
Abstract
Genomics can provide insight into the etiology of type 2 diabetes and its comorbidities, but assigning functionality to non-coding variants remains challenging. Polygenic scores, which aggregate variant effects, can uncover mechanisms when paired with molecular data. Here, we test polygenic scores for type 2 diabetes and cardiometabolic comorbidities for associations with 2,922 circulating proteins in the UK Biobank. The genome-wide type 2 diabetes polygenic score associates with 617 proteins, of which 75% also associate with another cardiometabolic score. Partitioned type 2 diabetes scores, which capture distinct disease biology, associate with 342 proteins (20% unique). In this work, we identify key pathways (e.g., complement cascade), potential therapeutic targets (e.g., FAM3D in type 2 diabetes), and biomarkers of diabetic comorbidities (e.g., EFEMP1 and IGFBP2) through causal inference, pathway enrichment, and Cox regression of clinical trial outcomes. Our results are available via an interactive portal (https://public.cgr.astrazeneca.com/t2d-pgs/v1/).
Similar content being viewed by others


Introduction
Diabetes mellitus is a complex, multifactorial metabolic disorder diagnosed via a single clinical feature, hyperglycaemia1,2. Among the major diagnoses of diabetes mellitus, type 2 diabetes (T2D) has the largest worldwide disease burden1. To date, large-scale genome-wide association studies (GWAS) for T2D3,4 have helped uncover important T2D biology, such as the link between C2CD4A and beta-cell dysfunction5,6. However, many T2D risk variants identified via GWAS have small effect sizes and tend to map to non-protein-coding regions, which makes it challenging to uncover their downstream biological effects. Polygenic risk scores (PRS), also known as polygenic scores (PGS), aggregate the small effects of these variants and can be utilised for risk prediction and stratification, including for T2D7,8.
Pairing PGS with molecular data can provide a powerful approach to understanding the downstream biological effects of polygenic risk to disease. This can help elucidate key pathways relevant to disease pathophysiology and potentially find therapeutic targets that may be missed by traditional disease-gene mapping approaches. For example, studies by Ritchie et al. and Steffen et al. tested the association between PGS and protein expression levels, uncovering molecular mechanisms underlying polygenic risk of cardiometabolic diseases9,10. Furthermore, PGS and proteomics information, coupled with causal inference methodology such as mediation, complements approaches that use individual genetic variants as instruments, such as Mendelian randomisation (MR). This is particularly crucial for proteins that lack the genetic variation (i.e., large enough effect size and/or frequency) needed to serve as MR instruments.
T2D is highly heterogeneous, with affected individuals having different degrees of insulin resistance and beta-cell dysfunction11. In addition, the vast majority of T2D patients have at least one additional comorbidity, such as hypertension, obesity or hyperlipidaemia12, resulting from the complex interplay between the pathophysiology of T2D, adiposity, and the other comorbidities. A single T2D PGS is unlikely to capture this heterogeneity. To address this, partitioned polygenic scores (pPS), derived from the genetic clustering of GWAS-identified T2D variants, have been developed to capture biological processes underlying T2D genetic risk13,14. These pPS improve the prediction of clinical outcomes in patients with T2D compared to a conventional genome-wide T2D PGS15. Based on these observations, we hypothesise that leveraging genome-wide and partitioned T2D PGS, cardiometabolic PGS, and large-scale proteomics data will identify proteins and pathways that lead to the development of T2D or comorbidity and discover molecular mechanisms where these processes intersect.
In this work, we interrogate how PGS for T2D (including five partitioned T2D scores13: beta cell, lipodystrophy, liver lipid, obesity, proinsulin) and its cardiometabolic complications (coronary artery disease - CAD, chronic kidney disease - CKD and adiposity ascertained as body mass index - BMI) perturb the plasma proteome using data from the UK Biobank Pharma Proteomics Project (UKB-PPP)16,17. We further perform causal inference in the UK Biobank and survival analysis for cardiorenal outcomes in randomised controlled trials (RCTs) using the PGS-associated proteins to identify potential therapeutic targets and biomarkers of T2D comorbidities. By leveraging a large-scale biobank and two RCTs, our study provides insights into the etiology of T2D and its comorbidities that may have translational potential.
Results
Study design
This study leverages proteogenomic data from three studies: the UK Biobank18 (UKB), Exenatide Study of Cardiovascular Event Lowering19 (EXSCEL), and Dapagliflozin Effect on Cardiovascular Events–Thrombolysis in Myocardial Infarction 5820 (DECLARE–TIMI 58). The UKB is a population-level biobank with 14 years of follow-up time, with proteomic data available as part of the UK Biobank Pharma Proteomics Project (UKB-PPP). EXSCEL and DECLARE-TIMI 58 were both cardiovascular outcome trials in patients with T2D with mean follow-up times of 3.2 and 4.2 years, respectively (see Methods for a full cohort description). We first tested T2D and cardiometabolic PGS for association with circulating proteins in the UKB, followed by further analyses to identify putatively causative proteins among the set of PGS-associated protein biomarkers. Then, to assess the association between PGS-associated proteins and common comorbidities, we used cardiorenal outcomes in EXSCEL and DECLARE-TIMI 58. See Fig. 1 for an overview of our workflow, Methods, and Supplementary Data 4 and 5 for descriptions of all PGS.

The centre of the flowchart corresponds to the primary objectives or aims of the study (in Blue). Branching off each objective are short summaries of the analyses corresponding to that objective (in Red).
Associations of T2D PGS with protein expression levels
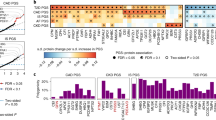
To determine how polygenic risk for T2D impacts the circulating proteome, we tested PGS for T2D, including the partitioned scores, for association with circulating proteins in the UKB-PPP. The genome-wide T2D PGS (PGST2D_gw) was associated with 648 proteins in the UKB discovery set (Table 1); of these, 617 replicated in the UKB replication set (FDR < 5%). The proteins that were among the top 1% in terms of variance (R2) explained by the PGST2D_gw include PON3, CKB, APOF, and IGFBP2 (Fig. 2A). The partitioned T2D scores and the PGST2D_gwas (i.e., the PGS derived from GWAS-significant variants) were associated with fewer proteins compared to the genome-wide T2D score (PGST2D_gw) (Supplementary Figs. 2–6 and Supplementary Data 6). Despite comprising fewer variants than PGST2D_gw, three of the partitioned T2D scores were significantly associated with proteins that were not associated with the PGST2D_gw: PGST2D_beta_cell (76% of its associations), PGST2D_liver_lipid (55%), and PGST2D_lipodsytrophy (8%). When comparing the beta coefficients of the different T2D PGS for the circulating proteins, we found that they were negatively correlated between PGST2D_gw and PGST2D_liver_lipid (Pearson’s r = − 0.19, p = 5.9×10-25) and between PGST2D_gw and PGST2D_proinsulin (Pearson’s r = − 0.07, p = 3.8 × 10−4, Fig. 2C). While their effects on the circulating proteome differ, the actual PGST2D_gw and PGST2D_liver_lipid scores are positively correlated (see Supplementary Fig. 7). Overall, this suggests that the partitioned T2D scores capture protein associations representing perturbations in specific T2D-related biological pathways that may be obscured when variant effects are aggregated in a genome-wide score (PGST2D_gw).

A Volcano plot of PGST2D_gw-protein beta coefficients (obtained from linear regression) and the unadjusted -log10 p-values (two-sided), with the colour indicating the magnitude of the -log10 p-values. Labelled proteins are among the top 1% in terms of variance (R2) explained by the PGST2D_gw. B Beta-beta plot of PGST2D_gw beta coefficients on circulating proteins with (y-axis) and without (x-axis) BMI adjustment. The diagonal is dashed grey, while the regression line is solid grey. Each point represents a protein; light blue points indicate replicated proteins that remained significant with the adjustment, red points indicate replicated proteins that were no longer significant after the adjustment, and dark blue points indicate proteins that did not significantly replicate prior to adjusting for BMI or pQTLs. C Pearson’s correlations of PGS beta coefficients from the regression on circulating protein levels. Red indicates pairs of PGS with positively correlated effect sizes; blue indicates negatively correlated effect sizes. “*” indicates correlations with a p-value < 0.05 and “**” indicates correlations with a p-value < 0.001 (a Bonferroni correction for 45 comparisons). P-values are unadjusted and two-sided t test as the test statistic follows a t distribution. D Bar plot indicating the overlap between proteins significantly associated with the T2D PGS and the other cardiometabolic PGS. The x-axis is the PGS label, and the y-axis is the percentage of PGST2D_gw-associated proteins that are also associated with another PGS (e.g., over 60% of proteins were also associated with the PGSBMI). E Beta-beta plot of PGST2D_gw effect sizes on circulating proteins with (y-axis) and without (x-axis) pQTL adjustment, with the same definitions as panel (B) albeit for a pQTL adjustment.
Impact of adiposity on T2D score-protein associations
T2D and adiposity are highly interconnected21, and as such, protein-T2D score associations can be confounded or mediated by adiposity. To explore which protein associations are independent of adiposity, we added one of three anthropometric measures capturing obesity as covariates to our PGS-protein models: body mass index (BMI), waist circumference (WC), and waist-hip ratio (WHR). Out of the 648 proteins significantly associated with the PGST2D_gw in the discovery set, 314 remained significant after adjustment for BMI (Fig. 2B), demonstrating the close interplay between T2D genetic risk, BMI, and circulating protein levels. Adjusting for WC or WHR instead of BMI produced similar results (see Supplementary Fig. 8 and Supplementary Data 7), with associations for 301 and 360 proteins remaining significant, respectively.
Next, to explore the extent to which adiposity is the mechanism by which the T2D scores exert their influence on the plasma proteome, we performed a mediation analysis with the T2D scores as the exposure, BMI as the mediator, and protein expression levels as the outcome (see Supplementary Data 7, Methods). For 94 of the 617 proteins significantly associated with the PGST2D_gw, the indirect effect and total effects were significant (p-value < 4.3 × 10−6), but the direct effect of the PGS was not. This suggests that for these proteins, such as ADM, LEP and TNF, BMI mediated the bulk of the PGST2D_gw’s effect on the circulating levels. For 518 proteins, the direct effect of PGST2D_gw remained significant, but BMI still mediated part of the PGST2D_gw’s effect on the proteins: the proportion mediated by BMI ranged from relatively small (0.05 for MANSC1) to relatively large (0.82 for FABP4), with a median of 0.38 (see Supplementary Fig. 8C). For 12 proteins, the indirect and direct effects were in the opposite direction. One of these proteins, TIMP4, has been previously reported to have a discordant relationship with adiposity and T2D21. Among the partitioned T2D scores, it was only PGST2D_obesity that the bulk of the effect was mediated via BMI.
Cardiometabolic score associations
We then tested a selection of cardiometabolic PGS representing common T2D comorbidities (CAD, CKD, and BMI) for their association with protein expression levels (Panel A from Supplementary Figs. 9–11). This analysis aimed to assess proteomic signatures that are specific to and shared across polygenic risk for T2D and its comorbidities. The proteins associated with the PGST2D_gw were frequently associated with one or more cardiometabolic PGS (see Fig. 2D). Overall, 64% were also significantly associated with the PGSBMI, 33% were associated with the PGSCKD, and 17% were also associated with the PGSCAD. The beta coefficients of the various comorbidity scores on the circulating protein levels were all positively correlated with that of the PGST2D_gw (p-value < 0.05, Fig. 2C), supporting the epidemiological observation that cardiometabolic diseases are highly interconnected22,23.
However, for a subset of proteins, the directions of the effect of the T2DT2D_gw and a comorbidity PGS on circulating levels were opposite (e.g., the effect of PGST2D_gw and PGSCKD on MANSC4 levels, see Supplementary Data 8), possibly indicating more complicated relationships such as compensatory responses21,24.
Genetic ancestry and PGS-protein associations
To examine whether our findings are robust and portable across ancestries, we compared and contrasted PGS-protein association patterns across five genetically predicted ancestry groups from the UKB-PPP cohort (see Methods). As over 92% of the UKB-PPP cohort is of European ancestry, we compared beta coefficients (rather than p-values) of the PGS-protein associations between the different ancestry groups. The correlations of the PGS-protein beta coefficients between genetically predicted European and non-European ancestries were less than 1 (Pearson’s r), reflecting the fact that translatability of PGS and, by extension, their proteomic associations, across ancestries is a potential concern (see Supplementary Figs. 12–15 and Supplementary Data 6). However, the beta coefficients were notably more correlated across the ancestry groups when we restricted our comparison to the statistically significant PGS-protein associations, suggesting that our replication strategy was effective and that significant associations are more likely to be robust and translatable across ancestries.
Polygenicity of PGS-protein associations and widespread effects of GCKR
A key motivation of our PGS-based approach is that it can help detect proteomic associations that may otherwise be undetectable via single-variant analysis (i.e., protein quantitative trait loci or pQTL analysis). To evaluate whether the PGS-protein associations we detected were driven by a single pQTL tagged by the PGS or represent the cumulative, polygenic effect of variants in the PGS, we additionally tested the PGS-protein associations by including previously reported pQTLs as covariates in the regression model. After adjusting for both cis and trans pQTLs (see Methods), most protein-PGST2D associations remained significant (612 out of 617; see Fig. 2E, Table 1, Supplementary Data 6), demonstrating that these associations were indeed polygenic in nature. This was largely true for all evaluated PGS. However, for the PGST2D_liver_lipid and the PGST2D_beta_cell, most protein associations were not significant after pQTL adjustment (Supplementary Fig. 2C and 4C). In the case of the PGST2D_liver_lipid, the index variant at the GCKR locus (rs1260326) explained the bulk of its association signature (280 out of 306 proteins), highlighting the pleiotropic effect of this variant on circulating proteins. In contrast, most proteins associated with the PGST2D_beta_cell score were explained by 7 different pQTLs (four from the ABO locus, one from the NCR3LG1 locus, and one from the FGFBP3 locus).
Causal inference using Mendelian randomisation and cis instruments
To assess if a PGS-protein association represents forward causality (i.e., the PGS perturbs a protein’s expression level that leads to disease) and, therefore, characterise putatively causal proteins for T2D and its key comorbidities (BMI, CAD, and CKD), we utilised two-sample Mendelian randomisation (MR) with statistically independent pQTLs as instruments. We applied four conventional MR methods (IVW, median, weighted median, and MR-Egger; see Methods) to those proteins with sufficient genetic instruments.
Six proteins had significant causal evidence with regards to T2D (median p-value across the four MR methods <0.05, FDR-adjusted) without evidence of pleiotropy (MR-Egger intercept p-value > 0.05): MANSC4, GLRX5, NUCB2, PAM, ARG1, and NCR3LG1, (see Fig. 3A and Supplementary Data 9), with PAM and MANSC4 significant after a more stringent Bonferroni correction (p-value < 9.2 × 10−6). For BMI, 14 proteins were significant using cis instruments (see Supplementary Fig. 16A), with NADK remaining significant after a Bonferroni correction (p-value < 8.7 × 10−6). For CAD, we identified eight proteins using cis instruments (FDR-adjusted p-value < 0.05; see Fig. 3D), with FES, LPA and PCSK9 significant after a Bonferroni correction. For CKD, we identified one protein using cis instruments, UMOD (with UMOD significant after a Bonferroni correction; see Supplementary Fig. 16D). Finally, we conducted an MR analysis for cardiovascular and renal outcomes in UKB participants with T2D. Using cis instruments, we identified two proteins: UMOD and CCN4 (FDR p-value < 0.05; see Supplementary Fig. 17A).

A Type 2 diabetes (T2D) Mendelian randomisation (MR) with cis instruments for each protein as the exposure. B T2D MR with both cis and trans instruments for each protein as the exposure. C Cis colocalization using T2D and protein quantitative loci (pQTL) genome-wide association study (GWAS) information. D Coronary artery disease (CAD) MR with cis instruments for each protein as the exposure. E CAD MR with both cis and trans instruments for each protein as the exposure. F CAD colocalization using CAD and pQTL GWAS information. In the MR plots, four conventional MR methods are displayed (simple median, weighted median, IVW, MR-Egger), plus an additional MR method called MR-Link-2. All proteins displayed in this figure had a median p-value across the four conventional MR methods < 0.05 (FDR-adjusted) and no pleiotropy as detected by MR-Egger (MR-Egger intercept p-value > 0.05). For panels (A, B, D, and E), the points represent the causal estimate obtained by each MR method (in the log-odds scale) and error bars represent the 95% confidence interval calculated using the standard error for each MR estimate. Note that MR-Link-2 estimates are on a different scale than the other MR methods but show consistency in the direction of effect. Finally, “*” signifies proteins with colocalization evidence. For panels A through F, the exposure (protein) GWAS had a sample size of 34,557 European-ancestry UK Biobank participants, and the outcome GWAS (T2D, CAD) had a sample size of 409,048 non-overlapping European-ancestry UK Biobank participants.
We also employed a pleiotropy-robust method designed for cis molecular data, MR-Link-225 (see Methods). As MR with only one locus can be underpowered and can present challenges for pleiotropy estimation, this MR method complements our conventional MR analysis. MR-Link-2 and the conventional MR were largely concordant, though MR-Link-2 did identify an additional 12 proteins that were not found using the other MR methods (see Supplementary Data 10). Ultimately, using cis instruments, we found evidence of a causal link for 44 proteins, 70% of which were associated with at least one PGS.
Mendelian randomisation with the addition of trans instruments
After adding trans instruments in our MR models, we found 4 more proteins with significant causal evidence for T2D (LGALS4, MENT, PDIA5, and PPM1B, FDR-adjusted p-value < 0.05), with LGAL34 significant after Bonferroni correction (p-value < 8.7 × 10−6). For BMI, we identified an additional 7 proteins (see Supplementary Fig. 16B), four of which were also significant after a Bonferroni correction (APOBR, PRSS53, GYS1, RAP1A). For CAD, six additional proteins were associated with CAD (FDR p-value < 0.05; see Fig. 3E), with FURIN and LMOD1 significant after a Bonferroni correction (p-value < 8.7 × 10−6). No additional proteins were added after the inclusion of trans instruments for CKD. For the comorbidities in UK Biobank participants with T2D, we found that SELE increased the risk of myocardial infarction.
Multivariable Mendelian randomisation with BMI and T2D
For BMI and T2D, due to their high intercorrelation (see above), we also employed multivariable MR (MVMR)26. By using genetic variants that impact both BMI and protein expression levels as MR instruments, we can estimate the effects of both exposures, even if they are related (i.e., through mediation). We identified 55 loci (cis regions of proteins) with sufficient instrument strength and without evidence of pleiotropy, to simultaneously evaluate both BMI and protein exposures on T2D (see Supplementary Data 11). Using this approach, we found 11 with nominal evidence (p-value < 0.05) that the BMI exposure (3 proteins), the protein exposure (7 proteins), or both (1 protein) lead to T2D risk, with one, FAM171B, significant after a multiple testing correction (FDR p-value < 0.05; see Supplementary Fig. 17B and C). Including trans instruments allowed us to evaluate 19 additional proteins with MVMR. Of these, 7 had nominal evidence (p-value < 0.05) that the BMI exposure (4 proteins, including TNF), the protein exposure (2 proteins), or both (1 protein) conferred T2D risk, with UROD significant after an FDR correction.
Colocalization of T2D and comorbidity GWAS signals with UK Biobank pQTLs
We performed statistical colocalization to test whether pQTL and T2D/comorbidity GWAS signals arise from the same causal variant, thus providing additional support for our MR analyses. The cis regions of 9 proteins colocalized with T2D GWAS signals (see Fig. 3C), 31 proteins for BMI GWAS signals (Supplementary Fig. 16C), 14 proteins for CAD (Fig. 3F), and 3 for CKD (Supplementary Fig. 16E). For trans pQTLs, 70 had evidence for colocalization with T2D GWAS signals, corresponding to 502 proteins, suggesting that trans effects plays a large part in the genetic risk for complex disease as previously proposed27 (see Supplementary Data 9). Similarly, 117 trans pQTLs had evidence for colocalization with BMI GWAS signals. For the comorbidities in UKB participants with T2D, one signal was colocalized (UMOD and CKD).
The mediation of PGS effects by circulating proteins in the UKB
An orthogonal approach to MR for inferring the causal pathway is mediation, which tests whether a protein mediates the effect of a PGS on incident disease risk. Mediation can be used to support MR findings, and it can also provide information on directionality for proteins that lack the requisite number of genetic instruments for MR. Among the UKB participants with proteomics data, 2081 were diagnosed with T2D during 14 years of follow-up time. In our mediation analysis, we modelled the PGS as the exposure, an individual protein as the mediator, and either incident T2D, CKD, or CAD as the outcome. Ultimately, 520 of the 617 PGST2D_gw-associated proteins significantly mediated the effect of the PGS on incident T2D risk. After adjusting for BMI, this is reduced to 238 proteins (Supplementary Fig. 18A and Supplementary Data 12). We also performed mediation using the partitioned scores (Supplementary Fig. 18B): 83 proteins mediated the effect of the PGST2D_lipodystrophy score (74 after BMI adjustment) and 6 mediated the effect of the PGST2D_beta_cell score (4 after BMI adjustment). Finally, we also performed mediation analysis for the PGSCKD with incident CKD and the PGSCAD with incident CAD, finding 470 (422 after BMI adjustment) and 33 (12 after BMI adjustment) mediating proteins, respectively (Supplementary Fig. 18A).
Reverse Mendelian randomisation with cardiometabolic traits
Next, we employed reverse MR to identify instances of reverse causation, i.e., where the cardiometabolic trait (T2D, CAD, CKD, BMI) alters the protein expression levels (Methods). If a PGS-associated protein is implicated in reverse MR, this suggests that the developing disease state affects the levels of the protein. After applying a Bonferroni correction as in our primary PGS-protein association analysis (p-value < 4.3 × 10−6 across 4 MR methods), our reverse MR analysis suggested that circulating levels of 38 proteins were influenced by T2D, including GDF15 (Supplementary Data 13), and several other proteins strongly associated with the PGST2D_gw (e.g., APOF, PON3, PRCP). In the case of GDF15, elevated serum levels have been reported in T2D and it is known to play a role in regulating food intake and metabolism28. However, none of the proteins identified in the forward MR analysis for T2D were implicated in our reverse MR. It is worth noting that proteins identified via reverse MR could still be causal for other comorbidities or influence T2D risk via feedback mechanisms.
For the other cardiometabolic disorders, we found 3 proteins influenced by CAD risk (MMP12, CNTN4, LGALS4), 8 by CKD risk, and 539 by BMI (Supplementary Data 13), using reverse MR analysis. The high number of proteins influenced by BMI genetic risk indicates that the levels of many circulating proteins are impacted by adiposity levels. For CKD and CAD, none of the proteins we identified in our forward MR were associated in the reverse MR, though this was the case for 7 of the 21 BMI-associated proteins (CXCL16, LGALS3, GALNT10, PSCA, IL12RB2, ITGAL, SERPINA7). However, when we overlaid our mediation results with the reverse MR results, a proportion of mediating proteins had evidence for reverse causality, ranging from 2% of proteins for CKD and PGSCKD to 7% for the PGST2D_gw.
Time-to-event analyses of PGS-associated proteins in EXSCEL and DECLARE
As PGS-associated proteins could have clinical relevance as biomarkers for diabetic comorbidities and, in some cases, could be causally linked, we tested their association with cardiovascular and renal clinical trial endpoints in the placebo arms of EXSCEL and DECLARE-TIMI 58 (DECLARE).
In EXSCEL, the baseline levels of 241 proteins were significantly associated with time to a major adverse cardiovascular event (MACE), time to hospitalisation for heart failure (HHF), or time to the renal outcome (see Methods). Of these 241 proteins, 50% were significantly associated with the PGST2D_gw, 72% were significantly associated with the PGSCKD, and 9% were significantly associated with the PGSCAD in the UKB PGS analysis. After adjusting for clinical risk factors, 118 proteins remained significant (see Fig. 4A and Supplementary Data 14). Notably, 3 proteins were also independent of NT-proBNP for time to HHF (MARCO, EGFR, and EFEMP1), as well as 20 proteins for MACE. In DECLARE, 64 proteins associated with an outcome in EXSCEL were available for replication, of which 20 were significantly replicated using the corresponding outcome in DECLARE (Fig. 4B and Supplementary Data 15). When adjusting for clinical risk factors, 6 proteins were significantly associated with either MACE or HHF (FDR-adjusted p-value < 0.05), while for the time to DECLARE’s composite renal outcome, IGFBP6 was nominally significant (p-value = 0.03; Fig. 4C–E). EFEMP1, SPON1, and CST3 remained significantly associated with the HHF endpoint after adjusting for both clinical risk factors and NT-proBNP.

A Summary of results in EXSCEL, with the x-axis corresponding to the three different models used (see Methods) and the y-axis corresponding to the number of proteins significant for each of the three outcomes (Bonferroni p-value < 0.05). B Summary of the replication results in DECLARE, with the x-axis corresponding to the three different models used and the y-axis corresponding to the number of proteins significant for each of the three outcomes (FDR p-value < 0.05). C Results from the survival analysis of the study-specific renal outcome in the placebo arms in EXSCEL and DECLARE. All displayed proteins replicated in DECLARE for the base model (with age, sex, age2, age*sex, and genetic PCs 1–10 as covariates). D Results from the survival analysis of the major adverse cardiovascular event (MACE) outcome in the placebo arms in EXSCEL and DECLARE. All displayed proteins replicated in DECLARE for the base model. E Results from the survival analysis of the hospitalisation for heart failure (HHF) outcome in the placebo arms in EXSCEL and DECLARE. For panels (C–E), the points represent the hazard ratios and error bars represent the 95% confidence interval obtained using the standard error for each hazard ratio. Note that all displayed proteins replicated in DECLARE for the base model. In panels (C–E), EXSCEL had a sample size of 1407 study participants from the placebo arm with available proteomics information, while DECLARE had a sample size of 497 study participants from the placebo arm with available proteomics information.
Both trials featured repeat measurements with proteins assayed at baseline and a second timepoint (12 months for EXSCEL, 6 months for DECLARE). We evaluated proteins using the same procedure as with the baseline measurements (see Supplementary Fig. 19). In general, while the results were similar between time points, hazard ratios were systemically larger for the renal outcome in EXSCEL and the composite renal outcome in DECLARE (see Supplementary Fig. 20A and B). Additional proteins were significant for MACE, HHF, and the trial-specific respective renal outcome (e.g., TFF3) at the second timepoint. For the renal outcome in EXSCEL and the composite renal outcome in EXSCEL, this trend is partially ameliorated through the adjustment of baseline measurements (see Supplementary Fig. 20G and H). Overall, larger hazard ratios at the second time point could be indicative of cumulative exposure to a causal protein or reverse causality.
PGS mediation in EXSCEL and DECLARE
Building on our endpoint analysis in the clinical trials, we sought to identify which protein biomarkers had evidence for causality. First, we evaluated whether any PGS were associated with clinical outcomes and, if so, whether they were associated with any circulating proteins in the trials (see Supplementary Data 16–18). We then performed mediation when both conditions were satisfied. In EXSCEL, we modelled PGSCAD as the exposure, PGSCAD-associated proteins as the mediator, and MACE as the outcome. We found evidence that the PGSCAD mediates its effect through the circulating protein levels of C9, LBP, ITIH4, APOM, and HS6ST2 (Supplementary Fig. 21A and Supplementary Data 19). In DECLARE, the PGSBMI and PGSCAD were significantly associated with HHF and MACE, respectively. In the DECLARE mediation analysis, we did not identify any proteins that mediated the PGSCAD’s effect. However, for the PGSBMI and HHF, we found evidence for 63 proteins mediating its effect (FDR-adjusted p-value < 0.05; see Supplementary Fig. 21B and Supplementary Data 20). When including BMI in the models, 17 proteins still appeared to mediate the PGSBMI effect, though at a nominal significance level (p < 0.05; Supplementary Fig. 21C and Supplementary Data 20).
Notable Pathways enriched across multiple PGS-protein sets
Next, to identify pathways underlying polygenic risk for cardiometabolic disorders, we performed pathway enrichment analysis of the PGS-associated proteins. The PGST2D_gw protein set was significantly enriched for 12 pathways after p-value adjustment (see Methods), of which 3 were also enriched in the PGSCAD protein set (Supplementary Fig. 22). Pathways that are significantly enriched in both the PGST2D_gw and PGSCAD protein sets may reflect shared causal mechanisms, such as those involving the complement system (Supplementary Figs. 23). Proteins in the complement and coagulation cascades pathway were identified as causal for cardiovascular disease in MR (C1R and C1S) and mediation (18 complement-related proteins mediated the effect of PGST2D_gw and 4 complement-related proteins mediated the effect of the PGSCAD). In the clinical trials, C9 mediated the effect of the PGSCAD on the MACE outcome. In terms of biomarkers, C2 and CD59 from the complement cascade and PLAUR from the coagulation cascade were associated with the time to cardiovascular outcomes in EXSCEL and DECLARE (Supplementary Fig. 23B and D).
While the insulin-like growth factor binding proteins (IGFBPs) pathway was not enriched in the PGSCAD or the PGSCKD associated protein sets, it was enriched in the PGST2D_gw, PGSBMI, and the PGST2D_liver_lipid protein association sets. In addition, its constituent proteins were identified in many of our analyses for both T2D and its comorbidities. IGFBP2 was among the most significant protein associations for the PGST2D_gw, while IGFBP4 and IGFBP6 were strongly associated with the PGSCKD (Fig. 5A). While causal effects for IGFBPs on the tested diseases were not supported in our MR analysis, IGFBP2 and IGFBP6 were implicated with T2D and CKD, respectively, using mediation. In the clinical trials, many IGF-related proteins were significantly associated with both cardiovascular and renal outcomes (Fig. 5B and C).

A PGS associations with IGF binding proteins. A single asterisk (*) indicates the association was nominally significant, while two (**) indicates significance using FDR and three (***) indicates significance using a Bonferroni correction from linear regression of circulating protein levels in the UK Biobank. B Associations of proteins in this pathway with clinical trial outcomes in EXSCEL using Cox proportional hazards regression, adjusting for demographic covariates and clinical risk factors (see Methods). C Associations of proteins in this pathway with clinical trial outcomes in DECLARE using Cox proportional hazards regression, adjusting for demographic covariates and clinical risk factors (see Methods). For panels B and C, the dashed line corresponds the p-value threshold where FDR < 5% (when applied to the proteins in this pathway). Note that panels (B and C) display unadjusted, two-sided p-values obtained from Cox proportional hazards regression.
Discussion
In this study, we defined the intersection of polygenic risk for T2D, its cardiometabolic comorbidities, and the plasma proteome by leveraging proteogenomic data across a population-scale biobank and two RCTs. As variants identified through T2D GWAS are often intergenic with small effect sizes, understanding their functional consequences can be challenging. By aggregating GWAS variants into PGS (including T2D, pathway-specific, and comorbidity PGS) and testing their association with large-scale proteomic data, our study provides an enhanced understanding of proteomic signature of T2D as well as causal proteins and pathways that may mediate T2D risk. Furthermore, our PGS-directed approach has the added benefit of evaluating more proteins for causality than analyses such as Mendelian randomisation (MR) that rely on individual variants (only 69% of the Olink-assayed proteins have sufficient pQTLs to perform MR). Our study has potential implications in highlighting biomarkers of T2D comorbidities, providing therapeutic targets for T2D, and uncovering the biological underpinning of cardiometabolic diseases.
Our study yielded insights into the proteomic consequences of polygenic risk for T2D and its comorbidities. The PGST2D_gwas, despite its derivation from the same summary statistics as the PGST2D_gw, was strongly associated with proteins identified by single locus methods, such as PAM, while the PGST2D_gw’s associations appeared to be more polygenic (Fig. 2). Given the causal links between adiposity and T2D, nearly all of the PGST2D_gw protein associations are at least partially mediated by BMI, though the contribution of BMI can range from relatively small (e.g., 7% of PGST2D_gw’s effect on MANSC4) to relatively large (e.g., 90% of the PGST2D_gw’s effect on LEP). Notably, the partitioned T2D scores, apart from PGST2D_obesity, were less influenced by adiposity (Supplementary Figs. 2–6). The partitioned T2D scores also seemed to capture unique aspects of T2D biology (189 proteins were associated with the partitioned scores but not the overall T2D score, i.e., PGST2D_gw), thus, serving as proof of concept for the development of such scores. For example, the effect of the PGST2D_beta_cell score on incident T2D was mediated by FAM3D, a causal relationship not detected by the overall PGST2D_gw. FAM3D is thought to be involved in glucose regulation29. Further, we found the beta coefficients of PGST2D_liver_lipid and the PGST2D_gw on the plasma proteome were frequently in the opposite direction, even though the scores themselves are positively correlated (Fig. 2 and Supplementary Fig. 7). This is consistent with the known discordant effects of PGST2D_liver_lipid and the PGST2D_gw on clinical biomarkers such as plasma triglycerides13. It seems plausible that the GCKR locus (index variant: rs1260236), captured by PGST2D_liver_lipid, drives a specific subtype of T2D with unique phenotypic consequences13,15.
To identify PGS-protein associations that are attributable to forward causation, and discover potential therapeutic targets, we performed causal inference using Mendelian randomisation (MR) and mediation for the relevant clinical endpoints (i.e., T2D, BMI, and the T2D comorbidities CAD and CKD). For T2D, we found evidence using both MR and cis colocalization for 5 proteins, although two of them, ABO and APOE, likely harbour pleiotropic effects. PAM’s involvement in the etiology of T2D has been described in previous studies30. Less is known about MANSC4 and NCR3LG1, but we note that MANSC4 has previously been identified via MR31. Among MR-significant proteins without colocalization evidence are NUCB2, ARG1, and LGALS4, all of which either play a role in glucose regulation32, insulin resistance33 or have been previously linked to diabetes34,35. For CAD, we identified 7 proteins with significant MR and cis colocalization evidence (see Fig. 3). Among these, LPA and PSCK9’s role in the etiology of cardiovascular disease is well-established36,37,38, while evidence in the literature for FURIN and FES continues to grow39,40,41. Notably, ERBB4, with significant MR estimates and cis colocalization for BMI (IVW: − 0.26, 95% CI: − 0.378 to − 0.138), has previously been linked to metabolic disorders and obesity42,43,44. With an alternative method, MR-Link-2, we identified additional proteins, such as FGF5 with CAD, and confirmed many of the associations we found with conventional MR. However, this approach also found more pleiotropic effects than our primary MR analysis (see Supplementary Data 9 and 10). As MR-Link-2 is designed to be pleiotropy-robust, we did not filter potentially pleiotropic instruments in advance as we did for the other MR analyses. In the case of some loci (such as LPA), the observed pleiotropic effect could be caused by statistically independent pQTLs in linkage disequilibrium25.
Of the 56 proteins identified via MR, 40 were significantly associated with at least one PGS. For the three traits evaluated via mediation (CKD, CAD, T2D), 49% of MR-identified proteins also significantly mediated the effect of a PGS on incident disease. Proteins with causal evidence based on MR, promising as biomarkers for cardiovascular/renal comorbidities, or among the top 5 (by variance explained) of mediating proteins for each score are summarised in Supplementary Data 21, along with their druggability and gene expression information obtained from DrugnomAI45,46.
We aimed to identify putative causal proteins and pathways that link T2D with its cardiovascular and renal comorbidities. To this end, we performed MR in patients with T2D as well as survival analysis and mediation analyses in EXSCEL and DECLARE-TIMI 58. With MR, we found that CCN4 (WISP1) and SELE conveyed risk for the development of cardiovascular outcomes while UMOD conveyed risk for renal comorbidities. UMOD’s role in CKD has previously been supported by MR47, while CCN4 and SELE (E-selectin) are thought to be involved in T2D progression48 and vascular inflammation49, respectively. Several proteins implicated in our survival analysis, such as TFF3 and EFEMP1, could potentially explain mechanisms underlying the development of comorbidities50,51. We found 6 proteins that mediated the PGSCAD’s effect on MACE in EXSCEL (C9, LBP, ITIH4, APOM, CES1, and HS6ST2), providing supporting evidence that they might serve as biomarkers for cardiovascular disease in patients with T2D52,53,54.
Pathway enrichment of PGS-associated proteins can inform whether the proteins act collectively via certain biological pathways and can help guide therapeutic development (Supplementary Fig. 22). Pathways shared across multiple cardiometabolic PGS-protein sets could also reveal mechanisms relevant to the development of these comorbidities. Pathways involving the complement system, notably, were enriched in both the PGSCAD and PGST2D_gw protein sets. For example, C1R and C1S, which activate the C1 complex in the classical complement pathway55, were significantly associated with the PGST2D_gw in addition to their MR and colocalization with CAD. Therapies targeting the complement system have been developed or proposed for a wide range of diseases, including inflammatory kidney disorders and cardiovascular disease56,57. The insulin-like growth factor binding proteins (IGFBPs) pathway was enriched in both the PGST2D_gw and PGSBMI protein sets. IGFBP2 was a biomarker for both cardiovascular and renal outcomes in EXSCEL and DECLARE-TIMI 58. Interestingly, lower levels of IGFBP2 are associated with incident T2D while higher levels are associated with incident CKD (see Fig. 5A), as has previously been observed58,59. While the exact physiological role of IGFBPs in the pathogenesis of cardiometabolic disease requires further investigation, we provided evidence of a shared role in both adiposity traits and cardiometabolic disease60,61,62.
Our study has limitations. First, the trans-ancestry portability problem is commonly observed in biomedical research, including with PGS, though we sought to mitigate this by employing multi-ancestry PGS and trans-ancestry analyses. Second, since our study included UKB data, we selected scores that did not make use of the UKB (e.g., relatively older and smaller GWAS) to avoid overfitting, which may attenuate our theoretical statistical power. Third, despite excluding subjects with prevalent cardiometabolic diagnoses, the PGS-related analyses were not immune to reverse causality as several proteins strongly associated with the PGST2D_gw were implicated in our reverse MR analysis. It is also possible that we are capturing instances of feedback mechanisms. Fourth, defining phenotypes in the UKB via electronic health records could have an impact on analyses due to potential issues such as misclassification error63. Fifth, regression analyses including both T2D and BMI could exhibit collider bias as T2D and adiposity are causally linked, and as such, we do not consider any model that is significant after BMI adjustment when it is not significant without the BMI adjustment. Collider bias could also impact the analyses in the clinical trials as all participants were selected on a particular characteristic64 (i.e., T2D with cardiovascular risk factors); however, adjusting for the risk factors as we have done could address this. Sixth, this study used data derived from circulating proteins, which may not necessarily represent the causative tissue for the diseases under examination. Seventh, while the cardiovascular outcomes in EXSCEL and DECLARE are aligned, the renal endpoints differ between the two trials (see Methods). This, in addition to differing eligibility criteria, could contribute to the effect size differences we observed in the survival analyses for the renal outcomes. Finally, the PGS, and by extension the mediation analyses using the PGS, could be impacted by horizontal pleiotropy as the PGS by design is not limited to variation reflecting a single exposure9. However, the partitioned scores help reduce this to a single mechanism. Our mediation sensitivity analyses likely reflect this as many of our mediation models were not robust to potential mediator-mediator confounding (see Methods, Supplementary Data 12). The same limitation applies to MR analyses of complex traits and protein levels using trans pQTLs, though we have sought to minimise this confounding by testing for pleiotropy in our analytical framework.
Overall, we leveraged data from both a population-based setting and clinical trials to elucidate the proteomic signatures of polygenic risk for T2D and its comorbidities; provide causal evidence for the associated proteins; identify proteins and biological pathways that connect T2D with its comorbidities; and provide evidence for existing therapeutic and potentially new target opportunities. We also developed an interactive portal that allows users to interrogate and download the results of our analyses (https://public.cgr.astrazeneca.com/t2d-pgs/v1/).
Methods
Cohort description
The UK Biobank (UKB) is a deeply phenotyped population-based cohort comprised of approximately 500,000 subjects with array genotyping, exome/whole genome sequencing data and linkage to electronic health care record data with over 14 years of follow-up time18. The UK Biobank Pharma Proteomics Project (UKB-PPP) is a private-public partnership that assayed 2923 unique proteins (2922 after excluding one protein without sufficient measurements) in a subset of 54,306 UKB participants using the Olink Explore proteomics platform16,17. The UK Biobank operates under approval from the North West Multi-centre Research Ethics Committee (MREC). All UKB participants provided informed consent.
Exenatide Study of Cardiovascular Event Lowering (EXSCEL) examined the cardiovascular effects of once-weekly exenatide, a glucagon-like peptide-1 (GLP-1) agonist, in T2D patients with a median follow-up time of 3.2 years19. In a subset of trial participants (N = 2823), both genotyping and SomaScan proteomics data were generated (see Supplementary Data 1). All participants provided written informed consent, and the trial protocol was approved by ethics committees at each of the trial’s participating sites. EXSCEL can be found on ClinicalTrials.gov (NCT01144338).
Dapagliflozin Effect on Cardiovascular Events–Thrombolysis in Myocardial Infarction 58 (DECLARE-TIMI 58) was a phase 3 RCT that examined the cardiovascular effect of dapagliflozin, an inhibitor of sodium-glucose co-transporter-2 (SGLT2), in patients with T2D with multiple risk factors for or established atherosclerotic cardiovascular disease with a median follow-up time of 4.2 years20. Similar to EXSCEL, for a subset of participants both genotyping and Olink proteomics data were available (N = 915). All participants provided written informed consent, and the trial protocol was approved by the institutional review board at each of the trial’s participating sites. DECLARE-TIMI 58 can be found on ClinicalTrials.gov (NCT01730534).
See Extended Methods in Supplementary Information for a description of genotyping and proteomics quality control. Supplementary Fig. 1 shows the proteomic intersection between the three cohorts with proteomics data. We note that a previous study has found that SomaScan and Olink measurements to be moderately correlated65.
UK Biobank phenotype definitions
For the UKB, we used ICD10 codes and clinically meaningful “Union” phenotypes constructed by merging relevant ICD10 codes (release from Feb 2022) as detailed in Wang et al. 66. For identifying prevalent cardiometabolic conditions that needed to be excluded from analyses, we used the following ICD10 codes: E10-E14 (any diabetes diagnosis), N18 (CKD), and I20-I25 (ischaemic heart disease, also known as CAD). Incident cases were defined using ICD10 codes (E11 for T2D, N18 for CKD, I25 for CAD) when the earliest date of diagnosis (determined by fields 41270, 40001, 40002) occurred after baseline (when a sample was donated for proteomics). Note that relatively few cases were diagnosed within 30 days of sample collection (Supplementary Data 2).
Randomised controlled trials (RCTs) phenotype definitions
We used three trial outcomes that were available in both trials, i.e., time to the composite cardiovascular outcome (major cardiovascular events or MACE, comprising of cardiovascular death, nonfatal myocardial infarction, or non-fatal ischaemic stroke), time to hospitalisation for heart failure (HHF), time to the renal outcome (for EXSCEL, two consecutive measurements of eGFR < 30 ml/min/1.73 m2 and for DECLARE-TIMI 58, a composite renal outcome comprising of a sustained decrease of 40% or more in eGFR to < 60 ml/min/1.73 m2, new end-stage renal disease, or death from renal causes). Note that the competing risk of death was addressed through censoring and the use of Cox regression67. DECLARE-TIMI 58 cardiovascular endpoints were adjudicated by independent adjudication committees. See Supplementary Data 3 for a description of phenotypes.
Polygenic score estimation
To avoid overfitting when estimating the PGS in the UKB, we retrieved genome-wide association study (GWAS) summary statistics from external studies43,68,69,70,71,72,73 that did not contain UKB participants (Supplementary Data 4), maximising both sample size and diversity. We trained genome-wide T2D, CAD, BMI, and CKD PGS using PRS-CS74 (when only one GWAS per trait was available) or PRS-CSx75 with these GWAS summary statistics. We ran PRS-CSx and PRS-CS with phi set to ‘auto’. For the partitioned T2D polygenic scores (pPS), we obtained the variants and cluster weights generated by Udler et al. corresponding to five distinct genetic clusters, i.e., beta cell, lipodystrophy, liver lipids, obesity, and proinsulin13. To enable comparisons between genome-wide and GWAS-significant PGS, we also generated a set of GWAS-significant T2D summary statistics using the clump procedure implemented in PLINK v1.9 (p-value < 5 × 10−8, R2 < 0.1, 250 kilobase window around each index variant). All PGS for UKB, UKB-PPP, EXSCEL, and DECLARE-TIMI 58 were estimated using post-QC imputed data and PLINK v2.00a4LM76 (see Supplementary Data 5).
PGS validation
To validate the PGS, we tested their associations with traits defined using ICD10 codes and/or biomarkers (Supplementary Data 2) in all unrelated UKB-PPP participants (resolved to the 2nd degree using KING 2.3.077) and using R and adjusting for age, age2, sex, age*sex, age2*sex, UKB centre, array, and PCs 1-20. We also used KING 2.3.0, with the 1000 Genomes Project (1KGP), as a reference, to predict genetic ancestry. KING first performs principal component analysis on the 1KGP cohort, followed by projecting the UKB-PPP samples into the 1KGP PC space. Then, it uses a support vector machine and the 1KGP super-population labels to train a model and predict the ancestry of the target cohort. We tested the PGS for association with its target trait in the genetically predicted subsets (European, South Asian, East Asian, African, and Admixed American, all with a posterior probability > 0.9) and in the entire multi-ancestry cohort. For the CAD, BMI, and T2D, PRS-CSx inferred three sets of PGS (a “European” PGS, an “East Asian” PGS, and a meta-analysis of the two). In these cases, we retained the PGS with the strongest association with CAD, BMI, and T2D in the full cohort analysis.
PGS and protein associations in UK Biobank
For testing PGS for association with protein expression levels, we modelled our analysis on the same internal replication structure as used in the UKB-PPP consortium pQTL GWAS. First, we restricted the analysis to unrelated participants (resolved to the 2nd degree) without a baseline diagnosis of diabetes or a relevant cardiometabolic condition (any Diabetes diagnosis, CAD, CKD) at data collection (UKB’s baseline timepoint; N = 44,381) to reduce confounding due to reverse causality as previously suggested by Ritchie et al. 9. Then, we stratified the cohort into the consortium-identified discovery subset consisting of European-ancestry participants (N = 29,496) and the replication subset of remaining pan-ancestry participants (N = 14,885). We tested each PGS for association with protein expression levels in the discovery subset using linear regression in R, using the same covariates (age, age2, sex, age*sex, age2*sex, batch, UKB centre, array, time to analysis, genetic PCs 1-20) as the UKB-PPP consortium pQTL GWAS16. Significant PGS-protein associations after a Bonferroni correction accounting for 10 scores and 2922 proteins (p-value < 1.7 × 10-6) were then moved forward to be tested in the replication subset. To account for the difference in sample sizes in the two subsets, we applied an FDR correction to our replication analysis separately to each PGS9. Associations were considered internally replicated with an FDR-adjusted p-value < 0.05.
To assess whether a PGS-protein association was driven by a single locus, we repeated the PGS-protein association analyses after adjusting for independent cis and trans pQTLs obtained from the UKB-PPP consortium’s pQTL GWAS. Similarly, we repeated PGS-protein association analyses after adjusting for BMI, waist-hip ratio, and waist circumference to describe the influence of adiposity on the PGS associations. Finally, to explore the impact of genetic ancestry on the transferability of PGS-protein associations, we tested PGS-protein associations after stratifying the UKB-PPP cohort by the KING-predicted ancestry labels described above. We then compared and contrasted PGS-protein association patterns in the genetically predicted European ancestry subset with that of the other four predicted super-populations. As the UKB-PPP cohort is over 92% European ancestry, we used beta coefficients rather than p-values and compared the beta coefficients of the PGS on circulating protein levels using Pearson’s r and the slope of the regression line fitted to the beta coefficients.
Mediation of PGS-protein associations by adiposity
For the PGST2D_gw-protein associations that were no longer significant, we performed mediation analysis to investigate if BMI mediated the effect of the PGS on the circulating protein levels. We used the 'mediate' function from mediation R package78 (4.5.0) to perform mediation with the PGS as the exposure, BMI as the mediator, and the circulating protein levels as the outcome. We performed a sensitivity analysis using the 'medsens' function from the same R package.
GWAS and pQTL summary statistics for Mendelian randomisation
We leveraged statistically independent pQTLs identifed by the UKB-PPP consortium from the European ancestry 35K discovery subset to use as instruments for our two-sample Mendelian randomisation (MR) analysis. The UKB-PPP pQTL GWAS16 used a minor allele count cut-off of 50, a minimum INFO score of 0.7, and a study-wide significant threshold p-value < 3.4 × 10−11. The pQTLs were defined as cis if they were within +/− 1MB of a gene’s transcription start site. For this same set of pQTLs, we also extracted summary statistics using the pan-ancestry combined UKB-PPP cohort for our two-sample trans-ancestry MR analysis.
We then generated trait-level GWAS summary statistics using the UKB after excluding the UKB-PPP participants to avoid sample overlap. Individuals were retained for the GWAS if they passed an internal set of quality control criteria including sex concordance, heterozygosity, ploidy, were assigned a continental PEDDY-predicted79 ancestry with a probability ≥ 0.90, and were not included in the UKB-PPP study. For the generation of summary statistics for Mendelian randomisation, we used E11 for T2D, N18 for CKD, the union term of 120-I25 for CAD, and body mass index (BMI) measured at baseline. For traits in UKB patients with T2D, we used the ICD10 codes in Supplementary Data 2, which contains a complete description of UKB phenotype definitions and cohort sample size (pre-QC). We utilised REGENIE for the GWAS, which employs a two-step approach as previously described80. For the first step, we used quality-controlled genotyped variants (MAF > 1%, genotyping rate >99%, HWE p-value > 10−15, < 10% missingness and LD pruning using 1000 variant windows, 100 sliding windows and r2 < 0.8), while for the second step we used imputed variants with a MAC > 50 and an INFO score > 0.7. For traits assessed in the full cohort (T2D, BMI, CKD, CAD), we performed each GWAS within each of the predicted continental ancestries, adjusting for age, sex, and genetic PCs 1-10. For traits assessed in UKB participants with T2D, we performed each GWAS within the predicted European-ancestry subset, adjusting for age, sex, and genetic PCs 1-10.
2-sample Mendelian randomisation (MR)
We performed a two-sample Mendelian randomisation (MR) analysis to conduct a proteome-wide scan for proteins suspected to play a potential causal role in the development of T2D or T2D comorbidity. For our primary MR analysis, we utilised the European ancestry GWAS data. To identify weak instruments, we calculated the F-statistic81 for each instrument (F-statistic = \({\beta }^{2}/{{Se}}^{2}\)) and removed instruments with an F-statistic < 10. We also removed instruments that were pQTLs for 5 or more proteins to reduce pleiotropy. We performed MR on all proteins with 3 or more cis pQTLs using the MendelianRandomization82 R package (version 0.7.0) and applying the simple and weighted median83, IVW84, and MR-Egger85 methods. We then repeated the analysis using both cis and trans instruments. We addressed violations of the pleiotropy assumption by flagging results with an MR-Egger intercept p-value < 0.059. We then took the median p-value across all MR methods to identify significant associations. For traits available in the entire UKB cohort, we reported both Bonferroni and FDR levels of significance, while for traits in patients with T2D (ICD10 code E11), we only applied an FDR correction due to the smaller sample sizes.
Multi-variable Mendelian randomisation
As T2D and adiposity are closely intertwined, we performed multivariable MR (MVMR) using the MVMR R package26, BMI GWAS data from the FinnGen cohort (r11)86 and pQTL data from the UKB-PPP discovery cohort (European ancestry) as exposures, and T2D GWAS data from the European-ancestry subset of the UKB (excluding UKB-PPP participants) as the outcome. The FinnGen study is a large-scale genomics initiative that has analyzed over 500,000 Finnish biobank samples and correlated genetic variation with health data to understand disease mechanisms and predispositions. The project is a collaboration between research organisations and biobanks within Finland and international industry partners. MVMR reports both instrument strength and pleiotropy estimations, and we retained analyses with the conditional F-statistic > 10 for both exposures and no evidence of pleiotropy (Q-statistic p-value > 0.05). Like our primary MR analysis, we repeated MVMR using cis only and cis plus trans instruments. We then applied an FDR correction on the results.
Mendelian randomisation using cis molecular data and MR-Link-2
We also performed cis MR analysis using MR-Link-225, a methodology developed explicitly for MR using GWAS information obtained cis molecular data. We again use the pQTL GWAS data from the UKB-PPP as the exposure and the UKB GWAS data as the outcome, though in this case taking the entire cis region as input. MR-Link-2 requires genotype data for LD, and for this, we used the UKB-PPP participants from the GWAS data (the European ancestry discovery cohort).
Mendelian randomisation sensitivity analyses including statistical colocalization
For sensitivity analyses, we repeated MR analyses using pan-ancestry summary statistics from the full UKB-PPP cohort and the meta-analysed trans-ancestry GWAS MR analyses (via a fixed-effected meta-analysis). We also conducted a colocalization analysis for any variant with GWAS p-value < 1 × 10−6 for both the trait and the protein using the coloc package87 (version 5.1.0.1) in R 4.2.2 on a +/− 250 kilobase window centred on the variant. We used the summary statistics-based method (coloc.abf) with the default priors (p1: 10−4, p2: 10-4, p12: 10-5) and considered the GWAS variant and the pQTL to have strong colocalization evidence following recommended criteria (PP.H3 + PP.H4 > 0.99 and PP.H4/PP.H3 > 5)9,87,88. For loci located in the cis region of an Olink-assayed protein, we also used the coloc.susie89 method with LD matrices computed with the imputed UKB-PPP data and PLINK v1.90b6.1876,90.
Mediation analyses of PGS, circulating proteins, and incident disease in UKB
In this framework, we set the PGS as the exposure, the protein as the mediator, and incident disease as the outcome. We utilised incident cases (prevalent cases were excluded), the same set of covariates as the UKB PGS-protein association analysis (see above), and the Medflex R package9,91 (version 0.6-10) to perform mediation analysis with natural effects models. We considered a mediation model to be significant if the mediation (indirect) p-value and the total p-value were both significant after a Bonferroni correction (p-value < 1.4 × 10−6); the direct effect p-value was allowed to be not significant as it is possible that the PGS’s effect is primarily mediated through the tested protein. As a sensitivity analysis, we performed mediation for both all participants and only European ancestry; we filtered out proteins that were not significant in both scenarios.
To evaluate robustness to potential mediator-mediator confounding, we employed the mediation R package (4.5.0)78. First, we again performed mediation with the PGS as exposure, the protein as a mediator, and the incident disease as the outcome using the 'mediate' function. Then, we performed the sensitivity analysis using the 'medsens' function, which performs a simulation-based sensitivity analysis. As the PGS is fixed at birth, it is assumed that there will not be any PGS-mediator confounding.
Reverse Mendelian randomisation
We obtained MR instruments for reverse MR from the European-ancestry T2D, CKD, BMI, and CAD GWAS data that we generated for our forward MR analysis, plus an additional model for CAD using only the ICD10 code I25. We performed LD pruning using PLINK v1.90b6.18’s clumping procedure with a 500 KB window size, a minimum p-value threshold of 5 × 10−8, and R2 < 0.001 using UKB-PPP as the LD reference. We matched each instrument with variants from the pQTL GWAS data. We then performed reverse MR using the MendelianRandomization R package in the same manner as described above.
Association and mediation of proteins with trial outcomes in EXSCEL and DECLARE-TIMI 58
We tested proteins for their association with the time to EXSCEL outcomes with the survival package in R 3.6.1 (https://github.com/therneau/survival) and replicated significant associations in DECLARE-TIMI 58. In both cases, we restricted analyses to the placebo arm. We used Cox proportional hazards regression and adjusted for age, sex, age*sex, and genetic PCs 1-10. For both EXSCEL and DECLARE-TIMI 58, two time points were available for proteomics: baseline plus 12 months for EXSCEL, and baseline plus 6 months for DECLARE. In both studies, we performed the proportional hazards regression analysis three times, using the baseline measurements, the repeat measurement, and the repeat measurement with baseline included as an additional covariate, respectively, as exposures. For EXSCEL, each SomaLogic aptamer was tested separately and reported. In the case of statistically significant associations in the clinical trial time-to-event analyses, (FDR p-value < 0.05 as well as a more stringent Bonferroni-corrected p-value in EXSCEL), we assessed the proteins using more comprehensive models that included clinical risk factors to evaluate their suitability as a biomarker (see “Description of risk factors” section below). For all models, we tested for the proportional hazards assumption using the cox.zph() function and flagged any model with a global p-value < 0.05 as violating model assumptions.
Description of risk factors included in clinical trial time-to-event analyses
In the case of statistically significant associations in the clinical trial time to event analyses, we assessed the proteins using more comprehensive models to evaluate their suitability as a biomarker. These models included both the above covariates along with additional risk factors specific to the outcome of interest. For heart failure outcomes, we also adjusted for coronary artery disease, atrial fibrillation, baseline eGFR, baseline UACR (diagnosis of albuminuria for EXSCEL), and prior heart failure92. For MACE, we also adjusted for hypertension, hypercholesterolaemia, BMI at baseline, smoking (current, or smoking cessation ≤3 months), and atherosclerotic disease (prior myocardial infarction, percutaneous coronary intervention/coronary artery bypass grafting, cerebrovascular accident/transient ischaemic attack, or peripheral arterial disease)93. Note, for DECLARE, the prior cardiovascular disease and multiple risk factors variables were used as they captured the MACE risk factors. For renal outcomes, we adjusted for atherosclerotic cardiovascular disease at baseline, heart failure at baseline, systolic blood pressure at baseline, T2D duration, HbA1c at baseline, eGFR at baseline, urine albumin-to-creatinine ratio (ACR) at baseline, and haemoglobin at baseline94. Note, for EXSCEL, certain analytes were only assessed intermittently, resulting in sample size loss when adjusting for these covariates. Consequently, we replaced ACR with a baseline diagnosis of albuminuria and performed a sensitivity analysis with and without haemoglobin. Despite the substantial difference in sample sizes, the hazard ratios were highly correlated (r = 0.91, p-value < 2.2 × 10−16), thus we omitted the haemoglobin adjustment for EXSCEL. Finally, for HHF and MACE, we included a third model that also adjusted for NT-proBNP to identify proteins that were independent of this biomarker as NT-proBNP was profiled in both EXSCEL and DECLARE.
Mediation analysis in EXSCEL and DECLARE
In the scenarios where a PGS is associated with both a clinical trial outcome and a protein (see Extended Methods), and the protein is in turn also associated with the same outcome, we performed mediation analysis using the Medflex R package (version 0.6-10) in the same manner as above, with the indirect p-value adjusted using the FDR approach. To maximise sample size for mediation, we made use of the full proteomics cohort and included the treatment arm as a covariate in addition to age, sex, age*sex, and genetic PCs 1-10.
Pathway enrichment
For all sets of proteins identified by the PGS-protein analyses, we used the gProfiler95 tool (https://biit.cs.ut.ee/gprofiler/gost) to test if these sets of encoded genes were enriched for KEGG, WikiPathways, or REACTOME pathways. Note that we restricted the statistical ___domain to the genes whose protein products are captured by the Olink panels.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data generated by this study (filtered for a nominal p-value of < 0.05) are available in the supplementary materials, many of which are also available via the web portal. Source data for the display figures has been provided. The UK Biobank has assigned the proteomics dataset to Category 1839 and ‘Field 30900’, details of which can be found here: https://biobank.ndph.ox.ac.uk/showcase/label.cgi?id=1839. Requests to access UK Biobank data can be made here: https://www.ukbiobank.ac.uk/enable-your-research/apply-for-access. Clinical trial data can be accessed following AstraZeneca’s data-sharing policies: https://www.astrazenecaclinicaltrials.com/our-transparency-commitments/. The PGS used in the study are available in the PGS Catalogue (https://www.pgscatalog.org/) under Publication ID PGP000701 and score IDs PGS005110-PGS005119. Source data are provided in this paper.
Code availability
This study was carried out using only publicly available software. Analysis scripts are available at https://github.com/astrazeneca-cgr-publications/plasma-proteomic-markers-prs-t2d-scripts.
References
Cole, J. B. & Florez, J. C. Genetics of diabetes and diabetes complications. Nat. Rev. Nephrol. 16, 377–390 (2020).
Meigs, J. B. The genetic epidemiology of Type 2 diabetes: Opportunities for health translation. Curr. Diab. Rep. 19, 62 (2019).
Suzuki, K. et al. Genetic drivers of heterogeneity in type 2 diabetes pathophysiology. Nature 627, 347–357 (2024).
Vujkovic, M. et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat. Genet. 52, 680–691 (2020).
Kuo, T. et al. Identification of C2CD4A as a human diabetes susceptibility gene with a role in β cell insulin secretion. Proc. Natl. Acad. Sci. USA 116, 20033–20042 (2019).
Kycia, I. et al. A common type 2 diabetes risk variant potentiates activity of an evolutionarily conserved Islet stretch enhancer and increases C2CD4A and C2CD4B expression. Am. J. Hum. Genet. 102, 620–635 (2018).
Yun, J.-S. et al. Polygenic risk for type 2 diabetes, lifestyle, metabolic health, and cardiovascular disease: a prospective UK Biobank study. Cardiovasc. Diabetol. 21, 131 (2022).
Ritchie, S. C. et al. Integrated clinical risk prediction of type 2 diabetes with a multifactorial polygenic risk score. Preprint at https://doi.org/10.1101/2024.08.22.24312440 (2024).
Ritchie, S. C. et al. Integrative analysis of the plasma proteome and polygenic risk of cardiometabolic diseases. Nat. Metab. 3, 1476–1483 (2021).
Steffen, B. T. et al. Proteomic analysis of diabetes genetic risk scores identifies complement C2 and neuropilin-2 as predictors of type 2 diabetes: the Atherosclerosis Risk in Communities (ARIC) Study. Diabetologia 66, 105–115 (2023).
Ahlqvist, E., Prasad, R. B. & Groop, L. Subtypes of Type 2 Diabetes Determined From Clinical Parameters. Diabetes 69, 2086–2093 (2020).
Iglay, K. et al. Prevalence and co-prevalence of comorbidities among patients with type 2 diabetes mellitus. Curr. Med. Res. Opin. 32, 1243–1252 (2016).
Udler, M. S. et al. Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: A soft clustering analysis. PLoS Med. 15, e1002654 (2018).
Kim, H. et al. High-throughput genetic clustering of type 2 diabetes loci reveals heterogeneous mechanistic pathways of metabolic disease. Diabetologia 66, 495–507 (2023).
DiCorpo, D. et al. Type 2 diabetes partitioned polygenic scores associate with disease outcomes in 454,193 individuals across 13 cohorts. Diabetes Care 45, 674–683 (2022).
Sun, B. B. et al. Plasma proteomic associations with genetics and health in the UK Biobank. Nature 622, 329–338 (2023).
Dhindsa, R. S. et al. Rare variant associations with plasma protein levels in the UK Biobank. Nature 622, 339–347 (2023).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Holman, R. R. et al. Effects of once-weekly exenatide on cardiovascular outcomes in Type 2 diabetes. N. Engl. J. Med. 377, 1228–1239 (2017).
Wiviott, S. D. et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 380, 347–357 (2019).
Coral, D. E. et al. A phenome-wide comparative analysis of genetic discordance between obesity and type 2 diabetes. Nat. Metab. 5, 237–247 (2023).
Mak, K.-H. et al. Prevalence of diabetes and impact on cardiovascular events and mortality in patients with chronic coronary syndromes, across multiple geographical regions and ethnicities. Eur. J. Prev. Cardiol. 28, 1795–1806 (2022).
Einarson, T. R., Acs, A., Ludwig, C. & Panton, U. H. Prevalence of cardiovascular disease in type 2 diabetes: a systematic literature review of scientific evidence from across the world in 2007-2017. Cardiovasc. Diabetol. 17, 83 (2018).
Hirano, T. Pathophysiology of diabetic dyslipidemia. J. Atheroscler. Thromb. 25, 771–782 (2018).
Graaf, A. van der et al. MR-link-2: pleiotropy robust cis Mendelian randomization validated in four independent gold-standard datasets of causality. Preprint at https://doi.org/10.1101/2024.01.22.24301400 (2024).
Zuber, V., Colijn, J. M., Klaver, C. & Burgess, S. Selecting likely causal risk factors from high-throughput experiments using multivariable Mendelian randomization. Nat. Commun. 11, 29 (2020).
Liu, X., Li, Y. I. & Pritchard, J. K. Trans effects on gene expression can drive omnigenic inheritance. Cell 177, 1022–1034 (2019).
Breit, S. N., Brown, D. A. & Tsai, V. W.-W. The GDF15-GFRAL pathway in health and metabolic disease: friend or foe? Annu. Rev. Physiol. 83, 127–151 (2021).
Moser, C. et al. FAM3D: A gut secreted protein and its potential in the regulation of glucose metabolism. Peptides 167, 171047 (2023).
Thomsen, S. K. et al. Type 2 diabetes risk alleles in PAM impact insulin release from human pancreatic β-cells. Nat. Genet. 50, 1122–1131 (2018).
Yuan, S. et al. Plasma proteins and onset of type 2 diabetes and diabetic complications: Proteome-wide Mendelian randomization and colocalization analyses. Cell Rep. Med. 4, https://doi.org/10.1016/j.xcrm.2023.101174 (2023).
Nakata, M. & Yada, T. Role of NUCB2/nesfatin-1 in glucose control: diverse functions in islets, adipocytes and brain. Curr. Pharm. Des. 19, 6960–6965 (2013).
Ren, Y. et al. Arginase: Biological and therapeutic implications in diabetes mellitus and its complications. Oxid. Med. Cell. Longev. 2022, 2419412 (2022).
Dieden, A. et al. Galectin-4 is associated with diabetes and obesity in a heart failure population. Sci. Rep. 13, 20285 (2023).
Li, X.-S., Yan, C.-Y., Fan, Y.-J., Yang, J.-L. & Zhao, S.-X. NUCB2 polymorphisms are associated with an increased risk for type 2 diabetes in the Chinese population. Ann. Transl. Med. 8, 290 (2020).
Sundararaman, S. S., Döring, Y. & van der Vorst, E. P. C. PCSK9: A multi-faceted protein that is involved in cardiovascular biology. Biomedicines 9, 793 (2021).
Kamstrup, P. R. Lipoprotein(a) and cardiovascular disease. Clin. Chem. 67, 154–166 (2021).
Alagarsamy, J., Jaeschke, A. & Hui, D. Y. Apolipoprotein E in cardiometabolic and neurological health and diseases. Int. J. Mol. Sci. 23, 9892 (2022).
Liu, Z.-W., Ma, Q., Liu, J., Li, J.-W. & Chen, Y.-D. The association between plasma furin and cardiovascular events after acute myocardial infarction. BMC Cardiovasc. Disord. 21, 468 (2021).
Yang, W., Cao, J., McVey, D. G. & Ye, S. Allele-specific epigenetic regulation of FURIN expression at a coronary artery disease susceptibility locus. Cells 12, 1681 (2023).
Karamanavi, E. et al. The FES gene at the 15q26 coronary-artery-disease locus inhibits atherosclerosis. Circ. Res. 131, 1004–1017 (2022).
Vulf, M. et al. NGR4 and ERBB4 as Promising diagnostic and therapeutic targets for metabolic disorders. Front. Biosci. Elite Ed. 15, 14 (2023).
Locke, A. E. et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206 (2015).
Zeng, F. et al. ErbB4 deletion predisposes to development of metabolic syndrome in mice. Am. J. Physiol. Endocrinol. Metab. 315, E583–E593 (2018).
Raies, A. et al. DrugnomeAI is an ensemble machine-learning framework for predicting druggability of candidate drug targets. Commun. Biol. 5, 1–16 (2022).
Finan, C. et al. The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med. 9, eaag1166 (2017).
Devuyst, O., Bochud, M. & Olinger, E. UMOD and the architecture of kidney disease. Pflugers Arch. 474, 771–781 (2022).
Maiese, K. Prospects and perspectives for WISP1 (CCN4) in diabetes mellitus. Curr. Neurovasc. Res. 17, 327–331 (2020).
Zhang, J., Huang, S., Zhu, Z., Gatt, A. & Liu, J. E-selectin in vascular pathophysiology. Front. Immunol. 15, 1401399 (2024).
Zhang, T., Zhang, Y., Tao, J., Rong, X. & Yang, Y. Intestinal trefoil factor 3: a new biological factor mediating gut-kidney crosstalk in diabetic kidney disease. Endocrine 84, 1–10 (2023).
Keshawarz, A. et al. Cardiovascular disease protein biomarkers are associated with kidney function: The Framingham Heart Study. PLOS ONE 17, e0268293 (2022).
Jasra, S. K., Badian, C., Macri, I. & Ra, P. Recognition of early myocardial infarction by immunohistochemical staining with cardiac troponin-I and complement C9*. J. Forensic Sci. 57, 1595–1600 (2012).
Huo, Y., Lai, Y., Feng, Q., Wang, Q. & Li, J. Serum ITIH4 in coronary heart disease: a potential anti-inflammatory biomarker related to stenosis degree and risk of major adverse cardiovascular events. Biomark. Med. 16, 1279–1288 (2022).
Pretorius, E., Mbotwe, S. & Kell, D. B. Lipopolysaccharide-binding protein (LBP) reverses the amyloid state of fibrin seen in plasma of type 2 diabetics with cardiovascular co-morbidities. Sci. Rep. 7, 9680 (2017).
Rossi, V., Bally, I., Lacroix, M., Arlaud, G. J. & Thielens, N. M. Classical complement pathway components C1r and C1s: purification from human serum and in recombinant form and functional characterization. Methods Mol. Biol. 1100, 43–60 (2014).
Emmens, R. W. et al. On the value of therapeutic interventions targeting the complement system in acute myocardial infarction. Transl. Res. J. Lab. Clin. Med. 182, 103–122 (2017).
Patriquin, C. J. & Kuo, K. H. M. Eculizumab and beyond: The past, present, and future of complement therapeutics. Transfus. Med. Rev. 33, 256–265 (2019).
Narayanan, R. P. et al. IGFBP2 is a biomarker for predicting longitudinal deterioration in renal function in type 2 diabetes. Endocr. Connect. 1, 95–102 (2012).
Haywood, N. J., Slater, T. A., Matthews, C. J. & Wheatcroft, S. B. The insulin like growth factor and binding protein family: Novel therapeutic targets in obesity & diabetes. Mol. Metab. 19, 86–96 (2018).
Stanley, T. L. et al. Relationship of IGF-1 and IGF-binding proteins to disease severity and glycemia in nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab. 106, e520–e533 (2021).
Barutaut, M. et al. Insulin-like growth factor binding protein 2 predicts mortality risk in heart failure. Int. J. Cardiol. 300, 245–251 (2020).
Wang, S., Chi, K., Wu, D. & Hong, Q. Insulin-like growth factor binding proteins in kidney disease. Front. Pharmacol. 12, 807119 (2021).
YOUNG, J. C., CONOVER, M. M. & FUNK, M. J. Measurement error and misclassification in electronic medical records: methods to mitigate bias. Curr. Epidemiol. Rep. 5, 343–356 (2018).
Tönnies, T., Kahl, S. & Kuss, O. Collider bias in observational studies. Dtsch. Ärztebl. Int. 119, 107–112 (2022).
Eldjarn, G. H. et al. Large-scale plasma proteomics comparisons through genetics and disease associations. Nature 622, 348–358 (2023).
Wang, Q. et al. Rare variant contribution to human disease in 281,104 UK Biobank exomes. Nature 597, 527–532 (2021).
Austin, P. C., Lee, D. S. & Fine, J. P. Introduction to the analysis of survival data in the presence of competing risks. Circulation 133, 601–609 (2016).
Mahajan, A. et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 50, 1505–1513 (2018).
Suzuki, K. et al. Identification of 28 new susceptibility loci for type 2 diabetes in the Japanese population. Nat. Genet. 51, 379–386 (2019).
Nikpay, M. et al. A comprehensive 1000 Genomes–based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 47, 1121–1130 (2015).
Ishigaki, K. et al. Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nat. Genet. 52, 669–679 (2020).
Wuttke, M. et al. A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat. Genet. 51, 957–972 (2019).
Akiyama, M. et al. Genome-wide association study identifies 112 new loci for body mass index in the Japanese population. Nat. Genet. 49, 1458–1467 (2017).
Ge, T., Chen, C.-Y., Ni, Y., Feng, Y.-C. A. & Smoller, J. W. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat. Commun. 10, 1776 (2019).
Ruan, Y. et al. Improving polygenic prediction in ancestrally diverse populations. Nat. Genet. 54, 573–580 (2022).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7 (2015).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Tingley, D., Yamamoto, T., Hirose, K., Keele, L. & Imai, K. mediation: R Package for causal mediation analysis. J. Stat. Softw. 59, 1–38 (2014).
Pedersen, B. S. & Quinlan, A. R. Who’s Who? detecting and resolving sample anomalies in human DNA sequencing studies with peddy. Am. J. Hum. Genet. 100, 406–413 (2017).
Mbatchou, J. et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet. 53, 1097–1103 (2021).
Rasooly, D. et al. Genome-wide association analysis and Mendelian randomization proteomics identify drug targets for heart failure. Nat. Commun. 14, 3826 (2023).
Yavorska, O. O. & Burgess, S. MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int. J. Epidemiol. 46, 1734–1739 (2017).
Bowden, J., Davey Smith, G., Haycock, P. C. & Burgess, S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40, 304–314 (2016).
Burgess, S., Butterworth, A. & Thompson, S. G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 37, 658–665 (2013).
Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44, 512–525 (2015).
Kurki, M.I. et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613, 508–518 (2023).
Giambartolomei, C. et al. A Bayesian framework for multiple trait colocalization from summary association statistics. Bioinforma. Oxf. Engl. 34, 2538–2545 (2018).
Guo, H. et al. Integration of disease association and eQTL data using a Bayesian colocalisation approach highlights six candidate causal genes in immune-mediated diseases. Hum. Mol. Genet. 24, 3305–3313 (2015).
Wallace, C. A more accurate method for colocalisation analysis allowing for multiple causal variants. PLOS Genet. 17, e1009440 (2021).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Steen, J., Loeys, T., Moerkerke, B. & Vansteelandt, S. medflex: An R package for flexible mediation analysis using natural effect models. J. Stat. Softw. 76, 1–46 (2017).
Segar, M. W. et al. Validation of the WATCH‐DM and TRS‐HFDM risk scores to predict the risk of Incident hospitalization for heart failure among adults with type 2 diabetes: A multicohort analysis. J. Am. Heart Assoc. 11, e024094 (2022).
Brady, W. & de Souza, K. The HEART score: A guide to its application in the emergency department. Turk. J. Emerg. Med. 18, 47–51 (2018).
Moura, F. A. et al. Risk assessment of kidney disease progression and efficacy of SGLT2 inhibition in patients with type 2 diabetes. Diabetes Care 46, 1807–1815 (2023).
Raudvere, U. et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198 (2019).
Acknowledgements
We thank the participants and investigators of the UK Biobank study who made this work possible (Resource Application Numbers 26041 and 65851). We are grateful to the research and development leadership teams at the 13 participating UKB-PPP member companies (Alnylam Pharmaceuticals, Amgen, AstraZeneca, Biogen, Bristol-Myers Squibb, Calico, Genentech, Glaxo Smith Klein, Janssen Pharmaceuticals, Novo Nordisk, Pfizer, Regeneron, and Takeda) for funding the study. We also want to acknowledge the participants and investigators of DECLARE-58 TIMI, EXSCEL, and the FinnGen studies. M.I. and S.C.R. were supported by core funding from the British Heart Foundation (RG/18/13/33946) NIHR Cambridge Biomedical Research Centre (BRC-1215-20014; NIHR203312) [*]. M.I. was also supported by the Cambridge BHF Centre of Research Excellence (RE/18/1/34212), BHF Chair Award (CH/12/2/29428) and by Health Data Research UK (Molecules to Health Records programme), which is funded by the Medical Research Council (UKRI), the National Institute for Health Research, the British Heart Foundation, Cancer Research UK, the Economic and Social Research Council (UKRI), the Engineering and Physical Sciences Research Council (UKRI), Health and Care Research Wales, Chief Scientist Office of the Scottish Government Health and Social Care Directorates, and Health and Social Care Research and Development Division (Public Health Agency, Northern Ireland. *The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care.
Author information
Authors and Affiliations
Contributions
D.P.L., A.N., D.S.P., S.C.R. and M.I. conceptualised this study. D.P.L. performed analyses and drafted the main text and supplementary materials. A.N., D.S.P., and M.I. supervised the study. D.M. and D.V. developed the web portal. M.G. and X.J. assisted with data curation. B.B.S., H.R., C.D.W. and S.P. contributed to the development of the UKB-PPP resource. R.J.M. and R.R.H. led the EXSCEL clinical trial. F.A.M., S.D.W. and M.S.S. led the DECLARE-TIMI 58 clinical trial (including several post-hoc analyses) and/or genotyping and proteomics data collection. M.S.U. developed the partitioned T2D polygenic scores. I.A.G.-N. and J.O. provided insight into the DECLARE-TIMI 58 trial and T2D biology. All authors read, commented on, and agreed upon the submitted manuscript.
Corresponding authors
Ethics declarations
Competing interests
D.P.L., M.G., D.M., D.V., X.J., I.A.G., S.P., J.O., A.N. and D.S.P. are employees of AstraZeneca and may hold AstraZeneca stock options. B.B.S. and H.R. are employees of Biogen and may hold stock options. C.D.W. is an employee of Janssen Pharmaceuticals, a Johnson & Johnson company, and may hold stock options. R.R.H. reports personal fees from Anji Pharmaceuticals, AstraZeneca and Novartis. R.J.M. received research support and honoraria from Abbott, American Regent, Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Boston Scientific, Cytokinetics, Fast BioMedical, Gilead, Innolife, Eli Lilly, Medtronic, Medable, Merck, Novartis, Novo Nordisk, Pfizer, Pharmacosmos, Relypsa, Respicardia, Roche, Rocket Pharmaceuticals, Sanofi, Verily, Vifor, Windtree Therapeutics, and Zoll. M.I. is a trustee of the Public Health Genomics (PHG) Foundation, a member of the Scientific Advisory Board of Open Targets and has research collaborations with Nightingale Health and Pfizer which are unrelated to this study. F.A.M. received consulting fees from Janssen. S.D.W. received grants from Amgen, AstraZeneca, Daiichi Sankyo, Eisai, Janssen, Merck, and Pfizer, and consulting fees from AstraZeneca, Boston Clinical Research Institute, Icon Clinical, and Novo Nordisk. M.S.S. received research grant support through Brigham and Women’s Hospital from Abbott, Amgen, Anthos Therapeutics, AstraZeneca, Boehringer Ingelheim, Daiichi-Sankyo, Eisai, Intarcia, Ionis, Merck, Novartis, and Pfizer, and consulting for Althera, Amgen, Anthos Therapeutics, AstraZeneca, Beren Therapeutics, Boehringer Ingelheim, Bristol-Myers Squibb, Dr. Reddy’s Laboratories, Fibrogen, Intarcia, Merck, Moderna, Novo Nordisk, Precision BioSciences, and Silence Therapeutics. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zhengming Chen, who co-reviewed with James Liu, Heinz Freisling, who co-reviewed with Laia Peruchet-Noray and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Loesch, D.P., Garg, M., Matelska, D. et al. Identification of plasma proteomic markers underlying polygenic risk of type 2 diabetes and related comorbidities. Nat Commun 16, 2124 (2025). https://doi.org/10.1038/s41467-025-56695-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56695-z
