-NRC starvation (Supplementary Fig. 1A) yielded phenotypes in line with previous reports4,19. Specifically, pre-NRC starvation caused developmental arrest, whereas post-NRC starvation resulted in precocious metamorphosis and a smaller body size (Supplementary Fig. 1B–D and Supplementary Data 1 for a complete list of genotypes and statistical results). We then collected RG samples for RNA sequencing from the three groups of larvae at 108 h AEL, with three biological replicates in each group (Supplementary Fig. 1E). Principal component analysis demonstrated that the data clustered into three distinct groups: the control, pre-NRC starvation, and post-NRC starvation (Supplementary Fig. 1F and Supplementary Data 2). Compared to the control, pre-NRC starvation reduced the expression of all genes involved in steroidogenesis, however, post-NRC starvation had little effect on their expression. Neither pre-NRC nor post-NRC starvation had a significant influence on the expression of juvenile hormone biosynthetic genes. These data strongly suggest that pre-NRC starvation blocked steroidogenesis through transcriptional downregulation (Supplementary Fig. 1G and Supplementary Data 2). Differential gene expression analysis revealed that 554 genes were exclusively downregulated by pre-NRC starvation, whereas only seven genes were exclusively downregulated by post-NRC starvation. In addition, the expression of 10 overlapping genes was decreased in both treatments (Fig. 1A and Supplementary Data 3).

A Transcriptome analysis revealed that 564 genes were downregulated when larvae were starved prior to the NRC (pre-NRC) compared to continuously feeding larvae, and 17 genes were downregulated when larvae were starved after the NRC (post-NRC). Expression levels of 554 genes were specifically decreased in response to pre-NRC starvation (ST). Abbreviation: pre-NRC ST, starved from 72 h after egg laying to 108 h; post-NRC ST, starved from 84 h after egg laying to 108 h. B The pre-NRC ST-regulated cohort (554 genes) includes 81 mitochondria-associated genes and 24 proteasome genes. C Danielsen et al., 2016, found that PG-specific RNAi targeting 1906 genes resulted in developmental timing defects. Of these, 1006 RNAi lines resulted in developmental arrest in third-instar larvae. In our study, we found that 554 genes were specifically downregulated upon pre-NRC ST. Overlap (pink) revealed that 82 pre-NRC ST-downregulated genes are associated with ST-induced developmental arrest. Abbreviation: L2P, pupariation of L2 larvae; Delay, developmental delays; P lethal, pupal lethality; Arrest, arrest in L1, L2, and L3. D The 82 overlapping genes were manually classified based on protein functions.
We then analyzed the 554 downregulated genes by term enrichment analysis. The most significantly enriched term was “mitochondria” (81 genes, ~14.6%), which are necessary for normal mitochondrial function, and “proteosome” (24 genes, ~4.3%), in the “cellular component” category. Mitochondria are central organelles required for energy metabolism, heme synthesis, and apoptosis26. The proteasome is a multi-subunit protease complex essential for degrading unstable proteins27. The 81 mitochondria-associated genes comprised several sub-categories, including mtDNA translation (23), mitochondrial metabolism (17), oxidative phosphorylation (17), mitochondrial transport (12), antioxidants (4), and others (8). With respect to the 24 “proteasome” genes, 14 encoded proteins acting in the proteosome core particle, while the remaining 10 function in the regulatory particle (Fig. 1B and Supplementary Data 4). In a prior genome-wide PG-specific RNAi screen, 1906 RNAi lines interfered with normal development. Specifically, RNAi knockdown of 1006 genes in the PG caused developmental arrest in different developmental stages, whereas RNAi targeting the remaining 900 genes led to other developmental defects8. In total, 82 of the 554 downregulated genes in the RG by pre-NRC starvation overlapped with the 1006 essential genes identified by the PG-specific RNAi screen, suggesting that these genes establish the context by which nutritional cues impinge on steroidogenesis in response to pre-NRC starvation (Fig. 1C and Supplementary Data 5).
Next, we categorized the functions of proteins encoded by the 82 downregulated genes (Fig. 1D). As expected, we found four Halloween genes, including shroud (Sro), Phm, Dib, and Sad (7). We also identified eight transcription factors, half of which have known roles in Halloween gene regulation, including Knirps (Kni), numerous disordered muscles (Nom aka Seance), Ouija board (Ouib), and Ventral veins lacking (Vvl)28,29,30,31. Importantly, we identified seven mitochondria-associated genes, comprising ~8.5% of the 82 downregulated essential genes. COX6B, Neb-cGP, and ATPsynC are structural components of the oxidative phosphorylation system32,33. mRPL47 is a component of the mitochondrial large ribosomal subunit, which is encoded by the mitochondrial genome34. TrpRS-m is a tryptophanyl-tRNA synthetase that functions within mitochondria to promote the formation of tryptophanyl-tRNA35. Tom20 and mge (a homolog of human Tom22) are important components of the protein translocase of the outer mitochondrial membrane, which is responsible for selective uptake of cytoplasmic proteins into mitochondria36,37. Among the cohort of 82 downregulated essential genes were 20 genes encoding proteasome subunits, comprising 83.3% of the 24 downregulated proteasome genes. The same cohort also harbored seven genes encoding the HSP60 Chaperonins, including Hsp60A, Chaperonin containing TCP1 subunit 1 (CCT1), CCT3, CCT5, CCT7, CCT8, and CG11287, which mediate proper protein folding for quality control38,39. Finally, we also identified three DNA polymerase genes, two immunity-related genes, and 19 enzyme-encoding genes. The composite analyses suggest that genes involved in mitochondria and proteasomes might link nutrition with steroidogenesis in response to pre-NRC starvation, which is indispensable for steroidogenesis in PG cells.
Pre-NRC starvation promotes mitochondrial tubulation to produce ATP
Since mitochondria play central roles in energy metabolism26, we examined how mitochondrial homeostasis controls steroidogenesis in the PG prior to the NRC. Mitochondria are highly dynamic organelles capable of shape changes, and they undergo fission and fusion to regulate the degree of mitochondrial fragmentation and tubulation40. Using a mitochondria-targeted GFP-based reporter (mito-GFP), we observed morphological changes in PG mitochondria from 72 h AEL to the white prepupal stage (WPP). Interestingly, we found that the ratio of fragmented mitochondria (small and round) progressively increased from 60% at 72 h to ~100% at 108 h, and then decreased from the early wandering stage (EW) to WPP (Fig. 2A, Supplementary Fig. 2A and Supplementary Data 1). We observed similar trends for Erk and Akt activity in PG cells, which were monitored by p-Erk staining and Akt-SPARK reporter41 (based on the technique of “separation of phases-based activity reporter of kinase, SPARK)”, respectively (Supplementary Fig. 2B, C and Supplementary Data 1). Our observation is consistent with previously described developmental profiles of insulin signaling and TORC1 activity in the PG42. We then examined whether and how the morphological dynamics of PG mitochondria were regulated by starvation, PTTH signaling, insulin signaling, and TORC1 activity prior to the NRC. Indeed, pre-NRC starvation gradually reduced the ratio of fragmented mitochondria, resulting in mitochondrial tubulation over time (Fig. 2B and Supplementary Data 1). Moreover, pre-NRC starvation significantly decreased PTTH signaling (p-ERK staining) (Fig. 2C and Supplementary Data 1), insulin signaling (InR-SPARK and AKT-SPARK reporters) (Fig. 2D, E and Supplementary Data 1), and TORC1 activity (p-S6K staining) (Fig. 2F and Supplementary Data 1). Furthermore, PG-specific inhibition of PTTH signaling via RNAi directed against Torso or expression of RasDN prior to the NRC significantly increased mitochondrial tubulation. Inhibition of insulin signaling via InR-RNAi or expression of Dp110DN had similar effects, and simultaneous inhibition of PTTH signaling and insulin signaling via Torso- and InR-RNAi caused comparable phenotypic defects. However, PG-specific inhibition of TORC1 activity by expression of TORDN or TSC1/2 had no effects on mitochondrial dynamics (Fig. 2G and Supplementary Data 1). These results showed that PTTH and insulin signaling, but not TORC1 activity, prevented mitochondrial tubulation and promoted mitochondrial fragmentation in PG cells, which were essential for fulfilling the NRC.

A The percentage of fragmented mitochondria in PG cells from 72 h AEL to WPP. From left to right, n = 30, 16, 27, 17, 24, and 40, respectively. B Mitochondrial morphology (green) was assessed in PG cells. Magnified images of the highlighted areas (red dotted box) are shown. Right: the percentage of fragmented mitochondria. Nucleus: DAPI (cyan). From left to right, n = 30, 23, 24, and 17, respectively. C–F Representative images of PTTH (C), p-ErK, green, 108 h: n = 24, pre-NRC ST: n = 27), insulin (D, E InR-SPARK and AKT-SPARK, green), and TORC1 signalings (F, p-S6K, green, 108 h: n = 9, pre-NRC ST: n = 8) in PG cells of control and pre-NRC ST larvae. G, H Representative images of mitochondrial morphology (green) in PG cells (168 h AEL) with inhibition of PTTH, insulin, TORC1 signalings G and mitochondria-associated genes H. Magnified images of the highlighted areas (red dotted box) are shown. G From left to right, n = 7, 7, 7, 6, 6, 13, 9, and 7, respectively. H From left to right, n = 32, 19, 19, 20, 12, 16, 17, and 19, respectively. I, J Images showing Marf (I Marf-EGFP, green) and p-Drp1 (J p-Drp1 Ser637 antibody, green) staining in PG cells of control and pre-NRC ST larvae at 108 h AEL. Mitochondria: mito-mCherry (red). Nucleus: DAPI (cyan). From left to right, n = 26, 15, 30, and 22, respectively in (I, J). K, L Images showing ATP-SPARK puncta (green) in PG cells of control and pre-NRC ST or Marf-expressing larvae at 108 h AEL. M, N Representative images of mitochondria morphology (M, green, From left to right, n = 20, 27, 24, and 19, respectively) and ATP-SPARK puncta (N, green) in PG cells (144 h AEL) with PG-specific expression of RasV12, Dp110CA, or Rheb. Magnified images of the highlighted areas (red dotted box) are shown. O, P Images showing p-AMPK levels (p-AMPK, green, 108 h: n = 17, pre-NRC ST: n = 19) O and AMPK-SPARK puncta (green) P in PG cells of controls and pre-NRC ST larvae at 108 h AEL. All Data are presented as mean ± SD. Different lowercase letters represent statistical significance by one-way ANOVA. The genotypes and statistical data are provided in Supplementary Data 1.
Next, we explored whether PG-specific RNAi against the seven mitochondria-associated genes affected mitochondrial morphology prior to the NRC. RNAi targeting COX6B, Neb-cGP, and TrpRs-m, but not the other four genes, significantly promoted mitochondrial tubulation (Fig. 2H and Supplementary Data 1). Mining of our RG transcriptome data (Supplementary Data 2) revealed that pre-NRC starvation significantly increased the expression of Marf, which encodes a homolog of human MFN2 (a dynamin family GTPase), a protein that mediates outer mitochondrial membrane tethering and thus mitochondrial fusion43,44. We also assessed in vivo protein levels of Marf using the Marf-EGFP reporter, which showed that pre-NRC starvation significantly increased Marf-EGFP fluorescence intensity in the PG (Fig. 2I and Supplementary Data 1). Drp1 is another dynamin family GTPase that promotes mitochondrial fission and fragmentation, however, phosphorylation of Drp1 at S637 (p-Drp1) antagonizes its activity45. Consistently, pre-NRC starvation significantly increased p-Drp1 staining in PG cells (Fig. 2J and Supplementary Data 1). Considering that Marf expression is known to result in mitochondrial tubulation43,44, our studies indicated that pre-NRC starvation promoted mitochondrial tubulation through increasing Marf expression and Drp1 phosphorylation at S637. In addition, Marf-EGFP intensity and p-Drp1 staining in PG cells gradually decreased from 72 h AEL to 108 h AEL, while they increased again at EW and WPP, respectively, with a significant increase of p-Drp1 at WPP (Supplementary Fig. 2D, E and Supplementary Data 1).
One would expect that impaired mitochondrial function should negatively impact energy homeostasis, and result in a reduction of ATP levels. To test this, we expressed an ATP-SPARK reporter46,47 in PG cells to image ATP abundance directly. To our surprise, we observed that pre-NRC starvation significantly increased the number and size of ATP-SPARK droplets (Fig. 2K), indicating that ATP levels were elevated by mitochondrial tubulation. Importantly, the PG-specific expression of Marf was sufficient to promote ATP production (Fig. 2L). The results together indicate that the rise in PG ATP levels was consistent with increased prevalence of mitochondrial fusion and tubulation. Notably, PG-specific expression of RasV12 or Dp110CA, but not Rheb, significantly reduced mitochondrial tubulation and ATP production induced by pre-NRC starvation (Fig. 2M, N and Supplementary Data 1). This confirmed that PTTH and insulin signaling, but not TORC1 activity, were involved in this process. AMP-activated protein kinase (AMPK) is an important intracellular energy sensor, which is activated by high AMP/ATP ratios5,6,7. AMPK activities can be tracked by p-AMPK staining and an AMPK-SPARK reporter41. We observed that pre-NRC starvation resulted in significant AMPK activation, showing a decline in cellular energy status (Fig. 2O, P and Supplementary Data 1). During development, p-AMPK level was high at 72 h, gradually decreased over time, and rebounded at WPP, while ATP level was high at 72 h and decreased over time (Supplementary Fig. 2F–H and Supplementary Data 1). Collectively, due to the reduction of PTTH signaling and insulin signaling in PG cells, pre-NRC starvation promotes mitochondrial tubulation to produce ATP for cell survival even in energy-constrained conditions.
Pre-NRC starvation prevents mitochondrial import of Dib
Among the seven mitochondria-associated genes that were downregulated by pre-NRC starvation in the PG, Tom20 and mge encode components of the mitochondrial outer membrane translocase complex, suggesting regulatory roles in mitochondrial import (Fig. 1D). Importantly, Dib and Sad are likely mitochondrial cytochrome P450 enzymes that catalyze the final two sequential hydroxylation steps in the ecdysone biosynthesis pathway48. We generated a Dib-mCherry knock-in reporter line via CRISPR/Cas9, which demonstrated that the signals produced by Dib-mCherry and mito-GFP overlapped in PG cells (Fig. 3A and Supplementary Data 1). In non-starvation conditions, Dib-mCherry was predominantly found in mitochondria; however, its mitochondrial localization was dynamic. Specifically, we saw a gradual increase of Dib-mCherry from 72 h to 108 h, a peak at EW, followed by a decline at WPP (Fig. 3A). Remarkably, pre-NRC starvation caused Dib-mCherry to form cytoplasmic aggregates, indicating that mitochondrial import was increasingly blocked as starvation conditions continued (Fig. 3B and Supplementary Data 1). We re-assessed the cellular localization of Dib-mCherry with the MitoTracker Green dye49, which yielded comparable results (Fig. 3C and Supplementary Data 1). In addition, we observed that pre-NRC starvation significantly increased the colocalization of Dib-mCherry aggregates with lysosomes (which can clear abnormal protein aggregates), as evidenced by lysotracker staining50,51 (Fig. 3D). Moreover, inhibition of PTTH signaling, insulin signaling or both, as well as TOR activity, decreased mitochondrial import of Dib-mCherry prior to the NRC (Fig. 3E and Supplementary Data 1).

A Colocalization of Dib-mCherry (red) and mitochondria (mito-GFP, green) were examined in PG cells from 72 h AEL to WPP. Right: Mander’s colocalization analyses. From left to right, n = 32, 60, 47, 40, 43 and 51, respectively. B Colocalization of Dib-mCherry (red) and mitochondria (mito-GFP, green) were examined in PG cells after 12 h, 24 h, and 36 h starvation from 72 h AEL. Right: Mander’s colocalization analyses. From left to right, n = 32, 54, 34, and 47, respectively. C, D Colocalization of Dib-mCherry (red) and mitochondria (C, mitotracker, green) or lysotracker (D green) were examined in PG cells of control and pre-NRC ST larvae at 108 h AEL. C n = 18 (108 h), 15 (pre-NRC ST). E, F Representative images of colocalization of Dib-mCherry (red) and mitochondria (mito-GFP, green) in PG cells (168 h AEL) with inhibition of PTTH, insulin, and TORC1 signalings E as well as mitochondria-associated genes and CCT genes F. E From left to right, n = 20, 21, 11, 31, 45, 21, 31, and 20, respectively. F From left to right, n = 25, 16, 13, 26, 32, 24, 16, 20, 20, 22, 23 and 23, respectively. G Representative images showing colocalization of Dib-mCherry (red) and mitochondria (mito-GFP, green) in PG cells (168 h AEL) with PG-specific expression of RasV12, Dp110CA, and Rheb. From left to right, n = 20, 61, 22 and 30, respectively. H, I Representative images showing colocalization of Dib-mCherry (red) and mitochondria (marked by mito-GFP or Tom20-GFP, green) in the PG (168 h AEL). Right: Mander’s colocalization analyses. From left to right, n = 30, 37, 9 and 25, respectively in (H, I). Two-tailed paired t test for two-column comparison. ***p < 0.001. J Developmental timing of phm > w1118 (n = 28) and phm > Marf (n = 15) under feeding conditions. K Body size of phm > w1118 and phm > Marf under feeding conditions. n = 58 (Phm > w1118), 51 (Phm > Marf). Two-tailed paired t test for two-column comparison. ***p < 0.001. All Data are presented as mean ± SD. Different lowercase letters represent statistical significance by one-way ANOVA. The genotypes and statistical data are provided in Supplementary Data 1.
We then examined whether the seven mitochondria-associated genes were required for mitochondrial import of Dib. PG-specific RNAi knockdown of Tom20 and mge decreased Dib-mCherry mitochondrial import and caused the accumulation of mCherry aggregates in the cytoplasm. RNAi knockdown of COX6B, Neb-cGP, TrpRS-m, and ATPsynC, but not mRPL47, had similar effects (Fig. 3F and Supplementary Data 1). As outlined earlier (Fig. 1D), pre-NRC starvation downregulated seven genes encoding the HSP60 Chaperonins, which mediate proper protein folding38,39. As expected, PG-specific RNAi against these genes also caused the formation of Dib-mCherry aggregates in the cytoplasm (Fig. 3F and Supplementary Data 1), indicating that the HSP60 chaperonins are required for mitochondrial import of Dib. Importantly, PG-specific activation of PTTH signaling or insulin signaling significantly rescued Dib importation into mitochondria. By contrast, TORC1 activity was not nearly as well as PTTH signaling and insulin signaling (Fig. 3G and Supplementary Data 1). Notably, PG-specific expression of Tom20 alone resulted in a comparable rescue (Fig. 3H and Supplementary Data 1). PG-specific Marf expression prior to the NRC resulted in mitochondrial tubulation and the presence of cytoplasmic Dib-mCherry aggregates, along with a delay in developmental timing and a larger pupal size (Fig. 3I–K and Supplementary Data 1). These data showed that prior to the NRC and under nutrient-replete conditions, PTTH signaling, insulin signaling, and TORC1 activity promote mitochondrial fission and fragmentation, which stimulate mitochondrial import of Dib and thus steroidogenesis, but not ATP production.
Pre-NRC starvation causes severe mitophagy in PG cells
It is well known that mitochondrial dysfunction stimulates mitophagy, which in turn eliminates damaged mitochondria52. We detected mitophagy occurrence in PG cells using mitochondria-targeted Keima (mt-Keima), a pH-dependent fluorescent protein probe53. Three hours of pre-NRC starvation was sufficient to acutely trigger mitophagy, which continued to increase as time progressed (Fig. 4A, Supplementary Fig. 3A and Supplementary Data 1). PG-specific inhibition of PTTH and insulin signaling, but not TORC1 activity, caused a significant rise in mitophagy occurrence prior to the NRC (Supplementary Fig. 3B, C and Supplementary Data 1); whereas activating these three pathways reduced mitophagy induction caused by pre-NRC starvation (Fig. 4B and Supplementary Data 1). Moreover, PG-specific RNAi knockdown of all the seven mitochondria-associated genes induced varying degrees of mitophagy, especially for COX6B, Neb-cGP, and TrpRS-m (Supplementary Fig. 3D, E and Supplementary Data 1).

A Mitophagy visualized by mt-Keima was assessed in PG cells of control and pre-NRC ST larvae at 108 h AEL. PG cells were imaged via sequential excitations (561 nm, red; 405 nm, green). n = 12 (108 h), 7 (pre-NRC ST). B, E Images showing mitophagy in PG cells with PG-specific expression of RasV12, Dp110CA, Rheb (B, 144 h AEL) and Cat (E, 168 h AEL). B From left to right, n = 26, 18, 19 and 21, respectively. E n = 48 (Control), 26 (Cat). C, D Images showing mitochondrial ROS (C, mitoSOX, red) and intracellular ROS (D, DCFH-DA, green) levels in PG cells of controls and pre-NRC ST larvae at 108 h AEL. Magnified images of the highlighted areas (yellow or red dotted box) are shown. F–H Images showing Pink1 (F, red, 108 h: n = 18, pre-NRC ST: n = 13, 108 h AEL), Parkin (G, green, 108 h: n = 17, pre-NRC ST: n = 18, 108 h AEL) and BNIP3 (H, green, 120 h: n = 11, pre-NRC ST: n = 11, 120 h AEL) protein levels in PG cells of controls and pre-NRC ST larvae. Mitochondria: mito-GFP (green, F) or mito-mCherry (red, G). Nucleus: DAPI (cyan). I Representative images of mitophagy in PG cells (168 h AEL) with knockdown of Parkin, Parkin+Pink1, or BNIP3. From left to right, n = 23, 24, 26 and 35. J Images of PG cells expressing Atg5-GFP (green) under feeding and pre-NRC ST conditions. n = 5 (108 h), 5 (pre-NRC ST). K Images of PG cells expressing Lamp1-GFP (green) and Atg8a-mCherry (red) underfeeding and pre-NRC ST conditions. n = 27 (108 h), 19 (pre-NRC ST). L Images of colocalization of Lamp1-GFP (green) and Atg8a-mCherry (red) in PG cells (144 h AEL) with PG-specific expression of RasV12, Dp110CA, and Rheb. n = 51 (Control), 41 (RasV12), 46 (Dp110CA), 8 (Rheb). M, N Mander’s colocalization analyses of corresponding images in (K, L), respectively. Two-tailed paired t test for two-column comparison. ***p < 0.001. O Images of mitophagy in PG cells (168 h AEL) with PG-specific expression of Atg1DN. n = 48 (Control), 23 (Atg1DN). P Images of colocalization of Lamp1-GFP (green) and Atg8a-mCherry (red) in PG cells (108 h AEL) with PG-specific expression of ParkinCA. Right: Mander’s colocalization analyses. n = 27 (Control), 39 (ParkinCA). Two-tailed paired t test for two-column comparison. ***p < 0.001. Q Developmental timing of phm > w1118 (n = 7) and phm>ParkinCA (n = 8) under feeding conditions. All Data are presented as mean ± SD. The genotypes and statistical data are provided in Supplementary Data 1.
Stress-activated mitophagy is linked to several factors involved in mitochondrial quality control, i.e., increasing reactive oxygen species (ROS), which are produced in and released from dysfunctional mitochondria54,55. We thus examined whether pre-NRC starvation promoted superoxide levels in PG cells using the mitoSOX dye and DCFH-DA staining to measure mitochondrial and cytosolic ROS levels, respectively. Although pre-NRC starvation was able to elevate both mitochondrial and cytosolic ROS levels, timing differed. Specifically, pre-NRC starvation caused an acute rise in mitochondrial ROS levels after just three hours of treatment, whereas cytosolic ROS took 12 hours to rise (Fig. 4C, D and Supplementary Fig. 4A). These data indicate that mitochondria are likely the major source of intracellular ROS generation. Clearance of ROS by expressing Catalase (Cat), which encodes an enzyme involved in hydrogen peroxide elimination56, in pre-NRC-starved PG cells effectively decreased mitophagy (Fig. 4E and Supplementary Data 1). Expressing superoxide dismutase 1 (SOD1) and superoxide dismutase 2 (SOD2) had similar but comparatively weaker effects (Supplementary Fig. 4B and Supplementary Data 1). Thus, the production of ROS was a consequence of pre-NRC starvation and a causative factor of mitochondrial dysfunction that was followed by an increase in mitophagy.
Most studies investigating mitophagy have focused on three canonical pathways, including Pink1/Parkin, Binp3/Nix, and Fundc152,57,58. We, therefore, mined the RG transcriptome data and analyzed the expression levels of genes associated with these pathways in response to pre-NRC starvation. We found that pre-NRC starvation caused the upregulation of Pink1 and CG5059 (homologous to human BNIP3), but not CG5676 (homologous to human FUNDC1) (Supplementary Data 2). Using Pink1-HA and BNIP3-GFP flies as reporters, we confirmed that the corresponding protein levels were also increased by pre-NRC starvation (Fig. 4F, H and Supplementary Data 1). In Pink1-dependent mitophagy, Pink1 recruits Parkin to damaged mitochondria, which ubiquitinates multiple target proteins in the outer mitochondrial membrane, which initiates autophagic degradation54. Although the mRNA levels of parkin were not elevated by pre-NRC starvation (Supplementary Data 2), we noticed a distinct aggregation of Parkin-EGFP (Fig. 4G and Supplementary Data 1). Importantly, PG-specific RNAi against Parkin, Pink1 and Parkin, or BNIP3 partially reduced the occurrence of pre-NRC starvation-dependent mitophagy (Fig. 4I and Supplementary Data 1), highlighting the regulatory roles of the two canonical pathways.
Mitophagy is a type of selective autophagy that specifically removes damaged mitochondria59. A previous report has shown that pre-NRC starvation promotes extensive autophagy in PG cells9. Consistent with this, we observed that pre-NRC starvation caused the accumulation of Atg5-GFP-positive protein aggregates and increased colocalization of Atg8a-mCherry and Lamp1-GFP (Fig. 4J, K, M, Supplementary Fig. 4C, D and Supplementary Data 1). PG-specific inhibition of PTTH signaling, insulin signaling, and TORC1 activity induced autophagy formation (Supplementary Fig. 4E, F and Supplementary Data 1), while stimulating these pathways reduced autophagy induction due to pre-NRC starvation (Fig. 4L, N and Supplementary Data 1). Further, PG-specific expression of Atg1DN or Atg8-RNAi reduced mitophagy occurrence caused by pre-NRC starvation (Fig. 4O, Supplementary Fig. 4G and Supplementary Data 1). These results show that autophagy plays an essential role in the removal of damaged mitochondria. Conversely, the activation of mitophagy by ectopically expressing ParkinCA prior to the NRC promoted autophagy formation and led to developmental arrest (Fig. 4P, Q and Supplementary Data 1). We conclude that pre-NRC starvation, which causes excessive mitophagy in PG cells, prevents steroidogenesis partially through the promotion of severe autophagy.
Pre-NRC starvation-induced mitophagy is coupled to proteasome dysfunction
Transcriptome analysis revealed that pre-NRC starvation downregulated 24 essential genes in the ubiquitin–proteasome system (Fig. 1), which is essential for removing improperly folded proteins (Fig. 3). We thus examined whether pre-NRC starvation reduced proteasome activity in PG cells, and for this, we used the FK2 antibody, which recognizes mono- and polyubiquitinated proteins60,61. Three hours of pre-NRC starvation acutely induced the formation of FK2 puncta, which progressively increased as starvation continued (Fig. 5A). When we expressed CL1-GFP, a GFP-reporter fused to the CL1 degron62, to analyze the activity of the ubiquitin-dependent proteasome pathway in the PG by different means, similar results were obtained (Fig. 5A). The two bioassays indicated that pre-NRC starvation caused proteasome dysfunction. PG-specific inhibition of PTTH signaling, insulin signaling, and TORC1 activity caused different degrees of proteasome dysfunction; however, activating PTTH signaling and insulin signaling, but not TORC1 activity, reduced proteasome dysfunction caused by pre-NRC starvation (Fig. 5B–D).

A Images of proteasomal activity (FK2 or CL1-GFP) in PG cells after 0 h, 3 h, 6 h and 12 h starvation from 72 h AEL. FK2, red; CL1-GFP, green. Magnified images of the highlighted areas (yellow dotted box) are shown. FK2: from left to right, n = 16, 12, 20 and 36, respectively. CL1-GFP: From left to right, n = 20, 33, 18 and 23, respectively. B, C Proteasomal activity visualized by FK2 (B, red) or CL1-GFP (C, green) was assessed in PG cells of phm > w1118, phm > Torso-RNAi, phm > RasDN, phm > InR-RNAi, phm > Dp110DN, phm > Torso + InR-RNAi, phm > TORDN and phm > TSC1/2 under control of Gal80ts. FK2: from left to right, n = 11, 12, 28, 11, 12, 15, 8 and 8, respectively. CL1-GFP: From left to right, n = 15, 26, 30, 26, 27, 7, 28 and 23, respectively. Magnified images of the highlighted areas (yellow dotted box) are shown. D, E Proteasomal activity visualized by FK2 (red) was assessed in PG cells of D phm > w1118 (n = 7), phm > RasV12 (n = 18), phm > Dp110CA (n = 9), phm > Rheb (n = 9), E phm > w1118 (n = 3), phm > Rpt3-RNAi (n = 3), phm > Rpn11-RNAi (n = 3), and phm > prosβ1-RNAi (n = 3). Magnified images of the highlighted areas (yellow dotted box) are shown. F, G Representative images of mitophagy (F, 561 nm, red; 405 nm, green) and mitochondria (G, green) in PG cells (132 h AEL) with knockdown of Rpt3, Rpn11 and prosβ1. F n = 17 (Control), 18 (Rpt3-RNAi), 11 (Rpn11-RNAi), 6 (Prosβ1-RNAi). G n = 16 (Control), 19 (Rpt3-RNAi), 19 (Rpn11-RNAi), 12 (Prosβ1-RNAi). H, I Images of proteasomal activity (FK2 antibody, red) H or CL1-GFP (green) I in PG cells of controls and PG-expressing ParkinCA under 29 °C. Magnified images of the highlighted areas (yellow dotted box) are shown. H n = 14 (Control), 9 (ParkinCA). J Proteasomal activity visualized by FK2 (red) was assessed in PG cells of phm > w1118 (n = 17), phm > COX6B-RNAi (n = 12), phm > Neb-cGP-RNAi (n = 14), phm > TrpRS-m-RNAi (n = 23), phm > ATPsynC-RNAi (n = 15), phm > mRPL47-RNAi (n = 4), phm > Tom20-RNAi (n = 15), and phm > mge-RNAi (n = 15). Magnified images of the highlighted areas (yellow dotted box) are shown. The genotypes and statistical data are provided in Supplementary Data 1.
We next assessed the potential crosstalk between mitochondria and proteasomes. PG-specific RNAi targeting three key proteasome genes, including Prosb1, Rpn11, and Rpt3, not only caused proteasome dysfunction, but also increased the rate of mitophagy to varying degrees. Furthermore, Prosb1-RNAi or Rpn11-RNAi eliminated most mitochondria in PG cells (Fig. 5E–G and Supplementary Data 1). Likewise, induction of mitophagy occurrence by PG-specific RNAi against mitochondria-associated genes or expression of ParkinCA caused proteasome dysfunction (Fig. 5H–J). Taken together, we conclude that pre-NRC starvation impaired mitochondrial homeostasis, which caused extensive autophagy and proteasome dysfunction, followed by a block in steroidogenesis and, thus, developmental arrest.
Post-NRC starvation does not impair mitochondrial homeostasis in PG cells
Since the transcriptome data revealed that post-NRC starvation downregulated only seven genes in the RG (Fig. 1), we hypothesized that nutrient deficiency did not significantly impact mitochondrial homeostasis in PGs after the NRC. As expected, post-NRC starvation did not affect mitochondrial dynamics, mitochondrial import of Dib, mitophagy occurrence, and proteasome activity in PG cells (Supplementary Fig. 5A–M and Supplementary Data 1), suggesting that nutrient uptake was dispensable for mitochondrial homeostasis in PG cells once larvae had passed the NRC. Likewise, PTTH signaling, insulin signaling, and TORC1 activity did not show any significant changes (Supplementary Fig. 5N–P and Supplementary Data 1). However, upon post-NRC starvation, PG-specific inhibition of PTTH signaling, insulin signaling or both, and TORC1 activity impaired mitochondrial homeostasis in PG cells (Fig. 6A–D and Supplementary Data 1), and moreover, significantly or dramatically caused developmental arrest with decreasing pupariation ratios (Fig. 6E, F and Supplementary Data 1). We conclude that although post-NRC starvation does not cause developmental arrest, PTTH signaling, insulin signaling, and TORC1 activity are indispensable for the animals to commit larvae to overcome post-NRC starvation, probably due to sufficient storages of their signal resources in other tissues (i.e., insulin storage in the corpora cardia24).

A–D PG-specific inhibition of PTTH signaling, insulin signaling, and TORC1 after the NRC impairs mitochondrial homeostasis. Representative images of mitochondrial morphology A, green, colocalization of Dib-mCherry (red) and mitochondria (mito-GFP, green) B, mitophagy C, and proteasomal activity D, red in the PG (168 h AEL) with knockdown of Torso, InR, and Torso + InR, or expression of RasDN, Dp110DN, TORDN and TSC1/2. A n = 24 (Control), 20 (Torso-RNAi), 21 (RasDN), 20 (InR-RNAi), 23 (Dp110DN), 24 (Torso+InR-RNAi), 21 (TORDN), 27 (TSC1/2). B n = 30 (Control), 29 (Torso-RNAi), 23 (RasDN), 34 (InR-RNAi), 19 (Dp110DN), 21 (Torso+InR-RNAi), 34 (TORDN), 23 (TSC1/2). C n = 28 (Control), 20 (Torso-RNAi), 19 (RasDN), 34 (InR-RNAi), 30 (Dp110DN), 27 (Torso+InR-RNAi), 28 (TORDN), 26 (TSC1/2). E, F PG-specific inhibition of PTTH, insulin, and TORC1 signalings after the NRC causes developmental arrest. Developmental timing with the indicated genotypes was examined under feeding conditions. n = 22 (Control), 8 (Torso-RNAi), 16 (RasDN), 12 (InR-RNAi), 15 (Dp110DN), 15 (Torso+InR-RNAi), 19 (TORDN), 11 (TSC1/2). Data are presented as mean ± SD. The genotypes and statistical data are provided in Supplementary Data 1.
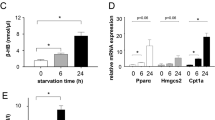
Because post-NRC starvation resulted in precocious metamorphosis and a smaller body size but had little effect on the expression of genes involved in steroidogenesis (Supplementary Fig. 1 and Supplementary Data 1), we investigated how post-NRC starvation accelerated steroidogenesis and promoted precocious metamorphosis. Strikingly, of the seven genes downregulated by post-NRC starvation (Fig. 7A), it appeared that only sit was involved in steroidogenesis8. As confirmed by quantitative real-time PCR (qPCR) analysis, post-NRC starvation, but not pre-NRC starvation, decreased sit expression in the RG (Fig. 7B and Supplementary Data 1). Similarly, sit expression in the RG was much lower during the wandering stages compared to the feeding stages (Fig. 7C and Supplementary Data 1). In line with the finding that Sit regulates cholesterol trafficking and lipid mobilization8, PG-specific RNAi knockdown of sit resulted in the accumulation of lipid droplets after the NRC but not prior to the NRC (Fig. 7D, E). Previous reports also showed that autophagy mobilizes cholesterol-rich lipid droplets in PG cells for steroidogenesis8,9,10, and as such we examined whether this was essential when post-NRC starvation was applied. Under normal conditions, the accumulation of lipid droplets in PG cells appeared to be slow during the feeding stages, with a peak in EW larvae, followed by a rapid decline at WPP (Fig. 7F, G and Supplementary Data 1). By contrast, we observed rapid lipid droplet accumulation when larvae were starved after the NRC (Fig. 7H, I and Supplementary Data 1). In Atg1 mutant larvae, starvation at 96 h AEL (body weight ~1.04 mg) was not able to induce precocious metamorphosis (Fig. 7J and Supplementary Data 1) but led to a smaller pupa (Fig. 7K, L and Supplementary Data 1). However, simultaneous PG-specific expression of NPC1-RNAi (functionally similar to sit) and Atg1-Atg13 resulted in precocious metamorphosis8. In contrast, upon post-NRC starvation, expression of sit or inhibition of Atg1 or both slightly delayed pupariation and decreased pupariation ratios (Fig. 7M and Supplementary Data 1). Taken together, we conclude that both i) the downregulation of sit to accumulate cholesterol-rich lipid droplets and ii) the activation of autophagy to mobilize lipid droplets are essential for post-NRC starvation to accelerate steroidogenesis and promote precocious metamorphosis.

A Heatmap represents expression profiles of the seven genes that were exclusively downregulated in post-NRC ST larvae. Numbers in the box represent log2 expression values (zero-centered) that resulted from the integrated data analysis of the indicated genes. B, C qPCR analysis of sit was performed in RGs of the feeding (n = 3), pre-NRC ST (n = 3), post-NRC ST (n = 3) larvae or feeding larvae at different developmental points, including 96 h (n = 3), EW (n = 3), and LW (n = 3). Different lowercase letters represent statistical significance by one-way ANOVA. D, E The projected confocal z-stack showing that loss of sit in the PG at 96 h E, but not 84 h D, caused accumulation of lipid droplets. F The projected confocal z-stack showing that lipid droplets visualized by Nile Red staining (Red) in w1118 PG cells at four developmental points. Magnified images of the indicated areas (yellow dotted box) are shown. n = 9 (96 h), 12 (108 h), 5 (EW), 5 (WPP). G Quantification of lipid droplets in the indicated time points shown in (F). Different lowercase letters represent statistical significance by one-way ANOVA. H The projected confocal z-stack showing lipid droplets visualized by Nile Red staining in w1118 PG cells of larvae after 0 h, 12 h, and 24 h starvation from 84 h AEL or at WPP. Magnified images of the indicated areas (yellow dotted box) are shown. n = 9 (84 h), 8 (84 h ST + 12 h), 9 (84 h ST + 24 h), 7 (WPP). I Quantification of lipid droplets in the indicated time points shown in (H). Different lowercase letters represent statistical significance by one-way ANOVA. J Developmental timing and body size of Atg1 mutant (Atg1−/−) larvae under feeding (n = 3) and starvation (n = 3) conditions. K, L Body size of Atg1 mutant (Atg1−/−) under feeding and starvation conditions. Atg1−/−: n = 47, Atg1−/− ST: n = 9. Two-tailed paired t test for two-column comparison. ***p < 0.001. M Developmental timing of tub-Gal80ts phm > w1118 (n = 17), tub-Gal80ts phm>sit (n = 13), tub-Gal80ts phm>Atg1-RNAi (n = 7) and tub-Gal80ts phm>sit+Atg1-RNAi (n = 16) under post-NRC ST conditions. All Data are presented as mean ± SD. The genotypes and statistical data are provided in Supplementary Data 1.
Discussion
This study shows that pre-NRC starvation selectively downregulates at least 81 mitochondria-associated genes in the Drosophila PG, which prevents mitochondrial fragmentation and Dib import, and causes severe mitophagy and proteasome dysfunction, resulting in the blockage of steroidogenesis followed by developmental arrest. However, post-NRC starvation did not impair mitochondrial homeostasis, but reduced sit expression and induced moderate levels of autophagy, leading to accelerated steroidogenesis and precocious metamorphosis. In short, nutrient status alters developmental fates via a switch in mitochondrial homeodynamics of PG cells to control steroid hormone production in Drosophila (Supplementary Fig. 6).
Here, we showed that pre-NRC starvation impaired mitochondrial homeostasis in PG cells. Pre-NRC starvation-induced mitochondrial tubulation-stimulated ATP production but reduced mitochondrial import of Dib, which is indispensable for steroidogenesis (Figs. 2 and 3). Mitochondria are highly dynamic organelles, which undergo constant fusion and fission, both of which are closely tied to mitochondrial function40. In nutrient-replete conditions, we observed that mitochondrial fragmentation in PG cells was stimulated by the nutrient-responsive pathways, indicating a positive regulatory role for steroidogenesis. Moreover, PG-specific expression of Marf not only prevented mitochondrial fragmentation, but also reduced steroidogenesis. Recent work showed that fragmented mitochondria are vital for a range of physiological events in specialized cells, i.e., mtDNA selection in the female germline63 and increasing Ca2+ uptake and synaptic transmission during axonal development64. Importantly, pre-NRC starvation-induced mitochondrial tubulation resulted in metabolic reprogramming, allowing cells to shift from steroidogenesis to ATP production even during metabolic stress, a strategy that possibly increases resistance to starvation conditions.
A key mechanism that interrupts ecdysone biosynthesis relies on inhibiting mitochondrial import of Dib. Increasingly longer starvation periods will eventually cause larval death before the NRC is reached, but re-feeding survivors will allow normal development to resume. Second, we found that mitophagy and proteasome dysfunction were linked (Figs. 4 and 5). In this study, we showed that mitophagy and proteasome dysfunction were two major cellular consequences of pre-NRC starvation; and moreover, mitophagy and proteasome dysfunction mutually reinforced each other, and accelerated the inhibition of steroidogenesis. Taken together, our approaches allowed us to unravel the cellular and molecular mechanisms by which pre-NRC starvation blocks steroidogenesis in the PG.
Post-NRC starvation did not impair mitochondrial homeostasis in PG cells, but accelerated steroidogenesis and caused precocious metamorphosis (Figs. 6, 7, and Supplementary Fig. 5). A number of studies have shown that Sit and autophagy are responsible for precocious metamorphosis caused by post-NRC starvation8,9,10. We showed here that the downregulation of sit and the activation of autophagy are both indispensable for this physiological event. Once the NRC is passed, developmental plasticity allows animals to survive in smaller body size with decreased reproductive capacity1. In addition, we propose that sit downregulation and autophagy initiation may be induced by 20E after the NRC, thus forming a positive feedback regulatory loop to accelerate this irreversible process. Both insect metamorphosis and mammalian puberty are governed by steroid hormones and neuroendocrine signals, highlighting a conserved maturation system across animals. Focusing on mitochondrial homeodynamics, we here discovered the molecular and cellular mechanisms of how nutrient status alters developmental fates in Drosophila. In future, it will be essential to examine whether the major or principal discoveries in this study apply to mammalian puberty and other systems.
Most studies investigating the impact of the NRC on steroidogenesis have concentrated on TORC1 activity, neglecting the roles of PTTH signaling and insulin signaling. We hypothesize that during Drosophila development, most environmental cues are integrated via the PTTH- and ILP-producing neurons, since both appear to be more important than TORC1 for the regulation of steroidogenesis prior to the NRC. Although PTTH and ILPs act mainly through the Ras/Raf/Erk pathway and the PI3K/Akt pathway, respectively, PTTH or ILP can activate either pathway, but to different degrees. Moreover, other extracellular signals, such as EGF, PDGF, and VEGF, also regulate steroidogenesis through these two pathways. In addition to insulin signaling and PTTH signaling, TORC1 can be activated by small nutrient components, such as amino acids, fatty acids, and sugar5,6,7. Our results clearly showed that PTTH signaling and insulin signaling play more important roles than TORC1 activity prior to the NRC (Figs. 2–5), whereas they are nearly equally important after the NRC (Fig. 6), showing that steroidogenesis is coordinately regulated by PTTH signaling, insulin stimulation, and TORC1 activation in a stage-specific manner.
Although the PG plays a central role in establishing the NRC, other tissues, including the fat body, midgut, and neuroendocrine system, are also crucial for regulating steroidogenesis and fulfilling the NRC24,25,65,66. We believe that larvae must accumulate sufficient levels of stored ILP (in the corpora cardiaca24) and nutrients (primarily in the fat body65), alongside the PG’s ability to produce adequate ecdysone, to overcome the post-NRC starvation and to cause precocious metamorphosis. Moreover, we propose that nutrient ingestion prior to the NRC is critical for survival, whereas feeding after the NRC mainly benefits future fitness65. Consequently, the PG must communicate with other tissues and organs to integrate systemically derived signals to appropriately navigate the NRC.
In summary, mitochondrial homeodynamics in the PG serves as the mechanistic foundation to switch the NRC in Drosophila, because pre- but not post-NRC starvation impairs mitochondrial homeostasis. This study has established a paradigm for exploring how a steroid hormone-producing endocrine gland accurately responds to environmental cues, allowing the coordination of intrinsic and extrinsic signals to control the juvenile-to-adult transitions in animals.
Methods
Fly stocks
For the list of fly stocks, please refer to Supplementary Table 1. Flies were normally raised on standard Drosophila medium with a 12 h/12 h light cycle at 25 °C unless stated otherwise. For efficient expression of RNAi transgenes, larvae were raised at 29 °C. For temperature control of gene expression using Gal80ts, larvae were first reared at 18 °C and then were shifted to 29 °C to allow gene expression. The genotypes of the fly strains generated in this paper and the experimental conditions that require clarification are shown in Supplementary Data 1.
Generation of transgenic flies
UAS-mt-Keima
The mt-Keima cDNA was amplified from the pCHAC-mt-mKeima plasmid (Addgene #72342) using PCR primers containing EcoRI and XhoI restriction enzyme sites and was subcloned into the pUAST vector after restriction digestion. The pUAST-mt-Keima construct was verified by DNA sequencing and microinjected into w1118 embryos. The pair of primers used are follows:
mt-Keima-F: 5’ GAATTCATGTCCGTCCTGACGCC
mt-Keima-R: 5’ CTCGAGTTAGCCCAGCAGGGAGTG
UAS-Sit-HA
The full-length open reading frame of Drosophila sit (NM_142806) was amplified from a larval whole body cDNA library. The PCR fragment was cloned into the pMD18-T vector (Takara). The sit-HA cDNA was amplified from the pMD18-T-Sit vector using PCR primers containing EcoRI and KpnI restriction enzyme sites and was subcloned into the pUAST vector after restriction digestion. The pUAST-sit-HA construct was verified by DNA sequencing and microinjected into w1118 embryos. The pairs of primers used are as follows:
sit-cDNA-F: 5’ ATGGCAGCCGTCAACGCAAC
sit-cDNA-R:5’ TTGTGCTCCTCCCTCTTA
UAS-sit-HA-F: 5’ GAATTCATGGCAGCCGTCAACGCAA
UAS-sit-HA-R: 5’ GGTACCTTAAGCGTAGTCTGGGACGTCGTATGGGTACAAGGCCTTCTGCTTCTCC
Dib-mCherry CRISPR knock-in flies
The CRISPR/Cas9 technology was used to establish the C-terminal mCherry tagged Dib knock-in fly which was performed by Qidong Fungene Biotechnology (http://www.fungene.tech). Guide RNA targets were designed with Chopchop67 and CCTop68. Following the stablished procedures69, Dib sgRNA template for in vitro transcription by T7 polymerase was generated by annealing two DNA oligonucleotides following by PCR amplification. In vitro transcription was performed with the T7 RiboMAX TM Kit (Promega, P1320). Transcripts were purified by phenol-chloroform extraction and isopropanol precipitation. Plasmid MLM3613 (Addgene #42251) was linearized with PmeI (New England Biolabs) and purified by ethanol precipitation. Cas9 mRNA was transcribed with mMESSAGE mMACHINE® T7 Transcription Kit (Ambion), polyadenylated with the E. coli Poly(A) polymerase Kit (NEB), and purified with the RNeasy Mini Kit (QIAGEN). Candidate DNA fragments and mCherry were amplified and linked to the pBluescirpt SK backbone with Gibson Assembly Kit (Cat# E2611L, NEB), and that produced the final donor construct. To avoid cleavage of the donor plasmid, the PAM site was replaced with a synonymous codon. Embryos were injected using standard protocols. The sgRNA (250 ng/ul), Cas9 mRNA (500 ng/ul) and donor plasmid (500 ng/ul) were mixed and injected into at least 300 embryos.
The primers used for gRNAs and PCR validation are as follows:
Dib-sgRNA1: 5’ AATCGCAGATCGATCAGCAC
Dib-sgRNA2: 5’ ATCGATCAGCACTGGAGCAT
Dib-mCherry-F1: 5’ TGCACCAAGCCGGCCAGGATAAG
Dib-mCherry-R1: 5’ GGAGCCGTACATGAACTGAG
Dib-mCherry-F2: 5’ ACGACGCTGAGGTCAAGAC
Dib-mCherry-R2: 5’ ACCGAACAACCTGGATCTTAATCC
Fluorescence microscopy
Larvae were dissected rapidly in PBS at room temperature. For RGs that expressed fluorescent-tagged proteins, tissues were mounted immediately in PBS for imaging. For RGs treated with dyes or immunohistochemical assay, the methods are described in the following sections.
Dye staining
Larvae were inverted to expose RGs and stained with Lysotracker (Thermo Fisher, Cat# L7526), mitoSOX (Thermo Fisher, Cat# M36008), DCFH-DA (Beyotime, Cat# S0033S), and mitotracker (Beyotime, Cat# C1048) in PBS or cell culture medium for 15-30 mins. After washing three times in PBS or cell culture medium, RGs were mounted on slides for imaging. Images were obtained using an Olympus Fluoview FV3000 confocal microscope.
Immunohistochemistry
Dissected tissues were fixed and stained with dyes or antibodies according to standard procedures23. The primary antibodies used were Rabbit anti-p-Erk (1:200, Abcam, Cat# ab32538), Rabbit anti-p-S6K (1:200, Cell Signaling Technology, Cat# 9209), Rabbit anti-p-Drp1(Ser637) (1:200, Affinity Biosciences, Cat# DF2980), Rabbit anti-p-AMPK (1:200, Cell Signaling Technology, Cat# 2535), Mouse anti-HA (1:200, Santa Cruz Biotechnology, Cat# sc-7392), Mouse anti-FK2 (1:200, Enzo Life Sciences, Cat# ENZ-ABS840-0100). Secondary antibodies used were Alexa Fluor 488 goat anti-rabbit IgG (1:200, A11008; Invitrogen), Alexa Fluor 594 goat anti-mouse IgG (1:200, A11032; Invitrogen). Nuclei were stained with DAPI at 1:2,000 (C1002; Beyotime). Confocal images were collected on an Olympus Fluoview FV3000 confocal microscope. Immunohistochemical staining was quantified by means of color image analysis using ImageJ.
RNA-seq
The RGs of control or starved w1118 larvae were dissected in PBS in batches of 10–40 at a time, and pooled into three biological replicates for every treatment. About 1,200 RGs were pooled per sample, which yielded ~1–5 μg of RNA required for sequencing. Total RNA was extracted using the miRNeasy Micro Kit (QIAGEN, Germany), then stored at ~80 °C. RNA quality, especially DNA contamination, was monitored on 1.5% agarose gels. RNA concentration was measured using NanoDrop 2000 (Thermo Fisher Scientific, USA). RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, USA). High-quality RNA samples from Drosophila RGs were sent to Biomarker Technologies Corporation (Beijing, China) for construction of cDNA libraries and sequencing.
Libraries were generated using NEBNext® Ultra™ small RNA Sample Library Prep Kit for Illumina® (NEB, USA) and NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (NEB, USA) following manufacturer’s recommendations, and index codes were added to attribute sequences to each sample. The libraries prepared were sequenced on an Illumina Hiseq platform and paired-end reads were generated. Raw sequencing data has been deposited at Gene Expression Omnibus (GEO) database (accession: GSE267248).
Paired fastq sequencing reads were aligned to the annotated D. melanogaster reference genome (BDGP release 6) using HISAT270, StringTie71, and Bowtie72. The resulting alignment files were reconstructed with Cufflinks. Expression levels were quantified as FPKM.
qPCR
Total RNA samples were prepared from the RGs of D. melanogaster. qPCR was performed using the IQ SYBR Green supermix (Bio-Rad). qPCR was carried out in triplicate using rp49 as an internal control. The primers used for qPCR are as follows:
sit-qPCR-F: 5’ GTCAACGCAACACAGGTGGA
sit-qPCR-R: 5’ ACGAAGAAGAGATAGAAGGCCAG
mRpS9-qPCR-F: 5’ CGATACATGAAACCGCTGATGGA
mRpS9-qPCR-R: 5’ CGCACTGTGACATCGGCTCTA
Nile Red staining
Larvae were inverted to expose RGs and fixed with 4% paraformaldehyde for 40 min. Dissected RGs were washed three times in PBS and stained with 0.2 μg/ml Nile Red (Sangon Biotech, Cat# A606340) in PBS for 1 h. After washing three times in PBS, RGs were mounted on slides for imaging. Images were obtained using an Olympus Fluoview FV3000 confocal microscope.
Developmental timing
Eggs were collected at 25 °C for 2–4 h on standard food in bottles. For UAS-Marf experiments, these bottles were kept at 25 °C to allow developing into the second instar, and 20-30 larvae per vial were transferred to and raised at 25 °C. For UAS-ParkinCA experiments, these bottles were first kept at 25 °C for 24 h to allow egg hatching and then transferred to 29 °C for 24 h to allow developing into the second instar, and 20-30 larvae per vial were transferred to and raised at 29 °C. The starvation experiments: controls were kept at 25 °C to allow developing into the second instar, and 20-30 larvae per vial were transferred to and raised at 25 °C. For the pre-NRC starvation cohort, 72 h-old larvae were transferred onto a piece of filter paper soaked in distilled water and raised at 25 °C. For the post-NRC starvation populations, 84h-old larvae were transferred onto a piece of filter paper soaked in distilled water and raised at 25 °C. The Atg1 mutant starvation, 96 h larvae were transferred onto a piece of filter paper soaked in distilled water and raised at 25 °C. Pupariation was recorded by counting pupa number every 12 h.
Quantification of body size
Images of pupae were captured using a Nikon SMZ25 fluorescence stereomicroscope. Pupae were measured for each experimental condition using ImageJ.
Quantification of percentage of fragmented mitochondria
The rate of fragmented mitochondria was quantified manually47. Briefly, for morphological quantification, each image was randomly assigned into three sections, and each section area was 400 × 400 pixel. Then the number of fragmented and tubular mitochondria were manually counted. The rate of fragmented mitochondria for all figures was recorded. The collected data were organized into Supplementary Data 1 or used to generate box plots.
Quantification of colocalization
Colocalization analysis was performed using the JaCoP plugin in ImageJ73. Consistently, identical threshold parameters were maintained across all images throughout the image analysis process. Mander’s Colocalization Coefficient was used.
Quantification of mitophagy
As described by Cornelissen et al., the metric (ratio of high [561/405] ratio area / total mitochondrial area) served as an indicator for mitophagy74. Mt-Keima fluorescence was imaged in two channels via two sequential excitation lasers (405 nm, green; 561 nm, red). High (561/405) ratio areas were segmented and measured with the Analyze Particles plugin in ImageJ. The total mitochondrial area was measured by calculating the area of the total emission at 405 nm excitation using the Analyze Particles plugin in ImageJ.
Quantification of lipid droplet
Images stained by Nile Red were merged into a single section for each RG using Olympus Fluoview FV3000 built-in software. The area of lipid droplets was quantified using ImageJ. A freehand selection option in ImageJ software was used to outline the RG. Nile Red stained RG images were thresholded for the lipid droplet signal. Then, the area of RG and the total area of the lipid droplets were calculated automatically in the software.
Statistics
All experiments were repeated at least three times, and Student’s t test or one-way ANOVA was used to determine statistical significance. Differences were considered significant at p < 0.05. Data are presented as mean ± SD. ***p < 0.001; **p < 0.01; *p < 0.05. The statistical data for all figures are listed in Supplementary Data 1.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA sequencing data in this study have been deposited in the NCBI Gene Expression Omnibus (GEO) database under accession GSE267248. Data points displayed on charts and graphs are detailed in Supplementary Data 1.
References
Nijhout, H. F. A developmental-physiological perspective on the development and evolution of phenotypic plasticity. In: Conceptual Change in Biology: Scientific and Philosophical Perspectives on Evolution and Development, A. C. Love, ed. (Dordrecht: Springer Netherlands), pp. 147–173 (2015).
Villamor, E. & Jansen, E. C. Nutritional determinants of the timing of puberty. Annu. Rev. Publ. Health 37, 33–46 (2016).
Norris, S. A. et al. Nutrition in adolescent growth and development. Lancet 399, 172–184 (2022).
Mirth, C. K. & Riddiford, L. M. Size assessment and growth control: how adult size is determined in insects. BioEssays 29, 344–355 (2007).
Yamanaka, N., Rewitz, K. F. & O’Connor, M. B. Ecdysone control of developmental transitions: lessons from Drosophila research. Annu. Rev. Entomol. 58, 497–516 (2013).
Koyama, T., Texada, M. J., Halberg, K. A. & Rewitz, K. Metabolism and growth adaptation to environmental conditions in Drosophila. Cell. Mol. Life Sci. 77, 4523–4551 (2020).
Pan, X., Connacher, R. P. & O’Connor, M. B. Control of the insect metamorphic transition by ecdysteroid production and secretion. Curr. Opin. Insect Sci. 43, 11–20 (2021).
Danielsen, E. T. et al. A Drosophila genome-wide screen identifies regulators of steroid hormone production and developmental timing. Dev. Cell 37, 558–570 (2016).
Pan, X., Neufeld, T. P. & O’Connor, M. B. A tissue- and temporal-specific autophagic switch controls Drosophila pre-metamorphic nutritional checkpoints. Curr. Biol. 29, 2840–2851.e2844 (2019).
Texada, M. J. et al. Autophagy-mediated cholesterol trafficking controls steroid production. Dev. Cell 48, 659–671.e654 (2019).
Yamanaka, N., Marqués, G. & O’Connor, M. B. Vesicle-mediated steroid hormone secretion in Drosophila melanogaster. Cell 163, 907–919 (2015).
Zhang, T. et al. Evolution of the cholesterol biosynthesis pathway in animals. Mol. Biol. Evol. 36, 2548–2556 (2019).
Caldwell, P. E., Walkiewicz, M. & Stern, M. Ras activity in the Drosophila prothoracic gland regulates body size and developmental rate via ecdysone release. Curr. Biol. 15, 1785–1795 (2005).
Colombani, J. et al. Antagonistic actions of ecdysone and insulins determine final size in Drosophila. Science 310, 667–670 (2005).
Mirth, C., Truman, J. W. & Riddiford, L. M. The role of the prothoracic gland in determining critical weight for metamorphosis in Drosophila melanogaster. Curr. Biol. 15, 1796–1807 (2005).
Layalle, S., Arquier, N. & Léopold, P. The TOR pathway couples nutrition and developmental timing in Drosophila. Dev. Cell 15, 568–577 (2008).
Rewitz, K. F., Yamanaka, N., Gilbert, L. I. & O’Connor, M. B. The insect neuropeptide PTTH activates receptor tyrosine kinase torso to initiate metamorphosis. Science 326, 1403–1405 (2009).
Ou, Q., Magico, A. & King-Jones, K. Nuclear receptor DHR4 controls the timing of steroid hormone pulses during Drosophila development. PLoS Biol. 9, e1001160 (2011).
Ohhara, Y., Kobayashi, S. & Yamanaka, N. Nutrient-dependent endocycling in steroidogenic tissue dictates timing of metamorphosis in Drosophila melanogaster. PLoS Genet. 13, e1006583 (2017).
Zeng, J., Huynh, N., Phelps, B. & King-Jones, K. Snail synchronizes endocycling in a TOR-dependent manner to coordinate entry and escape from endoreplication pausing during the Drosophila critical weight checkpoint. PLoS Biol. 18, e3000609 (2020).
Rewitz, K. F., Yamanaka, N. & O’Connor, M. B. Steroid hormone inactivation is required during the juvenile-adult transition in Drosophila. Dev. Cell 19, 895–902 (2010).
Li, K. et al. 20-Hydroxyecdysone (20E) Primary response gene E75 isoforms mediate steroidogenesis autoregulation and regulate developmental timing in Bombyx. J. Biol. Chem. 291, 18163–18175 (2016).
Liu, S. et al. Antagonistic actions of juvenile hormone and 20-hydroxyecdysone within the ring gland determine developmental transitions in Drosophila. Proc. Natl Acad. Sci. USA 115, 139–144 (2018).
Ghosh, S. et al. A local insulin reservoir in Drosophila alpha cell homologs ensures developmental progression under nutrient shortage. Curr. Biol. 32, 1788–1797.e1785 (2022).
Rodenfels, J. et al. Production of systemically circulating Hedgehog by the intestine couples nutrition to growth and development. Genes Dev. 28, 2636–2651 (2014).
Nunnari, J. & Suomalainen, A. Mitochondria: in sickness and in health. Cell 148, 1145–1159 (2012).
Wong, E. & Cuervo, A. M. Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb. Perspect. Biol. 2, a006734 (2010).
Danielsen, E. T. et al. Transcriptional control of steroid biosynthesis genes in the Drosophila prothoracic gland by ventral veins lacking and knirps. PLoS Genet. 10, e1004343 (2014).
Uryu, O. et al. Cooperative control of ecdysone biosynthesis in Drosophila by transcription factors Séance, Ouija Board, and molting defective. Genetics 208, 605–622 (2018).
Komura-Kawa, T. et al. The Drosophila zinc finger transcription factor Ouija Board controls ecdysteroid biosynthesis through specific regulation of spookier. PLoS Genet. 11, e1005712 (2015).
Zhao, Y. et al. Stop codon readthrough alters the activity of a POU/Oct transcription factor during Drosophila development. BMC Biol. 19, 185 (2021).
Ren, J. C., Rebrin, I., Klichko, V., Orr, W. C. & Sohal, R. S. Cytochrome c oxidase loses catalytic activity and structural integrity during the aging process in Drosophila melanogaster. Biochem. Biophys. Res. Commun. 401, 64–68 (2010).
Lovero, D. et al. Characterization of Drosophila ATPsynC mutants as a new model of mitochondrial ATP synthase disorders. PloS One 13, e0201811 (2018).
Marygold, S. J. et al. The ribosomal protein genes and Minute loci of Drosophila melanogaster. Genome Biol. 8, R216 (2007).
Maffezzini, C. et al. Mutations in the mitochondrial tryptophanyl-tRNA synthetase cause growth retardation and progressive leukoencephalopathy. Mol. Genet. Genom. Med. 7, e654 (2019).
Chen, P. L. et al. Vesicular transport mediates the uptake of cytoplasmic proteins into mitochondria in Drosophila melanogaster. Nat. Commun. 11, 2592 (2020).
Periasamy, A. et al. An increase in mitochondrial TOM activates apoptosis to drive retinal neurodegeneration. Sci. Rep. 12, 21634 (2022).
Gestaut, D., Limatola, A., Joachimiak, L. & Frydman, J. The ATP-powered gymnastics of TRiC/CCT: an asymmetric protein folding machine with a symmetric origin story. Curr. Opin. Struct. Biol. 55, 50–58 (2019).
Jin, M., Liu, C., Han, W. & Cong, Y. TRiC/CCT chaperonin: structure and function. Subcell. Biochem. 93, 625–654 (2019).
Kleele, T. et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 593, 435–439 (2021).
Li, Y. et al. Metabolic control of progenitor cell propagation during Drosophila tracheal remodeling. Nat. Commun. 13, 2817 (2022).
Koyama, T., Rodrigues, M. A., Athanasiadis, A., Shingleton, A. W. & Mirth, C. K. Nutritional control of body size through FoxO-Ultraspiracle mediated ecdysone biosynthesis. eLife 3, e03091 (2014).
Campello, S. & Scorrano, L. Mitochondrial shape changes: orchestrating cell pathophysiology. EMBO Rep. 11, 678–684 (2010).
Senyilmaz, D. et al. Regulation of mitochondrial morphology and function by stearoylation of TFR1. Nature 525, 124–128 (2015).
Chang, C. R. & Blackstone, C. Drp1 phosphorylation and mitochondrial regulation. EMBO Rep. 8, 1088–1089 (2007).
Zhang, Q. et al. Visualizing dynamics of cell signaling in vivo with a phase separation-based kinase reporter. Mol. Cell 69, 347 (2018).
Zhou, C. et al. TRABD modulates mitochondrial homeostasis and tissue integrity. Cell Rep. 43, 114304 (2024).
Warren, J. T. et al. Molecular and biochemical characterization of two P450 enzymes in the ecdysteroidogenic pathway of Drosophila melanogaster. Proc. Natl Acad. Sci. USA 99, 11043–11048 (2002).
Wang, X. et al. A red-emission fluorescent probe for visual monitoring of lysosomal pH changes during mitophagy and cell apoptosis. Analyst 145, 7018–7024 (2020).
Lamark, T. & Johansen, T. Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int. J. Cell Biol. 2012, 736905 (2015). (2012).
Ma, X. et al. CCT2 is an aggrephagy receptor for clearance of solid protein aggregates. Cell 185, 1325–1345.e1322 (2022).
Lazarou, M. et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314 (2015).
Kim, Y. Y. et al. Assessment of mitophagy in mt-Keima Drosophila revealed an essential role of the PINK1-Parkin pathway in mitophagy induction in vivo. FASEB J. 33, 9742–9751 (2019a).
Eiyama, A. & Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 33, 95–101 (2015).
Schofield, J. H. & Schafer, Z. T. Mitochondrial reactive oxygen species and mitophagy: a complex and nuanced relationship. Antioxid. Redox Signal. 34, 517–530 (2021).
Mackay, W. J. & Bewley, G. C. The genetics of catalase in Drosophila melanogaster: isolation and characterization of acatalasemic mutants. Genetics 122, 643–652 (1989).
Liu, L. et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14, 177–185 (2012).
Zhang, J. & Ney, P. A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 16, 939–946 (2009).
Tolkovsky, A. M. Mitophagy. Biochim. Biophys. Acta 1793, 1508–1515 (2009).
Fujimuro, M., Sawada, H. & Yokosawa, H. Dynamics of ubiquitin conjugation during heat-shock response revealed by using a monoclonal antibody specific to multi-ubiquitin chains. Eur. J. Biochem. 249, 427–433 (1997).
Haas, K. F., Woodruff, E. 3rd & Broadie, K. Proteasome function is required to maintain muscle cellular architecture. Biol. Cell 99, 615–626 (2007).
Bence, N. F., Sampat, R. M. & Kopito, R. R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292, 1552–1555 (2001).
Lieber, T., Jeedigunta, S. P., Palozzi, J. M., Lehmann, R. & Hurd, T. R. Mitochondrial fragmentation drives selective removal of deleterious mtDNA in the germline. Nature 570, 380–384 (2019).
Lewis, T. L. Jr., Kwon, S. K., Lee, A., Shaw, R. & Polleux, F. MFF-dependent mitochondrial fission regulates presynaptic release and axon branching by limiting axonal mitochondria size. Nat. Commun. 9, 5008 (2018).
Yamada, T., Hironaka, K.-i, Habara, O., Morishita, Y. & Nishimura, T. A developmental checkpoint directs metabolic remodelling as a strategy against starvation in Drosophila. Nat. Metab. 2, 1096–1112 (2020).
Tyson, J. J., Monshizadeh, A., Shvartsman, S. Y. & Shingleton, A. W. A dynamical model of growth and maturation in Drosophila. Proc. Natl Acad. Sci. USA 120, e2313224120 (2023).
Labun, K. et al. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 47, W171–w174 (2019).
Labuhn, M. et al. Refined sgRNA efficacy prediction improves large- and small-scale CRISPR-Cas9 applications. Nucleic Acids Res. 46, 1375–1385 (2018).
Bassett, A. R., Tibbit, C., Ponting, C. P. & Liu, J. L. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep. 4, 220–228 (2013).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019b).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Cornelissen, T. et al. Deficiency of parkin and PINK1 impairs age-dependent mitophagy in Drosophila. Elife 7, e35878 (2018).
Acknowledgements
We thank the Bloomington Drosophila Stock Center, the Tsinghua Fly Center, the Vienna Drosophila Resource Center, and the Core Facility of Drosophila Resource and Technology (CEMCS, CAS) for providing fly stocks. We thank Drs. Michael O’Connor, Thomas P. Neufeld, Lei Xue, Chao Tong, Hai Huang, Yunpeng Huang, Junzheng Zhang, and Wenfeng Chen for providing fly stocks. We thank the Core Facility of Drosophila Resource and Technology (CEMCS, CAS) and Qidong Fungene Biotechnology for transgenic fly generation. This work was funded by the National Natural Science Foundation of China (Grant No. 31930014 and 32220103003) and the Laboratory of Lingnan Modern Agriculture Project (Grant No. NT2021003) to S. Li, the National Natural Science Foundation of China (Grant No. 32222013 and 32070441) and the Guangdong Natural Science Funds for Distinguished Young Scholar (2022B1515020043) to S. Liu, and the National Natural Science Foundation of China (32200385) to J. Zhang.
Author information
Authors and Affiliations
Contributions
S. Li led the project. J.Z. conducted RG dissections for transcriptome analysis. J.Z., S. Liu, Y. Li., G.X., and H.D. conducted the experiments. J.Z., S. Liu, and S. Li analyzed the data. S. Liu, S. Li, and K.K.-J. conceptualized the study and wrote the manuscript, with the other authors providing editorial comments.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, J., Liu, S., Li, Y. et al. Nutrient status alters developmental fates via a switch in mitochondrial homeodynamics. Nat Commun 16, 1258 (2025). https://doi.org/10.1038/s41467-025-56528-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56528-z
