
Abstract
Atomically dispersed Ru-Cu dual-atom catalysts (DACs) with asymmetric coordination are critical for sustainable ammonia production via electrochemical nitrate reduction (NO3RR), but their rational synthesis remains challenging. Here, we report a pulsed discharge strategy that injects a microsecond pulse current into ruthenium (Ru) and copper (Cu) precursors supported by nitrogen-doped graphene aerogels (NGA). The atomically dispersed Ru and Cu dual atoms anchor onto nanopore defects of NGA (RuCu DAs/NGA) through explosive decomposition of the metal salt nanocrystals. The catalyst achieves 95.7% Faraday efficiency and 3.1 mg h−1 cm−2 NH3 yield at −0.4 V vs. RHE. In situ studies reveal an asymmetric RuN2-CuN3 active-site dynamic evolution during NO3RR. Density functional theory calculations demonstrate that asymmetric RuN2CuN3/C structure synergistically optimizes intermediate adsorption and reduces energy barriers of key steps. The pulsed discharge enables ultrafast synthesis of various DACs (e.g., PtCu, AgCu, PdCu, FeCu, CoCu, NiCu) with tailored coordination environments, offering a general-purpose strategy for the precise preparation of atomically dispersed dual-atom catalysts, which are traditionally challenging to synthesize.
Similar content being viewed by others
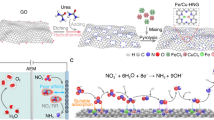


Introduction
The escalating concentration of nitrate (NO3−) in surface and groundwater has increasingly raised environmental and ecological concerns1,2,3. Addressing NO3− contamination by converting it into high value-added products, such as ammonia (NH3), presents a promising solution4,5. Ammonia is a widely utilized industrial chemical essential for human development, serving as a key component in fertilizers and various industrial processes. Traditionally, NH3 synthesis has relied primarily on the Haber-Bosch process, which operates under high temperatures and pressures, leading to substantial energy consumption and significant greenhouse gas emissions. In contrast, producing NH3 via the electrochemical reduction of NO3−, powered by renewable electricity, offers significant potential for economic and environmental sustainability6,7,8. Despite these advantages, the process from NO3− to NH3 involves multiple complex intermediate conversions and an eight-electron transfer process, posing considerable challenges to the activity and high Faradaic efficiency (FE) of newly developed electrocatalysts. Overcoming these hurdles is crucial for advancing this promising method of NH3 production and realizing its full potential for both economic and environmental benefits.
In recent years, atomically dispersed catalysts have garnered attention due to their highest atomic utilization and exceptional performance compared to conventional metal nanoparticles9,10,11,12,13. Advanced atomically dispersed catalysts have been developed for NO3− reduction reaction (NO3RR)14,15,16,17, yet challenges persist in achieving high activity and selectivity with single metal sites. For example, single copper (Cu) sites demonstrate high activity and Faradaic efficiency in alkaline media but require higher overpotentials and lead to nitrite (NO2−) accumulation, which adversely affects NH3 yield18,19,20,21. Various strategies, including surface modification, doping, and altering coordination structures, have been proposed to enhance catalyst performance22,23,24,25. Surface modification can introduce new active sites or improve the accessibility of existing ones, thereby increasing overall catalytic activity. Doping involves the integration of foreign atoms into the catalyst matrix, which can modify electronic properties and enhance adsorption capabilities. Altering coordination structures, such as creating asymmetric coordination environments, also play a critical role in improving catalytic performance. For instance, modifying the coordination environment of metal sites can optimize the binding strength of intermediates, reduce the energy barriers for key reactions, and ultimately lead to higher efficiency and selectivity in the NO3RR process. Collectively, these strategies aim to fine-tune the properties of catalysts at the atomic level to achieve desired performance characteristics. Concurrently, ruthenium (Ru) has demonstrated significant activity in NH3 production, particularly in facilitating intermediate adsorption and NO2− conversion26,27,28,29. Research further indicates that combining Cu with other metals can modulate the local electronic structure, thereby improving overall catalyst performance30,31,32. The concept of dual-atom catalysts (DACs), which consist of two paired metal atoms, has emerged as a promising approach to overcoming these challenges33,34,35,36. DACs leverage the synergistic effects between the metal pairs to reduce the energy barriers of complex reactions37,38. In this context, designing an atomically dispersed bimetallic catalyst with both Cu and Ru can potentially enhance intermediate adsorption and lower energy barriers, thereby improving overall catalytic efficiency.
In this study, we introduce a nitrogen-doped graphene aerogel supported Ru-Cu dual-atom catalyst (RuCu DAs/NGA), synthesized rapidly via a pulsed discharge method. This method introduces substantial energy into the metal salt precursors in microseconds, leading to the explosive decomposition of the nanocrystals and the formation of atomically dispersed RuCu dual sites with an asymmetric RuN2-CuN3 coordination structure. Extensive characterization confirms this unique coordination, and the RuCu DAs/NGA catalyst demonstrates optimized electrochemical performance in NO3RR, achieving a Faraday efficiency of 95.7% and an ammonia yield rate of 3.1 mg h–1 cm–2 at –0.4 V vs. RHE. Real-time monitoring of the RuCu active sites and reaction intermediates using in situ spectroscopic techniques, along with density functional theory (DFT) calculations, reveal that the asymmetric structure optimizes intermediates adsorption and lowers the energy barrier of key reaction steps. Furthermore, the versatility of our pulsed discharge method is highlighted by its successful application in synthesizing other M-Cu (M = Ag, Pt, Pd, Fe, Co, Ni) dual-atom catalysts on NGA, suggesting a general strategy for the precise preparation of DACs.
Results
Pulsed discharge synthesis of RuCu DAs/NGA
As a demonstration, we tailored RuCu dual atoms on NGA substrates by pulsed discharge strategy (Fig. 1a). At first, nitrogen-doped graphene hydrogel (NGH) was prepared by the hydrothermal assembly approach, and then soaked in the aqueous solutions of copper chloride (CuCl2) and ruthenium chloride (RuCl3) for 6 h. NGA (Supplementary Figs. 1, 2) supported RuCl3 and CuCl2 (RuCl3-CuCl2/NGA) could be obtained through a quick freeze-drying method. The RuCl3-CuCl2/NGA was packed into a copper discharge tube and compressed by copper plugs. The two ends of the copper tube were connected to the two electrodes of the high-power pulsed discharge system through copper strips (Supplementary Figs. 3, 4). Once the capacitor was fully charged, the air switch of the discharge system would be triggered to close quickly. As a result of the huge pulse current, RuCl3-CuCl2/NGA itself generated an instant thermal shockwave (the temperature beyond 3000 K on single layer graphene) and local plasma on single-layer graphene (Supplementary Figs. 5–8). Furthermore, applying a high-intensity current pulse engenders a potent electromagnetic field, thereby giving rise to several transient regions of elevated temperature on graphene39,40. Metal salt structures explosively break down rapidly owing to huge energy input, even directly into gaseous *Ru, *Cu, and *Cl ions from solid nanocrystals (Fig. 1b, Supplementary Figs. 9, 10, Supplementary Movie 1). The repeated pulsed discharge would cause the metal nanoclusters gathered on the graphene to repeatedly vaporize, prompting the metal atoms to rearrange in space to form a diatomic structure, which is similar to the joule heating method41. These gaseous metal atoms are anchored by N atoms on the graphene and form bimetallic atom pairs under the action of the sharp pulsed electromagnetic field. The micropore distribution of NGA and graphene aerogels (GAs) by N2 adsorption-desorption tests are shown in Fig. 1c, with holes with diameters ranging from 0.4 to 0.8 nm being the main pore defects on N-doped graphene. The inset (Fig. 1c) is a schematic diagram of N-doped graphene with different micropore types, the N atoms provide a large number of sites for the fixing of atomically dispersed melt atoms. These sub-nanopores on two-dimensional graphene are the critical space-confined strategy in our design for anchoring bimetallic atom pairs. The apparent activity of the catalyst could be improved due to its high specific surface area and microporous structure, which is conducive to mass transfer. Figure 1d shows the typical current waveform during pulsed discharge. The voltage amplitude was set at 8 kV, and the value approached 0 after several oscillating attenuations, lasting a total duration of about 600 μs. The first current peak lagged behind the first voltage peak by ~25 μs, a result of the combined action of capacitors and inductors in the discharge system. The main peak parameters of the current waveform are listed in Supplementary Table 1 during the pulsed discharge process. The current presented a typical underdamped waveform in this resistance-inductance-capacitance (RLC) circuit, indicating that the resistance in the circuit did not change significantly during the pulsed discharge. The energy barriers of metal salts would blast and decompose when the transient energy input, as illustrated in Fig. 1e. Because the current and the effect of Joule heat are almost synchronized41,42, the temperature rise time from 0 to peak on the metal salt nanocrystals is about tens of microseconds. When metal salt nanocrystals were inputted such a large amount of energy in a short period, they would decompose explosively and the metal ions anchored on NGA during the pulsed discharge (Supplementary Note). The magnetic pinching effect generated by the dynamic electromagnetic field inhibits the radial expansion of the formed ions, maintaining the relatively high-density plasma containing Ru and Cu ions on the NGA. Meanwhile, the air within the porous NGA structure would give rise to localized corona discharge plasmas comprising *O and *N ions.

a A schematic illustration of the preparation strategy by the pulsed discharge. b A schematic diagram of metal atoms loaded on the porous NGA by the pulsed discharge. c The micropore distribution of NGA and GAs (graphene aerogels), inset is a schematic diagram of N-doped graphene with different micropore types. d The total current curve in the circuit during discharge. e A schematic plot showing the brief formation process of RuCu DAs/NGA changes with energy input. f A research tendency of single atom catalysts to increase the variety of metal elements on the support and form asymmetric coordination structures. Source data for Fig. 1 are provided as a Source Data file.
During the cooling process, some nearby metal atoms together inevitably coalesce to form metal nanoclusters (Supplementary Fig. 11). Multiple repetitions of pulsed discharge treatment are necessary, as they induce repeated vaporization and dispersion of the metal atoms. The mixed Ru and Cu atoms could form atomic pairs on the defects of NGA caused by the trapping effect driven by thermal dynamics. Furthermore, the repeated pulse discharge is conducive to the full mixing of Ru and Cu metal atoms forming more evenly distributed Ru-Cu atom pairs on NGA after spatial transfer and re-arrangement. Following multiple discharge events, these conditions facilitate the formation of robust bonds between the metal atom pairs and the NGA carrier. Due to the current peak value being only kept for tens of microseconds and then quickly dropping to 0, the distinctive NGA-supported atomic dispersed metals structure could be retained at high cooling rates and stably exist (Supplementary Figs. 12, 13). Our experiments have demonstrated that RuCu DAs/NGA can be fully obtained after six cycles of pulsed discharges.
A significant development trend in single-atom catalysts is the incorporation of multiple metal elements on the support to form asymmetric coordination structures (Fig. 1f). Utilizing the rapid thermal shockwave and corona plasma effects of pulsed discharge, atomically dispersed catalysts of multiple metals can be rapidly synthesized. Additionally, the microsecond-scale heating and cooling characteristics on single-layer graphene create favorable conditions for the formation and stable existence of asymmetric coordination structures between the metal and support atoms. Compared to other synthesis methods for atomically dispersed catalysts, our pulsed discharge synthesis technique (Supplementary Fig. 14) stands out due to its exceptionally high instantaneous temperatures and extremely short duration, on the order of hundreds of microseconds.
After six pulsed discharge treatments, the still intact RuCu DAs/NGA sample was recovered from the discharge tube. Figure 2a displays the profile scanning electron microscope (SEM) image of RuCu DAs/NGA, the 3D porous structures (tens of microns) are similar to that of the initial NGA. The transmission electron microscopy (TEM) image still exhibits the features of curly porous (tens of nanometers) graphene (Supplementary Fig. 15). Figure 2b shows the high-angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) image of RuCu DAs/NGA, no nanoparticles could be found on graphene. The corresponding energy dispersive spectrum mapping results are illustrated in Fig. 2c, where the elements C (red), N (blue), Cu (yellow), and Ru (orange-red) are uniformly distributed (Supplementary Figs. 16–18). The high-resolution HAADF-STEM image reveals a significant number of evenly distributed bright spots on the graphene, as shown in Fig. 2d and Supplementary Figs. 15g, 19. These bright spots are thought to be atomically dispersed Ru and Cu atoms because the atomic numbers of metals are much higher than that of C, N, and other nonmetallic elements. Figure 2e is a partial enlargement image of Fig. 2d, due to the atomic number of Ru being much larger than that of Cu, the brightness of Ru atoms in the STEM dark field image is significantly higher than that of Cu atoms. Meanwhile, a large proportion of Ru-Cu metal atoms form pairs. Figure 2f depicts the corresponding two 3D intensity profiles in Fig. 2e, which exhibits the obvious intensity difference between Ru and Cu atoms, as well as the significant difference with C, N, and other atoms in NGA.

a SEM image of RuCu DAs/NGA. b HAADF-STEM image of RuCu DAs/NGA. c EDS mapping images of RuCu DAs/NGA, C (red), N (green), Cu (yellow), and Ru (orange red). d High magnification HAADF-STEM image (dark field). e The local magnified image of RuCu DAs/NGA. f The corresponding 3D intensity profiles for e. g The EELS map results of Ru-Cu pair in RuCu DAs/NGA, with the HAADF-STEM image on the top left, the spectrum corresponding to the box area on the top right, the Cu map at the bottom left, and the overlay image on the bottom right. Low pass filtering and principal component analysis were combined to denoise the raw EELS data. The highlighted energy loss range was used to produce the Cu-L map. h The distance frequency between adjacent Cu and Ru atoms, and the frequency of single sites and Ru-Cu dual sites. i A schematic plot of Ru-Cu pair atoms with different types anchored on the N-doped graphene. Source data for Fig. 2 are provided as a Source Data file.
To more accurately determine the bimetallic structures, we conduct electron energy loss spectroscopy (EELS) analysis to obtain more detailed information on the elemental composition and chemical environment. After being denoised by the combination of low pass filtering and principal component analysis, the EELS spectrum of the darker atom distinctly revealed the characteristic Cu-L edge feature (Fig. 2g). However, it is difficult to analyze the EELS spectrum for the brighter atom, because the characteristic signal of Ru-M edge was overwhelmed by the signals of C-K edge and N-K edge, preventing us from obtaining effective EELS data for the Ru atom43. In the atomic-resolution spectrum image (SI) HAADF of Ru-Cu atom pairs, a clear contrast between the two atoms is evident. The Cu EELS map extracted from the highlighted Cu-L energy loss range perfectly matches the Cu atom in the SI-HAADF image. Given that no other metal elements are present on NGA besides Ru and Cu, any metal atom observed with a brighter contrast than Cu in the atomic-resolution SI-HAADF can only be attributed to Ru. Repeated EELS tests (Supplementary Figs. 20, 21) confirmed the good reproducibility of this result. Furthermore, the frequency histogram of Ru and Cu adjacent atomic distance distribution and the frequency distribution histogram of single and double sites statistically in the HAADF-STEM images are shown in Fig. 2h. The average distance between Ru and Cu atoms is about 0.25 nm, and the two easily interact at this distance. The proportion of Ru-Cu dual sites is about 65.9%, indicating that the RuCu DAs/NGA sample prepared by pulsed discharge is worthy of the name. Notably, lots of N atoms provide rich sites for anchoring metal atoms. The contents of Ru and Cu are 0.39 at% (3.1 wt%) and 0.52 at% (2.6 wt%), respectively (Supplementary Fig. 15), which is consistent with the results (Ru 3.3 wt%, Cu 2.9 wt%) of inductively coupled plasma optical emission spectrometry (ICP-OES) test.
Cu SAs-DAs/NGA and Ru SAs-DAs/NGA (Supplementary Figs. 22, 23) were prepared using the same pulsed discharge parameters and metal salt sources as used for RuCu DAs/NGA. A portion of Cu and Ru atoms exist as atomic pairs (Cu pairs: 43.8%; Ru pairs: 40.5%) on Cu SAs-DAs/NGA and Ru SAs-DAs/NGA respectively. A tendency for Ru and Cu to agglomerate could be observed when the voltage was changed to 9 kV (Supplementary Fig. 24). This indicates that higher energy input causes the Ru-Cu atomic pairs to deviate from their original equilibrium positions, resulting in the formation of alloy nanocluster structures on N-doped graphene. Besides, the samples with different Cu/Ru ratios (2:1 and 1:2) were prepared by pulsed discharge at 8 kV. The HAADF-STEM images (Supplementary Fig. 25) of Ru1Cu2 Clu/NGA and Ru2Cu1 Clu/NGA reveal that changing the Cu/Ru ratio led to the agglomeration of atoms as well. These results indicate that the synthesis of uniformly distributed Ru-Cu diatomic pairs on NGA is suitable at current conditions.
Figure 2i presents a schematic plot of Ru-Cu dual atoms with different types anchored on the N-doped graphene, this is the main reason for the different lengths between Ru and Cu atoms. Furthermore, the varying angles formed between the RuCu and the NGA planes can also influence the interatomic distances within the Ru-Cu atom pairs (Supplementary Fig. 26). The intensity ratio of D ( ~ 1346 cm–1) to G (~1594 cm–1) for RuCu DAs/NGA is slightly higher than that for NGA in the Raman test (Supplementary Fig. 27), demonstrating that more defects were formed in graphene owing to Ru-Cu atoms dopant during pulsed discharge. The compression tests suggest that the doping of Ru and Cu atoms did not enhance the compressive strength of the NGA support, and the pulsed discharge process may have caused some damage to the NGA support (Supplementary Fig. 28). The X-ray diffraction (XRD) pattern (Supplementary Fig. 29) displays only one distinct wide peak at ~25°, attributed to the stacked graphene layers. No peaks of Ru or Cu crystals appear in the RuCu DAs/NGA. The results of XRD and Raman show that the NGA structure in RuCu DAs/NGA cannot be changed after pulsed discharge.
Analysis of the atomic bond structure
The atomic bond structure of RuCu DAs/NGA was investigated by X-ray photoelectron spectroscopic (XPS) and XAFS. The XPS spectra of RuCu DAs/NGA are shown in Supplementary Fig. 30, a distinct N 1 s peak was identified, which contains a C-N bond and a weak C-Ru/Cu bond. The Cu 2p3/2 spectrum has two peaks at 931.5 and 933.4 eV, respectively, assigned to Cu+ and Cu2+44,45. Additionally, the weak signals of Ru 3p were detected as well. The XAFS results of RuCu DAs/NGA and references are presented in Fig. 3 and Supplementary Fig. 31. Figure 3a shows the Cu K-edge X-ray absorption near-edge spectra (XANES) of RuCu DAs/NGA and references (Cu, CuO, Cu SAs/NGA, and Cu SAs-DAs/NGA). The absorption edge of RuCu DAs/NGA is closer to that of CuO than that of Cu foil, indicating that the oxidation state of Cu in RuCu DAs/NGA is closer to CuO. The k3-weighted Fourier transform (FT) from Cu K-edge extended X-ray absorption fine structure (EXAFS) spectra (Fig. 3b) present the peaks of RuCu DAs/NGA and Cu SAs/NGA are located at ~1.45 Å, which attributed to the Cu-N bond in first shell scattering46,47,48. Moreover, the secondary peak at 2.30 Å of RuCu DAs/NGA is close to the first shell ___location of the Cu foil (2.24 Å), which implies that the existence of the metallic diatomic coordination structure in RuCu DAs/NGA. Figure 3c displays the Cu K-edge EXAFS fitting result of RuCu DAs/NGA in the R space. The fitting and experimental results have a high degree of matching in different spaces (R space, k space, and q space, Supplementary Fig. 32). Correspondingly, the structural parameters were extracted from the Cu K-edge EXAFS fitting results (Supplementary Table 2). The coordination number of Cu was estimated to be 2.9 Cu-N (first peak) and 0.8 Cu-Ru (second peak) in the first shell according to the fitting results, with bond lengths of 1.93 Å and 2.59 Å, respectively. The Ru K-edge XANES of RuCu DAs/NGA and references (Ru, RuO2, Ru SAs/NGA, and Ru SAs-DAs/NGA) are shown in Fig. 3d. The absorption edge of RuCu DAs/NGA is located between RuO2 and Ru. The k3-weighted FT from Ru K-edge EXAFS spectra (Fig. 3e) exhibit the strong peaks of RuCu DAs/NGA and Ru SAs/NGA are located at about 1.50 Å, which correspond to the Cu-N bonds in the first shell. Similarly, the secondary peak at 2.36 Å of RuCu DAs/NGA is close to the peak position of the Ru foil (2.34 Å), indicating the presence of the metallic bond in RuCu DAs/NGA as well. Likewise, a good fitting result in R space is shown in Fig. 3f. Based on the fitting results, the coordination numbers of Ru-N and Ru-Cu are 2.2 and 0.8 in the first shell respectively. The bond lengths of Ru-N and Ru-Cu are 2.07 Å and 2.57 Å, respectively. Ultimately, a proposed asymmetric coordination structure (RuN2-CuN3/NGA) is depicted in Fig. 3g. According to the first derivative of the Cu absorption edge of RuCu DAs/NGA and references, the valence states of Cu and Ru in RuCu DAs/NGA were estimated to be 1.32 and 3.0, as shown in Fig. 3h. Based on the EELS results (Supplementary Fig. 33) and comparison with standard Cu spectra of different valence states, the valence state of Cu atoms paired with Ru is at least comparable to that of unpaired Cu atoms in RuCu DAs/NGA. Although limited by a low signal-to-noise ratio and a small sample size, this result cannot be fully consistent with that of the XAFS studies. However, it can still support the conclusion regarding the valence state analysis of Cu derived from the spectroscopic characterization to some extent. The Cu and Ru K-edge wavelet transform (WT) EXAFS results of RuCu DAs/NGA and references were utilized to distinguish backscattered atoms, as shown in Fig. 3i, j. The maximum intensity position of Cu for RuCu DAs/NGA is ~6.7 Å, which is closer to that of CuO (~6.2 Å) than that of Cu foil (~8.0 Å). Similarly, the maximum intensity position of Ru for RuCu DAs/NGA (~6.0 Å) is close to that of RuO2. The difference in intensity between the RuCu DAs/NGA and the references originates from the combined contribution of Ru-N, Cu-N, and Ru-Cu.

a The Cu K-edge XANES spectra of RuCu DAs/NGA and the references (Cu foil, Cu SAs/NGA and CuO). b Cu K-edge FT k3-weighted EXAFS spectra of RuCu DAs/NGA and references. c The Cu K-edge EXAFS fitting result of RuCu DAs/NGA in the R space. d The Ru K-edge XANES spectra of RuCu DAs/NGA and the references (Ru foil, Ru SAs/NGA, and RuO2). e Ru K-edge FT k3-weighted EXAFS spectra of RuCu DAs/NGA and references. f The Ru K-edge EXAFS fitting result of RuCu DAs/NGA in the R space. g The proposed atomic structure model of RuCu DAs/NGA (yellow, Ru; green, Cu; cyan, N; gray, C). h The valence states of Cu and Ru in RuCu DAs/NGA and references based on the first-order derivative of XANES spectra. i The WT-EXAFS results of Cu in RuCu DAs/NGA and references. j The WT-EXAFS results of Ru in RuCu DAs/NGA and references. Source data for Fig. 3 are provided as a Source Data file.
Based on the analysis of XAFS results for Cu SAs-DAs/NGA and Ru SAs-DAs/NGA, in addition to the single-atom Cu or Ru structures, a certain amount of Cu2 or Ru2 structures also exist in both samples (Supplementary Figs. 34, 35, Supplementary Table 3). This observation is consistent with the results obtained from the HAADF-STEM analysis. For the cluster samples (RuCu DAs/NGA(9 kV), Ru1Cu2 Clu/NGA, Ru2Cu1 Clu/NGA), the metal atoms (Cu and Ru) form strong coordination structures with the N atoms on the NGA support (Supplementary Figs. 36–41). The coordination number between the metal atoms themselves is relatively low, indicating that although these cluster catalysts are not as well-dispersed on the support as dual-atom catalysts, a substantial number of metal atoms are still exposed.
Generality of synthesis and structural analysis of CuM DAs/NGA (M = Pt, Ag, Pd)
Since the instantaneous temperature generated by the pulsed discharge is super high compared to the thermal decomposition temperature of ordinary metal salts, this method can rapidly prepare the atomical level dispersion for most metals. In addition to the Cu-Ru diatomic structure, the combinations of Cu and several other metals were designed to form asymmetric coordination structures of Cu-M/NC on NGAs in this research. The combination of Cu and other metals atomically dispersed dual catalysts on NGA provides different potential applications for different electrocatalytic scenarios. Here, the general pulsed discharge synthesis method was easily extended to the preparation of other metals dual atoms supported by NGA, such as PtCu DAs/NGA, AgCu DAs/NGA, PdCu DAs/NGA, etc. Figure 4a shows a TEM image of PtCu DAs/NGA, which exhibits 3D pleated graphene characteristics and is not supported with metal nanoparticles or clusters. After washing with water and freeze-drying, PtCu DAs/NGA is the broken lamellar graphene aerogel (inset Fig. 4a). The EDS mapping images of PtCu DAs/NGA are displayed in Fig. 4b, Pt and Cu elements are uniformly distributed on the N-doped graphene sheets. The contents of Pt and Cu are 0.5 at% (7.2 wt%) and 0.55 at% (2.6 wt%), respectively (Supplementary Fig. 42), which is consistent with the results (Pt 7.5 wt%, Cu 3.0 wt%) of ICP-OES test. Figure 4c presents a HAADF-STEM high magnification image of PtCu DAs/NGA, with the red oval dotted line surrounding the Pt-Cu bimetallic pair sites.

a TEM image of PtCu DAs/NGA. b EDS mapping images of PtCu DAs/NGA, C (red), N (green), Pt (yellow), and Cu (cyan). c High magnification HAADF-STEM image of PtCu DAs/NGA. d The Pt L3-edge XANES spectra of PtCu DAs/NGA and the references (Pt foil and PtO2). e Pt L3-edge FT k3-weighted EXAFS spectra with the fitting in R space of PtCu DAs/NGA and references. f TEM image of Ag-Cu DAs/NGA. g EDS mapping images of AgCu DAs/NGA, C (red), N (green), Ag (cyan), and Cu (yellow). h High magnification HAADF-STEM image of AgCu DAs/NGA. i The Ag K-edge XANES spectra of AgCu DAs/NGA and the references (Ag foil and Ag2O). j Ag K-edge FT k3-weighted EXAFS spectra with the fitting in R space of AgCu DAs/NGA and references. k TEM image of PdCu DAs/NGA. l EDS mapping images of PdCu DAs/NGA, C (blue), N (red), Pd (pink) and Cu (yellow). m High magnification HAADF-STEM image of PdCu DAs/NGA. n The Pd K-edge XANES spectra of PdCu DAs/NGA and the references (Pd foil and PdO). o Pd K-edge FT k3-weighted EXAFS spectra with the fitting in R space of PdCu DAs/NGA and references. p The oxidation states of metals in PtCu DAs/NGA, AgCu DAs/NGA, and PdCu DAs/NGA respectively. q The frequency of single sites and Cu-M (Pt, Ag, and Pd) dual sites. The optimized structures and differential charge densities of r PtCu DAs/NGA, s AgCu DAs/NGA, and t PdCu DAs/NGA (atomic model colors: dark cyan, Pt; orange, Cu; incanus, Ag; purple, Pd; blue, N; gray, C; white, H). Source data for Fig. 4 are provided as a Source Data file.
Further, the coordination structure of PtCu DAs/NGA was analyzed through XAFS testing. The Pt L3-edge XANES results of PtCu DAs/NGA and references (Pt foil and PtO2) are shown in Fig. 4d, the valence state of Pt in PtCu DAs/NGA is between 0 and +4 based on the intensity of white lines. The valence state of Cu in PtCu DAs/NGA was calculated to be 1.36 (Supplementary Fig. 43). The k3-weighted FT from Pt L3-edge EXAFS spectrum (Fig. 4e) exhibits the peak of PtCu DAs/NGA is located at ~1.63 Å, which corresponds to the Pt-N bond in the first shell. Besides, the second peak at 2.30 Å for PtCu DAs/NGA, is comparable to the first shell of Pt foil (2.60 Å), with proposing the metal-metal bond. The EXAFS fitting result in R space for PtCu DAs/NGA is inserted in Fig. 4e, and it can be seen that the experimental and fitting curves are in agreement. Equally, the characterizations and structures of AgCu DAs/NGA and PdCu DAs/NGA are shown in Fig. 4f–o and Supplementary Figs. 44–47. Supplementary Table 4 provides the best-fitting structural parameters, suggesting the local coordination structures of PtN2CuN3, AgN2CuN3, and PdN2CuN3 in the three samples. According to the first derivative of the Cu (Ag, Pd) absorption edge (or the white-line peak area of Pt) for PtCu DAs/NGA, AgCu DAs/NGA, PdCu DAs/NGA, and their references, the oxidation states of Cu are estimated to be 1.36, 1.32 and 1.42 in PtCu DAs/NGA, AgCu DAs/NGA, and PdCu DAs/NGA respectively (Fig. 4p). The oxidation states of Pt, Ag, and Pd in three catalysts were calculated to be 1.73, 0.36 and 1.75 respectively. The percentage of Pt-Cu, Ag-Cu, and Pd-Cu dual sites are 64.9%, 61.6%, and 65.4% respectively, demonstrating that interacting metals dual sites dominate as compared to single sites in PtCu DAs/NGA, AgCu DAs/NGA, and PdCu DAs/NGA (Fig. 4q).
The optimized structures and differential charge densities of PtCu DAs/NGA, AgCu DAs/NGA, and PdCu DAs/NGA are shown in Fig. 4r–s. The bond lengths of Pt-Cu, Ag-Cu, and Pd-Cu pairs are estimated to be 2.22 Å, 2.32 Å, and 2.31 Å respectively. Furthermore, the differential charge densities of PtN2CuN3/C, AgN2CuN3/C, and PdN2CuN3/C were calculated to elucidate the electrical properties of the asymmetric Cu-M sites. The asymmetric deployment of the modulated MN2-CuN3 leads to a significantly polarized surface charge distribution. Electron enrichment (cyan) near the MN2 site and electron deficiency near CuN3 can be attributed to electron transfer from the CuN3 site to the PtN2, AgN2, and PdN2 sites, respectively (Supplementary Fig. 48). The three extended studies demonstrate the universal strategy to synthesize unsymmetrical atomic interface structures for the pulsed discharge method. This combination of Cu and noble metals could open more catalytic possibilities for atomically dispersed diatomic catalysts. The pulsed discharge method is indeed designed as a versatile strategy for preparing asymmetric atomic interface structures by leveraging the high-energy environment to atomize metal precursors and the strong affinity of the NGA scaffold to anchor and stabilize the metal atoms. Based on our investigations, the pulsed discharge method is not limited to RuCu, PtCu, PdCu systems but can also be extended to other transition metal-based systems, such as FeCu DAs/NGA, CoCu DAs/NGA, and NiCu DAs/NGA (Supplementary Figs. 49–51). The results, confirmed by HAADF-STEM analysis, demonstrate the successful formation of dual-atom structures. This demonstrates the versatility and universality of our approach.
Electrocatalysis and in situ study
The reaction of nitrate reduction to the ammonia was evaluated in 0.1 M KNO3 and 0.1 M KOH conditions, and linear sweep voltammetry (LSV) curves are shown in Fig. 5a. RuCu DAs/NGA showed the lowest onset potential and the fastest current density decrease in the three catalysts. The Faradaic efficiency and ammonia evolution rate were studied by chronoamperometry with different operated potentials (Supplementary Fig. 52). Figure 5b presents the FE of the NH3 product (FENH3) at different potentials (–0.1 V to –0.6 V vs. RHE). Impressively, the FENH3 of RuCu DAs/NGA reached 95.7 % at –0.4 V vs. RHE, the performance is very competitive compared to other catalysts reported recently (Supplementary Table 5). The partial current density of NH3 (JNH3) on RuCu DAs/NGA has the optimal performance among the three catalysts from –0.1 V to –0.6 V vs. RHE (Fig. 5c), suggesting the enhanced NO3RR performance through the tailored asymmetric RuN2-CuN3 coordination structure strategy. The JNH3 reached –21.3 mA cm–2 on RuCu DAs/NGA at –0.3 V vs. RHE. The NH3 yield rate results (Fig. 5d) exhibit that RuCu DAs/NGA is more active and selective than the references. The NH3 yield rate of RuCu DAs/NGA reached 3.1 mg h–1 cm–2 at –0.4 V vs. RHE, which still presents an optimal performance compared to similar catalysts in recent reports (Fig. 5f, Supplementary Table 5). Moreover, the yield rate of RuCu DAs/NGA reached 1.63 mg h–1 cm–2 and 4.38 mg h–1 cm–2 at –0.3 V and –0.5 V vs. RHE respectively, which is better than the references (Ru SAs/NGA, Ru SAs-DAs/NGA, Cu SAs/NGA, Cu SAs-DAs/NGA, RuCu Clu/NGA(9 kV), Ru1Cu2 Clu/NGA, Ru2Cu1 Clu/NGA) and superior to similar catalysts as well. The activity contribution of single-atom sites and dual-atom sites were evaluated under controlled reaction conditions during the NO3RR. The role of single atomic sites in the electrochemical process would be more significant at higher negative potentials (Supplementary Figs. 53, 54). But the contributions of the NH3 yield rate are lower than 10% for single sites compared to Ru-Cu dual sites at different applied potentials.

a LSV curves of RuCu DAs/NGA, Ru SAs-DAs/NGA, Cu SAs-DAs/NGA, the mixture of Ru SAs-DAs/NGA and Cu SAs-DAs/NGA, Ru SAs/NGA, Cu SAs/NGA, and NGA measured in 0.1 M KNO3 and 0.1 M KOH electrolyte. b The FEs of NH3 production at different applied potentials for RuCu DAs/NGA, Ru SAs-DAs/NGA and Cu SAs-DAs/NGA. c Partial NH3 current densities of three catalysts at different potentials. d Potential-dependent yield rate of NH3 generation over RuCu DAs/NGA. e Cycling tests of RuCu DAs/NGA for NO3RR at −0.4 V vs. RHE. f Comparison of NH3 yield rate and potentials for RuCu DAs/NGA and various catalysts recently reported (See the details in Supplementary Table 6) g Potential-dependent in situ ATR-SEIRAS spectra (1000–2000 cm−1) during the electrochemical NO3RR process. h Cu and i Ru K-edge XANES spectra of RuCu DAs/NGA at different applied potentials (OCP, –0.1 V, –0.2 V, –0.3 V, −0.4 V, and −0.5 V) during the NO3RR process. j The valence states of Cu and Ru in RuCu DAs/NGA and references based on the first-order derivative of XANES spectra. k Cu and l Ru K-edge k3-weighted FT-EXAFS at different potentials during the NO3RR process. All the potentials have not been iR-corrected. Source data for Fig. 5 are provided as a Source Data file.
The ECSA of each sample is measured and analyzed the catalytic activity data accordingly (Supplementary Fig. 55). After normalizing the current density to the electrochemical surface area, it is evident that RuCu DAs/NGA still exhibits the highest activity, followed by Ru SAs-DAs/NGA, with Cu SAs-DAs/NGA showing the lowest activity (Supplementary Fig. 56). This further confirms that the synergistic effect between Ru and Cu in RuCu DAs/NGA can enhance the electrocatalytic NO3RR process. The 1H NMR spectra of 14NH4+ and 15NH4+ (Supplementary Fig. 57) display characteristic triplet and doublet peaks at chemical shifts of 6.8–7.0, respectively, while the peak at approximately 6.3 corresponds to the internal standard C4H4O4. This result directly confirms that the NH4+ in the product originates from NO3−, rather than from environmental impurities. To ascertain the endurance of RuCu DAs/NGA under the conditions of the NO3RR, a series of 10 continuous electrolysis cycles were executed. Figure 5e illustrates that RuCu DAs/NGA exhibits sustained high Faradaic efficiency and NH3 yield rates across these cycles, substantiating its exceptional durability. The current density loss was at the negligible operated potential of –0.4 V vs. RHE during the 24 h continuous NH3 testing from NO3RR (Supplementary Fig. 58). The characteristic features of Ru-Cu dual atoms are still prominent, and statistical analysis indicates that the proportion of Ru-Cu dual atoms remains at approximately 60.6%, which is near that of the original sample (Supplementary Fig. 59). Based on the XAFS results (Supplementary Figs. 60, 61) of RuCu DAs/NGA after the stability test, it can be observed that there is little alteration in the electronic structures of Ru and Cu. The EXAFS fitting results reveal that post-stability testing, the coordination structure of the catalyst remains as RuN2-CuN3/C. This indicates that after prolonged NO3RR tests for NH3 production, the RuCu DAs/NGA catalyst retains its structural stability.
We tested the NO3RR performance at different nitrate concentrations (0, 1 mM, 2 mM, 10 mM, 100 mM, Supplementary Fig. 62). The current density showed a clear dependence on nitrate concentration. This indicates that the RuCu DAs/NGA exhibits higher activity at higher nitrate concentrations, suggesting that RuCu DAs/NGA is a promising catalyst for electrocatalytic nitrate reduction reactions. In a 0.1 M KOH solution without nitrate, there was only a slight change in current density at different potentials31,49,50. This indicates that RuCu DAs/NGA do not significantly facilitate HER under these conditions. Further tests were conducted to assess the Faradaic efficiency and production rate of NH3 for RuCu DAs/NGA at varying nitrate concentrations. It was evident that both the Faradaic efficiency and production rate of NH3 were significantly dependent on nitrate concentration. RuCu DAs/NGA exhibited higher Faradaic efficiency and the production rate for NH3 at higher nitrate concentrations, further eliminating the possibility of HER occurring under these conditions. This underscores the effectiveness of RuCu DAs/NGA as a catalyst for NO3RR, demonstrating enhanced performance with increased nitrate availability. This is attributed to the propensity of Ru-Cu atomic pairs to favor NO3RR, thereby suppressing HER. The decrease in activity of RuCu Clu/NGA(9 kV), Ru1Cu2 Clu/NGA and Ru2Cu1 Clu/NGA could be attributed to the agglomeration of metal atoms (Supplementary Fig. 63), which reduces the number of active sites, thereby resulting in weaker NO3RR performance than the 8kV-treated sample (RuCu DAs/NGA). This suggests that increasing the catalyst loading, which leads to the aggregation of metal atoms into nanoclusters, does not directly enhance the activity of catalysts. Instead, exposing more active sites can enhance the catalytic performance. Furthermore, an increase in Ru content improves the activity of catalysts more significantly than an increase in Cu content, indicating that ruthenium atoms play a more critical role in the electrocatalytic NO3RR.
To further elucidate the intermediates involved in NO3RR on various electrocatalysts, in situ ATR-SEIRAS measurements were conducted (Supplementary Fig. 64). Figure 5g presents the results for the RuCu DAs/NGA at open circuit potential (OCP), –0.1 V, –0.2 V, –0.3 V, –0.4 V, –0.5 V, and –0.6 V vs. RHE. A progressively increasing intensity of the NO3− vibrational band at ~1245 cm–1 was observed from OCP to –0.6 V, indicating the ongoing consumption of NO3− during electrolysis. A broad peak centered around 3400 cm−1 (Supplementary Fig. 65), identified as the *NH2 species, was consistently observed51,52. Additionally, the peak intensities of hydrogenation intermediates (*NH2 at ~1168 cm–1, *NH2OH at ~1115 cm–1), NH4+ (NH3 at ~1460 cm–1), and deoxidation intermediates (*NO2 at ~1630 cm–1) progressively increased53,54. These findings suggest that the RuCu DAs/NGA catalyst is highly effective in activating NO3−, subsequently facilitating the formation of substantial amounts of hydrogenation intermediates that are eventually converted into NH4+ (NH3). The detection of *NH2OH and *NH2 species indicates the co-occurrence of both indirect and direct reduction pathways during the NO3RR process on RuCu DAs/NGA. Additionally, a significant enhancement of the H-O-H stretching vibrations at approximately 1660 cm–1 and 3484 cm–1 was observed, indicating the dissociation of H2O into OH– and H+ species, which are stabilized on the RuCu DAs/NGA catalyst.
To study the structure-activity relationship of RuCu DAs/NGA on the atomic level, in situ XAFS tests were carried out during the electrochemical catalytic NO3RR process. Figure 5h, i present the in situ Cu K-edge and Ru K-edge XANES results of RuCu DAs/NGA at OCP, –0.1 V, –0.2 V, –0.3 V, –0.4 V, and –0.5 V vs. RHE. The absorption edges of both Cu and Ru in RuCu DAs/NGA tend to gradually move towards lower energy, together with the reduced intensity of their white lines, which implies the decrease in Cu and Ru valence states with the lower applied potentials. According to the results (Supplementary Fig. 66) of the first derivative of the absorption edge for RuCu DAs/NGA, the specific valence states of Cu were decreased from 1.27 to 0.73 when the operated potentials were reduced. Similarly, the valence states of Ru decreased from 2.87 to 2.62 continuously (Fig. 5j). In fact, Cu and Ru disclose similar trends in oxidation states owing to NO3− being adsorbed on the active site of the catalyst in the electrolyte, resulting in the interaction on the unpaired outermost d orbitals of Cu and Ru with the N 2p orbitals of NO3−. The oxidation states of both Cu and Ru would continue to decrease when lower reaction potentials were applied. The Cu-Ru dual active sites would interact with the adsorbed NO3− more easily to form Cu/Ru-N/O bonds at lower potentials, which leads to electron redistribution among Cu, Ru, N, and O atoms. The Cu K-edge FT-EXAFS spectra (Fig. 5k) of RuCu DAs/NGA show that the Cu-N peak shifts from 1.50 Å (OCP) to 1.41 Å (–0.3 V), which was considered as the compressing of Cu-N bonds. The Ru K-edge FT-EXAFS spectra (Fig. 5l) of RuCu DAs/NGA exhibit that the Ru-N peak moves from 1.56 Å (OCP) to 1.53 Å (–0.3 V), with the shrinking of Cu-N bonds as well. In addition, the Ru-Cu bonds also tend to shift to the left during the NO3RR process. Because the metal atoms are likely not in the same plane as graphene, it is possible to have both metal and metal-N bonds clamped. Besides, the Ru-Cu bonds also tend to move to the left slightly during the NO3RR process. The above bond lengths analysis is consistent with the specific fitting results (Supplementary Figs. 67, 68, and Supplementary Table 6). Due to the metal atoms are likely not on the same plane as graphene, it is possible to be pinched for both the Ru-Cu bonds and the Cu/Ru-N bonds. In brief, the high performance of RuCu DAs/NGA in the electrocatalytic NO3RR is due to the joint effect of the RuN2-CuN3 coordination moieties. During the NO3RR process, the local coordination structure (coordination number and bond length, etc.) near the Ru-Cu sites changed slightly, and a new stable structure would be formed. The detection of the reaction intermediates absorbed by the Ru-Cu active sites might be the main reason.
Theoretical study of NO3RR
To understand the fundamental mechanism of the NH3 product reduced from NO3− for RuCu DAs/NGA, the DFT method was used to investigate the whole process of the NO3RR on the asymmetrical RuN2CuN3/C moieties (Supplementary data 1). The differential charge densities (Fig. 6a) of RuN4/C, CuN4/C, and RuN2CuN3/C were calculated to elucidate the electrical properties of Ru and Cu sites and to study the synergistic interactions of the two asymmetric coordination metal atoms as well. The RuN4 site makes it easier to obtain electrons (electron-rich, green area) than the CuN4 site, showing a stronger reducing ability. Therefore, it can be predicted that RuN4 would have a stronger adsorption capacity for intermediates than CuN4. Through the asymmetric deployment of the modulated RuN2-CuN3, the surface charge distribution appears significantly polarized. Electron enrichment near RuN2 and electron deficiency near CuN3 are possibly attributed to the electron transfer from the Cu site to the Ru site. To reveal the underlying reason for the interactions between RuN2-CuN3 sites and the reactive species, the projected densities of state (PDOS) of CuN4/C, RuN4/C, and RuN2CuN3/C concentrating on d orbitals of Ru and Cu were simulated (Supplementary Fig. 69 and Fig. 6b). According to the d band center theory55, the d orbitals of Cu in CuN4 are further away from the Fermi level compared to Ru in RuN4, the adsorption of intermediates on CuN4 is weaker than that on RuN4. The modulated RuN2-CuN3 has a strong synergistic effect, the 4 d orbital of Ru is closer to the Fermi level, and it has a stronger adsorption effect for the reaction intermediates, which would be higher catalytic activity on RuN2-CuN3. Furthermore, another CuN3 site with a slightly weak adsorption ability may promote the desorption of intermediates, which is conducive to enhancing the reaction rate.

a The differential charge densities of RuN4/C, CuN4/C, and RuN2CuN3/C. b The projected density of states of RuN2CuN3/C. c Electrochemical NO3RR pathways and relative free energy on RuN4/C, CuN4/C, and RuN2CuN3/C at U = 0 vs. RHE. d The energy barriers of the key steps on RuN4/C, CuN4/C, and RuN2CuN3/C. Atomic model color: red, Ru; yellow, Cu; green, O; cyan, N; blue, C; light pink, H. Source data for Fig. 6 are provided as a Source Data file.
All NO3RR pathways and relative free energy on RuN4/C, CuN4/C, and RuN2CuN3/C at U = 0 vs. RHE are illustrated in Fig. 6c and Supplementary Figs. 70–72. NO3− is adsorbed first and discharged on the metal sites, forming *NO3, then transformed into *NO3H. After that, *NO3H converts into *NO2 with the departure of the OH−. Subsequently, as protons in H2O continue to be added to the intermediate and take O atoms away (forming OH−), *NH3 is gradually formed. Finally, *NH3 is desorbed to yield NH3 leaving the metal sites. The evolution process of intermediates from NO3− to NH3 product on the RuN2-CuN3/C structure is: *NO3 → *HNO3 → *NO2 → NOOH → NO → *NOH → *NHOH → *NH2OH → *NH2 → *NH3. The first six intermediates (*NO3, *HNO3, *NO2, *NOOH, *NO, and *NOH) are co-adsorbed by Ru and Cu together, and this optimized configuration reduces the energy barrier of the reaction. After that, the intermediates (*NHOH and *NH2OH) are adsorbed by Ru single atom. The N atom of *NH2 is absorbed by Ru and Cu atoms. Finally, *NH3 is absorbed by Ru single atom. These steps imply that the Ru atom plays a more important role in the RuN2-CuN3/C structure during electrocatalytic NO3RR. Ru and Cu atoms cooperate to reduce *NO3 to *NO at the early stages of the reaction. It is mainly Ru atom that reduces *NO to *NH3 at the later stages of the reaction. The synergistic effect between Ru and Cu atoms can reduce the reaction free energy and optimize the adsorption and desorption of intermediates, thereby facilitating the NO3RR process.
The whole reaction path and the relative free energies of each step are shown in Supplementary Table 7. The NO3RR reaction steps can be divided into two parts, namely the reactant or product absorption/desorption process (non-electron gain/loss reaction) and the electron gain/loss reaction steps. From NO3− to *NO3H, since the charge transfer number is 0, the process can be considered as the adsorption of the product by the metal sites. According to the energy position of *HNO3, the adsorption of *HNO3 by RuN2CuN3 is the strongest, followed by RuN4 and CuN4 is the worst. A large amount of reactants would accumulate in the RuN2CuN3 active site, and the increase of reactant concentration is beneficial to the reaction. From *HNO3 to *NH3, the intermediates in the elementary reaction would get an e− at each step. For the elementary reaction of gaining or losing electrons, the smaller the energy barrier is, the easier the reaction is. For RuN2CuN3, RuN4, and CuN4, the respective maximum energy barrier is 0.252 eV (*NHOH → *NH2OH), 0.5 eV (*NO → *NOH), and 0.994 eV (*NO → *NOH) in their electron gain/loss steps (Fig. 6d). Note that the energy barrier of *NO → *NOH on RuN2CuN3 is 0.242 eV. Compared with Cu, Ru reduces the energy barrier of the key step of *NO → *NOH, while the energy barrier of this step is further decreased in the Ru-Cu dual atoms structure. Thus, *NO → *NOH is no longer the key step in determining the reaction rate on RuN2CuN3, while *NHOH → *NH2OH is the critical step. The most critical intermediate is *NO in NO3RR, and reducing the energy barrier of its hydrogenation step is the key to the action of these catalysts. The asymmetric RuN2CuN3 structure exhibits the best effect in the three catalysts. Based on the relative free energy positions of *NO and *NOH, the adsorption of the Cu site to intermediates is too weak, and the Ru site could enhance the adsorption of these intermediates, but the adsorption of *NO is too strong and may not be conducive to the reaction. In the RuN2CuN3 structure, Ru maintains the strong adsorption of *NOH, but weakens the adsorption of *NO, so the energy barrier of this step is reduced. From the differential charge density of the key intermediates (*NO, Supplementary Fig. 73), *NO gets more charge in the Ru-containing system. The main difference between Ru-Cu and single Ru systems is the adsorption configuration, NO is adsorbed horizontally, and Ru-Cu acts together in the RuN2CuN3 system.
For the last step *NH3 → * + NH3 is the desorption process of the product, the adsorption of the intermediate at the Cu site is weak, and the product is easier to desorption. The desorption ability of Ru containing system is slightly larger, but it is also less than 1 eV, which can be carried out at room temperature (20 ~ 25 °C). The last step is not critical unless desorption is large reach to poison the metal sites, where the barrier has not yet reached the conditions for poison. Under actual reaction conditions, the product NH3 is highly soluble in water, and the desorption energy of less than 1 eV will not poison the metal site. Additionally, the system containing Ru has high selectivity for the NH3 path, almost only along the path of NH3 production. While CuN4/C may produce NO2− and NO (low energy barrier), NH3 selectivity is not high enough. This is consistent with the NO3RR test results.
Discussion
In conclusion, we successfully designed bimetallic atoms utilizing nanopore defects on NGA and rapidly synthesized atomically dispersed RuCu diatomic catalysts (RuCu DAs/NGA) through a pulsed discharge method. Our correlation analysis suggests an asymmetric coordination structure of RuN2CuN3 on NGA for RuCu DAs/NGA. Notably, this pulsed discharge technique can be adapted to efficiently prepare a variety of diatomic catalysts, including PtCu, AgCu, and PdCu DAs/NGA. The RuCu DAs/NGA catalyst exhibits remarkable electrocatalytic activity and selectivity for ammonia production via NO3RR. Both experimental investigations and theoretical calculations demonstrate that the tailored asymmetric RuN2CuN3/NGA structure fosters strong cooperativity, optimizing and regulating each elementary reaction effectively. The implications of this research extend beyond academic interest; the scalability and practical applications of our RuCu DAs/NGA catalyst are significant for sustainable NH3 production through NO3− reduction. The ability to rapidly prepare bimetallic catalysts with such optimized structures not only enhances catalytic performance but also holds promise for industrial applications where efficient and sustainable processes. We envision that the pulsed discharge strategy can facilitate the development of advanced catalysts applicable to various energy conversion and catalytic scenarios, thereby contributing to the ongoing efforts in sustainable chemistry and environmental remediation.
Methods
Chemicals
Copper chloride (CuCl2, 99%, Alfa Aesar), ruthenium chloride (RuCl3, 99%, Alfa Aesar), palladium chloride (PdCl2, 99%, Alfa Aesar), hydrogen hexachloroplatinate (H2PtCl4•xH2O, 99.995%, Alfa Aesar), argentum nitricum (AgNO3, 98%, Alfa Aesar), ferric chloride (FeCl3, 99%, Alfa Aesar), cobalt chloride (CoCl2, 99%, Alfa Aesar), nickel chloride(NiCl2, 99%, Alfa Aesar), mammonia (analytical grade, Alfa Aesar), ammonium hydroxide (NH3•H2O, 25-28%, Alfa Aesar), KOH (Sigma Aldrich), Nafion D-521 dispersion (5 wt%, Alfa Aesar).
Preparation of RuCu DAs/NGA and other samples
The single-layer graphene oxide (GO) was prepared by a modified Hummers method. GO (30 mg) and NH3•H2O (200 mg) were fully dispersed in deionized water (15 mL). The evenly dispersed mixture was poured into a small hydrothermal reactor (20 mL), heated to 180 °C, and kept there for 6 h. After cooling to room temperature (20–25 °C), the NGH was formed. After cleaning NGA several times with deionized water, it was immersed in an aqueous solution of copper and ruthenium chloride (Cu and Ru 3 at%) for 3 h. NGA-supported CuCl2 and RuCl3 nanocrystals (CuCl2-RuCl3/NGA) were obtained after rapid freezing with liquid nitrogen and freeze-drying. The prepared CuCl2-RuCl3/NGA was filled into a copper discharge tube using two threaded copper plugs to hold the ends (Supplementary Movie 2). Then the discharge tube containing CuCl2-RuCl3/NGA was connected in series to the circuit of the high-power pulsed discharge system. The charging voltage was set to 8 kV, and the air switch was automatically triggered after the capacitor finished charging. The pulse current would pass through the copper discharge tube and CuCl2-RuCl3/NGA, and CuCl2-RuCl3/NGA would be completely transformed into RuCu DAs/NGA after six repeated pulsed discharge processing. The preparation process of PtCu DAs/NGA, AgCu DAs/NGA, PdCu DAs/NGA, FeCu DAs/NGA, CoCu DAs/NGA, and NiCu DAs/NGA was similar to that of RuCu DAs/NGA. The pulsed discharge characteristics are presented in Supplementary Note. The NGH was soaked in an ethanol solution of copper acetyl acetone (Cu(acac)2, or Ru(acac)3) for 2 h, washed with distilled water, and freeze-dried to obtain nitrogen-doped graphene supported copper acetyl acetone (Cu(acac)2/NGA, Ru(acac)3/NGA). It is placed in a tube furnace and pyrolyzed in a nitrogen atmosphere for 2 h at a heating temperature of 800 °C to obtain the Cu SAs/NGA and Ru SAs/NGA. Besides, Cu SAs-DAs/NGA, Ru SAs-DAs/NGA, Ru1Cu2 Clu/NGA, and Ru2Cu1 Clu/NGA were prepared by pulsed discharge at 8 kV, the metal salts were changed only compared with RuCu DAs/NGA. RuCu Clu/NGA(9 kV) was prepared by pulsed discharge at 9 kV.
Characterizations
The microstructure of synthesized materials was examined by field-emission SEM (JEOL JSM-7200F) and TEM (FEI Tecnai G2 F20). Atomic-resolution HAADF-STEM imaging was performed using an FEI Themis Z microscope operated at 200 kV. Raman spectra were recorded on a LabRAM HR Evolution system with 532 nm laser excitation. XRD patterns were collected on a Bruker D8 Advance diffractometer (Cu Kα radiation, 5°/min scan rate). Elemental composition analysis was conducted via ICP-OES (Agilent 730). Surface area measurements were derived from N2 adsorption-desorption isotherms using the Brunauer-Emmett-Teller (BET) method (BSD-660M analyzer). XPS data were acquired using an ESCALAB 250Xi system with Al Kα radiation. EELS mapping was implemented on a Nion HERMES-100 microscope (60 kV, 32 mrad convergence angle). Synchrotron-based XAFS measurements were performed at the Shanghai Synchrotron Radiation Facility in fluorescence mode using pelletized samples (13 mm diameter, 1 mm thickness).
Electrocatalytic evaluation
Nitrate reduction experiments employed a three-electrode configuration with a CHI 760E workstation at ambient conditions. The electrochemical cell consisted of: (1) catalyst-coated carbon paper (1 cm2 geometric area, 0.4 mg cm−2 loading) as the working electrode, (2) graphite counter electrode, and (3) Hg/HgO reference electrode, separated by a Nafion membrane. Catalyst ink was prepared by homogenizing 5 mg catalyst in 0.75 mL isopropanol, 0.24 mL H₂O, and 10 μL Nafion solution (5 wt%) through 4-h ultrasonication. Electrochemical testing utilized 0.1 M KNO₃ + 0.1 M KOH catholyte (Ar-purged for 15 min) with linear sweep voltammetry (5 mV s–¹ scan rate) and chronoamperometric measurements at 350 rpm stirring. Uncompensated cell resistance (1.9 ± 0.2 Ω) was determined via a multimeter. Product quantification employed UV-Vis spectroscopy: NH3 detection used Nessler’s reagent (420 nm absorbance), while nitrate analysis involved p-sulfanilamide-based colorimetry (540 nm). Reference electrode calibration followed standard protocols in H₂-saturated 0.1 M KNO₃. All potentials are reported versus the RHE without iR compensation.
Current densities were normalized to the electrode’s geometric surface area (1 cm²). The Hg/HgO reference electrode was calibrated in H2-saturated 0.1 M KNO₃ using a symmetric Pt electrode system. Chronoamperometric measurements were conducted at multiple potentials for 1-h intervals under continuous stirring (350 rpm), with simultaneous collection of catholyte and anolyte for post-reaction analysis. Nitrogenous species (NO₃⁻, NO₂⁻, NH₃) were quantified via standardized colorimetric protocols.
Ammonia detection: 0.05 mL electrolyte was diluted to 5 mL, treated with 0.1 mL Nessler’s reagent, and analyzed at 420 nm after 20-min incubation.
Nitrate determination: A reagent solution containing 10 g p-sulfanilamide (dissolved in 100 mL H₂O + 25 mL H₃PO₄) and 0.5 g N-(1-naphthyl)ethylenediamine dihydrochloride was prepared in 250 mL total volume. Samples (0.05 mL electrolyte + 0.5 mL H₃PO₄) were diluted to 10 mL, mixed with 0.2 mL reagent, and measured at 540 nm after 20-min stabilization.
Calibration curves for nitrite and ammonia were generated by correlating UV-Vis absorbance (540/420 nm) with standard solution concentrations. All reported potentials were referenced to the RHE using the equation:
Where the initial pH of 0.1 M KOH + 0.1 M KNO3 is about 13.5 ± 0.2.
The Faradaic efficiency of product i is calculated as follows:
Where, FEi: the Faradaic efficiency of product i, Qtotal: the total charge (C); Ni: moles of product i; ni: electron transfer number per molecule in the product i; F: Faraday constant (96485 C mol–1).
The NH3 yield is calculated as follows:
Where C (μg mL−1) is the measured NH3 concentration, V (mL) is the volume of NH3 cathodic reaction electrolyte, M is the relative molecular mass of products, m is the amount of catalyst supported, t (h) the reduction time and S (1 cm2) is the geometric area of the carbon paper in the electrolyte.
ECSA analysis
The ECSA was determined by cyclic voltammetry in the non-Faradaic potential window (0.1–0.3 V vs. RHE) using 0.1 M KNO₃/KOH electrolyte. Double-layer capacitance (Cdl = j/v) was derived from the linear slope of capacitive current density versus scan rate (20–100 mV s⁻¹). The roughness factor (Rf) was calculated as Rf = Cdl/Cdlref, where Cdlref = 0.04 mF cm⁻² (reference value for smooth electrodes in alkaline media). ECSA was then defined as ECSA = RfS, with S = 1.0 cm² (geometric area)25,56,57. This metric quantifies active site density and surface roughness, enabling comparative analysis of RuCu DAs/NGA, Ru SAs-DAs/NGA, and Cu SAs-DAs/NGA catalysts.
Isotopic tracer studies
To confirm the nitrate-to-ammonia conversion pathway, 15N-labeled K15NO3 was employed as the nitrogen source. Post-electrolysis, 0.5 mL catholyte was acidified to pH ~2 with 0.5 M H2SO4, followed by the addition of 0.1 mL 0.04 wt% C4H4O4 for NMR sample stabilization. 1H NMR spectra (700 MHz) of 14NH4+ and 15NH4+ products were acquired, with isotopic shifts verifying the exclusive nitrate origin of ammonia8,28,31,58,59,60.
XAFS data processing
EXAFS data processing was performed using the IFEFFIT package with ATHENA and ARTEMIS modules. Raw spectra underwent post-edge background subtraction and edge-step normalization to extract χ(k) oscillations. Fourier transformation of χ(k) data (Hanning window, Δk = 1.0 Å⁻¹) generated radial distribution functions, isolating contributions from distinct coordination shells. Quantitative fitting employed the EXAFS equation:
Where S02 is the amplitude reduction factor, Fj(k) is the effective curved-wave backscattering amplitude, Nj is coordination number, Rj is bond distance, λ is the mean free path in Å, σj2 is the Debye-Waller factor, ϕj(k) is the phase shift. Theoretical parameters (Fj(k), λ(k), ϕj(k)) were calculated via FEFF8.2 simulations.
For Cu foil reference samples, coordination numbers were fixed to crystallographic values to determine S02, which was subsequently constrained during the fitting of atomically dispersed catalysts. Bond distances (Rj), disorder parameters (σj2), and energy shift (ΔE0) were optimized freely. This protocol ensures physically meaningful structural parameter extraction while minimizing overfitting.
In situ ATR-SEIRAS test
In situ ATR-SEIRAS measurements were conducted using a Thermo-Fisher Nicolet iS50 spectrometer. The electrochemical experiments employed a three-electrode cell setup with an electrolyte comprising 0.1 M KNO3 and 0.1 M KOH. The spectral resolution was set at 4 cm–1, and spectra recorded at open circuit potential (OCP) served as reference points. Measurements were taken across a potential range from 0 V to –0.6 V vs. RHE. To enhance signal sensitivity, a monocrystalline silicon substrate with a gold-plated surface was utilized. The scanning range spanned from 4000 cm–1 to 400 cm–1.
In situ XAFS characterization
Operando synchrotron XAFS measurements were performed using a custom-designed spectroelectrochemical cell. The three-electrode configuration comprised: Working electrode (catalyst-coated carbon paper, 1.5 cm diameter active area); Counter electrode (graphite rod); Reference electrode (Hg/HgO). The polycarbonate cell incorporated a lateral copper current collector contacting the catalyst layer, with electrolyte (0.1 M KNO₃/KOH) introduced under quiescent conditions. A precision-machined cap maintained fixed 5 mm electrode spacing during potential application via a CHI 760E workstation. XAFS spectra were acquired in fluorescence mode across multiple electrode regions to verify catalyst homogeneity, with beam positioning controlled by motorized XYZ stages. This configuration enabled real-time monitoring of Ru/Cu coordination changes during nitrate reduction at designated potentials.
Theoretical calculation methods
Spin-polarized DFT simulations were implemented in the Vienna Ab-initio Simulation Package (VASP)60,61 to investigate the properties of the prepared materials with the projector-augmented wave (PAW) method62. The Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional63 and DFT-D3 dispersion corrections were employed. A plane-wave cutoff energy of 450 eV and Γ-centered k-mesh were applied. Catalytic models (RuN2CuN3, RuN4, and CuN4) were constructed on a graphene (001) slab with 20 Å vacuum spacing.
Structural relaxations proceeded until energy and force residuals reached 10⁻⁵ eV and 0.02 eV/Å, respectively. Adsorption energies (ΔEads) were computed as:
Where Etotal, Esubstrate, and Eadsorbate denote energies of the composite system, pristine substrate, and isolated adsorbate. Gibbs free energy (G) incorporated vibrational contributions:
With ZPE = zero-point energy and TΔS = entropic term (298 K).
AIMD simulations verified thermal stability in the NVT ensemble. Systems underwent: (1) Temperature ramping (0 → 298 K over 1.49 ps); (2) Equilibration (298 K, 10 ps, 1 fs timestep); (3) Trajectory analysis for structural integrity.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are available within the article and its Supplementary Information files. Source data are provided with this paper.
References
Suryanto, B. H. R. et al. Challenges and prospects in the catalysis of electroreduction of nitrogen to ammonia. Nat. Catal. 2, 290–296 (2019).
Van Langevelde, P. H., Katsounaros, I. & Koper, M. T. Electrocatalytic nitrate reduction for sustainable ammonia production. Joule 5, 290–294 (2021).
Sun, J. et al. A hybrid plasma electrocatalytic process for sustainable ammonia production. Energy Environ. Sci. 14, 865–872 (2021).
Han, S. et al. Ultralow overpotential nitrate reduction to ammonia via a three-step relay mechanism. Nat. Catal. 6, 402–414 (2023).
Foster, S. L. et al. Catalysts for nitrogen reduction to ammonia. Nat. Catal. 1, 490–500 (2018).
Christensen, C. H., Johannessen, T., Sørensen, R. Z. & Nørskov, J. K. Towards an ammonia-mediated hydrogen economy? Catal. Today 111, 140–144 (2006).
Chen, J. G. et al. Beyond fossil fuel-driven nitrogen transformations. Science 360, eaar6611 (2018).
Huang, Y. et al. Pulsed electroreduction of low-concentration nitrate to ammonia. Nat. Commun. 14, 7368 (2023).
Li, P., Jin, Z., Fang, Z. & Yu, G. A single-site iron catalyst with preoccupied active centers that achieves selective ammonia electrosynthesis from nitrate. Energy Environ. Sci. 14, 3522–3531 (2021).
Shang, H. et al. Engineering unsymmetrically coordinated Cu-S1N3 single atom sites with enhanced oxygen reduction activity. Nat. Commun. 11, 3049 (2020).
Gan, T. & Wang, D. Atomically dispersed materials: Ideal catalysts in atomic era. Nano Res. 17, 18–38 (2024).
Cheng, Y. et al. Design strategies towards transition metal single atom catalysts for the oxygen reduction reaction – A review. Nano Res. Energy 2, e9120082 (2023).
Pan, Y. et al. Structural regulation with atomic-level precision: from single-atomic site to diatomic and atomic interface catalysis. Matter 2, 78–110 (2020).
Li, P. et al. Pulsed nitrate-to-ammonia electroreduction facilitated by tandem catalysis of nitrite intermediates. J. Am. Chem. Soc. 145, 6471–6479 (2023). 11.
Xu, J. et al. Breaking local charge symmetry of iron single atoms for efficient electrocatalytic nitrate reduction to ammonia. Angew. Chem. Int. Ed. 62, e202308044 (2023).
Wu, Z.-Y. et al. Electrochemical ammonia synthesis via nitrate reduction on Fe single atom catalyst. Nat. Commun. 12, 2870 (2021).
Ke, Z. et al. Selective NOx− electroreduction to ammonia on isolated Ru sites. ACS Nano 17, 3483–3491 (2023).
Gao, Q. et al. Synthesis of core/shell nanocrystals with ordered intermetallic single-atom alloy layers for nitrate electroreduction to ammonia. Nat. Synth. 2, 624–634 (2023).
Xue, Y. et al. Electrocatalytic hydrogenation boosts reduction of nitrate to ammonia over single-atom Cu with Cu(I)-N3C1 sites. Environ. Sci. Technol. 56, 14797–14807 (2022).
Wang, Y. et al. Unveiling the activity origin of a copper-based electrocatalyst for selective nitrate reduction to ammonia. Angew. Chem. Int. Ed. 59, 5350–5354 (2020).
Liu, Y. et al. Efficient tandem electroreduction of nitrate into ammonia through coupling Cu single atoms with adjacent Co3O4. Nat. Commun. 15, 3619 (2024).
Shao, J. et al. Electrochemical synthesis of ammonia from nitric oxide using a copper–tin alloy catalyst. Nat. Energy 8, 1273–1283 (2023).
Daiyan, R. et al. Nitrate reduction to ammonium: from CuO defect engineering to waste NOx-to-NH3 economic feasibility. Energy Environ. Sci. 14, 3588–3598 (2021).
Liu, K. et al. The atomic interface effect of single atom catalysts for electrochemical hydrogen peroxide production. Nano Res. 16, 10724–10741 (2023).
Fan, K. et al. Active hydrogen boosts electrochemical nitrate reduction to ammonia. Nat. Commun. 13, 7958 (2022).
Tao, H. et al. Nitrogen fixation by Ru single-atom electrocatalytic reduction. Chem 5, 204–214 (2019).
Peng, O. et al. Swinging hydrogen evolution to nitrate reduction activity in molybdenum carbide by ruthenium doping. ACS Catal. 12, 15045–15055 (2022).
Chen, F. et al. Efficient conversion of low-concentration nitrate sources into ammonia on a Ru-dispersed Cu nanowire electrocatalyst. Nat. Nanotechnol. 17, 759–767 (2022).
Gao, W. et al. Alloying of Cu with Ru enabling the relay catalysis for reduction of nitrate to ammonia. Adv. Mater. 35, 2202952 (2023). 19.
Zhou, Y. et al. Optimizing intermediate adsorption over PdM (M=Fe, Co, Ni, Cu) bimetallene for boosted nitrate electroreduction to ammonia. Angew. Chem. Int. Ed. 62, e202319029 (2023). 24.
Zhang, S. et al. Fe/Cu diatomic catalysts for electrochemical nitrate reduction to ammonia. Nat. Commun. 14, 3634 (2023).
Qikun, H. et al. Ammonia electrosynthesis from nitrate using a ruthenium-copper cocatalyst system: A full concentration range study. J. Am. Chem. Soc. 146, 668–676 (2023).
Bai, J. et al. Simultaneous integration of Fe clusters and NiFe dual single atoms in nitrogen-doped carbon for oxygen reduction reaction. Nano Res. 17, 2291–2297 (2024).
Pei, J. et al. Integrating host design and tailored electronic effects of yolk-shell Zn-Mn diatomic sites for efficient CO2 electroreduction. Angew. Chem. Int. Ed. 63, e202316123 (2024).
Chen, C. et al. Dual-metal single-atomic catalyst: The challenge in synthesis, characterization, and mechanistic investigation for electrocatalysis. SmartMat 3, 533–564 (2022).
He, H. et al. Recent advances in electrocatalysts for efficient hydrogen evolution reaction. Rare Met. (2024). https://doi.org/10.1007/s12598-024-02649-1.
Zhang, W. et al. Emerging dual-atomic-site catalysts for efficient energy catalysis. Adv. Mater. 33, 2102576 (2021).
Zhao, T. et al. Homonuclear dual-atom catalysts embedded on N-doped graphene for highly efficient nitrate reduction to ammonia: from theoretical prediction to experimental validation. Appl. Catal. B Environ. 339, 123156 (2023).
Liu, K. et al. Ultra‐fast pulsed discharge preparation of coordinatively unsaturated asymmetric copper single‐atom catalysts for CO2 reduction. Adv. Funct. Mater. 34, 2312589 (2024).
Shao, T. et al. Atmospheric-pressure pulsed discharges andplasmas: mechanism, characteristics andapplications. High. Volt. 3, 14–20 (2018).
Yao, Y. et al. High temperature shockwave stabilized single atoms. Nat. Nanotechnol. 14, 851–857 (2019).
Yang, C. et al. Overcoming immiscibility toward bimetallic catalyst library. Sci. Adv. 6, eaaz6844 (2020).
Qi, H. et al. Highly selective and robust single-atom catalyst Ru1/NC for reductive amination of aldehydes/ketones. Nat. Commun. 12, 3295 (2021).
Jia, Y. et al. Electronic oxide-support strong interactions in the graphdiyne-supported cuprous oxide nanocluster catalyst. J. Am. Chem. Soc. 145, 1803–1810 (2023).
Zheng, T. et al. Copper-catalysed exclusive CO2 to pure EtOH conversion via single atom alloying. Nat. Nanotechnol. 16, 1386–1393 (2021).
Su, X. et al. Complementary Operando spectroscopy identification of in-situ generated metastable charge-asymmetry Cu2-CuN3 clusters for CO2 reduction to ethanol. Nat. Commun. 13, 1322 (2022).
Cai, Y. et al. Insights on forming N, O-coordinated Cu single-atom catalysts for electrochemical reduction CO2 to methane. Nat. Commun. 12, 586 (2021).
Yang, T. et al. Coordination tailoring of Cu single sites on C3N4 realizes selective CO2 hydrogenation at low temperature. Nat. Commun. 12, 6022 (2021).
Zhang, X. et al. Regulating intermediate adsorption and H2O dissociation on a diatomic catalyst to promote electrocatalytic nitrate reduction to ammonia. Energy Environ. Sci. 17, 6717–6727 (2024).
Wang, Y. et al. Enhanced nitrate-to-ammonia activity on copper-nickel alloys via tuning of intermediate adsorption. J. Am. Chem. Soc. 142, 5702–5708 (2020).
Xu, Y. et al. Sulphur-boosted active hydrogen on copper for enhanced electrocatalytic nitrate-to-ammonia selectivity. Angew. Chem. Int. Ed. 63, e202400289 (2024).
Li, X. et al. Sub-nm RuOx clusters on Pd metallene for synergistically enhanced nitrate electroreduction to ammonia. ACS Nano 17, 1081–1090 (2022).
Shilong, J. et al. Descriptors for the evaluation of electrocatalytic reactions: d-band theory and beyond. Adv. Funct. Mater. 32, 2107651 (2024).
Daniela, C. M. et al. Improved synthesis of graphene oxide. ACS Nano 4, 4806–4814 (2010).
Andersen, M. Revelations of the d band. Nat. Catal. 6, 460–461 (2023).
Yang, F. et al. Bismuthene for highly efficient carbon dioxide electroreduction reaction. Nat. Commun. 11, 1088 (2020).
Watzele, S. et al. Determination of electroactive surface area of Ni-, Co-, Fe-, and Ir-based oxide electrocatalysts. ACS Catal. 9, 9222–9230 (2019).
WenSheng, G. et al. Alloying of Cu with Ru enabling the relay catalysis for reduction of nitrate to ammonia. Adv. Mater. 35, 2202952 (2023).
Gao, Q. et al. Breaking adsorption-energy scaling limitations of electrocatalytic nitrate reduction on intermetallic CuPd nanocubes by machine-learned insights. Nat. Commun. 13, 2338 (2022).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No. 12372332 to X.G., 12002048 to X.G. and 22375019 to W.C.), the Beijing Natural Science Foundation (Grant No. 2212018 to X.G.), Beijing Institute of Technology Research Fund Program for Young Scholars (Grant No. 2022CX01011 to W.C.). This research benefited from resources and supports from the Electron Microscopy Center at the University of Chinese Academy of Sciences and the BL14W1 in the Shanghai Synchrotron Radiation Facility.
Author information
Authors and Affiliations
Contributions
X.G., W.C. and P.C. conceived the idea, designed the research and wrote the paper. K.L. carried out the sample synthesis, characterization and wrote the paper. Z.S., K.Y., and X. Z. performed NO3RR measurements. W.C. carried out the in situ synchrotron radiation XAFS measurements and data analysis. F.Z., X.L. and Q.Z. helped with the spherical aberration electron microscopy test and discussion. X.P. performed the EELS measurements and analysis. B.Z., Z.C., and Y.W. helped with the modification of the paper. All the authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Guandao Gao, Chao Wang, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, K., Sun, Z., Peng, X. et al. Tailoring asymmetric RuCu dual-atom electrocatalyst toward ammonia synthesis from nitrate. Nat Commun 16, 2167 (2025). https://doi.org/10.1038/s41467-025-57463-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57463-9
