
Abstract
Microbes drive the biogeochemical cycles of earth systems, yet the long-standing goal of linking emerging genomic information, microbial traits, mechanistic ecosystem models, and projections under climate change has remained elusive despite a wealth of emerging genomic information. Here we developed a general genome-to-ecosystem (G2E) framework for integrating genome-inferred microbial kinetic traits into mechanistic models of terrestrial ecosystems and applied it at a well-studied Arctic wetland by benchmarking predictions against observed greenhouse gas emissions. We found variation in genome-inferred microbial kinetic traits resulted in large differences in simulated annual methane emissions, quantitatively demonstrating that the genomically observable variations in microbial capacity are consequential for ecosystem functioning. Applying microbial community-aggregated traits via genome relative-abundance-weighting gave better methane emissions predictions (i.e., up to 54% decrease in bias) compared to ignoring the observed abundances, highlighting the value of combined trait inferences and abundances. This work provides an example of integrating microbial functional trait-based genomics, mechanistic and pragmatic trait parameterizations of diverse microbial metabolisms, and mechanistic ecosystem modeling. The generalizable G2E framework will enable the use of abundant microbial metagenomics data to improve predictions of microbial interactions in many complex systems, including oceanic microbiomes.
Similar content being viewed by others
Introduction
Microbiomes are known to impact ecosystem functioning across the earth system, but accurately predicting ecosystem functioning by leveraging increasingly available genomic data1,2,3,4,5,6 remains a grand challenge7,8,9. For example, traditional process-based ecosystem-scale biogeochemical models have often applied simple parameterizations of biological and biochemical reactions10,11,12, ignoring the microbial controls on these processes. Further, those models that do explicitly represent microbial controls typically adopt parameter values obtained from lab-cultivated microorganisms that may differ from those at a particular site13. As a result, these models do not leverage the increasing wealth of microbial information from the uncultivated majority14 that may help provide appropriate parameter space and reduce uncertainties of model projections of carbon-climate feedbacks under global change15,16.
Constructing ecosystem models that explicitly resolve soil microbial community dynamics is challenging due to the complexity of microbial mechanisms and uncertainty in parameterization. To tractably reduce data dimensionality and complexity, trait-based microbial models are valuable in testing hypotheses of structure-function relationships of microbial communities across temporal and spatial scales17,18. However, most trait-based models are limited to a single process (e.g., nitrification19, litter decomposition20) or represent only a few microbial functional groups in an ecosystem21, vastly under-representing the metabolic diversity residing in complex systems like soils. In addition, critical microbial traits (e.g., growth rate, substrate uptake kinetics) are parameterized from discrete laboratory ecophysiology experiments, using cultivated microbial species that are not reflective of native functional biodiversity22. These issues result in a biased landscape of microbial traits23,24,25 and represent an important limitation to the expansion of microbially explicit models, adding significant uncertainty to predictions of ecosystem function13.
Microbial trait information inferred from meta-omics data, including metagenomics, can better represent the metabolic potential contained in complex systems26, potentially reducing the uncertainty of ecosystem predictions. For example, genome-based codon usage bias can be used to estimate minimum generation times (i.e., maximum achievable growth rates) of bacteria and archaea27,28,29. Additionally, workflows (e.g., the one we apply here, microTrait30) are emerging to extract a range of genome-inferred microbial fitness traits (e.g., energetic, resource acquisition, stress tolerance). Leveraging such genome-inferred traits could expand trait parameterization beyond cultivated organisms, to include the uncultivated microbial majority, particularly as genome-resolved microbiome data from uncultivated organisms now exceed data available from cultivated organisms. It is reasonable to assume that genomes from microorganisms derived directly from a given environment will better represent the microbial ecophysiological traits that underpin biogeochemical processes in those environments, potentially improving model prediction through better constraints.
Here we established a general genome-to-ecosystem (G2E) framework that integrates genome-inferred traits into an ecosystem model (ecosys), and then applied this framework to project ecosystem functioning at a well-studied permafrost site. Stordalen mire, like many permafrost sites31, is undergoing rapid changes associated with recent warming-induced permafrost thaw, thaw pond expansion, changing biology (vegetation, microbiota, viruses), and increasing greenhouse gas (GHG) emissions3,32,33,34,35. Ecosys is a mechanistic ecosystem model tested in many high-latitude sites and regions, including the Stordalen mire in northern Sweden36,37,38. Extensive research at this site provides thousands of phenotypically-linked metagenome-assembled genomes (MAGs) across the permafrost thaw gradient3. From these resources, we derived microbial kinetic traits and their distributions and evaluated their impacts on resolving uncertainties in ecosystem-scale predictions of methane (CH4) emissions. Although our work involves comparing CH4 emissions between simulations and field observations, our goal is not to perfectly fit the available CH4 observations at the site, as is often pursued through extensive parameter calibration. Instead, our goal is to explicitly link genomic data with ecosystem-scale process rates by integrating genome-inferred microbial traits into an ecosystem model. This goal, if achieved, would pave the way for the broad integration of increasingly available genomic data and genome-inferred microbial physiology and functioning data to inform ecosystem-scale functional predictions.
Results and discussion
A multi-scale genome-to-ecosystem framework
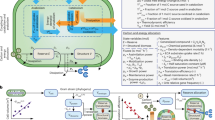
To integrate genome-inferred microbial traits into ecosys, we developed our G2E framework as follows (Fig. 1; “Methods”). First, we predicted traits from a collection of metagenome-assembled genomes (1529 MAGs and 647 representative genomes3) from Stordalen mire peat soil samples that spanned three sub-habitats (well-drained palsas underlain by continuous permafrost, intermediate-thaw bogs, and fully thawed fens). Traits were predicted using the microTrait workflow30, which extracts fitness traits from microbial genome sequences using literature-contextualized and selected profile-hidden Markov Models. MicroTrait defines microbial functional groups based on shared metabolic traits (e.g., hydrogenotrophic methanogenesis) across the genomes. Second, genomic traits were translated into relevant ecosys model parameters using DEBmicroTrait (where DEB = dynamic energy budget), a model built from allometric scaling laws and biophysical constraints based on the DEB theory39. From the Stordalen Mire MAGs, this resulted in eleven (Supplementary Table 1) microbial functional group-specific maximum (or potential) specific respiration rates and half-saturation constants (Rmax, Km, Supplementary Table 2), which are key parameters in the Michaelis-Menten rate law kinetics used in ecosys40. Of the eleven microbial functional groups, we focus on the five dominant groups that are known to directly control CH4 emissions and are well represented in our data: obligately aerobic heterotrophic bacteria, obligately anaerobic fermenters, acetoclastic methanogens, hydrogenotrophic methanogens, and (aerobic) methanotrophs. Third, we used ecosys to compute microbial respiration rates considering vertically-resolved and dynamic gradients in oxygen, water, carbon, nutrients, and temperature (“Methods”). Baseline ecosys simulations were run from 1901 to 2003 for spin-up as initial conditions (“Methods”)37. Finally, an ensemble of 1300 simulations of the years 2004–2009 was performed by sampling from the genome-inferred trait distributions for all microbial functional groups (Supplementary Table 2; Morris method, “Methods”). Sensitivity analyses were performed on the ensemble simulations. Together, this G2E framework allows genome-inferred traits to be integrated into mechanistically modeled microbial processes, which then determine simulated ecosystem predictions.

Genome sequencing data from field soil samples is annotated and traits are derived using a two-step workflow (i.e., microTrait and DEBmicroTrait). The products are inferred microbial traits including maximum specific respiration rates and half-saturation constants (Rmax, Km), which are used to parameterize a mechanistic-based ecosystem model (ecosys) that represents a functionally diverse microbial community. The diverse microbial community structure has been established in previous studies59,72 with literature-derived microbial kinetic traits. A complete list of functional groups and reactions is shown in Supplementary Table 1. ecosys then simulates depth-resolved hydrological, thermal, and plant processes and produces ecosystem-scale outputs that are compared against field observations. Detailed descriptions of these components can be found in “Methods”. HMM profile-hidden Markov Models, SOM soil organic matter, DOM dissolved organic matter, T temperature, N nitrogen, P phosphorous, Rn net radiation, LE latent heat flux, H sensible heat flux, G ground heat flux.
Stordalen mire as a case study for G2E
Genome-inferred microbial traits
We first assessed whether predicted trait magnitudes were consistent with experimental data from the literature for the five dominant functional groups across the Stordalen mire permafrost thaw gradient. Though genome-inferred Rmax and Km values spanned a wide range within each microbial functional group, the values partially overlapped with published experimental data and had smaller variability (“Methods”) (Fig. 2A, B and Supplementary Fig. 1). For some parameters (e.g., Rmax of fermenters, Rmax and Km of hydrogenotrophic methanogens), the differences were statistically indistinguishable (p > 0.05) (Supplementary Table 3). For others (e.g., Km of fermenters, Rmax and Km of aerobic heterotrophs, acetoclastic methanogens, and methanotrophs), statistically significant deviations were found (p < 0.05) (Supplementary Table 3), which either represent estimation errors or more likely reflect the fact that the organisms used to derive the literature estimates are not representative of those found at Stordalen mire27,41. Since literature values are inferred from different organisms, environments, and experimental protocols, it is challenging to interpret and apply them to other environments42. For example, phylogenetic analysis of the genomes shows that methanotrophs at Stordalen mire belong to the families of Methylococcaceae, Methylocystaceae, Beijerinckiaceae, and Hyphomicrobiaceae, whereas the measured kinetic traits from prior literature are primarily derived from the Methylococcaceae34. In addition, our genome-inferred half-saturation constants of aerobic heterotrophs were deliberately constrained to growth on glucose and acetate to be consistent with ecosys reaction stoichiometry.

Genome-inferred and published literature-derived data of A maximum specific respiration rate (Rmax) and B half-saturation constant (Km). Numbers (n) of genomes and published experimental data from the literature (Rmax, Km) are shown beside each box. The gray number indicates the count of genomes-inferred traits and the black number denotes the number of traits obtained from the literature (left value corresponds to Rmax and right value corresponds to Km). Genome-inferred C Rmax and D Km of dominant genomes in each sub-habitat. Dominant genomes are defined as the minimal set of genomes whose total relative abundance is ≥80%. Community-aggregated traits represent the trait properties weighted by the relative genome abundance (“Methods”). Numbers (n) of dominant genomes in each sub-habitat are shown on the right. Units of the literature-derived kinetic parameters are converted to compare with the genome-inferred values. Microbial traits of the other functional groups related to nitrogen cycling are shown in Supplementary Fig. 2. For aerobic heterotrophs, Rmax from pure culture studies is not reported, so we estimated the values by using maximum growth rate (h−1) information from pure culture studies divided by a general growth yield of 90 [gcell (mol substrate)−1]73,74. In each box in A–D, the boundaries represent the 25th and 75th percentile (interquartile range), the middle line represents the median value, and the whiskers represent the minimum and maximum values within 1.5 times the interquartile range. Data points outside of this range are considered outliers and are shown individually. For each functional group, genome-inferred Km and Rmax values are plotted against each other (Supplementary Fig. 3). Note that the default parameters in ecosys are from literature and included in the “Literature” group of this analysis.
The kinetic traits of microbes are influenced by their biophysical properties, substrate preferences, and associated stoichiometric and thermodynamic consequences. Substrate properties and thermodynamics vary across metabolic pathways due to differences in vegetation, organic matter, and water saturation across the permafrost thaw gradient sub-habitats43. We explored these genome-inferred traits across microbial functional groups with differing substrate preferences and across sub-habitats. While we found that, as expected, genome-inferred traits were significantly different (p < 0.05) between the five dominant functional groups (Supplementary Table 4), no differences (p > 0.05) of inferred traits from dominant genomes (defined as the minimal set of genomes whose total relative abundance is \(\ge \!\)80%) were observed between the three sub-habitat types (fen, palsa, bog) for functional groups whose sample size were sufficient to make statistical inferences (i.e., obligately aerobic heterotrophs and fermenters) (Fig. 2C, D and Supplementary Table 5). Note that although the genome relative abundance of these functional groups does vary across these three sub-habitats3, the lack of significant differences in trait distributions implies a similar trait space for each functional group within this permafrost site.
To scale trait distributions from individual organisms within each functional group to the sub-habitat level, we used a community-aggregated trait approach44,45. This approach provides a means to simplify ecosystem model parameterization by selecting a single trait parameter for a particular functional group within the same sub-habitat rather than using the full trait distribution. This community-aggregated trait value is the aggregation of the traits of all genomes in the community weighted by the relative abundance of the genomes that carry that trait (i.e., Rmax or Km)46 (“Methods”). We tested the approach by analyzing whether simulated GHG emissions differed between simulations employing the dominant and community-aggregated trait values (see below).
Microbial traits affect CH4 emissions
Given reasonable yet diverse genome-inferred trait distributions, we next asked whether (i) trait values obtained directly from genomes within this study would result in satisfactory estimates of ecosystem GHG emissions, and (ii) microbial trait distributions would impact modeled ecosystem GHG emissions. To address these questions, we used genome-inferred microbial traits as input parameters for ecosys, and projected simulations of CH4 emissions (Fig. 3A, B) and net ecosystem carbon exchange (NEE, Supplementary Fig. 4) in Stordalen mire for the two sub-habitats, fen, and bog, where CH4 emissions are highest47. Importantly, simulated CH4 emissions closely match the observed field emissions which indicates the validity of the trait values (Fig. 3A, B). Due to the variance in predicted traits distributions, parameterizing the model with genome-inferred microbial traits drives substantial variability in simulated CH4 emissions annually (144% and 152% in the bog and fen, respectively; Supplementary Fig. 6), seasonally (up to 281 mgC m−2 d−1), daily (up to 658 mgC m−2 d−1), and across both sub-habitats (Fig. 3A–D). Effects of trait variation on microbial respiration rates persist despite other dynamic environmental controls (e.g., soil temperature, oxygen, water).

Modeled and observed daily CH4 emissions in the A fen and B bog sites. For the palsa site, the model accurately captured the observed NEE and CH4 emissions (Supplementary Fig. 7). Blue-shaded areas show modeled daily mean emissions (2004–2009) due to the full range of microbial traits and solid lines show the best fit of the modeled emissions. Orange circles represent daily averaged CH4 emissions observed for each day of the year (DOY) across the simulation period when at least two years of data are available37. Given the absence of Autumn data in 2005, 2006, and 2008, DOY average Autumn CH4 emissions may not be representative of true inter-annual variability. Full-time series of results are provided in Supplementary Fig. 5. Simulated daily CH4 emissions over the simulation period are evaluated by calculating Pearson correlation coefficient (R) and root mean square error (RMSE) when quality control measurements are available (Fen: R = 0.51, RMSE = 80 mgC m−2 d−1, n = 359; bog: R = 0.44, RMSE = 22 mgC m−2 d−1, n = 392). Modeled daily CH4 emissions averaged over each season in C fen and D bog sites. The boxes represent the median and the first and third quartiles of daily emissions for each season (n = 1300) during 2004–2009. The whiskers represent the minimum and maximum values within 1.5 times the interquartile range. E Sensitivity analysis of annual CH4 emissions attributed to each microbial trait measured by Mean Elementary Effect (“Methods”) for dominant microbial functional groups. Results for all microbial traits including traits related to nitrogen cycling are provided in Supplementary Fig. 8, which shows a much lower sensitivity of CH4 emissions to these traits compared with the five dominant microbial functional groups. Each solid square bar depicts the interannual mean of 2004–2009 (n = 6), and the error bars represent the standard deviation around the mean. Rmax maximum specific respiration rate, HM hydrogenotrophic methanogen, AM acetoclastic methanogen, Aero_Heter aerobic heterotroph, DOC dissolved organic carbon. The substrate is shown as a superscript for some functional groups. Note that a positive Mean Elementary Effect of a microbial trait for CH4 emissions implies more CH4 is emitted when increasing that microbial trait. Km also contributes to the variation of CH4 emissions through its relationship with Rmax (Supplementary Fig. 3).
We used a Morris sensitivity analysis to identify the most significant microbial traits regulating annual CH4 emissions and found that the mean elementary effects (change of annual CH4 emissions due solely to a change in one microbial parameter relative to the annual CH4 emissions) range from 0 to ~60% (Fig. 3E). In both fen and bog, Rmax of hydrogenotrophic methanogens47 has the largest mean elementary effects, followed by Rmax of fermenters (Fig. 3E). In the fen, the elementary effect of acetoclastic methanogens is comparable to that of fermenters, but in the bog, where acetoclastic methanogens are less prevalent47,48, the acetoclastic methanogen elementary effect is relatively small. These results demonstrate that the range of inferred microbial traits can lead to large variations in ecosystem responses, with the most important traits being Rmax of hydrogenotrophic methanogens and fermenters, i.e., the organisms catalyzing substrate production for methanogenesis and those performing the process of methanogenesis itself. Since our comparisons between genome-inferred and literature-based trait values and the impacts of trait values on modeled CH4 emissions were focused on two sub-habitats at one site, it would be valuable to apply our approach at other sites to characterize when genome-inferred traits provide added value to modeled ecosystem carbon cycling. Importantly, our results also show a large variation of predicted NEE after ten years of simulation (Supplementary Fig. 4) due to microbial trait distributions, especially in the bog site. These modeling results indicate that interactions between microbial activity and belowground nutrient transformations may impact vegetation dynamics over multi-decadal time scales, and represent a model-inspired hypothesis to be tested at long-term study sites49.
Evaluation of trait-inferring strategies
Though the observed CH4 emissions were captured by simulations, the simulated CH4 emissions have large uncertainties (Fig. 3). To reduce these uncertainties, we evaluated three trait-inferring strategies (Fig. 4): (1) using all present genomes in a sub-habitat; (2) using the dominant genomes based on abundance; (3) using a community-aggregated approach where traits are weighted by relative genome abundance. We found that community-aggregated traits performed best for improving CH4 emission predictions, relative to traits derived from all present genomes or dominant genomes (Fig. 4). Specifically, with respect to the all-present-genomes approach, using community-aggregated traits increased correlation accuracy (R) by 19% with decreased bias (RMSE) by 12% in the fen, and increased R by 24% and decreased RMSE by 54% in the bog. Since only the community-aggregated traits included relative-abundance-weighting, this result suggests that using trait-relative abundance may be appropriate for simulating the observed fluxes. Since ecosystem output is impacted by the expression of traits rather than their genomic abundances, this finding is consistent with past work at this site that demonstrated the close link between the relative abundance of a key microbial lineage and CH4 production pathways47. The community-aggregated traits, without additional parameter tuning, provide comparable CH4 emission predictions compared to the default microbial trait parameters (non-genomic) (Supplementary Table 6). The good performance of community-aggregated traits indicates it is a promising approach to simplify microbiome kinetic trait parameters and provides a more credible mechanistic basis for ecosystem modeling50. Future work to incorporate absolute abundance and microbial activity indices into the trait-inference approach might lead to more accurate trait parameterization and further reduction in CH4 emission uncertainty.

A fen; B bog. The different trait-inference approaches are (1) trait ranges of present genomes in a sub-habitat, (2) trait ranges of dominant genomes (ranges shown in Fig. 2 as Dominant), and (3) community-aggregated trait values (shown in Fig. 2). Simulations for all three approaches are averaged across the simulation period for better visualization. Each box boundary represents the 25th and 75th percentile (interquartile range), the middle line represents the median value, and the whiskers represent the minimum and maximum values within 1.5 times the interquartile range. Data points outside of this range are considered outliers and are shown individually. For approaches (1) and (2), the variation of CH4 emissions mainly comes from trait variation as shown in Fig. 2. Comparison of individual years, where variation comes solely from trait variability, is shown in Supplementary Fig. 6. The number of simulations (n), which depends on the number of trait combinations, is shown for each approach. To evaluate these three strategies, Pearson correlation coefficient (R) and root mean square error (RMSE) were calculated based on the observed daily CH4 emissions. For strategies that involved multiple simulations, the mean R (\(\bar{R}\)) and mean RMSE (\(\overline{{RMSE}}\)) are used.
For the past two decades, scientists have been sampling and sequencing the microbes of earth systems. Nevertheless, the resulting understanding has not led to quantitative estimation of microbial impacts on ecosystem-scale biogeochemistry, nor to improvements in ecosystem model predictive capability. Here, we describe a genome-to-ecosystem (G2E) framework that integrates genome-inferred microbial traits into an ecosystem-scale model to explicitly link genomic data and ecosystem-level process rates. This G2E framework enabled us to perform a genome-to-ecosystem analysis in a rapidly thawing permafrost system. We quantitatively demonstrated the large impact of genomically observable variations in microbial capacity on ecosystem CH4 emissions. Our findings highlight the sensitivity of CH4 emissions to microbial trait parameterization, emphasizing the importance of accurate parameterization of microbial traits for ecosystem predictions.
This G2E framework provides three important advances for ecosystem science: (i) it leverages information contained in microbial genomes, which are records of organismal life-history strategies, to infer microbial trait values relevant to ecosystem models51; (ii) it bridges discoveries in microbial physiology and functioning to enable model evaluation of their impacts on ecosystem scale processes under a changing climate; and (iii) it integrates theory, observations, and numerical models across multi-disciplinary teams, providing a powerful framework to understand and predict ecosystem responses to change52. Although this study focuses on CH4 emission processes in a terrestrial permafrost ecosystem, the G2E framework, including the microbial trait inferences from genomes and its integration into complex ecosystem models, is generalizable. In particular, the models involved in the G2E framework represent many microbial traits and biogeochemical processes. With the increasing availability of meta-omics data and interest in mechanistic models, the G2E framework can be applied to other terrestrial and oceanic ecosystems. Further application of the G2E framework to diverse sites and ecosystems could help unlock the full potential of genomics data across larger landscape scales for improvements in understanding and prediction of microbial-climate feedback. The G2E framework developed here provides a tractable path to use process-rich models to synthesize and generalize how microbial traits influence ecosystem functioning and to improve ecosystem model predictability.
Methods
Study and sampling sites
Stordalen mire is a peatland situated in northern Sweden (68.35° N, 19.05° E), 10 km southeast of Abisko Scientific Research Station (ANS). The climate in this area is subarctic with annual mean temperature of 0.07 °C and mean precipitation of 308 mm y−1 (1986–2006)53. The three sub-habitats of the study site include well-drained palsa underlain by permafrost, intermediate-thaw bog with variable water table depth, and fully thawed and inundated fen. Significant changes in climate and landscapes have been recorded32,54. The mire has been closely monitored since the 1970s and a comprehensive dataset55 has been generated including terrestrial gas fluxes53, microbial sequencing3,47, subsurface geochemistry56, and plant data57.
The ecosys model
Ecosys is a mechanistic land model that fully couples carbon, energy, water, and nutrient cycles solved at an hourly step58. The carbon, nitrogen, and phosphorus transformations are dominated by the plant-microbe-soil carbon interactions that are driven by the energetics and kinetics of aerobic and anaerobic oxidation-reduction reactions. Ecosys represents a functionally diverse microbial community that represents microbial activities including respiration, growth, nutrient uptake, and mortality. The CH4 transformation is represented by methanogenesis and methanotroph processes based on the activities of anaerobic fermenters, acetolactic methanogens, hydrogenotrophic methanogens, and autotrophic methanotrophs. The rate of microbial respiration is a function of microbial biomass, soil moisture, temperature, O2 concentration, organic matter concentration, and nutrient availability. ecosys represents many essential metabolic handoffs, although not all are known to exist in real systems. For example, ecosys considers soil organic matter to be a substrate for hydrolysis by all active heterotrophic microbial communities. The resulting hydrolysis products are transferred to dissolved organic carbon, nitrogen, and phosphorus pools. Fermenters uptake dissolved organic carbon to produce acetate, CO2, and H2. Acetate is the substrate for respiration by acetoclastic methanogens, producing CH4 and CO2. The fermenter products CO2 and H2 serve as substrates for CO2 reduction by hydrogenotrophic methanogens, generating CH4 and H2O. Functional groups and reactions represented in ecosys are listed in Supplementary Table 1. A link to the detailed description of ecosys and code is given in “Code availability”.
Modules in ecosys that represent microbial processes (e.g., methanogenesis, methane oxidation) have been rigorously tested against lab incubation studies59,60. Ecosys has been broadly tested in several high-latitude ecosystems by comparing model estimates of energy and carbon fluxes with field observations38,49,61 as well as other ecosystems (publication list in ref. 62). Specifically, ecosys has been tested against the active layer depth, water table depth, and net ecosystem exchange in a thawing gradient in Stordalen mire, which is the test site in this work36,37. Hence, ecosys is an appropriate model to test the sensitivity of ecosystem responses to microbial processes.
Database assembly and processing
A database of metagenome-assembled genomes (MAGs) (n = 1529) and representative genomes (n = 647) from the permafrost thaw gradient was used in this study3. Those MAGs are based on 214 samples across habitats, depths, and sampling times, and are estimated to be >70% complete and <10% contaminated according to CheckM63. The relative abundance of each recovered representative genome within each sample is listed in Supplementary Data 1. Specifically, the relative abundance of a representative MAG as defined here was calculated as the trimmed mean coverage of the MAG divided by the total of trimmed mean coverages of all representative MAGs, ignoring reads that were not mapped to any representative. For more details about MAGs and sampling processing, please refer to ref. 3.
We acknowledge that employing RNA sequencing data would provide a higher resolution of the active microbial lineages and their expressed genes. Nevertheless, such data is unavailable for most samples analyzed in this manuscript. Furthermore, integrating RNA sequencing data with biogeochemical models presents challenges because of the complex and poorly characterized relationships between transcripts and biogeochemical reaction rates9,64,65. Therefore, integration of transcript data and ecosys is not possible in this study, despite its potential significance.
Assignment of genomes to functional groups using microTrait pipeline
Microbial traits (i.e., maximum growth rate) for each genome were inferred with microTrait30, a pipeline that infers ecological traits from genomic sequences. The traits represented in the microTrait hierarchy were mapped to functional groups represented in ecosys using a custom script (Supplementary Data 2). The minimum generation time for each genome was estimated based on codon usage bias, as shown in Supplementary Data 3.
DEBmicrotrait model
Trait predictions (i.e., maximum specific respiration rate, Rmax, and half saturation constant, Km) were based on a genome-informed, trait-based dynamic energy budget model (DEBmicroTrait)39. DEBmicroTrait partitions microbial biomass into generalized chemical compounds (reserve and structural biomass) with fixed macromolecular composition and chemical potential. This partitioning allows for a coarse-grained description of the energy fluxes during microbial growth, that is the catabolic and anabolic reactions through which microbes obtain and then utilize energy, respectively66. The coupling between catabolism and anabolism exists both in the sequential assimilation, partitioning, and dissipation of substrate and reserve compounds, with maintenance taking priority over growth and constitutive extracellular enzyme production. Rmax, derived from carbon and energy allocation under unlimited substrate concentration, is given as the sum of dissipative losses during microbial growth. To constrain carbon and energy fluxes, DEBmicroTrait integrates microTrait information into a workflow combining genome inference and biophysical modeling.
For instance, basal maintenance respiration is directly proportional to cell volume67. DEBmicroTrait further assumes that cells have evolved optimal protein densities in cellular compartments that maximize reaction rates68. The elemental composition of reserve and structural biomass influences substrate demand and follows allometric trends in cellular biomolecule composition with volume, but organisms can differ in their protein synthesis efficiency. Codon-usage bias sets an upper bound on protein translation power, while the number of ribosomal RNA operons predicts translation efficiency. In order to estimate Km, we assumed that the respiration rate follows an empirical Michaelis-Menten-like description with a half-saturation constant that reflects the cellular nutrient demand in terms of cell size and transporter protein density on the cell surface69. As a result, the modeled half saturation constant is a function of the specific respiration rate as shown in Supplementary Fig. 3. This relationship is then written in the ecosys model to make sure the sampling procedure in the sensitivity analysis follows the trade-off rule between half-saturation constant and respiration rate. The inferred traits including Rmax and Km were shown in Supplementary Data 4. A detailed description of the DEBmicroTrait model is available39.
Weighted traits based on relative abundance
Representative genomes were grouped into functional groups based on their shared functions. The relative abundance of each functional group within each sample was calculated as the sum of the relative abundance of all the representative genomes that belong to the same functional group. The relative abundance of functional groups varied between samples, and the largest variation was due to sub-habitat (i.e., fen, bog, and palsa), followed by depth and time. Thus, we defined genomes within the same functional group within the same sub-habitats as one community and normalized the relative abundance so that the relative abundance of the community equaled one. We assumed that the relative abundance was a measure of biomass within the community and the “biomass ratio” hypothesis is valid (i.e., ecosystem functioning is controlled by the trait values of the dominant contributors to the community biomass)45. Then the most abundant genomes within one community were identified, where the most abundant genomes (i.e., dominant genomes) were defined as the minimal set of genomes whose total relative abundance was \(\ge \!\)80%. The community-aggregated trait was calculated as the aggregation of trait values of all the genomes in the community, weighted by the relative abundance of the genomes carrying each value, to quantify the community-level properties70. The community-aggregated traits of the three sub-habitats are shown in Supplementary Data 5. We also identified the presence or absence of genomes by whether the relative abundances were above 0. The trait range of the present and dominant genomes in one sub-habitat was shown in Supplementary Data 6.
Traits synthesized from literature
We compiled the kinetic trait values including Rmax and Km from pure culture studies where microbes were cultivated under optimal conditions (i.e., abundant substrate concentrations, optimal pH, and temperature). The units of Rmax and Km from the literature experiments were converted to compare with the genome-inferred trait values. Supplementary Data 7 presents the synthesized range of traits of different microbial functional groups, along with the corresponding references.
Morris method and sensitivity analysis
The method of Morris was used to estimate the sensitivity of CH4 emissions to the genome-inferred microbial traits. This method71 derives the global sensitivity by sampling a set of local partial derivatives, known as elementary effects (EE), across the parameter spaces. Each parameter space is scaled to [0,1] and partitioned into (m-1) equally-sized intervals. Unlike a local sensitivity analysis, which calculates the partial derivative of the target function with respect to the reference value of the parameter, the Morris method randomly selects multiple reference values that span the entire parameter range. So the calculation of EE (Eq. 1) is repeated a number of times for each parameter, and the mean value (mean EE) is used as sensitivity indices
where f (x) is the target function, and in this case, is the simulated CH4 emissions by ecosys, \(\varDelta\) is a multiple of 1/(m − 1). In this study, each parameter space was partitioned to 100 intervals and a total of 1300 simulations were performed to calculate the mean elementary effects (EE).
In this study, the mean EE of daily CH4 emission was first calculated, and then summed up for each year to get the mean EE of annual CH4 emission. To assess the magnitude of effects, the mean EE of annual CH4 emission is divided by the average annual emission of a total of 1300 runs. The mean and standard deviation of mean EE of CH4 emission for fen and bog were calculated and shown in Fig. 3E.
Statistical analysis
To evaluate the performance of the ecosys, we computed the correlation coefficient (R) and root mean square error (RMSE) by comparing them with observed daily CH4 emissions. The correlation coefficient (R) is defined as
where \({x}_{i}\) is the simulated daily CH4 emission and \(\bar{x}\) is the overall mean, \({y}_{i}\) is the observed daily CH4 emissions and \(\bar{y}\) is the overall mean. n is the total number of daily CH4 emission data that is quality controlled.
Root mean square error (RMSE) is defined as
We used non-parametric Kruskal–Wallis one-way analysis to test the significance of the difference in trait values between the genome-inferred and literature data (Supplementary Table 3), the genome-inferred trait among different functional groups (Supplementary Table 4), and the genome-inferred trait among different sub-habitats (Supplementary Table 5) for the same functional group. The statistical results were considered significant if p < 0.05. All statistical testing was carried out in a Python environment.
Model experiment
The baseline (spin-up) simulation begins in 1901, when we initialize soil properties (e.g., soil texture, bulk density, carbon and nitrogen stocks), seed the vegetation, and run the model forward from 1901 to 200337. Simulations with different trait parameters, sampled based on Morris methods, were then performed over 2004–2009 when field observations of CH4 and CO2 emissions were available. A total of 1300 simulations over 2004–2009 were performed and used for analysis, each with its own microbial trait parameter set. The simulation results and observations are shown in Supplementary Data 8. Although not shown, we conducted two other simulation experiments in which we initialized the microbial trait parameters on Jan. 1, 2000, and 2003, and analyzed the same period (2004–2009) as the original design. We found consistent results as shown in Fig. 3 indicating the disequilibrium caused by the parameter changes was small. We also performed two simulations (with the best-fit and community-aggregated traits) each in the fen and bog starting in 1901 to assess the potential impacts of initial condition differences on emissions over the observational record (Supplementary Fig. 9). Both simulations generally capture the observed seasonal dynamics for CH4 emissions and NEE. Noticeable differences between these two simulations were found in the fen CH4 emissions, with the observed daily CH4 emissions falling between them.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data generated in this study are provided in the Supplementary Data and have been deposited at https://doi.org/10.5281/zenodo.14219121. The metagenome-assembled genome (MAG) dataset is available at NCBI under BioProject PRJNA386568, with information about these MAGs provided in Supplementary Data 3 of a previous publication in ref. 3.
Code availability
The microTrait model30 is available for download at https://github.com/ukaraoz/microtrait. The DEBmicroTrait model39 is available for download at https://github.com/giannamars/DEBmicroTrait.jl. The ecosys model, documentation38,62 and simulation runs are available for download at https://github.com/jinyun1tang/ECOSYS and https://doi.org/10.5281/zenodo.14219121. Figure 1 was generated by Adobe Illustrator. All the other figures were generated by the programming language Python (3.11).
References
Paoli, L. et al. Biosynthetic potential of the global ocean microbiome. Nature 607, 111–118 (2022).
Anantharaman, K. et al. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat. Commun. 7, 13219 (2016).
Woodcroft, B. J. et al. Genome-centric view of carbon processing in thawing permafrost. Nature 560, 49–54 (2018).
Hultman, J. et al. Multi-omics of permafrost, active layer and thermokarst bog soil microbiomes. Nature 521, 208–212 (2015).
Bahram, M. et al. Structure and function of the global topsoil microbiome. Nature 560, 233–237 (2018).
Bourquin, M. et al. The microbiome of cryospheric ecosystems. Nat. Commun. 13, 3087 (2022).
Rynearson, T. A. Navigating in a sea of genes. Science 358, 1129–1130 (2017).
Hanemaaijer, M. et al. Systems modeling approaches for microbial community studies: from metagenomics to inference of the community structure. Front. Microbiol. 6, 213 (2015).
Grossart, H.-P., Massana, R., McMahon, K. D. & Walsh, D. A. Linking metagenomics to aquatic microbial ecology and biogeochemical cycles. Limnol. Oceanogr. https://doi.org/10.1002/lno.11382(2020).
Riley, W. J. et al. Barriers to predicting changes in global terrestrial methane fluxes: analyses using CLM4Me, a methane biogeochemistry model integrated in CESM. Biogeosciences 8, 1925–1953 (2011).
Sokol, N. W. et al. Life and death in the soil microbiome: how ecological processes influence biogeochemistry. Nat. Rev. Microbiol. https://doi.org/10.1038/s41579-022-00695-z (2022).
Zhuang, Q. et al. Methane fluxes between terrestrial ecosystems and the atmosphere at northern high latitudes during the past century: a retrospective analysis with a process-based biogeochemistry model. Global Biogeochem. Cycles https://doi.org/10.1029/2004GB002239 (2004).
Sulman, B. N. et al. Multiple models and experiments underscore large uncertainty in soil carbon dynamics. Biogeochemistry 141, 109–123 (2018).
Solden, L., Lloyd, K. & Wrighton, K. The bright side of microbial dark matter: lessons learned from the uncultivated majority. Curr. Opin. Microbiol. 31, 217–226 (2016).
Wieder, W. R., Bonan, G. B. & Allison, S. D. Global soil carbon projections are improved by modelling microbial processes. Nat. Clim. Chang. 3, 909–912 (2013).
Tang, J. & Riley, W. J. Weaker soil carbon–climate feedbacks resulting from microbial and abiotic interactions. Nat. Clim. Chang. 5, 56–60 (2014).
Zakharova, L., Meyer, K. M. & Seifan, M. Trait-based modelling in ecology: a review of two decades of research. Ecol. Modell. 407, 108703 (2019).
Allison, S. D. & Goulden, M. L. Consequences of drought tolerance traits for microbial decomposition in the DEMENT model. Soil Biol. Biochem. 107, 104–113 (2017).
Bouskill, N. J., Tang, J., Riley, W. J. & Brodie, E. L. Trait-based representation of biological nitrification: model development, testing, and predicted community composition. Front. Microbiol. 3, 364 (2012).
Allison, S. D. A trait-based approach for modelling microbial litter decomposition. Ecol. Lett. 15, 1058–1070 (2012).
Wieder, W. R., Grandy, A. S., Kallenbach, C. M., Taylor, P. G. & Bonan, G. B. Representing life in the earth system with soil microbial functional traits in the MIMICS model. Geosci. Model Dev. 8, 1789–1808 (2015).
Steen, A. D. et al. High proportions of bacteria and archaea across most biomes remain uncultured. ISME J. 13, 3126–3130 (2019).
Button, D. K. Kinetics of nutrient-limited transport and microbial growth. Microbiol. Rev. 49, 270–297 (1985).
Button, D. K., Robertson, B., Gustafson, E. & Zhao, X. Experimental and theoretical bases of specific affinity, a cytoarchitecture-based formulation of nutrient collection proposed to supercede the michaelis-menten paradigm of microbial kinetics. Appl. Environ. Microbiol. 70, 5511–5521 (2004).
Madin, J. S. et al. A synthesis of bacterial and archaeal phenotypic trait data. Scientific Data 7, 170 (2020).
Sharon, I. & Banfield, J. F. Microbiology. Genomes from metagenomics. Science 342, 1057–1058 (2013).
Weissman, J. L., Hou, S. & Fuhrman, J. A. Estimating maximal microbial growth rates from cultures, metagenomes, and single cells via codon usage patterns. Proc. Natl. Acad. Sci. USA 118, e2016810118 (2021).
Vieira-Silva, S. & Rocha, E. P. C. The systemic imprint of growth and its uses in ecological (meta)genomics. PLoS Genet. 6, e1000808 (2010).
Long, A. M., Hou, S., Ignacio-Espinoza, J. C. & Fuhrman, J. A. Benchmarking microbial growth rate predictions from metagenomes. ISME J. 15, 183–195 (2021).
Karaoz, U. & Brodie, E. L. microTrait: a toolset for a trait-based representation of microbial genomes. Fronti. Bioinform. 2, 918853 (2022).
Callaghan, T. V. et al. A new climate era in the sub-Arctic: accelerating climate changes and multiple impacts. Geophys. Res. Lett. https://doi.org/10.1029/2009GL042064C (2010).
Varner, R. K. et al. Permafrost thaw driven changes in hydrology and vegetation cover increase trace gas emissions and climate forcing in Stordalen mire from 1970 to 2014. Philos. Trans. A Math. Phys. Eng. Sci. 380, 20210022 (2022).
Johansson, T. et al. Decadal vegetation changes in a northern peatland, greenhouse gas fluxes and net radiative forcing. Glob. Chang. Biol. 12, 2352–2369 (2006).
Singleton, C. M. et al. Methanotrophy across a natural permafrost thaw environment. ISME J. 12, 2544–2558 (2018).
Trubl, G. et al. Soil viruses are underexplored players in ecosystem carbon processing. mSystems 3, e00076–18 (2018).
Chang, K.-Y. et al. Methane production pathway regulated proximally by substrate availability and distally by temperature in a high‐latitude mire complex. J. Geophys. Res. Biogeosci. 124, 3057–3074 (2019).
Chang, K.-Y. et al. Large carbon cycle sensitivities to climate across a permafrost thaw gradient in subarctic Sweden. Cryosph. Discuss. 1, 53 (2018).
Grant, R. F., Humphreys, E. R. & Lafleur, P. M. Ecosystem CO2 and CH4 exchange in a mixed tundra and a fen within a hydrologically diverse Arctic landscape: 1. Modeling versus measurements. J. Geophys. Res. Biogeosci. 120, 1366–1387 (2015).
Marschmann, G. L. et al. Predictions of rhizosphere microbiome dynamics with a genome-informed and trait-based energy budget model. Nat. Microbiol. https://doi.org/10.1038/s41564-023-01582-w(2024).
Grant, R. F. & Roulet, N. T. Methane efflux from boreal wetlands: theory and testing of the ecosystem model Ecosys with chamber and tower flux measurements. Global Biogeochem. Cycles 16, 2–1 (2002).
Steven, B., Pollard, W. H., Greer, C. W. & Whyte, L. G. Microbial diversity and activity through a permafrost/ground ice core profile from the Canadian high Arctic. Environ. Microbiol. 10, 3388–3403 (2008).
Fink, J. W., Held, N. A. & Manhart, M. Microbial population dynamics decouple growth response from environmental nutrient concentration. Proc. Natl. Acad. Sci. USA 120, e2207295120 (2023).
Fofana, A. et al. Mapping substrate use across a permafrost thaw gradient. Soil Biol. Biochem. 175, 108809 (2022).
Shipley, B., Vile, D. & Garnier, E. From plant traits to plant communities: a statistical mechanistic approach to biodiversity. Science 314, 812–814 (2006).
Garnier, E. et al. Plant functional markers capture ecosystem properties during secondary succession. Ecology 85, 2630–2637 (2004).
Fierer, N., Barberán, A. & Laughlin, D. C. Seeing the forest for the genes: using metagenomics to infer the aggregated traits of microbial communities. Front. Microbiol. 5, 614 (2014).
McCalley, C. K. et al. Methane dynamics regulated by microbial community response to permafrost thaw. Nature 514, 478–481 (2014).
Hodgkins, S. B. et al. Changes in peat chemistry associated with permafrost thaw increase greenhouse gas production. Proc. Natl. Acad. Sci. 111, 5819–5824 (2014).
Bouskill, N. J., Riley, W. J., Zhu, Q., Mekonnen, Z. A. & Grant, R. F. Alaskan carbon-climate feedbacks will be weaker than inferred from short-term experiments. Nat. Commun. 11, 5798 (2020).
Wieder, W. R. et al. Explicitly representing soil microbial processes in Earth system models. Global Biogeochem. Cycles 29, 1782–1800 (2015).
Martiny, J. B. H., Jones, S. E., Lennon, J. T. & Martiny, A. C. Microbiomes in light of traits: a phylogenetic perspective. Science 350, aac9323 (2015).
Blankinship, J. C. et al. Improving understanding of soil organic matter dynamics by triangulating theories, measurements, and models. Biogeochemistry 140, 1–13 (2018).
Bäckstrand, K. et al. Annual carbon gas budget for a subarctic peatland, Northern Sweden. Biogeosciences 7, 95–108 (2010).
Holmes, M. E. et al. Carbon accumulation, flux, and fate in stordalen mire, a permafrost peatland in transition. Global Biogeochem. Cycles https://doi.org/10.1029/2021gb007113(2022).
Bolduc, B. et al. The IsoGenie database: an interdisciplinary data management solution for ecosystems biology and environmental research. PeerJ 8, e9467 (2020).
Hodgkins, S. B. et al. Soil incubations reproduce field methane dynamics in a subarctic wetland. Biogeochemistry 126, 241–249 (2015).
Hough, M. et al. Coupling plant litter quantity to a novel metric for litter quality explains C storage changes in a thawing permafrost peatland. Glob. Chang. Biol. 28, 950–968 (2022).
Grant, R. F. et al. Mathematical modelling of arctic polygonal tundra with ecosys: 1. Microtopography determines how active layer depths respond to changes in temperature and precipitation. J. Geophys. Res. Biogeosci. 122, 3161–3173 (2017).
Grant, R. F. Simulation of methanogenesis in the mathematical model ecosys. Soil Biol. Biochem. 30, 883–896 (1998).
Grant, R. F. Simulation of methanotrophy in the mathematical model ecosys. Soil Biol. Biochem. 31, 287–297 (1999).
Grant, R. F., Humphreys, E. R., Lafleur, P. M. & Dimitrov, D. D. Ecological controls on net ecosystem productivity of a mesic arctic tundra under current and future climates. J. Geophys. Res. https://doi.org/10.7939/R3X921R5S (2011).
Mekonnen, Z. A., Riley, W. J., Randerson, J. T., Grant, R. F. & Rogers, B. M. Expansion of high-latitude deciduous forests driven by interactions between climate warming and fire. Nat. Plants 5, 952–958 (2019).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Moran, M. A. et al. Sizing up metatranscriptomics. ISME J. 7, 237–243 (2013).
Reed, D. C., Algar, C. K., Huber, J. A. & Dick, G. J. Gene-centric approach to integrating environmental genomics and biogeochemical models. Proc. Natl. Acad. Sci. USA 111, 1879–1884 (2014).
Calabrese, S., Chakrawal, A., Manzoni, S. & Van Cappellen, P. Energetic scaling in microbial growth. Proc. Natl. Acad. Sci. USA 118, e2107668118 (2021).
Lynch, M. & Marinov, G. K. The bioenergetic costs of a gene. Proc. Natl. Acad. Sci. USA 112, 15690–15695 (2015).
de Groot, D. H., van Boxtel, C., Planqué, R., Bruggeman, F. J. & Teusink, B. The number of active metabolic pathways is bounded by the number of cellular constraints at maximal metabolic rates. PLoS Comput. Biol. 15, e1006858 (2019).
Tang, J. & Riley, W. J. Competitor and substrate sizes and diffusion together define enzymatic depolymerization and microbial substrate uptake rates. Soil Biol. Biochem. 139, 107624 (2019).
Piton, G. et al. Using proxies of microbial community‐weighted means traits to explain the cascading effect of management intensity, soil and plant traits on ecosystem resilience in mountain grasslands. J. Ecol. 108, 876–893 (2020).
Morris, M. D. Factorial sampling plans for preliminary computational experiments. Technometrics 33, 161–174 (1991).
Grant, R. A review of the Canadian ecosystem model—ecosys. Modeling Carbon and Nitrogen Dynamics for Soil Management. https://doi.org/10.1201/9781420032635.ch6 (2001).
Kayser, A., Weber, J., Hecht, V. & Rinas, U. Metabolic flux analysis of Escherichia coli in glucose-limited continuous culture. I. Growth-rate-dependent metabolic efficiency at steady state. Microbiology 151, 693–706 (2005).
Neubauer, P., Häggström, L. & Enfors, S. O. Influence of substrate oscillations on acetate formation and growth yield in Escherichia coli glucose limited fed-batch cultivations. Biotechnol. Bioeng. 47, 139–146 (1995).
Acknowledgements
We thank the EMERGE Biology Integration Institute Coordinators (members listed in Supplementary Information) for project guidance and management. This research is a contribution of the EMERGE Biology Integration Institute, funded by the National Science Foundation, Biology Integration Institutes Program, Award # 2022070 (V.I.R., R.K.V., S.R.S., M.B.S., E.L.B., and the EMERGE Coordinators). Additional support for individual contributors included the following. Z.L. was additionally supported by Lawrence Livermore National Laboratory under the auspices of the U.S. Department of Energy under contract DE-AC52-07NA27344. W.J.R. was supported by the Belowground Biogeochemistry Scientific Focus Area and U.K. was supported by the Watershed Function Science Area, both funded by the US Department of Energy, Office of Science, Office of Biological and Environmental Research under contract no. DE-AC02-05CH11231. G.L.M. was supported by the LLNL “Microbes Persist” Soil Microbiome Scientific Focus Area SCW1632 and an associated KBase award SCW1746. N.J.B. was supported by the US Department of Energy, Office of Science (BER), Early Career Research Program (#FP00005182). B.J.W. was supported by an Australian Research Council Future Fellowship (#FT210100521). J.T. was supported by the Laboratory Directed Research and Development Program of Lawrence Berkeley National Laboratory. We thank the Swedish Polar Research Secretariat and SITES for the support of the work done at the Abisko Scientific Research Station. SITES is supported by the Swedish Research Council’s grant 4.3-2021-00164. This research used resources of the National Energy Research Scientific Computing Center (NERSC) which is a US Department of Energy Office of Science user facility. This research used the Lawrencium computational cluster resource provided by the IT Division at the Lawrence Berkeley National Laboratory (Supported by the Director, Office of Science, Office of Basic Energy Sciences, of the US Department of Energy under Contract no. DE-AC02-05CH11231).
Author information
Authors and Affiliations
Consortia
Contributions
Z.L., W.J.R., and E.L.B. conceptualized this study. Z.L., W.J.R., G.L.M., E.L.B., J.T., U.K., I.A.S., Q.W., K.C., R.F.G., E.K., P.M.C., K.C.W., and B.J.W. contributed to the methodology. R.F.G. developed the ecosys model. Z.L., W.J.R., G.L.M., J.T., U.K., Q.W., and E.L.B. contributed to the investigation. Z.L., W.J.R., N.J.B., and G.L.M. contributed to the visualization. W.J.R., S.R.S., E.L.B., R.K.V., M.B.S, K.C.W, and the EMERGE Biology Integration Institute Coordinators contributed to the funding acquisition. W.J.R., E.L.B., and the EMERGE Biology Integration Institute Coordinators contributed to the project administration. W.J.R. supervised this study. Z.L., W.J.R., J.T., N.J.B., G.L.M., Q.W., and E.L.B. contributed to the writing of the original draft. All co-authors have reviewed and edited this manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Z., Riley, W.J., Marschmann, G.L. et al. A framework for integrating genomics, microbial traits, and ecosystem biogeochemistry. Nat Commun 16, 2186 (2025). https://doi.org/10.1038/s41467-025-57386-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57386-5
