
Abstract
Chromatin topology can impact gene regulation, but how evolutionary divergence in chromatin topology has shaped gene regulatory landscapes for distinctive human traits remains poorly understood. CTCF sites determine chromatin topology by forming domains and loops. Here, we show evolutionary divergence in CTCF-mediated chromatin topology at the ___domain and loop scales during primate evolution, elucidating distinct mechanisms for shaping regulatory landscapes. Human-specific divergent domains lead to a broad rewiring of transcriptional landscapes. Divergent CTCF loops concord with species-specific enhancer activity, influencing enhancer connectivity to target genes in a concordant yet constrained manner. Under this concordant mechanism, we establish the role of human-specific CTCF loops in shaping transcriptional isoform diversity, with functional implications for disease susceptibility. Furthermore, we validate the function of these human-specific CTCF loops using human forebrain organoids. This study advances our understanding of genetic evolution from the perspective of genome architecture.
Similar content being viewed by others


Introduction
Humans and our closest extant relatives, such as chimpanzees and other great apes, share strikingly similar coding gene repertoires, yet exhibit pronounced phenotypic differences1,2. The major genetic differences between humans and these primates reside in the non-coding regions of the genome3,4. It is postulated that alterations in gene regulation serve as a driving force for evolutionary innovation during speciation5. However, the molecular mechanism by which these genetic changes in non-coding regions dictate the symphony of gene regulatory programs, and in turn forge the unique evolutionary innovations in humans, is still largely enigmatic.
An increasingly important dimension to this enigma is that the spatial organization of the genome within the nucleus plays a central role in determining which genes are accessible for transcription and which remain dormant6. In the three-dimensional (3D) genome, regulatory landscapes are partitioned into preferentially self-insulated domains where enhancers communicate with promoters7. Changes in the 3D genome architecture can have cascading effects, rewiring gene regulatory networks and potentially resulting in various diseases8. Thus, a natural hypothesis is that evolutionary variation in 3D genome architectures may provide a compelling lens through which to decipher the emergence of unique human traits and functionalities.
The 3D genome organization depends on the interplay between the CCCTC-binding factor (CTCF), its DNA-binding sequences, and an extruding cohesin complex7,9,10. Phase separation, which forms dynamic, membraneless compartments that concentrate regulatory factors, may complement loop extrusion in organizing genome architecture11,12. Within this framework, CTCF acts as a polar blocking factor, hindering cohesin translocation along the chromatin by binding its DNA sites with a convergent orientation, thereby forming CTCF-mediated chromatin loops (CTCF loops)10,13. These interconnected CTCF loops structure chromatin into CTCF-mediated chromatin domains (CCDs), resembling the topologically associating domains (TADs) defined by Hi-C13,14. Endowed with insulating properties, these CTCF loops direct the targeted interactions between enhancers and their cognate promoters to compose precise transcriptional programs by preventing ectopic functional interactions across CTCF-defined insulated neighborhoods7,9. Remarkably, even subtle alterations, such as point mutations at a single CTCF site, can be sufficient to drastically alter the process of chromatin looping, thereby affecting the precise packaging of the underlying chromosomal segments into insulated domains15,16. This pivotal role of CTCF sites positions them as inherited structural codes within the 3D genome, configuring chromatin loops and CCDs. Notably, the evolutionary divergence of CTCF sites is a fundamental mechanism of genome evolution, as evident even in primate evolutionary trajectories17,18,19,20,21. Thus, CTCF-mediated divergence in chromatin topology, rooted in the genetic variation of CTCF sites, may provide a framework for understanding the partitioning of regulatory landscapes and how this can be instrumental in sculpting developmental phenotypes and fostering evolutionary innovation.
Recent evidence has demonstrated the impact of rearranged TADs, driven by lineage-specific genomic rearrangements, on species adaptation through the induction of prominent changes in gene expression patterns22,23. In the primate lineage, which exhibits high genomic synteny, TAD boundaries show a marked depletion of structural variations, reflecting evolutionary constraints that preserve their essential roles in chromatin organization and gene regulation24,25. This observation suggests that the regulatory landscape within the TAD may undergo changes contributing to primate evolution, possibly through mechanisms other than overt rearrangements of TADs. Supporting this hypothesis, Luo et al. recently identified human-specific TADs and chromatin loops that are notably enriched in enhancer-enhancer interactions26. These loops influence the regulation of genes critical for neuron development and plasticity, particularly in the subplate, a transient zone implicated in human neural circuit formation26. Furthermore, a recent study involving consecutive deletions of CTCF sites at the HoxD cluster in mouse gastruloids has brought to light that even modest perturbations of chromatin folding via CTCF sites can exert substantial influence on transcriptional patterns—both in terms of levels and timing—thereby shaping the developmental process27. Despite these insights, the significance of subtle changes resulting from CTCF-mediated divergence in chromatin topology within the TAD in shaping regulatory landscapes has been underappreciated, primarily due to limitations in the resolution of existing methods to precisely map CTCF-mediated chromatin topology.
In this study, we performed a comprehensive analysis of the evolutionary divergence in CTCF-mediated chromatin topology across humans, chimpanzees, gorillas, and macaques using chromatin interaction analysis by paired-end-tag sequencing (ChIA-PET)13 in different cell types. We identified human-specific CCD boundaries that reconfigure the transcriptional concordances, highlighting the potential impact of evolutionary divergence in CCDs on the genetic architecture of disease susceptibility. Furthermore, we revealed that evolutionarily divergent CTCF loops govern the connectivity of species-specific enhancers to their cognate genes in a concordant yet constrained manner, emphasizing the role of CTCF loop divergence in shaping species-specific gene expression patterns. In particular, we demonstrated the role of evolutionary divergence in CTCF loops in shaping transcriptional isoform diversity and validated the function of the human-specific CTCF loops using human forebrain organoids. These molecular insights contribute significantly to a broader understanding of genetic evolution.
Results
Mapping chromatin topological variation in humans and non-human primates
Our study aims to elucidate chromatin topological variation by comparing the 3D genomes of distinct primates, providing insights into their critical role in shaping distinctive human traits (Fig. 1a). To achieve this goal, we employed in situ ChIA-PET13 to map chromatin interactions in B-lymphoblastoid cells from humans, chimpanzees, gorillas, and macaques, focusing on interactions mediated by the protein factors CTCF and RNA polymerase II (RNAPII) (Fig. 1b). Direct comparison of CTCF-mediated chromatin interactions facilitates the identification of variations in chromatin topology resulting from divergent CTCF binding sequences between primates. In addition, the gene regulatory insights provided by RNA Pol II ChIA-PET mapping allow us to investigate alterations in enhancer-promoter interactions, which may be influenced by changes in chromatin topology (Fig. 1b). To broaden the scope of our study, we also characterized chromatin topological variation in primary neuronal cells from human and macaque fetal cortical tissue using CTCF and RNAPII in situ ChIA-PET (Figs. 1c and S1a–e). The ChIA-PET libraries derived from these two cell types across species showed high reproducibility, with an average Pearson’s correlation coefficient of 0.93 (Fig. S2a–c). After consolidating replicates, we obtained genome-wide CTCF- and RNAPII-mediated chromatin interactions of high quality across species (Supplementary Data 1), enabling a comprehensive analysis of chromatin topological variations across species (Fig. 1d, e).

a Overview of the study design. B-lymphoblastoid cells from humans, chimpanzees, gorillas, and macaques, as well as neuronal cells from humans and macaques (left), were used to detect CTCF- and RNAPII-mediated chromatin interactions (middle). The chromatin architectures of these species were compared to identify evolutionary divergence in chromatin topology (right). The million years ago (MYA) values are derived from established phylogenetic data to illustrate the evolutionary relationships between the species analyzed. Representative track views and chromatin contact maps illustrating CTCF- and RNAPII-mediated chromatin interactions in B-lymphoblastoid cells (b) and neuronal cells (c) from humans and non-human primates within identical syntenic genomic regions. Track views display the profiles of CTCF peaks and loops, as well as H3K27ac peaks and RNAPII loops for each respective species. Human reference genes and CTCF sites are shown at the top of the panel. The synteny track denotes the syntenic genomic regions between humans (blue) and each corresponding non-human primate (orange). In loop tracks, loop intensities are indicated by color gradients. The maximum intensity in each peak track is given on the left. Coordinates for syntenic genomic regions are provided. Genome-wide comparison of CTCF loops across species in B-lymphoblastoid cells (d) and neuronal cells (e). Pearson correlation coefficients (r) are provided.
Given that the immune systems of primates have been profoundly shaped by differing living habits, and the high-order cognitive functions of the brain are highly developed in humans28,29, our robust ChIA-PET datasets from these two representative cell types can facilitate the identification of divergence in chromatin topology and its association with disease susceptibility, such as autoimmune diseases and neurodevelopmental disorders.
Cross-species comparisons of CTCF-mediated chromatin contact domains
Our previous study demonstrated that CTCF-mediated chromatin interactions, through continuous intra-chromosomal connectivity, delineate chromatin contact domains (CCDs) similar to the TADs identified by Hi-C13. Based on the connectivity and contact frequency of CTCF loops, we identified an average of 1,199 CCDs across the genomes of the four species in B-lymphoblastoid cells (Fig. S3a, b). These CCDs manifested convergent CTCF sites at their boundaries with an average span of 1.8 Mb across species (Fig. S3c). Notably, the CCDs detected in B-lymphoblastoid and primary neuronal cells were largely similar within each species (Fig. S3d), consistent with our previous study indicating that CCDs are conserved between different cell types13. Given the insulating role of CCD boundaries in genome organization, we compared CCD boundaries between humans and non-human primates based on changes in insulation scores between human boundaries and their syntenic counterparts in non-human primates (Fig. S3e, see “Materials and methods”). Our analysis revealed that the human genome shares 71.2% of its CCD boundaries with chimpanzees, 66.8% with gorillas, and 55.5% with macaques (Fig. S3e, f), reflecting the relative evolutionary distances between humans and these non-human primates. This finding highlights that evolutionary divergence in CCD boundaries exists along the primate evolutionary trajectory, extending beyond the previously documented conservation of CCD boundaries30.
Through a three-way comparison with chimpanzees, gorillas, and macaques, we categorized human CCD boundaries into distinct categories. Initially, we classified human CCD boundaries into 1532 conserved across the primates and 1,040 that vary among the non-human primates, designated as non-human primate variable boundaries (Fig. S3g). Those variable boundaries were further subdivided into 112 human-specific boundaries, 390 ape-specific boundaries, and 538 miscellaneous boundaries with ambiguous variation patterns (Figs. 2a and S3g). Notably, we identified only 24 syntenic regions in humans that represent human-specific lost CCD boundaries compared to non-human primates. These syntenic regions, mainly in gene-poor regions, were excluded from downstream analyzes.

a Examples of CCD boundaries across species, showing insulation scores (IS), CTCF sites, peaks, and loops. Dashed lines indicate human CCD boundaries and their corresponding positions in non-human primates. b Heatmaps comparing the insulating function of variable boundaries (top) between humans and non-human primates, with conserved boundaries (bottom) as control. c Box plots of CTCF loop intensities across human-specific (HS), ape-specific (APE), miscellaneous (Misc), and conserved (Consv) boundaries. The box plot shows the median (central line), interquartile range (box), and whiskers extending to 1.5× the interquartile range. ****P < 0.0001; ns, not significant as determined by one-tailed Wilcoxon test. d Heritability enrichment for autism spectrum disorder susceptibility across variable and human-specific (HS) boundaries in 50 kb bins. The boundary bin is highlighted, with a red arrowhead indicating CTCF site orientation. Error bars show standard errors, and asterisks indicate significant enrichment after Bonferroni correction using LDSC (See Methods). e Heatmap showing unsupervised hierarchical clustering of normalized H3K27ac signals from 32 human primary tissues at variable boundaries in 50 kb bins. f H3K27ac intensity profiles at variable and conserved (Consv) boundaries with flanking regions, using data from three human brain regions. CTCF loop anchors (Random) serve as controls. Dashed lines denote the positions of boundaries or anchors. g Schematic outlining criteria for gene pairing to assess transcriptional concordance between human boundaries and their macaque counterparts (See Methods). h Smooth curve plots depicting transcriptional correlations between gene pairs as a function of genomic distance across expressions from human brain regions and their macaque counterparts. The gray shading indicates the 90% confidence intervals. P values were calculated using the one-tailed Wilcoxon test. i Scatter plots of transcriptional correlations for gene pairs separated by human-specific boundaries, using RNA-seq data from 34 human and 23 macaque medial frontal cortical tissues. Gray lines show linear regression of transcript levels for gene pairs, with shading indicating the 90% confidence intervals. Pearson correlation coefficients (r) and P values were calculated by two-sided t-test. The source data, n numbers, and exact P values for (c, d, i) are provided in the Source Data file.
As expected, we observed only sporadic CTCF loops crossing the non-human primate variable boundaries in humans, in contrast to their more prominent presence in non-human primates (Fig. 2b). Further analysis across multiple human cell lines confirmed the insulating function of these boundaries in humans, without notable cell type specificity (Fig. S3h). Notably, non-human primates exhibited a significantly higher intensity of CTCF loops crossing the syntenic counterparts of the human-specific boundaries than ape-specific and miscellaneous subcategories, as well as conserved categories (Fig. 2c), highlighting the evolutionary significance of human-specific boundaries in the human lineage.
We next investigated the genetic properties of CCD boundaries across primates in an evolutionary context. Our analysis revealed distinct characteristics of the non-human primate variable boundaries in humans, including markedly lower conservation and significant enrichment of human-specific nucleotide substitutions (Fig. S3i, j).
Taken together, our results indicate that there is evolutionary variation in CCD boundaries during primate evolution, associated with genetic divergence at these boundaries.
Human-specific CCD boundaries contribute to complex trait and disease heritability
Alterations in TAD boundaries have previously been linked to changes in phenotypic traits and increased disease risk31,32. Given that CCDs are conserved between cell types (Fig. S3d)13, and in particular that the human-specific CCDs identified in B-lymphoblast cells across primates are largely shared (102 of 112) in human primate neuronal cells, we sought to determine whether evolutionary variation in CCD boundaries is associated with complex traits and disease susceptibility in humans.
To explore this, we investigated the heritability enrichment of a curated set of 44 traits and disease susceptibility at the non-human primate variable boundaries using linkage disequilibrium score regression (LDSC)33,34,35. We found that genetic variation within 300 kb of these boundaries significantly contributed to the heritability of complex traits and disease susceptibility, particularly for neuropsychological, metabolic, and cardiopulmonary traits (Fig. S4a). Notably, the heritability associated with autism spectrum disorder (ASD) showed the strongest enrichment at these boundaries (Fig. S4a). We next sought to determine whether there were specific patterns of heritability for ASD across these boundaries and their corresponding CCDs. Subsequent analysis revealed a distinct heritability pattern for ASD in the vicinity of the boundaries, particularly within the 100–150 kb regions adjacent to the boundaries and within the CCDs themselves (Fig. 2d). Notably, human-specific CCD boundaries exhibited higher heritability for ASD within these regions compared to the non-human primate variable boundaries (Fig. 2d). These findings provide insights into the functional significance of human-specific CCD boundaries in shaping the complex genetic architecture underlying ASD susceptibility.
To test whether tissue-specific cis-regulatory elements are enriched near the non-human primate variable boundaries in humans, reflecting ASD heritability patterns (Fig. 2d), we investigated H3K27ac enrichment profile of various primary tissues, including blood, liver, digestive tissues, and brain tissues from distinct anatomical regions. Our results showed a pronounced enrichment of H3K27ac signals from brain tissues close to these boundaries and prominently within their associated CCDs compared to other tissue types (Fig. 2e). Furthermore, brain tissue from different anatomical regions consistently showed increased H3K27ac signals near these boundaries and within their associated CCDs, more so than conserved boundaries and their corresponding CCDs, or random regions (Figs. 2f and S4b).
Collectively, our results suggest that human-specific boundaries, which possess potential functional significance in gene regulation, contribute to the heritability of complex traits and disease susceptibility.
Human-specific CCD boundaries diverge transcriptional concordance
CCD boundaries act as insulators, ensuring appropriate enhancer-promoter interactions within CCDs while preventing chaotic regulatory influences from enhancers in neighboring CCDs7. Based on this notion, we interrogated the functional impact of evolutionary variation in CCD boundaries on gene transcriptional concordance using transcriptional data from various paired human and macaque brain tissues (Fig. 2g, see “Materials and methods”). Our results illustrated that gene pairs separated by the human-specific boundaries into adjacent CCDs exhibited random transcriptional correlations (Fig. 2h, see “HS”). In contrast, the corresponding macaque gene pairs located within the same CCDs displayed moderate transcriptional correlations, which decreased with increasing genomic distance between the gene pairs (Fig. 2h, see “HS”). Furthermore, gene ontology (GO) enrichment analysis revealed that these gene pairs are significantly associated with synaptic transmission and signaling in neurons (Fig. S4c). Similarly, APE boundaries exerted a comparable influence on transcriptional concordance between humans and macaques, although to a lesser extent than HS boundaries (Fig. 2h, see “APE”). For miscellaneous and conserved boundaries, we detected no marked differences in transcriptional correlation patterns between humans and macaques, both showing nearly random transcriptional behavior (Fig. 2h, see “Misc” and “Consv”). As representative examples, specific gene pairs associated with synaptic transmission and signaling showed distinct transcriptional correlations between humans and macaques, with the separation by human-specific boundaries occurring in humans but not in macaques (Figs. 2i and S4d–f).
In summary, the disruption of transcriptional coordination across HS-specific boundaries in humans facilitates more independent and precise regulation of genes, potentially leading to transcriptional innovation and fine-tuning of gene expression during evolution, thereby enhancing regulatory flexibility in human-specific contexts.
Unveiling the human-specific CCD boundary between the PCDH-β and PCDH-γ loci
The PCDH genes, which belong to a family of cell adhesion molecules, are essential for neuronal development and synaptic connectivity in the brain and are associated with several neurological disorders36,37. They are arranged in a tandem array and categorized into PCDH-α, -β, and -γ clusters37. The transcriptional regulation of the PCDH gene clusters depends on a process of alternative promoter selection, leading to the combinatorial diversification of PCDH proteins in individual neurons38,39,40,41. Previous studies have suggested that this meticulous regulation, resulting in an extensive repertoire of PCDH isoforms, relies predominantly on CTCF-mediated chromatin interactions42,43,44.
We identified a human-specific CCD boundary between the PCDH-β and PCDH-γ loci (Fig. 3a). In human primary neuronal cells, this boundary exhibited a strong insulating role, characterized by a significant increase in CTCF binding intensity (Fig. 3b) and a significant decrease in CTCF looping intensity between the PCDH-β and PCDH-γ loci compared to its macaque counterpart (Fig. 3c, d). Consistently, Hi-C data from the macaque germinal zone and cortical plate both showed pronounced chromatin interactions between the PCDH-β and PCDH-γ loci, forming a distinct architectural stripe across the syntenic counterpart of the human-specific boundary (Fig. S5a). Together, the presence of the human-specific boundary between the PCDH-β and PCDH-γ loci, which may contribute to the decreased physical contact between these loci.

a Track views illustrating CTCF loops within the PCDH-α, PCDH-β, and PCDH-γ loci in human and macaque neuronal cells. The red pentagon and dashed lines mark the human-specific (HS) boundary and its corresponding position in macaques. Transcription start sites (TSSs) for PCDH genes are shown. α1-14, PCDHA1 to PCDHA14; αC1 and αC2, PCDHAC1 and PCDHAC2; β1-16, PCDHB1 to PCDHB16; γA1-12, PCDHGA1 to PCDHGA12; γB1-7, PCDHGB1 to PCDHGB7; γC3-5, PCDHGC3 to PCDHGC5. b Volcano plot showing changes in CTCF binding intensity at the CTCF sites within the PCDH locus between human and macaque neuronal cells. The red dot represents the human-specific boundary. The x-axis denotes the log2 fold-change in binding intensity, and the y-axis indicates the FDR-adjusted p-values. c Box plot comparing CTCF-mediated chromatin interaction intensities between PCDH-β and PCDH-γ loci in human (n = 1287) and macaque (n = 1042) neuronal cells. The central line indicates the median, the box spans the interquartile range, and the whiskers extend to 1.5× the interquartile range. **** P < 0.0001 (one-tailed Wilcoxon test). d Contact maps depicting normalized CTCF-mediated chromatin interactions between PCDH-β and PCDH-γ loci in human (left) and macaque (right) neuronal cells at a 5-kb resolution. e Contact map showing different chromatin interaction patterns between the PCDH-β and PCDH-γ loci in human versus macaque neuronal cells. f RNAPII-mediated chromatin interactions at the PCDH-α, PCDH-β, and PCDH-γ loci in neuronal cells with enhancers (E1 to E5) were defined by H3K27ac peaks. g Heatmap of RNAPII-mediated chromatin interactions between PCDH promoters and enhancers (E1 to E5) in human and macaque neuronal cells. h Hierarchical clustering of combinatorial transcription of PCDH genes in individual human and macaque neuronal cells, grouped into nine clusters based on co-transcriptional profiles of PCDH genes. Species origin of each cell is indicated at the bottom. i Circos plots depicting the combinatorial transcription of PCDH genes in individual primary neuronal cells from humans (left) and macaques (right). The source data and exact P values for (b) and (c) are provided in the Source Data file.
To uncover the evolutionary origin of the human-specific boundary between the PCDH-β and PCDH-γ loci, we compared the DNA sequences of the CTCF site at this boundary in humans and macaques. Although the DNA sequence of the CTCF site is identical in both species, CTCF binding intensity in macaques is likely reduced due to a four-nucleotide deletion located 16 base pairs downstream of the CTCF site (Fig. 3b), as evidenced that the flanking sequences can modulate CTCF binding affinity45. Furthermore, using DNA methylation data from primary neurons obtained from human and macaque prefrontal cortical tissue46,47, we observed a significant reduction in DNA methylation at the human-specific boundary compared to its macaque counterpart (Fig. S5b, c), Thus, these changes in the DNA sequence surrounding the CTCF site and the level of DNA methylation facilitate the establishment of the human-specific boundary between the PCDH-β and PCDH-γ loci. Notably, although the DNA sequences of the CTCF sites within the PCDH-β locus are identical between humans and macaques, the human CTCF sites within the PCDH-β locus showed a trend of increasing DNA methylation compared to macaques (Fig. S5d). This increased methylation likely limited CTCF looping between the PCDH-β and PCDH-γ loci in humans, thereby strengthening the insulation effect of the human-specific boundary.
Collectively, our results demonstrate the presence of the human-specific CCD boundary between the PCDH-β and PCDH-γ loci.
Human-specific CCD boundary results in reduced combinatorial diversification of PCDHs in human neurons
Within the PCDH gene locus, CTCF sites are predominantly located within 500 bp upstream of individual PCDH promoters (Fig. S6a). Notably, we found that human PCDH-β promoters exhibited significantly reduced CTCF-mediated chromatin interactions compared to macaques (Fig. S6b, c), which is attributed to the presence of the human-specific boundary between the PCDH-β and PCDH-γ loci (Fig. 3a). In contrast, CTCF-mediated chromatin interactions proximal to the PCDH-γ promoters remain comparable between the two species (Fig. S6b, c). Given this configuration, we hypothesize that divergence in CTCF looping proximal to the PCDH-β promoters between species may influence alternative promoter selection for PCDH-β genes, contributing to distinct combinatorial diversification during evolution.
To test this hypothesis, we investigated RNAPII-mediated interactions within the PCDH locus using human and macaque primary neuronal cells. We identified five enhancers within this locus for both species based on H3K27ac and RNAPII binding profiles. Two of these enhancers (E1 and E2) resided within the PCDH-α locus, and the rest (E3 to E5) within the PCDH-γ locus (Fig. 3f). Notably, the PCDH-β locus appeared devoid of any enhancer activity (Fig. 3f). In both species, enhancers E1 and E2 primarily interacted with the PCDH-α promoters (Fig. 3f, g). Enhancers (E3 to E5) within the PCDH-γ locus exhibited stronger interaction intensities with PCDH-γ promoters in macaques compared to humans (Fig. 3f, g). Additionally, these enhancers formed more pronounced interactions with PCDH-β promoters in macaques, a pattern that is manifestly reduced in humans. This suggests that enhancer activity at the PCDH-γ locus in macaques supports broader promoter interactions, including with PCDH-β promoters, which may contribute to species-specific differences in PCDH gene regulation. (Fig. 3f, g). This observation aligns with the insulating effect of the human-specific boundary between the PCDH-β and PCDH-γ loci in humans, an effect that is notably absent in macaques. Our subsequent analysis revealed a stronger correlation in macaques, in contrast to humans, between RNAPII-mediated chromatin interactions at the PCDH-β and PCDH-γ promoters and those associated with CTCF sites proximate upstream of these promoters (Fig. S6d). Together, our analysis reveals that the human-specific CCD boundary, in synergy with divergent CTCF loops between the PCDH-β and PCDH-γ loci, reduces the connection of the PCDH-β promoters to distal enhancers within the PCDH-γ locus in humans.
We next sought to investigate alterations in the combinatorial repertoire of PCDH isoforms between humans and macaques. To this end, we analyzed 5159 and 7182 human and macaque excitatory neurons, respectively, using 5′ scRNA-seq with ultra-deep sequencing (Figs. S1e and 6e). At the single-cell level, PCDH-β isoforms exhibited markedly higher transcript levels in macaques compared to humans (Fig. S6g), consistent with strong RNAPII-mediated chromatin interactions connecting PCDH-β promoters to enhancers within the PCDH-γ locus in macaques (Fig. 3f, g). Moreover, through unsupervised clustering based on the transcription levels of PCDH isoforms, we clustered individual excitatory neurons from humans and macaques into distinct groups (Figs. 3h and S6f), highlighting the inherent difference in the combinatorial patterns of PCDH isoforms between the species. To quantify combinatorial diversification, we constructed co-occurrence networks for PCDH isoforms in human and macaque excitatory neuron populations by incorporating co-occurrence frequencies and observed proportions of PCDH isoforms (Fig. 3i). Our quantitative analysis revealed that the combinatorial diversification was significantly reduced in human excitatory neurons compared to macaques (Fig. S6h).
In summary, our results suggest that the human-specific CCD boundary between the PCDH-β and PCDH-γ loci, in conjunction with divergent CTCF loops, modulates the connection of the PCDH-β promoters to distal enhancers within the PCDH-γ locus, contributing to reduced combinatorial diversification of PCDH isoforms in humans during evolution (Fig. S6i).
Evolutionary divergence in CTCF loops aligns with changes in enhancer activity, directing the connectivity of species-specific enhancers to their cognate genes
We explored evolutionary divergence in CTCF loops between humans and non-human primates using B-lymphoblastoid and primary neuronal cells. Principal component analysis (PCA) of genome-wide CTCF loops revealed substantial distinctions between humans and non-human primates, consistent with their relative evolutionary distances (Fig. 4a). In a comparative analysis between humans and non-human primates, we identified 2133 human-specific gained (2.7%) and 2418 lost (3.1%) CTCF loops in B-lymphoblastoid cells (Fig. S7a, c). Additionally, we detected 5873 human-specific gained (7.3%) and 6708 lost (8.3%) CTCF loops between human and macaque primary neuronal cells (Fig. S7b). These species-specific changes in CTCF looping predominantly reflect cell type specificity, with little sharing observed between cell types (Fig. S7d). We verified the authenticity of the identified human-specific gained and lost CTCF loops through comparisons of interaction intensity and CTCF binding intensity at their anchors across species and cell types (Figs. 4b, c and S7e). In humans, the CTCF binding sequences at the anchors of these CTCF loops showed notably diminished conservation compared to the conserved CTCF loops (Fig. S7f) and a significant enrichment of human-specific nucleotide substitutions (Fig. S7g). In cross-species comparisons, CTCF-binding sequences from human-specific gained loops were more conserved than their macaque counterparts, whereas sequences from human-specific lost loops were less conserved than those in macaques (Fig. S7h). Collectively, our findings highlight the remarkably unique evolutionary divergence of CTCF loops in humans, driven by genetic variation between humans and non-human primates.

a Principal component analysis showing genome-wide differences in CTCF-mediated chromatin interactions among B-lymphoblastoid and neuronal cells across species. b Aggregate peak analysis (APA) of CTCF loops in B-lymphoblastoid cells, comparing human-specific gained, lost, and conserved loops between humans and non-human primates. c APA of CTCF loops in neuronal cells, comparing human-specific gained, lost, and shared loops between humans and macaques. d Overview for analyzing human-specific CTCF loops in demarcating RNAPII-mediated enhancer-promoter interactions. APA plots of RNAPII-mediated chromatin interactions associated with the anchors of human-specific gained, lost, and conserved/shared CTCF loops in B-lymphoblastoid cells (e) and in neuronal cells (f). Hierarchical clustering heatmaps of motif enrichment at the anchors of the human-specific gained, lost, and conserved/shared CTCF loops in B-lymphoblastoid cells (g) and neuronal cells (h). Enrichment profiles of human-specific gained enhancers at the anchors of human-specific gained CTCF loops (i) and human-specific lost enhancers at the anchors of human-specific lost CTCF loops (j) in B-lymphoblastoid (top) and neuronal cells (bottom), shown at a 10-kb bin resolution. Gray bars show the median of the background distribution with standard deviation error bars. Box plots of RNAPII loop interaction intensities at the anchors of human-specific gained (left) and lost (right) CTCF loops in humans, compared to non-human primates from B-lymphoblastoid cells (k) and macaques from neuronal cells (l). Box plots showing transcript levels of genes whose promoters interact with anchors of human-specific gained (left) and lost (right) CTCF loops through RNAPII loops in humans, compared to syntenic genes in non-human primates (B-lymphoblastoid cells, m) and macaques (neuronal cells, n). o Schematic illustrating a concordant yet constrained model involving the interplay between evolutionary variation in CTCF loops and divergence in enhancer activities. For (k–n), in box plots, the central line indicates the median, the box spans the interquartile range, and the whiskers extend to 1.5× the interquartile range. ****P < 0.0001, **P < 0.01, and *P < 0.05, as determined by one-tailed Wilcoxon test. Source data, n numbers, and exact P values are provided in the Source Data file.
Next, we investigated the functional impact of human-specific gained and lost CTCF loops in directing enhancer connectivity to target genes through RNAPII looping and the enrichment of transcription factor binding motifs at the anchors of these loops (Fig. 4d). Our analysis revealed that human-specific gained CTCF loops encompassed pronounced RNAPII-mediated chromatin interactions in both B-lymphoblastoid (Fig. 4e) and primary neuronal cells (Fig. 4f) compared to non-human primates. Consistently, the anchors of human-specific gained CTCF loops exhibited significant enrichment of H3K27ac in both cell types (Fig. S7i). However, RNAPII-mediated chromatin interactions were largely absent in humans within the regions of the human-specific lost CTCF loops (Fig. 4e, f). These findings indicate an evolutionary concordance between the divergence in CTCF loops and RNAPII looping activity, particularly at the anchor of the human-specific gained CTCF loops. Consistent with this, we observed a significant enrichment of cell type-specific motifs at the anchors of human-specific gained CTCF loops in B-lymphoblastoid (Fig. 4g) and primary neuronal cells (Fig. 4h), respectively. Furthermore, human-specific enhancers were significantly enriched within the human-specific gained CTCF loops (Fig. S7j), primarily located within 10 kb downstream of the CTCF site at their anchors (Fig. 4i). These human-specific enhancers proximal to the anchors of human-specific gained CTCF loops exhibited significantly stronger chromatin interactions with their target genes through RNAPII loops in both B-lymphoblastoid (Fig. 4k) and primary neuronal cells (Fig. 4l) compared to non-human primates. These target genes exhibited enhanced transcription in humans (Fig. 4m, n) and were significantly associated with specific cellular functions and identities in each cell type (Fig. S7l,m). Conversely, regions corresponding to the anchors of human-specific lost CTCF loops showed a significant reduction in active enhancers in both cell types compared to non-human primates (Figs. 4j and S7k). In addition, the human-specific lost enhancers, co-occurring proximal to the anchors of the human-specific lost CTCF loops, exhibited weakened chromatin interactions with their target genes (Fig. 4k, l) and reduced transcription of the target genes compared to non-human primates (Fig. 4m, n).
Taken together, our results indicate that the evolutionary divergence in CTCF loops is associated with changes in enhancer activity proximal to their anchors during evolution, playing a critical role in directing the connectivity of species-specific enhancers to their cognate genes in a concordant yet constrained manner (Fig. 4o). Through this mechanism, evolutionarily divergent CTCF loops contribute to shaping distinctive human transcriptional landscapes, thereby being associated with complex human traits and disease susceptibility.
Human-specific CTCF loops modulate human-specific enhancers, contributing to disease susceptibility
Given our findings elucidating the mechanism by which evolutionarily divergent CTCF loops orchestrate species-specific enhancers interacting with cognate genes, as well as their anchors' association with disease-linked QTLs (Fig. S7n, o), we next sought to explore the implication of this mechanism in relation to complex human traits and disease susceptibility.
To this end, we found that human-specific CTCF loops encompass the BLK locus in B-lymphoblastoid cells, a configuration absent in non-human primates (Fig. 5a). The BLK gene, predominantly expressed in B-cell lineages, exhibits expression variations that have been implicated in the susceptibility to systemic lupus erythematosus (SLE)48,49. These distinctive human CTCF loops originated from an evolutionarily gained CTCF site upstream of the BLK promoter (Fig. 5a, b). Multiple sequence alignment of this CTCF site and its syntenic counterparts in other species revealed two human-specific nucleotide substitutions at the most conserved positions within the CTCF motif (Fig. 5b). Notably, a naturally occurring insertion variant, rs558245864 (C > CG), found in human populations, disrupts this site at a position with a highly conserved nucleotide within the CTCF motif (Fig. 5b), resulting in the abolition of CTCF binding intensity (Fig. S8a). In HG00514, an individual homozygous for the variant rs558245864, this variant abolished CTCF binding at the human-specific CTCF site and completely eliminated CTCF loops originating from this site (Fig. S8b). Remarkably, HG00514 exhibited a CTCF-loop configuration at the BLK locus similar to that of non-human primates but distinct from GM12878, which carries the homozygous reference alleles at the CTCF site (Fig. 5a, see “Human” and Fig. S8b). Furthermore, we found that the variant rs558245864 is strongly associated with three other variants (rs2736337, rs2736340, and rs13277113) within the same linkage disequilibrium block (Fig. 5c). Remarkably, these three variants have been previously associated with susceptibility to autoimmune diseases48,49,50,51. Therefore, our analysis established a causal relationship between genetic variation, the disruption of human-specific CTCF loops, and SLE susceptibility.

a Human-specific CTCF loops encompass the BLK locus, originating from a human-specific CTCF site in B-lymphoblastoid cells. Dashed red lines indicate the human-specific CTCF site and its corresponding positions in non-human primates. b DNA sequence alignment of the human-specific CTCF site, with human-specific single-nucleotide substitutions highlighted. The insertion variant rs558245864 (C > CG) is marked. c Regional association plot showing rs558245864 and its linkage to GWAS single-nucleotide polymorphisms (SNPs) for SLE susceptibility. The surrounding SNPs of rs558245864 are colored according to their degree of linkage disequilibrium (r2) with rs558245864. P values for r2 (shown on left y-axis) were calculated using chi-square test with 1 degree of freedom. Recombination rates are shown in the sky-blue curve. d Visualization of RNAPII loops at the BLK locus from human (GM12878) and macaque B-lymphoblastoid cells. CTCF loops for GM12878 in the same region are shown in (a, see “Human”). Dashed red lines indicate the human-specific CTCF site and its corresponding position in macaques. The BLK promoter and the human-specific enhancer (HSE) are highlighted in yellow and blue, respectively. e Contact maps at a 1-kb resolution representing combined CTCF- and RNAPII-mediated chromatin interactions in B-lymphoblastoid cells from individuals GM12878 (top) and HG00514 (bottom) at the BLK locus. The red pentagon labeled with “B” marks the human-specific CTCF site. The genotypes at the CTCF site corresponding to each individual are shown in parentheses beside their IDs. The blue and green boxes indicate the reduced and increased chromatin interactions in HG00514 compared to GM12878, respectively. f BLK transcript levels in B-lymphoblastoid cells from individuals with different genotypes at the rs558245864 variant. In the box plots, the central line indicates the median, the box spans the interquartile range, and the whiskers extend to 1.5× the interquartile range. P values are provided (one-tailed Wilcoxon test). g BLK transcript levels in B lymphocytes from a healthy individual (C/C) and SLE patients (C/CG or CG/CG) with rs558245864. Data shown as mean ± standard deviation. P values are provided (one-tailed Student's t-test). The source data, n numbers, and exact P values for (f) and (g) are provided in the Source Data file.
Intriguingly, the variant rs558245864 represents the highest allele frequency in East Asian populations (Fig. S8d). Furthermore, we found that the variant rs558245864 appeared in Neanderthals with either homozygous or heterozygous alleles, but was absent in Denisovans (Fig. S8e). These findings suggest that the variant rs558245864 might undergo a unique evolutionary path in human populations.
We then sought to unravel the functional influence of these human-specific CTCF loops on the transcriptional regulatory changes at the BLK locus in the context of both evolutionary divergence and population variation. In the evolutionary divergence scenario, our findings revealed the presence of a human-specific enhancer located ~3.5 kb downstream of the human-specific CTCF site (Fig. S8c). This enhancer exhibited a robust RNAPII-mediated chromatin interaction with the BLK promoter, which was notably absent in macaques (Fig. 5d). These observations align with the mechanism we elucidated above (Fig. 4o). Furthermore, the human BLK promoter was predominantly involved in RNAPII-mediated chromatin interactions, which were largely confined within the human-specific CTCF loops (Fig. 5d, a, see “Human”). Conversely, in macaques, devoid of the constraint imposed by these CTCF loops, RNAPII-mediated chromatin interactions prominently shifted from the BLK promoter to its upstream region (Fig. 5d, a, see “Macaque”). In the context of population variation, the human-specific CTCF site in GM12878 contributed to the formation of a distinct architectural stripe, emphasizing its robust insulator function (Fig. 5e). Conversely, this architectural stripe was conspicuously absent in HG00514, as the site is compromised by the variant rs558245864 (Fig. 5e). Moreover, the BLK promoter in HG00514 exhibited prominently reduced RNAPII-mediated chromatin interactions with its cognate enhancers, including the human-specific enhancer proximal to the human-specific CTCF site, compared to GM12878 (Fig. 5e). Based on our genetic analysis linking the variant rs558245864 with SLE susceptibility, we analyzed its functional consequences in SLE patients using 4C-seq on B cells derived from peripheral blood mononuclear cells. Our analysis revealed that the human-specific enhancer proximal to the human-specific CTCF site displayed strong interactions with the BLK promoter in a healthy individual carrying homozygous reference alleles at the CTCF site (Fig. S8f). However, these interactions were significantly attenuated in an SLE patient with homozygous alleles for the variant rs558245864 (Fig. S8f).
In human populations, BLK transcription showed a negative correlation with the degree of CTCF site disruption by the variant rs558245864 (Fig. 5f). The same pattern of change was also observed in SLE patients with homozygous or heterozygous variants of rs558245864 (Fig. 5g).
Taken together, our results demonstrate the central role of human-specific CTCF loops in dictating the chromatin interaction of human-specific enhancers with cognate promoters in the context of evolutionary divergence and population variation, with functional links to disease susceptibility.
Human-specific CTCF loops modulate human-specific enhancers, shaping alternative isoform usage
The extensive usage of alternative transcript isoforms substantially contributes to transcriptome complexity, serving as a significant source of phenotypic diversity throughout evolution, particularly in intricate brain tissues52,53,54. We hypothesize that, beyond the evolutionary role of human-specific CTCF loops in directing gene activities outlined above, these loops may also possess a largely unexplored yet critical function in shaping alternative isoform usage through adaptive evolution.
To validate this premise, we identified three human-specific CTCF loops at the DLGAP1 locus in human primary neuronal cells compared to macaques (Fig. 6a, b). DLGAP1, primarily expressed in neurons, functions as a scaffolding protein by interacting with other synaptic proteins, such as SHANK and DLG, in the postsynaptic density, a specialized region of the neuronal synapse where neurotransmitter receptors and signaling molecules are clustered55,56,57. Mutations or dysregulation of DLGAP1 have been associated with neuropsychiatric disorders, including ASD and schizophrenia58,59,60,61. The identified human-specific CTCF loops arise from an evolutionarily acquired CTCF-binding sequence within an intron of DLGAP1 (Fig. S9a, b). Of these three loops, the two strongest loops (with PET counts of 82 and 48) exclusively encompassed the transcription start site (TSS) of the DLGAP1-206 isoform, whereas only one weak loop (with a PET count of 18) covered both TSSs of the DLGAP1-206 and DLGAP1-201 isoforms (Fig. 6a). These findings suggest that these human-specific CTCF loops may potentially influence the alternative isoform usage of DLGAP1 in humans compared to non-human primates.

a CTCF loops at the DLGAP1 locus in human and macaque neuronal cells. Human-specific loops, originating from a human-specific CTCF site (Fig. S9b), are shown with interaction intensities. Two loops, encompassing the DLGAP1−206 promoter, are highlighted in yellow. Dashed red lines indicate the human-specific CTCF site in humans and its corresponding position in macaques. b 5-kb resolution contact maps depicting CTCF-mediated chromatin interactions at the DLGAP1 locus in human (left) and macaque (right) neuronal cells. Zoomed-in views show interactions centered on the DLGAP1−206 promoter, with red circles marking significant differences (P < 0.05, as determined by DEseq2, see “Methods”) between species, corresponding to the human-specific CTCF loops shown in (a). c Paternally inherited variants at the human-specific CTCF site shown in (a) were detected in families with autism spectrum disorder. In family SF0171486, a 10-nucleotide deletion is observed in the patient’s sibling, whereas in family 12997, both the patient and the sibling both carry a C > A single nucleotide variant (SNV). d RNAPII loops connect eight human-specific enhancers to the DLGAP1−206 TSS, all within the human-specific CTCF loops in human neuronal cells. e Uniform Manifold Approximation and Projection (UMAP) visualization of single-cell DLGAP1 transcript levels in human (left) and macaque (right) neuronal cells, with annotations for NPC (neural progenitor cells), IPC (intermediate progenitor cells), and EN (excitatory neurons). f Frequency distribution of the predominant DLGAP1 isoforms, DLGAP1-201 and DLGAP1−206, in neuronal cells derived from humans (top) or macaques (bottom), as determined by single-cell isoform sequencing. A macaque-specific exon (MSE) is highlighted. g Heatmap of chromatin accessibility at the human-specific (HS) CTCF site, enhancers, and the DLGAP1−206 TSS during neural differentiation in the human fetal prefrontal cortex. h Diagram illustrating that human-specific CTCF loops act in a concordant yet constrained manner to restrict distinctive human enhancers to interact with specific isoform promoters through RNAPII looping, resulting in divergent isoform usage between species.
By screening whole-genome sequencing data from 32,414 individuals from 8665 ASD families, we identified two privately inherited variants in two ASD families: a 10-nucleotide deletion and a single-nucleotide variant, both of which disrupted the evolutionarily acquired CTCF-binding sequence at the DLGAP1 locus (Fig. 6c). This finding further supports that the human-specific CTCF loops originating from this CTCF site could exert a functional impact on DLGAP1.
To further explore this notion, we focused on RNAPII-mediated chromatin interactions at the DLGAP1 locus in primary neuronal cells from humans and macaques. Our analysis revealed that the TSSs of DLGAP1-201 and DLGAP1-206 each associate with distinct RNAPII-mediated interaction domains (RIDs) for transcriptional activities (Fig. S9c). Interestingly, the RID associated with the TSS of DLGAP1-206 was encompassed by the two most dominant human-specific CTCF loops, whereas the RID linked to the TSS of DLGAP1-201 was situated within the weaker human-specific CTCF loop (Fig. S9c). Furthermore, our comparative analysis revealed that the RID corresponding to the TSS of DLGAP1-206 manifested significantly stronger RNAPII-mediated chromatin interactions in humans compared to macaques (P < 0.001), whereas the RID related to the DLGAP1-201 TSS maintained a comparable interaction intensity between humans and macaques (P = 0.651) (Fig. S9d). Consistent with this observation, we discovered that the RID for DLGAP1-206 is densely populated with brain tissue-specific enhancers (Fig. S9e). Eight of these enhancers were unique to humans compared to macaques, showcasing intense interactions with the DLGAP1-206 TSS within the RID (Fig. 6d). Overall, these findings highlight that human-specific CTCF loops confine concordantly emerged human-specific enhancers interacting with the alternative TSS of DLGAP1 during evolution, consistent with the mechanism we elucidated above (Fig. 4o).
In our analysis of scRNA-seq data from primary neuronal cells, we found that DLGAP1 showed distinct transcriptional patterns between humans and macaques during the development of excitatory neurons (Figs. 6e and S9f), extending beyond the overall higher transcript levels of DLGAP1 in humans than in macaques (Fig. S9g). To accurately quantify alternative isoform usage, we performed single-cell isoform sequencing (scISO-seq) on human and macaque primary neuronal cells. We focused on excitatory neurons expressing DLGAP1 in both species to determine the usage ratios of DLGAP1-201 and DLGAP1-206 isoforms. The result revealed a more prominent utilization of the DLGAP1-206 isoform in humans compared to macaques (P = 0.0074) (Fig. 6f). We also developed a single-cell TSS sequencing (scTSS-seq) method to quantify alternative TSS usage with ultra-deep sequencing (Fig. S9h). This analysis confirmed our scISO-seq-based quantification, emphasizing the significant usage of the DLGAP1-206 isoform in human excitatory neurons (P < 2.2e-16) (Fig. S9i). Collectively, these findings substantiate the hypothesis that human-specific CTCF loops orchestrate concordantly emerged human-specific enhancers, which in turn shape the alternative TSS usage of DLGAP1.
During human fetal brain development, we observed consistent accessibility of the human-specific CTCF site from neural progenitor cells to excitatory neurons along the developmental trajectory (Fig. 6g). Additionally, eight human-specific enhancers and their interacting TSS of DLGAP1-206 increasingly gained accessibility (Fig. 6g). Consistent with this notion, DLGAP1-206 transcription steadily increased during human fetal brain development (Fig. S9j). These findings indicate a dynamic and orchestrated transcriptional regulation of DLGAP1-206 throughout human fetal brain development.
In conclusion, our results provide evidence for the pivotal role of human-specific CTCF loops in directing human-specific enhancers to orchestrate transcriptional isoform usage during evolution (Fig. 6h).
Functional validation of human-specific CTCF loops using human forebrain organoid
To validate the functional impact of human-specific CTCF loops at the DLGAP1 locus, we applied CRISPR-Cas9 genome editing to delete the human-specific CTCF site responsible for these loops in human-induced pluripotent stem cells (hiPSCs) (Figs. 7a and S10a). We then generated human forebrain organoids from both wild-type (WT) and knock-out (KO) hiPSCs. Through immunostaining of canonical markers, we characterized the cytoarchitectures of the organoids during development (Fig. S10b), confirming the successful induction of organoids from both WT and KO hiPSCs. The induced organoid models, faithfully mimicking the cellular composition and cytoarchitectural organization of the developing dorsal forebrain62,63, are ideal for investigating the functional significance of human-specific CTCF loops in fetal brain development.

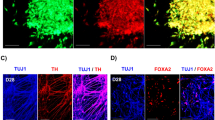
a Diagram illustrating CRISPR-Cas9 depletion of the human-specific (HS) CTCF site, as shown in Fig. 6a, in human induced pluripotent stem cells (hiPSCs) to generate forebrain organoids. b Hematoxylin and eosin staining of wild-type (WT) and knocked-out (KO) forebrain organoids at day 60, highlighting ventricular zones (VZs). Scale bar, 200 µm. Experiments were repeated six and four times for WT and KO conditions, respectively. c Immunofluorescence staining for NCAD and SOX2 in WT and KO organoids at day 35. Scale bar, 200 µm. Experiments were repeated three times for each group. d Immunofluorescence staining for TGIF1 in WT and KO organoids at day 60. Scale bar, 20 µm. Experiments were repeated four and three times for WT and KO conditions, respectively. e UMAP visualization of single-cell clusters (n = 32,095) from WT and KO organoids at days 14 and 60, showing cell annotations: G2M/S (cells in G2, M, and S phases of the cell cycle), NEC (neuroepithelial cells), NPC (neural progenitor cells), IPC (intermediate progenitor cells), EN (excitatory neurons), and IN (inhibitory neurons). f Dissimilarity matrix plot showing transcriptional differences between WT and KO organoids during differentiation, with altered alignment in the NEC and NPC stages (marked by a black arrow). Cell densities for each cellular stage along the differentiation pseudotime are shown. g Box plots comparing normalized transcript levels of TGIF1 in individual cells from WT and KO organoids at days 14 and 60, as measured by scRNA-seq. A total of 268 cells for WT and 137 cells for KO at Day 14 and 170 cells for WT and 187 cells for KO at Day 60 were included in the comparison. P values are provided (one-tailed Wilcoxon test). In the box plots, the central line indicates the median, the box spans the interquartile range, and the whiskers extend to 1.5× the interquartile range. h Transcriptional profiles of marker genes across differentiation pseudotime in WT and KO organoids, with colors for cell types. Black lines trace the regression of the transcript levels along the pseudotime, and the light gray shadows denote the 95% confidence intervals. The source data for (g) are provided in the Source Data file.
Initially, we observed that KO organoids consistently exhibited more ventricular zones (VZs) than WT during development (Fig. 7b, c and Fig. S10c, d), suggesting that the human-specific CTCF site deletion at the DLGAP1 locus could influence VZ development in organoids. Given that DLGAP1 primarily functions as a scaffolding protein that facilitates the assembly of the postsynaptic density of neurons and may not directly influence VZ development57,60, we hypothesize that the effect on VZ development may be mediated by other genes adjacent to the DLGAP1 locus. The gene TGIF1, located proximal to the CTCF site and upstream of DLGAP1 (Fig. 7a), emerged as a potential mediator. TGIF1, a conserved transcriptional repressor, is crucial for fetal brain development64,65, and its mutation or dysregulation has been associated with holoprosencephaly, a structural abnormality of the brain66,67,68,69,70. Notably, TGIF1 was detected in the VZ of both WT and KO organoids (Fig. 7d), underscoring its potential role in human forebrain organoid development.
To explore the impact of the human-specific CTCF site on forebrain development through TGIF1, we performed scRNA-seq on WT and KO organoids at day 14, when organoids predominantly contain progenitors and initiate VZ formation, and at day 60, when cortical excitatory neurons are present71. Our scRNA-seq analysis revealed six distinct cell clusters, delineating the developmental trajectory from neuroepithelial cell (NEC), neuronal progenitor cell (NPC), intermediate progenitor cell (IPC), and progressing separately towards excitatory glutamatergic neuron (EN), and inhibitory neuron (IN) (Figs. 7e and S10e–g). Subsequently, we utilized the time warping algorithm to align the differentiation pseudotime of WT and KO organoids, aiming to identify perturbations in the developmental dynamics of KO organoids72. Our analysis revealed a marked divergence in the developmental trajectory of KO organoids, particularly at the NEC and NPC stages (Fig. 7f), revealing accelerated differentiation in the KO organoids. This finding is consistent with a prominent decrease in NECs in KO organoids at day 14 compared to their WT counterparts (Fig. S10h). Furthermore, the TGIF1 transcript was significantly increased in KO organoids at day 14 compared to WT (Fig. 7g), with a steady increase at the NEC stage and a peak at the NPC stage (Fig. 7h, see “TGIF1”). Given that TGIF1 functions as a transcriptional repressor to maintain the undifferentiated state of NPCs and inhibit premature neuronal differentiation73,74,75,76, we investigated the transcriptional profiles of its target genes, such as FOXG1, PAX6, and SOX2, which are critical for NPC differentiation64,70,74,77,78,79, throughout the development of both WT and KO organoids. Our analysis revealed that the transcript levels of these target genes were markedly reduced at the NPC stage in KO organoids compared to WT, aligning with the elevated transcriptional activity of TGIF1 at the same stage in KO (Figs. 7h and S10i). Moreover, the gene ontology enrichment analysis of genes exhibiting differential expression between WT and KO organoids reinforced the disrupted developmental dynamics in KO organoids (Fig. S10j, k). Thus, our results demonstrate that the removal of the human-specific CTCF site alters the transcriptional profile of TGIF1, resulting in a disruption of organoid development.
Based on the findings above, we hypothesize that the removal of the human-specific CTCF site in KO organoids may grant the TGIF1 promoter to access enhancers that were confined within the human-specific CTCF loops originating from this CTCF site in WT (Fig. 7a). We then measured the chromatin interactions between the TGIF1 promoter and human-specific enhancers within these distinctive human CTCF loops in both WT and KO organoids (Fig. 8a). As a result, the TGIF1 promoter significantly increased chromatin interactions with the target enhancers in KO organoids compared to WT (Fig. 8b, see “TGIF1”). Collectively, our results suggest that the human-specific CTCF loops at the DLGAP1 locus modulate the transcriptional activity of TGIF1 by confining its connection to the human-specific enhancers.

a Diagram showing primers for the quantitative chromosome conformation capture (3C-qPCR) assays to assess interactions between human-specific enhancers (HSE1, HSE2) and promoters of TGIF1, DLGAP1-206, and DLGAP1-201 in WT and KO organoids. b Comparison of relative interaction intensity between anchor points, as described in (a), and promoters of TGIF1, DLGAP1-206, and DLGAP1-201 in WT and KO organoids. The data are presented as mean ± standard deviation, n = 3 or 4 for each condition. P values are provided (one-tailed Student’s t-test). c Bar plots showing fold change in DLGAP1-201 and DLGAP1-206 transcript levels between WT and KO organoids. Data are shown as mean ± standard deviation, with three organoids used as replicates for each time point. d Comparison of the isoform ratio (DLGAP1−206:DLGAP1−201) in WT and KO organoids. The P value was calculated using one-tailed Student’s t-test. e Representative action potential traces in neurons from WT and KO organoids at day 60. f Traces of miniature excitatory postsynaptic currents (mEPSCs) in excitatory neurons from WT and KO organoids at day 60, with CNQX used to inhibit receptor activity. Dot plots showing reduced amplitude (g) and frequency (h) of mEPSCs in KO organoid excitatory neurons at day 60 compared to WT. Each point represents an individual neuron, with black lines indicate the median. Data from two independent organoid batches (biological replicates). P values are provided (one-tailed Student’s t-test). i Diagram summarizing the pleiotropic effects of human-specific CTCF loops in human forebrain organoid development and function. HSEs human-specific enhancers, Es enhancers. The source data for (b, d, g, h) are provided in the Source Data file.
Next, we sought to validate the impact of the human-specific CTCF site on the alternative isoform usage of DLGAP1, as demonstrated in the cross-species comparison (Fig. 6), in both WT and KO organoids. Compared to WT, the DLGAP1-201 transcription in KO organoids decreased ~2-fold on days 70 and 80, corresponding to the peak presence of ENs during forebrain organoid development (Fig. 8c). Notably, DLGAP1-206 exhibited a more pronounced decline in transcription, decreasing ~4- to 5-fold in KO organoids compared to WT (Fig. 8c). Furthermore, WT and KO organoids represented distinct ratios for the usage of DLGAP1-201 and DLGAP1-206 isoforms, with a significantly decreased usage of DLGAP1-206 in KO organoids (Fig. 8d). These findings suggest that the deletion of the CTCF site has a more profound effect on the transcriptional activity of DLGAP1-206 than DLGAP1-201. We also found that in KO organoids, the DLGAP1-206 promoter exhibited significant reductions in chromatin interactions with its two cognate enhancers, due to the absence of the constraints imposed by the human-specific CTCF loops (Fig. 8a, b, see “DLGAP1-206”). The DLGAP1-201 promoter, located distal from these two enhancers, displayed a substantial reduction in chromatin interactions with one enhancer, while a moderate decrease in chromatin interactions with the other enhancer (Fig. 8a, b, see “DLGAP1-201”). Together, our results demonstrate that the deletion of the human-specific CTCF site at the DLGAP1 locus resulted in the reduced alternative usage of DLGAP1-206 in KO organoids, corroborating our previous findings that human primary neuronal cells used more DLGAP1-206 in the presence of the CTCF site compared than macaques (Fig. 6).
To assess the impact of altered alternative isoform usage of DLGAP1 on synaptic transmission in KO organoids, we conducted patch-clamp experiments on sections derived from both WT and KO organoids with batch replications to recode miniature excitatory postsynaptic currents (mEPSCs) in ENs (Fig. 8e, f). Our comparative quantification revealed that ENs from KO organoids exhibited a significant discernible reduction in both amplitude and frequency of mEPSCs compared to their WT counterparts (Fig. 8g, h). These findings suggest that the altered alternative isoform usage of DLGAP1 in KO organoids leads to a significant impact on synaptic transmission, resulting in reduced excitatory synaptic activity.
Taken together, our results demonstrate the pleiotropy of human-specific CTCF loops originating from a human-specific CTCF site, in shaping human forebrain organoid development and function (Fig. 8i). Disruption of human-specific CTCF loops by removal of a single human-specific CTCF site not only alters the transcriptional profile of TGIF1, perturbing the differentiation process in KO organoids but also changes the alternative isoform usage of DLGAP1, manifesting in diminished excitatory synaptic activity in KO organoids.
Discussion
In this study, we have uncovered significant evolutionary divergence in CTCF-mediated chromatin topology across humans and non-human primates. Our discovery provides a valuable framework for understanding how these divergences translate into the evolution of regulatory landscapes among primates, thereby shedding light on the molecular origins of unique human traits.
Our study elucidates two layers of evolutionary divergences in chromatin topology at the CCD and CTCF loop scales during primate evolution, involving distinct mechanisms that contribute to the sculpting of regulatory landscapes. Despite extensive gene conservation in primates, the evolutionary acquisition of CCD boundaries leads to a reshuffling of gene groupings through newly formed neighboring CCDs, with a broad influence on the rewiring of the transcriptional landscape. This mechanism enables a significant remodeling of cellular pathways, ultimately contributing to the emergence of phenotypic novelties, as observed in vertebrate lineage evolution80. Notably, human-specific CCDs show strong associations with ASD exhibiting the most significant heritability enrichment. As a neurodevelopmental disorder, ASD appears to be influenced by a polygenic component without any major genes73,74,75,76,81. Thus, this mechanism contributes to the genetic architectures of ASD during primate evolution, providing critical insights into the genetic basis of complex human traits and diseases. Divergent CTCF loops signify an evolutionary synergy with species-specific enhancer activities, critically dictating the connectivity of these enhancers to their cognate genes in a concordant yet constrained manner. Although numerous human-specific enhancers have been identified through species comparison82, their functional consequences might depend on their local divergent CTCF loops to exert regulatory effects. The mechanism based on divergent CTCF loops involves in fine-tuning expression of target genes in a more precise manner, instead of broad changes of regulatory landscape through the divergent of CCD boundaries. Our study advances the understanding of primate genomic evolution by highlighting the dual functional impact of evolutionary changes at the levels of both CCDs and CTCF loops. However, it remains to be seen how these layers of evolutionary divergence harmonize in shaping the evolution of regulatory landscape and whether they undergo the same selection pressure for adaptation.
We provided compelling evidence that the evolutionary divergence in CTCF loops plays a pivotal role in shaping transcriptional isoform diversity. This role extends from the previously established function as an insulating neighborhood governing transcriptional activation or repression by constraining enhancer accessibility. Importantly, mounting evidence indicates that the extensive usage of alternative transcript isoforms substantially contributes to transcriptome complexity, serving as a significant source of phenotypic diversity throughout evolution83,84,85. However, the driving force behind the contribution to transcriptional isoform diversity remains largely unknown. Our findings on the role of CTCF loops provide insights into understanding the chromatin topological variation as the genetic basis for divergence in isoform usage during the evolution of phenotypic differences.
Our study demonstrates that the divergent CTCF site associated with human-specific CTCF loops exhibits pleiotropic effects. This pleiotropy arises from the ability of these CTCF loops to exert local effects on multiple genes involved in various cellular processes and phenotypic traits. Our results extend beyond the conventional understanding of pleiotropy, which is limited to single genes or enhancers86,87, and expand our understanding of the intricate chromatin regulatory networks orchestrated by CTCF loops in shaping phenotypic traits and disease susceptibility in humans. In this study, we report a singular case of pleiotropy in evolutionarily divergent CTCF loops. However, it remains to be seen how universal pleiotropy in chromatin topology is during evolution—a direction worth pursuing in the future.
Our study shows that evolutionary divergence in CCDs can be conserved between cell types, whereas evolutionary divergence in CTCF loops exhibit cell type specificity in regulating transcriptional activity. However, given the limited cell types included in our study, further tissue- or cell-type-specific investigations are needed to fully elucidate the distinct cell-type specificity at the CCD and loop levels in chromatin topology divergence. Furthermore, the potential impact of sex differences on chromatin topology divergence should be explored to better understand the role of sex in regulatory divergence.
In summary, our study demonstrates that evolutionary divergence in CTCF-mediated chromatin topology acts as an essential driving force behind transcriptional innovation during primate evolution, shedding light on the topological mechanism of human distinctive traits and diseases in an evolutionary context.
Methods
Ethics statement
All human embryo research was reviewed and approved by the Guangzhou Women and Children’s Medical Center Ethics Committee (reference number: 2021/392B00). Human embryos resulting from medication abortion were donated voluntarily by patients following thorough counseling and in-depth consent procedures. Donors provided explicit written informed consent after receiving full information about the experimental purpose of the donation, potential research applications, and confirmation that no compensation would be provided. All donated embryos were anonymized before use.
Peripheral blood samples from systemic lupus erythematosus (SLE) patients were collected at The First Affiliated Hospital of Sun Yat-sen University, with the approval from the Ethics Committee of Zhongshan School of Medicine, Sun Yat-Sen University (reference number: 087). All samples were obtained with written informed consent from the participants after full counseling in accordance with the criteria set by the Declaration of Helsinki.
The macaque embryo was collected from Guangdong Landao Biotechnology Co. Ltd, according to the guidelines and approval from the Institutional Animal Care and Use Committee (approval No. LDACU 20211221-01).
Human fetal brain tissues
Human fetal brain tissues were obtained from seven normal embryos of unidentified gender. The embryos were staged using color Doppler ultrasonography depending on crown-rump length. Two normal embryos at post-conception week (PCW) 9, female, without known defects or diseases, were used for neuronal cell culture in this study. Meanwhile, normal embryos at PCW2, PCW9, PCW11, PCW13, and PCW18 were used for quantitative PCR (qPCR) experiments.
Macaque fetal brain tissues
The macaque fetal ages were determined by measuring the crown-rump lengths using ultrasonography and comparing these measurements with the standard growth chart for macaques88. One macaque embryo at day 51 (E51) of undefined gender (which later was identified as male through RNA-seq analysis) was obtained by cesarean section, aligning with the stage of fetal brain development analogous to PCW9 in human fetal embryos89.
Peripheral blood samples from SLE patients
Approximately, 3–6 mL of peripheral blood samples were collected from female SLE patients into EDTA anticoagulant tubes. These peripheral blood samples were kept on ice and processed within 2 h.
B-lymphoblastoid cell lines
Human B-lymphoblastoid cell lines GM12878 (Cat. No. GM12878) and HG00514 (Cat. No. HG00514) were ordered from Coriell Institute for Medical Research. Chimpanzee B-lymphoblastoid cell line EB176(JC) (Cat. No. 89072704) and gorilla B-lymphoblastoid cell line EB(JC) (Cat. No. 89072703) were purchased from the European Collection of Authenticated Cell Cultures (ECACC). The macaque B-lymphoblastoid cell line LCL8664 (Cat. No. CRL-1805) was ordered from the American Type Culture Collection (ATCC). All B-lymphoblastoid cell lines were maintained in RPMI 1640 (Thermo Fisher Scientific, Waltham, MA, USA, Cat. No. 72400146) medium supplemented with 15% (30% for EB176) of heat-inactivated fetal bovine serum (Thermo Fisher Scientific, Cat. No. 10099141) and 1% of penicillin/streptomycin (Thermo Fisher Scientific, Cat. No. 15140163). The cells were grown at a cell density of 0.3–1.0 × 106 mL in a 37 °C incubator with 5% CO2. The cells were harvested when the cell density reached 0.6–0.8 × 106 mL.
Human induced pluripotent stem cells
Human induced pluripotent stem cells (hiPSCs) were purchased from the Chinese Academy Sciences Cell Bank (Cat. No. DYR0100). The cells were maintained with mTeSR Plus complete medium (Stemcell, Vancouver, Canada, Cat. No. 100-0276) in 6-well plates pre-coated with 1% Matrigel (Corning, Corning, NY, USA, Cat. No. 354234) diluted in DMEM/F-12 (Thermo Fisher Scientific, Cat. No. C11330500BT) in a 37 °C incubator with 5% CO2. The medium was completely replaced every day.
Processing of human and macaque fetal brain tissues
The whole fetal brain was separated from the embryo and kept in ice-cold HBSS++ buffer, consisting of HBSS (Ca2+ free, Mg2+ free, Thermo Fisher Scientific, Cat. No. 14170112), 1% of penicillin-streptomycin, and 10 mM HEPES pH 7.4 (1 M, Thermo Fisher Scientific, Cat. No. 15630106), before transfer to the lab for further processing. The fetal brain tissue was dissected in a sterile hood within 2 h after the embryo collection. Briefly, the fetal brain was transferred to a 6-cm dish with 5 mL fresh ice-cold HBSS++ buffer, and the choroid membranes were removed from the fetal brain under a stereomicroscope. After rinsing away the blood with sterile HBSS++ buffer, prefrontal cortex tissues were dissected from human (PCW9) and macaque (E51) fetal brain tissues for immediate tissue digestion and cell isolation. The isolated cells were then applied for primary neuron culture following the procedures as described below. As for qPCR, the dissected prefrontal lobe cortex tissues were cut into small blocks of 100–200 mg in weight and frozen in liquid nitrogen for subsequent processing.
Neuronal cell isolation and culture
The prefrontal cortex tissues were digested according to a previously described method90 with some minor modifications. Briefly, the prefrontal cortex tissues from human PCW9 and macaque E51 fetal brains were transferred into a new sterile 3.5-cm dish with 100 µL fresh sterile ice-cold HBSS++ buffer. After discarding the HBSS++ buffer, the tissues were sliced into sizes less than 1 mm3 using a sterile surgical blade. Immediately, the minced prefrontal cortex tissues were digested in a 15-mL tube with 2 mL of 20-unit/mL papain (Worthington, Lakewood, NJ, USA, Cat. No. LK003178) and 200 µL 200-Knuitz-units/mL DNase I (Worthington, Cat. No. LK003172), both of which were dissolved in HBSS with Ca2+ and Mg2+ (Thermo Fisher Scientific, Cat. No. 14025092). The tube was then rotated 30 rounds per minute (rpm) at 30 °C for 30 minutes (min).
After incubation, the tube was centrifuged at 150 × g for 1 min at room temperature (RT), and the supernatant was removed as much as possible with a P1000 pipette. The tissue/cell pellet was suspended using 2 mL RT Neurobasal++ medium, containing Neurobasal medium (Thermo Fisher Scientific, Cat. No. 21103049) supplemented with 1× B27 (Thermo Fisher Scientific, Cat. No. 17504044) and 1× GlutaMAX (Thermo Fisher Scientific, Cat. No. 35050-061). Single cells were released by triturating the tissue/pellet by pipetting up and down ten times with 1-mL LoBind tip (Thermo Fisher Scientific, Cat. No. TFLR1121000-Q) in 45 s without introducing bubbles. The tube was settled for 1 min, and the supernatant containing the released single cells was transferred to a new 15-mL tube without disturbing the remaining tissues at the tube bottom. Another 2 mL Neurobasal++ was added into the tube with the remaining tissues. The trituration, as described above, was repeated two more times to achieve maximum single-cell release. The supernatant containing the released single cells was combined and centrifuged at 800 × g for 5 min at RT. After discarding the supernatant fraction, 6 mL complete medium, which was comprised of Neurobasal medium supplemented with 1× B27, 1× GlutaMAX, 1× penicillin/streptomycin, and 5 ng/mL human FGF2 (Novoprotein, Wuhan, China, Cat. No. GMP-C046), was added to resuspend the pelleted cells. An aliquot of 10 µL cell suspension was used to assess the cell viability. Usually, the isolated cells showed a cell viability of more than 98%.
Next, the isolated single cells were seeded in the 6-well plate wells that were pre-coated with 0.1 mg/mL Poly-D-Lys (Sigma-Aldrich, St. Louis, MO, USA, Cat. No. P6407) dissolved in sterile miliQ water. Each well was seeded with 1.2 million cells and added with 2 mL of complete medium. For culturing the primary neuronal cells that were used for immunostaining, ~10,000 cells were seeded into each well of the 24-well plate with a 12-mm coverslip (Electron Microscopy Science, Hatfield, PA, USA, Cat. No. 72196-12) pre-coated with Poly-D-Lys. The seeded cells were maintained in a 37 °C incubator with 5% CO2 by half medium exchange every 2 days since day 5 after seeding cells. The primary neuronal cells were collected on day 20 and day 15 for human and macaque cells, respectively. Those primary neuronal cells were applied for subsequential experiments.
Fluorescent immunocytochemistry of primary neuronal cells
The fluorescent immunocytochemistry of primary neuronal cells was conducted as previously described86,87,91. Briefly, the cells on each coverslip were rinsed once with 1 mL RT PBS (PH7.4, Ca2+ free, Mg2+ free throughout this study) before fixing by 1 mL 4% paraformaldehyde for 10 min at RT. The coverslips were rinsed twice with 1 mL PBS for 5 min with 50 rpm horizontal rotation. Later, the cells were permeabilized in 200 µL permeabilization buffer (0.3% Triton X-100 and 0.5% goat serum (BOSTER, cat.no. AR1009) in PBS) for 20 min at RT. After rinsing 3 × 10 min in PBS with 0.1% Tween 20 with 50 rpm horizontal rotation, the cells were blocked in 200 µL blocking buffer (10% goat serum in PBS) for 1 h at RT. The cells were then incubated with primary antibodies diluted in permeabilization buffer at 4 °C overnight, and secondary antibodies diluted in permeabilization buffer for 2 h at RT in dark. The primary antibodies applied in this study were against PSD95 (mouse, 1:50, Thermo Fisher Scientific, Cat. No. MA1-045) and CAMKII (rabbit, 1:100, Abcam, Waltham, MA, USA, ca.no. ab52476), two markers for mature excitatory neurons92,93. The secondary antibodies (diluted at 1:500) included goat anti-mouse antibody, Alexa fluor 555 (Thermo Fisher Scientific, Cat. No. A-21424), and goat anti-rabbit antibody, Alexa fluor 488 (Thermo Fisher Scientific, Cat. No. A-11034). DAPI (1:15,000, Sigma-Aldrich, Cat. No. D9542) was added together with the secondary antibodies to stain the nuclei. Imaging was performed with the Cari Zeiss LSM880 confocal microscope. Images were processed in ImageJ (Fiji)94.
Cleavage under targets and tagmentation (CUT&Tag) assays for primary neuronal cells and B-lymphoblastoid cells
CUT&Tag was performed as previously described95,96. Briefly, one well of primary neuronal cells cultured in the 6-well plate was rinsed with 2 mL RT PBS and dissociated with 1 mL trypsin-EDTA (0.05%) (Thermo Fisher Scientific, Cat. No. 25200072) for 3 min at RT. The dissociated cells were transferred to a 15-mL tube with 10 mL PBS and were centrifuged at 800 × g for 5 min at RT. The cells were rinsed once in 10 mL PBS and then fixed in 2 mL RT PBS with 0.1% formaldehyde for 2 min. A volume of 200 µL 2-M glycine was added to quench the crosslink, and the cells were centrifuged at 800 × g for 5 min at 4 °C. Approximately 0.1–0.5 million fixed primary neuronal cells or 0.5 million native HG00514 cells were resuspended in 500 µL Wash buffer (0.02 M HEPES pH 7.5, 100 mM NaCl, 0.5 mM spermidine, and 1× proteinase inhibitor) and attached to Concanavalin A beads (Bangs Laboratories, Fishers, IN, USA, Cat. No. BP531). The cells on Concanavalin A beads were incubated at RT with 1 µL primary antibody against CTCF (rabbit, Abclonal, Wuhan, China, Cat. No. A1133) or H3K27ac (rabbit, Active motif, Carlsbad, CA, USA, Cat. No. 39133) in 50 µL Wash buffer supplemented with 0.05% digitonin (Sigma-Aldrich, Cat. No. 300410), 2 mM EDTA, and 0.1% bovine serum albumin (BSA) for 1 h at RT, and then with 1 µL guinea pig anti-rabbit secondary antibody (Sigma-Aldrich, Cat. No. SAB3700894) in 50 µL Wash buffer + 0.05% digitonin for 1 h at RT. After washing three times with 200 µL ice-cold Wash buffer + 0.05% digitonin, the cells were then incubated for 1 h at RT with 0.5 µL pA-Tn5 (Vazyme, Nanjin, China, Cat. No. S603) pre-loaded with annealed adapter complex (see Supplementary Data 2) in 50 µL Wash-300 buffer (0.02 M HEPES pH 7.5, 300 mM NaCl, 0.5 mM spermidine, 0.01% digitonin, and 1× proteinase inhibitor). After rinsing three times with 200 µL Wash-300 buffer, the cells were resuspended in 300 µL Wash-300 buffer with 10 mM MgCl2 and were incubated at 37 °C for 1 h to perform DNA tagmentation. After quenching the tagmentation with 10 µL 0.5 M EDTA, 10 µL 10% SDS, and 2.5 µL 20 mg/mL proteinase K, the cells were vortexed at full speed for 2 s and incubated at 55 °C for 1 h to digest proteins and to reverse crosslink. The DNA was purified with phenol-chloroform-isoamyl alcohol (Thermo Fisher Scientific, Cat. No. 15593049) and precipitated with three volumes of absolute ethanol. After incubation at −80 °C for 30 min, the precipitated DNA was centrifuged at 20,000 × g for 20 min at 4 °C and rinsed once with 80% ethanol. After brief air-drying, the DNA was dissolved in 26 µL EB buffer. Subsequently, the DNA was PCR amplified for 12–13 cycles with N5 and N7 index primers (see Supplementary Data 2) to construct libraries. The amplified libraries were purified with 1.2× AMPure beads for sequencing.
Preparation of single-cell suspension from primary neuronal cells
The primary neuronal cells were collected at the indicated time points. For each collection, cells cultured in a well of the 6-well plate were rinsed with 2 mL RT PBS and dissociated with several drops of Trypsin-EDTA (0.05%) for 3 min at RT. After adding 2 mL RT PBS to the well to stop cell dissociation, we slowly pipetted the cells 20 times with a 1-mL LoBind tip and transferred the cells to a 15-mL tube. Next, 10 mL PBS at RT was added to the 15-mL tube, and the tube was centrifuged at 200 × g for 5 min at RT to pellet the cells. The cell pellet was then resuspended in 2 mL PBS and filtered twice with 35-µm filters (Falcon, Cat. No. 352235). Subsequently, the cell suspension was adjusted to a volume of 6.2 mL with PBS. We added 1.8 mL debris removal solution (Miltenyi, Bergisch Gladbach, Germany, Cat. No. 130-109-398) to the cell suspension and mixed well the suspension with a 5-mL pipette without introducing any bubbles. Later, we added 4 mL PBS on top of the cell suspension without disturbing the cells and centrifuged the tube with a swinging rotator at 3000 × g for 10 min at 4 °C to pellet cells. After centrifugation, the cell pellet was resuspended in 3 mL PBS, and quality control was performed to assess the cell debris ratio and cell viability. In case of low cell viability (<70%), the cells were pelleted at 800 × g for 5 min at RT and resuspended with 100 µL dead cell removal beads (Miltenyi, Cat. No. 130-090-101) according to the manufacturer’s instructions for depletion of dead cells. The enriched live cells were resuspended in PBS with 0.04% BSA, and quality control was conducted to evaluate the cell viability (>90%), cell debris (<5%), clusters (<3%), and cell density (1200–1500 cells/µL). The cells were kept on ice for <30 min until subsequent processing for single-cell transcriptome analysis.
Genotyping rs558245864 at the BLK locus in SLE patients
DNA was extracted from 100 µL peripheral blood of each SLE patient and healthy individual using the QIAamp DNA blood mini kit (Qiagen, Cat. No. 51104). The PCR primers were designed at the surrounding sequences of the rs558245864 site to amplify DNA fragments from the target region. Sanger sequencing was performed to genotype rs558245864 polymorphism. The primers for the target region amplification are listed in Supplementary Data 2.
B cell isolation from peripheral blood of SLE patients
Peripheral blood mononuclear cells were enriched from the peripheral blood of the SLE patients and normal individuals by routine gradient centrifugation using the lymphocyte separation medium (Tianjin Haoyang Biological Manufacture Co., Tianjin, China, Cat. No. LDS10750125). Next, the B cell isolation was carried out by BD IMag protocol according to the instructions of the manufacturer. Briefly, the enriched peripheral blood mononuclear cells were rinsed with 1 mL 1× IMag buffer (BD, NJ, USA, Cat. No. 552362). After washing, every 107 cells were suspended in 50 µL pre-homogenized anti-human CD19 magnetic particles (BD, Cat. No. 551520) and incubated at RT for 30 min. After incubation, the cells were diluted into 1–8 × 107 mL with 1× IMag buffer, and the tube was placed into the magnetic rack for eight to 10 min. After discarding the supernatant by pipetting, CD19+ B cells were kept in the tube and resuspended in PBS after rinsing twice with 1 mL 1× IMag buffer. The obtained CD19+ B cells were used for subsequential experiments.
Circular chromosome conformation capture combined with sequencing (4C-seq) assays with the B cells from the SLE patients
To quantify the chromatin interactions between the BLK promoter and the adjacent enhancers, a circular chromosome conformation capture combined with sequencing (4C-seq) assay was conducted in B cells derived from an SLE patient with the homologous rs558245864 variant (CG/CG) at the BLK locus, according to the method previously described97 with some modifications. Briefly, 10 million patient B cells were crosslinked in 5 mL PBS with 10% fetal bovine serum and 1% methanol-free formaldehyde for 10 min at RT with 15 rpm vertical rotation, quenched with 0.125 M glycine, and centrifuged at 500 × g for 5 min at 4 °C. The cells were rinsed twice in 20 mL cold PBS.
The fixed cells were resuspended in 1 mL cold lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 5 mM EDTA, 0.5% NP-40, 1% Triton X-100, 1× fresh proteinase inhibitor) and incubated on ice for 10 min to isolate nuclei. The nuclei were rinsed once with 500 µL 1.2× rCutSamrt buffer (NEB, cat.no. B6004SIVAL), and then resuspended in 500 µL 1.2× rCutSamrt buffer. A volume of 15 µL 10% SDS was added to the nuclei, each time with 5 µL, to achieve a final SDS concentration of 0.3%. The sample was then incubated at 37 °C for 1 h with 20 rpm vertical rotation before quenching with 50 µL 20% Triton X-100. Later, 300 U Csp6I (Thermo Scientific, cat.no. ER0211) was added to the sample before incubating the tube for overnight at 37 °C with 20 rpm vertical rotation. The next day, quality control was performed to ensure that the chromatin was digested to around 1300 bp. Then, the sample was incubated at 65°C for 20 min to inactivate the enzyme, and transferred to a 50 mL falcon tube, and resuspended in 7 mL 1× T4 DNA ligase buffer. 400 U T4 ligase was added to the tube, and the sample was incubated overnight at 16 °C with 10 rpm vertical rotation. The ligated DNA was typically >10,000 bp. The ligation product was then treated with 60 µL proteinase K and incubated at 65 °C for 4 h or overnight to decrosslink the DNA. The sample was treated with 30 µL RNase A and incubated for 45 min at 37 °C before purified with phenol chloroform isoamyl alcohol. Then DNA was precipitated with 0.3 M sodium acetate pH 5.6, and equal volume of ice-cold isopropanol. After rinsing with 10 mL cold 70% ethanol, the obtained DNA was dissolved in 150 µL 10 mM Tris-HCl pH 7.5 and further digested at 37 °C overnight by adding 50 µL 10× DpnII restriction buffer, 50 U DpnII enzyme (NEB, Cat. No. R0543S), and nuclease-free water to 500 µL. Quality control was conducted to ensure that the DNA was digested to around 800 bp. The digested DNA was purified with 1.8× AMPure beads, eluted in 1 mL EB, and transferred to a new 50 mL tube. A second ligation was conducted by adding into the DNA with 1.4 mL of 10× ligation buffer, 400 U T4 ligase, and nuclease-free water to 14 mL. The mixture was incubated at 16 °C overnight with 10 rpm vertical rotation. The ligated DNA was extracted with an equal volume of phenol-chloroform isoamyl alcohol and was precipitated with 0.3 M sodium acetate pH 5.6 and an equal volume of ice-cold isopropanol. The pellet DNA was rinsed with 15 mL cold 70% ethanol, air dried, dissolved in 100 µL 10 mM Tris-HCl pH 7.5, further purified with 1.8× AMPure beads, and finally eluted in 30 µL EB.
To construct 4 C libraries, a 16-cycle inverse PCR was performed in 4 tubes, each with 200 ng ligated DNA as template and primers for the viewpoint (VP). The PCR products were purified with 1× AMPure beads and eluted in 50 µL EB. A second round 20-cycle PCR was conducted to construct the sequencing library. The amplified libraries were purified with 0.9× AMPure beads, eluted in 30 µL EB, and subjected to sequencing.
B cells derived from a normal individual with homozygous reference allele (C/C) at the rs558245864 variant position were applied as the control. The viewpoint (VP) was situated in the human-specific enhancer close to the BLK promoter (Fig. S8f). Primers for the 4C library construction are shown in Supplementary Data 2.
Chromatin immunoprecipitation sequencing (ChIP-seq) library construction for B-lymphoblastoid cells
Chromatin immunoprecipitation was performed using the Covaris truChIP-seq chromatin shearing kit (Covaris, Woburn, MA, USA, Cat. No. 520154). Briefly, EB(JC) cells were crosslinked with 1% methanol-free formaldehyde. The nuclei were isolated and subjected to sonication in a milliTUBE-1 mL with AFA fiber (Covaris, Cat. No. 520135) using the Covaris sonicator (Covaris, model S220) with default parameters (PIP 140, duty factor 5%, CPB 200, setpoint temperature at 6 °C). Sixty microgram sheared chromatin (corresponding to ten million cells) was incubated overnight with 5 µg antibodies against CTCF or H3K27ac coated on protein-G beads (Thermo Fisher Scientific, Cat. No. 10009D). The immunoprecipitated DNA was eluted from the protein-G beads and purified by DNA clean & concentrator-5 columns (Zymo, Cat. No. D4014). Then 1–10 ng purified enriched DNA was end-repaired, ligated to index adapters, and PCR-amplified with the ultralow V2 DNA-seq library preparation kit (Nugen, Cat. No. 0344NB) to construct libraries for sequencing.
In situ chromatin interaction analysis with paired-end-tag sequencing (ChIA-PET) library construction
The in situ ChIA-PET library construction for B-lymphoblastoid cell lines was performed as previously described98 with minor modifications. Briefly, 100 million B lymphocytes cultured to 0.6–0.8 × 106 mL were harvested by spinning at RT for 5 min at 200 g and rinsed once with 40 mL RT PBS. The cells were well resuspended with 41 mL RT PBS before being added with 37% formaldehyde to 1% final concentration. The cell suspension was vertically rotated at 20 rpm for 20 min at RT. After quenching with 3 mL 2 M glycine, the cells were incubated for 5 min with 20 rpm vertical rotation at RT. The cells were then centrifuged at 720 × g for 5 min at 4 °C and vertically rotated at 20 rpm for 10 min twice in 45 mL RT PBS to remove the residual formaldehyde. The cells were then crosslinked again with 42 mL 1.5 mM EGS in PBS and vertically rotated at 20 rpm for 40 min at RT. The crosslink was then quenched with 3 mL 2 M glycine and rinsed twice with 45 mL PBS for 10 min vertical rotation at 20 rpm.
As to the primary neuronal cells derived from human and macaque fetal brain tissues, the cell crosslink process was slightly modified to achieve the highest cell recovery and signal specificity. Briefly, the culture medium was removed from the primary neuronal cells cultured in 6-well plates, and each well was rinsed once with 2 mL RT PBS. Then, the cultured cells in each well were crosslinked with 2 mL 1.5-mM EGS (Sigma-Aldrich, Cat. No. E3257) in PBS for 10 min while rotating horizontally at 50 rpm at RT. In each well, formaldehyde was added to 1% final concentration, and then the cells were horizontally rotated at 50 rpm for another 15 min at RT. To quench formaldehyde and EGS, 200 µL of 2-M glycine was added to each well and the cells were rotated horizontally at 50 rpm for 5 min at RT. The cells were then transferred on ice, and each well was rinsed three times with 2 mL of ice-cold PBS. The dual-crosslinked cells were harvested by scraper and then subjected to the same procedure of standard in situ ChIA-PET experiments. The antibody against CTCF or RNAPII (BioLegend, San Diego, CA, USA, Cat. No. 664911) was applied in the experiments.
CRISPR/Cas9-mediated genome editing in hiPSCs
To knock out the human-specific CTCF binding site at the DLGAP1 locus (Fig. 7a), we designed an sgRNA sequence targeted to the CTCF binding site using the E-CRISP design tool99. The sgRNA sequence was cloned into a Cas9 vector (pSpCas9(BB)-2A-Puro, Addgene, Cat. No. 62988) with the Bbsl site to obtain the sgRNA-Cas9 plasmids. The hiPSCs were digested into single cells with Accutase (Thermo Fisher Scientific, Cat. No. A1110501), and ~5 × 104 cells were seeded into a well of 24-well plate pre-coated with Matrigel, incubating with 200 µL mTeSR plus for 24 h in the presence of 10 µM Y-27632 (MedChemExpress, Monmouth Junction, NJ, USA, Cat. No. HY-10071). Two hundred microliters opti-MEM (Thermo Fisher Scientific, Cat. No. 11058021) with 1 µg of plasmids were pre-mixed with two µL of lipofectamine stem transfection reagent (Thermo Fisher Scientific, Cat. No. STEM00001) and equilibrated to RT for fifteen min. The mixed opti-MEM was used to replace the mTeSR plus medium in the well to achieve transient transfection of sgRNA-Cas9 plasmids into hiPS cells. The cells were incubated at a 37 °C incubator with 5% CO2 for 8 to 12 h before replacing back to the 500 µL RT mTeSR plus medium. The cells were recovered in the mTeSR plus for 48 h. We then added a final concentration of 1 µg/mL puromycin (Sangon Biotech, Cat. No. A610593-0025) into the medium and cultured the cells for 48 h to select transfected cells. During the selection, the medium was fully replaced every 24 h. Next, the cells were recovered in mTeSR plus for 24 h and dissociated into single cells with Accutase. The digested cells were seeded on a 10-cm dish pre-coated with Matrigel and grown in mTeSR plus medium until colonies appeared. The medium was added with a final concentration of 10 µM Y-27632 for the first 24 h and then was fully replaced every 24 h. Individual colonies were picked with a P200 pipette under a microscope inside the sterile laminar flow cabinet. Each colony was transferred to a well of 24-well plate pre-coated with Matrigel and cultured in 500 µL mTeSR plus with a full medium exchange every day. To verify the knockout of the CTCF binding site, DNA was extracted from cells in each colony when the colony represented a size large enough, and the target DNA region was PCR amplified for Sanger sequencing. The Sanger sequencing confirmed that the CTCF binding site on each allele was successfully deleted in the selected colony (Fig. S10a). The obtained knock-out (KO) clone was applied for dorsal forebrain organoid differentiation. The sequences of the sgRNA and the primers for genotyping are listed in Supplementary Data 2.
Dorsal forebrain organoid differentiation
The dorsal forebrain organoids were cultured using STEMdiff dorsal forebrain organoid differentiation kit (Stemcell, Cat. No. 08620) according to the instructions of the manufacturer. Briefly, wild-type (WT) or KO hiPS cells displaying ~70% confluent and <10% differentiation were used for organoid differentiation. Cells with any trace of differentiation were removed by pipette tips. Then, cells in each well were rinsed once with RT PBS and dissociated into single cells with 500 µL gentle cell dissociation reagent (Stemcell, Cat. No. 100-0485) for 10 min at 37 °C. Five microliter DMEM/F-12 was added to the cells to quench cell dissociation. The dissociated cells were resuspended by slowly pipetting 3–5 times with 1-mL LoBind tips, and 10 µL cells were used for quality control to measure cell density and viability (>98%). A total of 4.5 × 106 cells were centrifuged at 30 × g for 5 min at RT and then resuspended in 1.5 mL seeding medium to achieve a cell density of ~3 × 106 cells/mL. Next, one mL of the single cell suspension was added to a well of the AggreWell800 24-well plate (Stemcell, Cat. No. 34811) pre-rinsed with anti-adherence rinsing solution (Stemcell, Cat. No. 07010) and pre-added with 1 mL seeding medium, resulting in 10,000 cells/microwell. The AggreWell plate was centrifuged at 100 × g for 3 min at RT in a swinging bucket and incubated in a 37 °C incubator with 5% CO2. From day 1 to day 5, 1.5 mL of the medium was replaced every day without disturbing the aggregated cells (embryoid bodies). On day 6, the embryoid bodies were dislodged from the AggreeWell plate and filtered through a 37-µm reversible strainer to remove single cells. The embryoid bodies were rinsed into a 50-mL tube and transferred with a 1-mL wide bore tip to a 6-well ultra-low adherent plate (Stemcell, Cat. No. 38071) with 2 mL forebrain organoid expansion medium per well. On average, we placed about 30 embryoid bodies per well. The embryoid bodies were incubated in a 37 °C incubator with 5% CO2, with all medium in each well replaced every 2 days until day 25. On day 25, the organoid expansion medium was replaced with 2 mL forebrain organoid differentiation media, and a full-medium replacement was performed every 2 days from day 27 to 43. Since day 43, the forebrain organoid differentiation medium was replaced with 2 mL dorsal forebrain organoid maintenance medium, and the medium was fully replaced every 2 to 3 days. The dorsal forebrain organoids were maintained for up to 100 days. Samples for RNA-seq, scRNA-seq, immunohistochemistry, qPCR, tagmentation mediated-chromatin conformation capture-qPCR, and patch clamp were collected at the indicated time points.
Preparation of single-cell suspension from dorsal forebrain organoids
The organoids were digested into single cells according to previously described100,101 with minor modifications. Briefly, day-14 or day-60 organoids of each genotype (WT and KO) were transferred with a Pasteur tube to a new well of the 6-well plate, respectively. The medium was removed, and the organoids were rinsed once with 1 mL RT PBS. After removing PBS, 50 µL of papain in HBSS with Ca2+ and Mg2+ was added to the organoids, and the organoids were cut into <1 mm3 with a sterile surgical blade. A volume of 950 µL papain and 100 µL DNase I in HBSS with Ca2+ and Mg2+ was added to the minced organoids, and then the minced organoids were shaken at 70 rpm for 15 (day-14 organoids) or 30 (day-60 organoids) min in a 37 °C incubator with 5% CO2. The minced organoids were then triturated with a 1-mL LoBind tip by pipetting slowly 12 times without introducing any bubbles. Then, the minced organoids were incubated for another 10 min at 70 rpm in a 37 °C incubator with 5% CO2. We then further triturated the organoids with a 5-mL pipette by pipetting slowly ten times without introducing any bubbles. The released cells and tissue debris were transferred to a new sterile 15-mL tube and rested for 2 min to allow undigested cell clusters to sediment to the tube bottom. The supernatant containing single cells was transferred without disturbing the sediments into a new sterile 15-mL tube pre-loaded with 8 mL 3.75 mg/mL ovomucoid (Sigma-Aldrich, Cat. No. 6056) and 0.075% BSA in HBSS without Ca2+ or Mg2+. The tube was inverted ten times to mix the cells and was then centrifuged at 300 × g for 7 min at RT to pellet cells. The cell pellet was then resuspended in 500 µL PBS with 0.04% BSA and filtered twice with 35-µm filters before quality control for cell viability (>95%), debris (<5%), clusters (<3%), and cell density (1100–1500 cells/µL). The cells were kept on ice for <30 min until subsequential processing.
Single-cell transcriptome analysis for primary neuronal cells and differentiated dorsal forebrain organoids
The single-cell transcriptome analysis of the primary neuronal cells and organoids was performed using chromium next GEM single cell 5′ reagent kits v2 (10x genomics, Pleasanton, CA, USA, Cat. No. PN-1000265) according to the manufacturer’s instructions (Rev C). Briefly, the prepared single-cell suspension was loaded in chip K to generate single-cell emulsion, aiming for the recovery of about 10,000 cells per run. After reverse transcription, cDNA cleanup, and amplification, quality control was performed to measure the cDNA concentration with the Qubit HS kit (Thermo Fisher Scientific, Cat. No. Q33230) and the size distribution with Qsep100 capillary electrophoresis system (BiOptic, Taiwan, China, Qsep100). In total, 50 ng cDNA was used for library construction via fragmentation, end repair, ligation to adapters, and indexed PCR amplification. The amplified library was applied for double-size selection using 0.6×–0.8× volume of SPRIselect beads (Beckman, Brea, CA, USA, Cat. No. B23318). Finally, the purified scRNA-seq library was ready for sequencing.
Single-cell isoform sequencing library construction for primary neuronal cells
The 5′ scRNA-seq cDNA that was produced in the above-mentioned single-cell transcriptome analysis was used for single-cell isoform sequencing (scISO-seq) library construction. Briefly, 50 ng of the 5′ scRNA-seq cDNA derived from human and macaque primary neuronal cells was amplified with PR1 primer and TSO primer (see Supplementary Data 2) for six cycles with the high-fidelity 2× PCR master mix (New England Biolabs, Cat. No. M0541). The amplified cDNA was then purified with 0.6× Ampure beads and eluted into 30 µL EB buffer. Next, the purified cDNA (~2 µg) was subjected to size selection using BluePippin size selection system (Sage Science, BluePippin) with 1.5% gel (Sage Science, Beverly, MA, USA, Cat. No. BDF1510) under the “1.5% agarose high-pass” program for collection of desired cDNA with fragment size > 1900 bp. The size-selected cDNA fragments were eluted at RT for 2 h, usually resulting in a yield of 200–300 ng cDNA fragments with length > 1900 bp. The recovered cDNA was further amplified for 5 cycles with PR1 and TSO primers using the high-fidelity 2× PCR master mix. The amplified cDNA (500–1000 ng) was purified with 0.6× AMPure beads and eluted in 40 µL EB for PacBio sequencing library construction.
The PacBio sequencing library was generated using SMRTbell template prep kit (PacBio, Menlo Park, CA, USA, Cat. No. 100-938-900) according to the instructions of the manufacturer. Briefly, 500 ng cDNA was subjected to damage repair, end repair, and A-tailing with the DNA repair mix and end-repair mix. Then, the cDNA was ligated with SMRTbell adapters and purified with 1.3× SMRTbell beads. The eluted DNA was treated with the nuclease mix to remove linear DNA molecules. The obtained SMRTbell libraries were purified with 1.3× SMRTbell beads and then applied for quality control for the DNA fragment size profile using the Agilent Bioanalyzer 2100 system. Each library was sequenced on one cell using the PacBio Sequel II system.
Relative quantification of DLGAP1 TSS usage in primary neuronal cells
DLGAP1-201 and DLGAP1-206 were primary transcripts of DLGAP1 in human and macaque primary neuronal cells, according to our analysis. To relatively quantify DLGAP1 TSS usage, we developed a single-cell TSS sequencing (scTSS-seq) method to enrich DLGAP1 TSS sequences from the 5′ scRNA-seq cDNA generated by the 10× genomics platform (Fig. S9h). Briefly, 50 ng of the 5′ scRNA-seq cDNA derived from human and macaque primary neuronal cells were first amplified by PCR for 12 cycles with the PR1 primer on the 5′-end of each cDNA fragment and a specific 3′-end primer that targets the unique exons of DLGAP1-201 or DLGAP1-206 transcript isoform at the 5′ end of the mRNA (P1R primers). The PCR products were purified with appropriate volumes of AMPure beads according to the desired DNA fragment sizes. Next, the purified DNA was applied for a ten-cycle nested-PCR amplification using the PR1 primer and another 3′-end primer that targets an inner position of the unique exons at the 5′ end of the DLGAP1-201 or DLGAP1-206 transcript isoform (P2R primers). Later, the custom PR2 primers overlapping with the P2R primers (P2R_PR2 primers) were used with the PR1 primer to amplify the purified nest-PCR products by six-cycle PCR. After DNA purification, the index primers (10× genomics, Cat. No. PN-1000215) were used to generate the sequencing libraries according to the instructions of the manufactory. The sequence information for the specifically designed 3′-end primers for human and macaque DLGAP1-201 and DLGAP1-206 is included in Supplementary Data 2.
Relative quantification of DLGAP1 TSS usage in differentiated dorsal forebrain organoids
We relatively quantify the DLGAP1 TSS usage in the WT and KO dorsal forebrain organoids using the method described above with some modifications. Instead of using cDNA from 5′ scRNA-seq, we applied RNA extracted from WT and KO dorsal forebrain organoids at the indicated time points by TRIzol (Thermo Fisher Scientific, Cat. No. 15596018). After removing gDNA with gDNA remover (Abclonal, Cat. No. RK20429), cDNA was generated through template-switch reverse transcription by the M-MLV transcriptase (Vazyme, Cat. No. N721), using the template-switch oligo with a 10-bp unique molecular identifier (TSO-UMI) and the primer designed for the first shared exon between DLGAP1-201 and DLGAP1-206 isoforms (RT_DLGAP1_E5 primer, targeting exon 5 of DLGAP1-201). The cDNA was PCR amplified for eight cycles with the high-fidelity 2× PCR master mix, using PR1 primer (same sequence as the 5′ end of TSO-UMI) and RT_DLGAP1_E5 primer. After purification, the amplified cDNA was divided into two parts to construct DLGAP1-201 and DLGAP1-206 TSS enrichment libraries, according to the above-described scTSS-seq protocol for human DLGAP1 TSS enrichment. All the primers for reverse transcription and cDNA amplification are listed in Supplementary Data 2.
Organoid fixation, paraffin embedding, sectioning, and hematoxylin and eosin staining
WT and KO organoids collected at the indicated time points were fixed in 4% paraformaldehyde (Sangon, Cat. No. E672002-0100) for >24 h at 4 °C. Then, the organoids were dehydrated in a dehydration box sequentially with 75% ethanol for 4 h, 85% ethanol for 2 h, 90% ethanol for 2 h, 95% ethanol for 1 h, anhydrous ethanol for 30 min for two rounds, alcohol benzene for 10 min, and xylene for two rounds of 10 min. The dehydrated organoids were embedded into paraffin wax pre-melted at 65 °C for 1 h and poured into an embedding frame. The organoids in wax were frozen at −20 °C until the wax solidified. The organoids embedded in paraffin were then sectioned into 4-µm slices with a pathology slicer (Leica, model RM2016) pre-cooled to −20 °C. The slices were flattened in 40 °C water, attached to glass slides, dried for 30 min at 60 °C, and stored at RT. The hematoxylin and eosin (H&E) staining of the organoid sections was performed with the H&E dye solution set (Servicebio, Cat. No. G1003). The H&E-stained sections were scanned using the Zeiss AxioScan Z1.
Fluorescent immunohistochemistry of the organoid sections
Fluorescent immunohistochemistry of the organoid sections was performed as previously described62. Briefly, the dewaxed paraffin sections were subjected to antigen retrieval, permeabilization, blocking, and then sequential staining with primary and secondary antibodies. Antibodies applied here included primary antibodies against NCAD (mouse, 1:200, CST, Danvers, MA, USA, Cat. No. 14215S), SOX2 (rabbit, 1:200, Abcam, Cat. No. ab97959), Ki-67 (mouse, 1:400, CST, Cat. No. 9449T), TBR1 (rabbit, 1:500, Abcam, Cat. No. ab31940), FOXG1 (rabbit, 1:200, Abcam, Cat. No. ab18259), TGIF1 (rabbit, 1:200, Abclonal, Cat. No. A16984), and MAP2 (chicken, 1:1500, Abcam, Cat. No. ab5392). Secondary antibodies were labeled at a dilution factor of 1: 500, including goat anti-mouse IgG (H+L) highly cross-absorbed secondary antibody, Alexa Fluor 555, goat anti-rabbit IgG (H+L) highly cross-absorbed secondary antibody, Alexa Fluor 488, and goat anti-chicken IgY (H+L) secondary antibody, Alexa Fluor 647 (Thermo Fischer Scientific, Cat. No. A-21449). DAPI was added together with the secondary antibodies to 5 µM to visualize nuclei. The organoid sections were mounted for microcopy on glass slides using ProLong glass antifade mountant (Thermo Fisher Scientific, Cat. No. P36984) and were imaged with an Olympus BX63 microscope. Images were processed in ImageJ (Fiji).
Electrophysiology for organoids
The dorsal forebrain organoids derived from hiPS WT and KO genotypes at day 60 were subjected to electrophysiological recordings. To ensure the reproducibility of our electrophysiological experiments across different genetic backgrounds, we also generated a KO cell line from the H9 human embryonic stem cells using the same approach as described for the hiPS KO experiment. The electrophysiological recordings in organoids were conducted using the patch clamp technique following a previously described method62,63 with minor modifications. In brief, the organoids were quickly fixed to a block of 4% agarose and submerged in ice-cold oxygenated (95% O2 and 5% CO2) artificial cerebrospinal fluid containing 126 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 1 mM MgSO4, 2 mM CaCl2, 26 mM NaHCO3, and 10 mM glucose (pH 7.3). The organoids were then sectioned into 250-µm slices using a vibratome (Leica VT-1000S). Immediately after sectioning, the slices were incubated in continuously oxygenated artificial cerebrospinal fluid at 32 °C for at least 30 min before recording.
Whole-cell patch-clamp recordings were performed at RT. Neuronal cells were selected under an upright infrared Nikon microscope with a ×40 water-immersion objective (Nikon, FN-1). Patch clamp was conducted using an EPC 10 amplifier (HEKA, EPC10) with patch master software (HEKA, V2x90.1). To record current-evoked firing, the organoid slices were transferred to a slice chamber and continuously perfused with the artificial cerebrospinal fluid (2 mL/min). The current-evoked firing was recorded using recording electrodes of borosilicate glass (4–6 MΩ) filled with potassium-gluconate-based internal resistance solution containing 130 mM K-gluconate, 10 mM KCl, 0.2 mM EGTA, 10 mM HEPES, 4 mM ATP, 0.5 mM GTP, and 10 mM Na-Phosphocreatine (pH 7.2–7.3, osmolarity 290 mOsm). Cells were held at a resting membrane voltage of −70 mV, and action potential (AP) signals were recorded after a stabilization period of at least three min. AP signals were recorded in each sweep with a 500 ms stimulation current ranging from −20 pA to 200 pA, incremented by 20 pA per sweep. The AP numbers occurring during each current step were recorded.
Miniature excitatory postsynaptic currents (mEPSCs) were monitored at RT under the voltage-clamp mode. To this end, the organoid slices were perfused with the bath solution (120 mM Cs-methyl sulfonate, 10 mM HEPES, 10 mM Na-phosphocreatine, 5 mM lidocaine N-ethyl bromide (QX-314), 4 mM ATP, 0.5 mM GTP, 10 μM bicuculline, 50 μM D-AP5, and 0.5 μM TTX, pH 7.2–7.3, osmolarity 290 mOsm) at 3 mL/min. To detect AMPA receptor signals, recording electrodes (4–6 MΩ) were filled with the internal resistance solution. The signals of mEPSCs were recorded for 5 min at a holding potential of −70 mV. To further determine if the recorded signals in the voltage clamp were glutamate-dependent, mEPSC signals were recorded for an additional 5 min with the AMPA receptor blocker CNQX (20 µM, Sigma-Aldrich, Cat. No. C127) added to the bath solution after a baseline recording of 2 min at minimum.
Cells were excluded if input resistance changed > 15% and/or series resistance changed > 10% throughout the experiment. Only cells with a series resistance < 20 MΩ and an input resistance > 100 MΩ were included in the quantification. The amplitude and frequency of mEPSCs were analyzed using the Mini Analysis Program (Synaptosoft, v6.0.7) with default parameters.
RNA preparation and RNA-seq library construction
Total RNA was extracted with TRIzol and dissolved in nuclease-free water. The RNA quality was controlled by Qsep100 capillary electrophoresis system. One microgram RNA was subjected for library construction with the Total RNA-seq library prep kit (Vazyme, Cat. No. NR603), according to the instructions of the manufactory.
Reverse transcription and real-time qPCR assays
Real-time qPCR was conducted to assess the relative expression levels for DLGAP1-201 and DLGAP1-206 in fetal brain tissues and organoids and for BLK in SLE patient B cells. One microgram RNA from each indicated sample was transferred to a nuclease-free tube, depleted of genomic DNA, and reverse-transcribed with ABScript III RT master mix for qPCR with gDNA remover. The obtained cDNA was used as the template in the qPCR by 2x universal SYBR green fast qPCR mix using the Bio-Rad real-time PCR system. The expression level of GAPDH was used as a control. All primers for qPCR are listed in Supplementary Data 2.
Assessment of the chromatin interactions between human-specific enhancers (HSEs) at the DLGAP1 locus and TSSs
To investigate the chromatin interactions between HSEs at the DLGAP1 locus and the TSSs of TGIF1, DLGAP1-206, and DLGAP1-201, we developed a tagmentation-mediated chromatin conformation capture (3C) method. Briefly, ~20 organoids at day 14 or day 60 were dissociated into single cells, following the procedure outlined in the “Preparation of single-cell suspension from dorsal forebrain organoids” section. These single cells were dual-crosslinked, employing the approach used for primary neuronal cells in the “In situ ChIA-PET library construction” method. Subsequently, ~2 million dual-crosslinked cells underwent AluI (New England Biolabs, Cat. No. R0137) digestion, A-tailing, proximal ligation to biotinylated linkers, and sonication, following the corresponding steps of in situ ChIA-PET method, with reagent quantities adjusted accordingly. The sonicated chromatin was de-crosslinked using 0.4% SDS and 1 mg/mL proteinase K, with horizontal rotation at 1100 rpm for 6–14 h at 65 °C. After de-crosslinking, DNA was purified by DNA clean & concentrator-10 columns (Zymo, Cat. No. D4011). One microgram of the purified DNA was then incubated with 30 µL Dynabeads MyOne streptavidin T1 (Thermo Fisher, Cat. No. 65601) to capture DNA fragments ligated to biotinylated linkers during proximal ligation. Following a thorough wash step, DNA attached to the beads underwent an on-bead tagmentation by 30 µL Tn5 transposase (Vazyme, Cat. No. S111) pre-loaded with annealed adapter complex A-B (refer to Supplementary Data 2).
Using the on-bead tagmented DNA as the template, three-round PCR amplification was performed to enhance the chromatin interaction signals anchored at the VP loci. VP primers were designed in proximity to the AluI restriction digestion sites around the TSSs of TGIF1, DLGAP1-206, and DLGAP1-201 (see Supplementary Data 2). In the initial round, a 15-cycle PCR was executed using the N5 index primer and VP primer. The resulting DNA was purified with Ampure beads (1.2×) and applied in the subsequent 5-cycle PCR, using the P1 primer and VP_PR7 primer. After Ampure bead purification, the PCR product was further amplified in a 5-cycle PCR, using the P1 primer and N7 index primer.
To quantify the chromatin interactions between the HSEs and the TSSs of TGIF1, DLGAP1-206, and DLGAP1-201, the amplified 3C DNA was used as the template in qPCR with the corresponding VP primer and one HSE primer (Figs. 8a, 3C-qPCR primers). The enrichment of chromatin interactions was normalized against the enrichment of a control genomic region lacking AluI restriction digestion sites. HSE primers were designed in proximity to the AluI restriction digestion sites in the HSEs defined in Fig. 6d. Refer to Supplementary Data 2 for the HSE primers.
Sequencing
All sequencing libraries, except for the otherwise indicated ones, were sequenced on the Novaseq 6000 (Illumina) using a pair-end 150 bp mode.
Alignment of syntenic regions across primate reference genomes
To investigate the difference in transcription factor binding and mediated interactions among primates, alignment of the syntenic regions between primate genome reference sequences is required. Firstly, we downloaded the reference genome sequences for humans (hg38), chimpanzees (panTro5), gorillas (gorGor4), and macaques (rheMac8) from ENSEMBL Genome Browser (https://www.ensembl.org). Inconsistencies in the coordinates across these genomes posed a challenge to the direct comparison of their regions. To identify the syntenic regions that align with the human genome and at least one of the non-human primate genomes, we performed liftOver and CrossMap (v0.5.1)102 to lift the coordinates of non-human primate genomes to the human genome with specific chain files. These chain files of pre-computed whole-genome BLASTZ alignments documented the chained and netted pairwise genome alignments between human and chimpanzee, gorilla, or macaque genomes, which we downloaded from UCSC Genome Browser. This approach allows us to obtain syntenic regions across diverse species for the distinguishment of differences in chromatin structures and transcriptional regulation.
Construction of orthologous transcripts
To perform cross-species comparisons between humans and non-human primates, we need to filter the gene annotations to obtain a shared list of genes between human and non-human primates. However, discrepancies in transcript IDs from different species make the comparison of gene expression levels across species problematic. To address this issue, we performed the XSAnno pipeline103 to construct orthologous transcripts across primates. Firstly, we use the “AnnoConvert” to call liftOver to lift the exons of all human transcripts to each of the non-human primate reference genomes. The liftOver parameter “-minMatch” was set based on bootstrapping to 0.98 for chimpanzees, 0.98 for gorillas, and 0.91 for macaques, respectively. Subsequently, the lifted exons from the whole genome alignment annotation were aligned to the reference genomes of both species using BLAT. The interspecies percent identity (PID) and the percentage of aligned length (PL) were selected as criteria to filter exons without unique or highly conserved orthologs by running the “BlatFilter” function. Both the inter-species PID and PL were set to 0.95 for humans-chimpanzees and humans-gorillas, and to 0.9 for humans-macaques. Finally, to eliminate potential bias introduced by the unmappable exons during differential expression analysis, we used simNGS (v1.6)104 to generate simulated RNA-seq data for all exons and used DEseq2 (1.30.1) R package105 to identify differentially expressed exons. After deleting the differential expressed exons, we identified 46,524 human-chimpanzee orthologous genes, 35,457 human-gorilla orthologous genes, 41,298 human-macaque orthologous genes (rheMac10, downloaded from ENSEMBL Genome Browser), and 39,599 human-macaque orthologous genes (rheMac8). These constructed orthologous genes were then utilized to compare the gene expression levels between humans and each of the non-human primates.
Processing of RNA-seq data
The RNA-seq data from the human, chimpanzee, gorilla, and macaque B-lymphoblastoid cell lines, and data from human and macaque primary neuronal cells were aligned to their respective reference genomes using hisat2 (v2.1.0)106. Following the alignment, we removed the low-quality mapping (MAPQ < 20) using “samtools view” function of samtools (v1.3.1)107. The remaining reads were subjected to coverage calculation to evaluate the depth of sequence coverage across the genomes, using “bamCoverage” function of deepTools2 (v3.4.3)108 with the parameter “--binSize 1”. In order to quantify the gene expression levels, we then counted reads on exons of each gene using “htseq-count” with reference gene annotations. The gene count data generated in the previous step was subsequently employed to calculate the fragments per kilobase of exon model per million mapped fragments (FPKM) metric, which serves as a representative measure of gene expression. It is worth noting that for comparisons involving humans and non-human primates, the gene annotations relied on orthologous genes generated by XSAnno.
Preprocessing of scRNA-seq data
To process scRNA-seq data obtained from human and macaque primary neuronal cells as well as human dorsal forebrain organoids, we employed the 10x Genomics Cell Ranger Suite (v3.1.0)109. This suite was utilized for the alignment of demultiplexed FASTQ reads to their respective reference genomes (RheMac10 for macaques and GRCh38 for humans). The unique molecular identifier (UMI) counts for each cell barcode were tallied using the “cellranger count” function, leading to the generation of gene count matrices. It’s important to highlight that for scRNA-seq dataset from the human dorsal forebrain organoids, the annotation file for GRCh38 genes was employed, while the human-macaque orthologous genes, as earlier described through the XSAnno methodology, were utilized for both the human and macaque primary neuronal cells. These gene count matrices, representing the count of UMIs associated with each gene for every cell, were imported into the R environment for subsequent analyzes.
To ensure the integrity and reliability of the data, a series of stringent quality control measures were applied to the raw gene count matrices. Cells that exhibited an excessive presence of UMIs (i.e., over 20%) derived from mitochondrial genes were systematically excluded. Furthermore, cells that fell short of detecting a minimum of 200 genes were prudently discarded. The identification and removal of potential doublets were accomplished through “doubletFinder_v3,” utilizing an anticipated doublet rate of 5%. Afterward, the meticulously filtered gene count matrices were analyzed as follows using the Seurat R package (v3.2.3)110. Firstly, the “FindVariableFeatures” function, deploying the “vst (variance-stabilizing transformation) “ method, was applied to identify the top 2000 most variable genes within each sample. Subsequently, gene count matrices underwent normalization and scaling using the “NormalizeData” and “ScaleData” functions, respectively. Principal component analysis (PCA) was conducted on the selected variable genes and the top 30 principal components (PCs) were retained via the “RunPCA” function, contributing to dimensionality reduction. To facilitate the visualization of all cells in a two-dimensional space, “runUMAP” function was performed to reduce the dimensionality of the PCA embeddings. The construction of a shared nearest neighbor (SNN) graph was accomplished through the “FindNeighbors” function. Finally, the “FindClusters” function was applied, employing SNN modularity optimization based on the original Louvain clustering algorithm to identify distinct cell clusters. This preprocessing methodology ensures that the scRNA-seq data is thoroughly prepared for downstream analyzes
Processing of ChIP-seq and CUT&Tag data
The CTCF and H3K27ac ChIP-seq from human (CTCF: GSM822312, GSM935611, and GSM733752; H3K27ac: GSE50893), chimpanzee (CTCF: ERP002246; H3K27ac: GSE60269), gorilla (this study), and macaque (CTCF: ERP002246; H3K27ac: GSE60269) B-lymphoblastoid cell lines, as well as CUT&Tag data from human and macaque primary neuronal cells, were aligned to their respective reference genome using “bwa mem” of BWA (v0.7.15)111 with default parameters. Following alignment, the low-mapping quality (MAPQ < 20) reads were filtered by “samtools view”. Subsequently, we removed PCR duplicates using “MarkDuplicates” function of Picard (v1.107) (https://broadinstitute.github.io/picard/). The obtained uniquely aligned reads were subjected to coverage calculation, to comprehensively assess the read coverage across the genome, using “bamCoverage” function of deepTools with the parameter “--binsize 1”. Subsequently, the “macs2 callpeak” function of MACS2 (2.2.7.1)112 was employed to identify the CTCF or H3K27ac peaks with default parameters. The identified peaks, representing CTCF or H3K27ac binding, were harnessed to further investigate CTCF or RNAPII-mediated chromatin interactions. Additionally, these H3K27ac peaks played a pivotal role in the identification of HSEs, shedding light on distinct regulatory elements that are unique to individual species.
Identification of human-specific enhancers (HSEs) in B-lymphoblastoid cell lines and primary neuronal cells
To delineate HSEs in B-lymphoblastoid cells, we performed differential enhancer analysis on H3K27ac ChIP-seq data from humans and non-human primates. In order to mitigate the influence of sample-specific peaks, we retained only those peaks consistently identified across different replicates of each sample, denoting them as consensus peaks. Consequently, we identified a total of 43,500, 44,518, 42,254, and 40,984 consensus H3K27ac peaks in the human, chimpanzee, gorilla, and macaque B-lymphoblastoid cell lines, respectively. Notably, due to discrepancies in genomic coordinates, we selectively identified a subset of H3K27ac peaks that exhibited syntenic alignment across species. To do this, we lifted those non-human primate consensus H3K27ac peaks to the human reference genome using liftOver with parameters “-minMatch” set to 0.9 for chimpanzees and gorillas, and 0.85 for macaques. Simultaneously, the human consensus H3K27ac peaks were reciprocally lifted to the non-human primate reference genomes. This procedure yielded a total of 75,648 syntenic H3K27ac peaks conserved across the four species. Subsequently, we counted the ChIP-seq reads on syntenic H3K27ac peaks using “htseq-count” function of HTseq (v0.11.2)113. Combining the count matrices of all replicates, we further obtained 66,191 syntenic peaks with a total of >100 overlapping reads per peak. The identification of differential enhancers between humans and chimpanzees, gorillas, or macaques was carried out with DEseq2 (1.30.1) R package using the thresholds of “|log2(fold change)| > 2 and FDR < 0.01”. Ultimately, our analysis yielded 3880 human-specific gained enhancers and 2445 human-specific lost enhancers in B-lymphoblastoid cells.
Similarly, we performed the differential enhancer analysis using H3K27ac CUT&Tag data from human and macaque primary neuronal cells to identify HSEs. Following the data processing, we obtained a total of 44,904 and 45,000 consensus H3K27ac peaks in humans and macaques, respectively. These peaks were further refined to 46,283 syntenic H3K27ac peaks that located on syntenic genomic regions between the human and macaque genomes. Subsequently, we counted unique mapping reads on these syntenic peaks and filtered out 45,984 syntenic peaks with a total read count of overlapping reads > 50. DEseq2 (1.30.1) R package was applied to identify the HSEs using the thresholds of “|log2(fold change)| > 1 and FDR < 0.01”. Finally, we delineated 4749 human-specific gained enhancers and 3767 human-specific lost enhancers in primary neuronal cells.
Processing of ChIA-PET data
The ChIA-PET data was conducted per the established pipeline outlined previously13. Briefly, the paired-end tag (PET) sequences were scrutinized for the presence of bridge linker sequences. Only those PETs harboring these requisite linkers were deemed suitable for further analytical exploration. The linkers were meticulously excised, and the sequences on each side were subsequently mapped to the reference genomes, using the “bwa mem” function of BWA (v0.7.15). Solely those PETs achieving unique alignments, with mapping quality (MAPQ) ≥ 30, were retained. In order to eliminate potential redundancies, duplicate PETs were effectively removed using the “MarkDuplicates.jar” function of Picard (v1.107). Moreover, to obviate potential bias stemming from sex-based differences in human and non-human primate samples, PETs originating from chromosomes X and Y were excluded from subsequent analyzes.
Each PET was classified as either a self-ligation PET (sPET, where both ends correspond to the same DNA fragment) or an inter-ligation PET (iPET, where the two ends correspond to different DNA fragments within the same chromatin complex). This classification was made based on the genomic span between the two ends of the PET: sPETs possessed a genomic span of less than 8 kb, while iPETs, indicative of long-distance interactions of particular interest, were characterized by a span exceeding 8 kb. To accurately reflect interaction frequencies between fragments and to define interaction regions, the ends of the iPETs were extended to 250 bp along the reference genome. The iPETs that exhibited overlaps at both ends (after extension) were aggregated into iPET clusters. The count of iPETs within a cluster served as an indicator of interaction frequency between the two genomic regions, which were deemed anchors. To gauge the reproducibility of the CTCF or RNAPII ChIA-PET libraries, the genome coverage of iPETs was computed using the “bamCoverage” function in deepTools (v3.4.3), at a resolution of 10 kb. Pearson correlation coefficient was calculated between each pair of replicates to evaluate reproducibility (Fig. S2a). Subsequently, uniquely mapped and non-redundant iPETs from all replicates of CTCF or RNAPII libraries for the same cell type were combined to generate the respective iPET clusters. Among them, the GM12878 CTCF (4DNES7IB5LY9) and RNAPII (4DNESZ25MOZV) ChIA-PET datasets were downloaded from 4D Nucleome Data Portal (4DN, https://data.4dnucleome.org/).
To identify chromatin interactions mediated by CTCF or RNAPII, several filters were applied to the iPET clusters. Clusters containing fewer than 4 iPETs were eliminated, and only clusters featuring a genomic span of less than 2 Mb between the two anchor regions were retained as CTCF- or RNAPII-mediated chromatin loops. Further refinement was undertaken separately for CTCF and RNAPII loops. For CTCF loops, only those displaying CTCF motifs and CTCF binding peaks on both anchors were preserved. Ultimately, this yielded 105,430, 104,319, 131,459, and 118,762 CTCF loops in GM12878, EB176, EBJC, and LCL8664, respectively. Additionally, 108,066 and 82,052 CTCF loops were identified in human and macaque primary neuronal cells, respectively. For RNAPII loops, a prerequisite was the presence of H3k27ac peaks on both anchor regions. Overall, we delineated 68,807, 91,959, 109,459, and 100,860 RNAPII loops in GM12878, EB176, EBJC, and LCL8664, respectively. Meanwhile, 168,016 and 131,936 RNAPII loops were recognized in human and macaque primary neuronal cells, respectively. These CTCF and RNAPII loops were used to explore changes in chromatin structures and transcriptional regulation during primate evolution.
Comparison of CTCF loop identification between Peakachu and ChIA-PET pipeline
We compared the loops identified by Peakachu114 and our ChIA-PET pipeline as described above. First, the raw CTCF ChIA-PET data for each species and cell type were converted into 5 kb resolution contact matrices using the hicConvertFormat tool from the hicExplorer package, providing the foundational data for subsequent chromatin loop predictions. Next, chromatin loops for each cell type were predicted using Peakachu, with the parameters: peakachu score_genome -r 5000 -m CTCF-ChIAPET-peakachu-pretrained.150 million.10 kb.pkl. To ensure the reliability of the identified loops, we applied stringent filtering to the predicted loops using the command peakachu pool -r 5000 -t 0.975. Loops with PET counts below 8 were further excluded. Ultimately, Peakachu identified 19,404, 17,149, 16,634, and 16,813 CTCF loops in B cells from humans, chimpanzees, gorillas, and macaques, respectively. Additionally, 24,469 and 14,077 CTCF loops were identified in primary neuronal cells from humans and macaques, respectively. For comparison, loops identified by the ChIA-PET pipeline were subjected to the same filtering criteria, excluding loops with PET counts below 8. Using this approach, we identified 52,230, 52,542, 68,054, and 60,299 CTCF loops in B cells from humans, chimpanzees, gorillas, and macaques, respectively, and 60,722 and 39,357 loops in primary neuronal cells from humans and macaques, respectively. The results of the comparative analysis are presented in Figs. S2b and 2c.
Identification of matin contact domains
CTCF-mediated chromatin loops delineated in B-lymphoblastoid cells were used to identify chromatin contact domains (CCDs). We initially divided each chromosome into 10-kb bins. Pairs of such bins within the same chromosome were then combined to form a contact matrix. Subsequently, we meticulously extracted the sub-matrix corresponding to each CTCF loop and populated it with PET counts specific to that particular loop. The amalgamation of these filled sub-matrices yielded a comprehensive contact matrix, where the contact map served to depict the coverage of CTCF loops for each genomic bin along the chromosome. In order to determine CCDs in chromosomes, we employed the insulation score (IS) as a metric to gauge the intensity of CTCF interactions for each genomic bin. The ISs were computed across the entire genome using the “matrix2insulation.pl” script from the cworld::dekker package (https://github.com/dekkerlab/cworld-dekker). It is noteworthy that we determined the minimum IS value for each chromosome by identifying the 0.01 quantile of IS values specific to that chromosome. The positions corresponding to the lowest IS points were designated as the boundaries of candidate CCDs. The genomic region encompassed between these designated boundaries was regarded as a candidate CCD. To further refine the candidate CCDs, we scrutinized the maximum coverage of CTCF loops within each candidate. Only those candidate CCDs exhibiting a maximum coverage surpassing the 25% quantile threshold were retained as valid CCDs. Furthermore, we meticulously adjusted the boundaries of CCDs to align with the nearest CTCF sites positioned on the CTCF anchors. Finally, we recognized 1286, 1064, 1342, and 1105 CCDs in human, chimpanzee, gorilla, and macaque B-lymphoblastoid cell lines, respectively (Fig. S3b). The genomic span between the two boundaries of each CCD was scrutinized, revealing an average span of ~1.8 Mb. Furthermore, the analysis of the orientation of CTCF sites situated at the boundaries of each CCD revealed that roughly 60.5% of these CTCF sites were convergent, with this proportion displaying a uniform distribution across species (Fig. S3c).
Identification of human-specific CCDs
To discern human-specific CCDs, we embarked on a comprehensive analysis, comparing the IS of CCD boundaries between humans and chimpanzees, gorillas, or macaques in B-lymphoblastoid cells and primary neuronal cells, separately. The results unveiled substantial variability in the range of IS values across different chromosomes among these species. However, the direct identification of human-specific CCD boundaries by contrasting the differences in ISs between human CCD boundaries and their syntenic loci in non-human primates proved to be an intricate challenge. To eliminate background differences in ISs across species, we meticulously standardized the IS values for entire chromosomes, ensuring that the scores were confined within the range of −1 to 1 for each chromosome. Subsequently, we lifted the CCD boundaries from humans to the chimpanzee reference genome using liftOver, concurrently reciprocating the process for the CCD boundaries from chimpanzees to the human reference genome, with a parameter setting of “-minMatch = 0.9”. This strategic step facilitated a direct comparison between the IS values of human CCD boundaries and their syntenic loci within the chimpanzee genome. Notably, those boundaries located in non-syntenic regions were directly recognized as human- or chimpanzee-specific boundaries. The differential IS, termed delta IS, was calculated by subtracting the IS values of syntenic loci in the chimpanzee genome from those of human CCD boundaries. When the delta IS plummeted below −0.85, the corresponding boundary was classified as a human-specific boundary. Conversely, when the delta IS exceeded 0.85, the CCD boundary was denoted as a chimpanzee-specific boundary (Fig. S3e). This analytical approach was extended to identify human-specific CCDs in comparisons with gorillas or macaques.
Overall, 1040 of the human CCD boundaries are specific relative to at least one of the chimpanzees, gorillas, and macaques, referred to as non-human primate variable boundaries (Variable). While the remaining 1532 boundaries are conserved across four species, referred to as conserved boundaries (Consv), serving as the control for subsequent investigations. In a bid to illustrate the insulating effects of these human-specific boundaries, we implemented an approach akin to aggregate peak analysis. This analytical technique facilitated the portrayal of CTCF loop intensity within a given CCD and, of equal importance, the demonstration of CTCF loops occurring between adjacent CCDs. For these Variable or Consv boundaries within human B-lymphoblastoid cells, we initially identified their syntenic loci within non-human primate genomes using liftOver. Subsequently, we selected each pair of CCDs adjacent to every Variable or Consv boundary and systematically subdivided each CCD into 100 bins. This process culminated in the generation of a 200 × 200 matrix for each pair of adjacent CCDs. Within these matrices, we quantified the intra-pair PETs between every two bins and aggregated them, ultimately yielding contact maps as illustrated in Fig. 2b. Concurrently, we evaluated the intensity of CTCF loops crossing the boundaries of HS, APE, Misc, or Consv type in humans (Fig. 2c). This analysis underscored the formidable insulating impact of HS, APE, and Misc boundaries in human B-lymphoblastoid cells, with almost no CTCF loops crossing these boundaries, in stark contrast to the scenario observed in other species.
Genetic characteristics around human-specific CCD boundaries
To assess the conservation of the identified variable boundaries across different cell types, we determined the insulation scores for these boundaries using CTCF ChIA-PET data from a variety of human cell types.
Specifically, we acquired the hic files for CTCF ChIA-PET data from HFFc6 (human foreskin fibroblast cell line, accession number: 4DNESCQ7ZD21), WTC-11 (human induced pluripotent stem cell line, accession number: 4DNESHWX9JLY), and H1-hESC (human embryo stem cell line, accession number: 4DNESR9S8R38) from the 4DN database. Additionally, the hic files from primary neuronal cells were generated in-house. These hic files were then converted to hicpro format using the “hicConvertFormat” function of HiCExplorer (v3.5.1)115 with a resolution of 1 kb. Subsequently, we calculated the insulation scores within a range of ±500 kb around the variable or conserved (Consv) boundaries for each cell type using “genome.wide.insulation” function of GENOVA (v0.9.98)116. Subsequently, we plotted the heatmaps and mean profiles using “insulation.heatmap” function of GENOVA (v0.9.98). These profiles revealed that both variable and conserved boundaries defined in B-lymphoblastoid cells consistently exhibit low insulation scores in human different cell types (Fig. S3h).
To explore the genomic sequence features at the variable boundaries, we gathered annotations for the Genomic Evolutionary Rate Profiling (GERP) score, human-specific bases (extracted from “Age of Base”), and transposons from the RepeatMasker database (https://www.repeatmasker.org/). Using the “computeMatrix” function of deepTools (v3.4.3), we quantified specific bases, all short interspersed nuclear elements (SINE), and all long interspersed nuclear elements (LINE) within a range of ±500 kb separately around all the variable and Consv boundaries. Additionally, we randomly selected 1,000 CTCF sites from regions outside the identified CCD boundaries as controls and referred to them as Random. The results revealed that the sequences of variable boundaries are not evolutionarily conserved and are enriched with human-specific bases compared to both Consv boundaries and Random control (Fig. S3i,j).
To characterize the distinct profiles of H3K27ac histone modifications in various cell types at variable boundaries, we collected genomic coverage for H3K27ac ChIP-seq from 33 primary tissues or cell lines in the Roadmap Epigenomics database (https://egg2.wustl.edu/roadmap/). We employed CrossMap (v0.5.1) to lift the coverage signals of these H3K27ac ChIP-seq data to the hg38 reference genome. Subsequently, we used the “computeMatrix” function of deepTools to calculate the H3K27ac profiles within a region spanning from 150 kb upstream to 550 kb downstream of variable and Consv boundaries. Heatmaps were generated to visualize the mean H3K27ac intensity at all variable CCD boundaries for each cell/tissue type (Fig. 2f). Additionally, we presented detailed H3K27ac modifications at the CCD boundaries in eight primary tissues, including six brain tissues from different anatomical regions, T memory cells, and the rectal smooth muscle. The heatmap and curves of mean H3K27ac intensity were plotted at both variable and Consv boundaries. In parallel, we selected 1000 random CTCF sites located outside the identified CCD boundaries as controls, naming them as Random. This analysis demonstrated that the variable boundaries effectively constrained the H3K27ac signal to the interior of the CCDs and that the signal intensity gradually decreased with increasing distance from the boundary (Fig. 2g and Fig. S4b). Notably, this enrichment effect was more pronounced for H3K27ac signals in human brain regions compared to other tissues.
Heritability enrichment across categorized CCDs
Heritability enrichment was estimated using LD Score Regression (LDSC), which calculates the proportion of heritability explained by single-nucleotide polymorphisms (SNPs) within a specific annotation, relative to the proportion of SNPs within that annotation33. To compute the heritability, we defined regions upstream (150 kb) and downstream (550 kb) of the CCD boundaries, segmenting these regions into 15 bins of 50 kb each. Heritability enrichment was then calculated for different types of CCD boundaries using S-LDSR across 44 traits. LDSC software was downloaded from http://www.github.com/bulik/ldsc. Summary statistics for the traits used in the heritability enrichment calculations were obtained from Finucane HK et al.33. A detailed outline of the analysis step is provided below.
In order to facilitate the correspondence of the boundary regions with the available trait annotation information, we aligned the coordinates of the human-specific boundaries from the hg38 reference genome to the hg19 reference genome using liftOver. An essential step in our analysis involved the creation of an annotation for the human-specific CCD boundaries, a process initiated using the “make_annot.py” function. To provide the required reference for LD score calculations, precomputed European LD scores were obtained from the Broad Institute’s resources (https://data.broadinstitute.org/alkesgroup/LDSCORE/eur_w_ld_chr.tar.bz2). Subsequently, LD scores were meticulously calculated for the annotation with reference to Single Nucleotide Polymorphisms derived from HapMap3 (https://data.broadinstitute.org/alkesgroup/LDSCORE/w_hm3.snplist.bz2) using “ldsc.py” function. Finally, we ran “ldsc.py” on summary statistics for the specified traits using baseline-LD model annotations and the annotation of interest. In this analysis, we assigned a total of 44 traits and grouped them into five categories covering the neuropsychological, metabolic, immunologic, cardiopulmonary, and hematologic features. This methodological approach ensures a systematic examination of the genetic correlations associated with human-specific CCD boundaries and their relationship to a broad spectrum of traits, shedding light on the potential genetic underpinnings of these unique genomic regions in human complex traits and diseases (Figs. 2d and S4a).
Co-expression correlation analysis of gene pairs situated on opposite sides of the human-specific CCD boundaries
To investigate the influence of human-specific CCD boundaries on gene expression, we conducted a comparative analysis of the gene pairs situated on opposing sides of variable boundaries in distinct brain regions of humans and macaques. In the initial step, we collected gene expression data from human and macaque primary brain tissues. Gene expression profiles for 16 anatomical brain regions in humans were sourced from the BrainSpan database (http://www.brainspan.org/), while RNA-seq data for corresponding brain regions in macaques were obtained from the National Center for Biotechnology Information (NCBI, PRJNA448973). Following the processing of the RNA-seq data, we calculated the expression profiles of all macaque reference genes in divergent brain regions using the “mrfQuantifier” function of RSEQtools (v0.6)117. Consequently, we obtained expression profiles of the human and macaque brain regions.
To compare gene expression correlations across various brain regions, homologous gene pairs common to humans and macaques were identified and selected. To do this, all human genes within a 300-kb range on either side of variable boundaries were paired. Only orthologous gene pairs in humans and macaques, as delineated by XSAnno annotation, were retained for subsequent analyzes. The remaining homologous gene pairs were distributed across two CCDs separated by variable boundaries in humans, while they resided within the same CCD in macaques. Similarly, homologous gene pairs situated on both sides of the conserved boundaries were chosen to serve as controls. These control gene pairs were segregated by conserved boundaries into two CCDs in both humans and macaques (Fig. 2h). Consequently, two sets of gene pairs were obtained: 3999 pairs on both sides of variable boundaries and 4933 pairs on both sides of conserved boundaries to calculate gene expression correlations.
Leveraging the gene expression profiles from various brain regions in humans and macaques, we computed Pearson correlation coefficients for each gene pair. The expression correlations of selected gene pairs within the medial prefrontal cortex were plotted against the genomic span between the two gene TSSs. Notably, the correlations of gene pairs on both sides of human-specific CCD boundaries declined as the distance between TSSs increased (Fig. 2i). Finally, we selected three gene pairs, each pair positioned on both sides of a human-specific (HS) boundary, and generated their expression plots across three brain regions. After computing their expression correlations, we observed a lower degree of correlation in humans compared to macaques (Figs. 2i and S4d–f).
Comparison of DNA methylation profiles between human and macaque neuronal cells within the PCDH gene cluster
Scanning the human-specific CCD boundaries, we found a human-specific CCD boundary located upstream of the PCDH-γ gene cluster in B-lymphoblastoid cells. To confirm its human-specific nature, we scrutinized non-human brain tissues for the presence of this boundary. The contact matrices from the germinal zone and cortex plate of macaques (GSE163177) were acquired. Contact matrices corresponding to the syntenic region of the PCDH gene cluster were extracted and visualized using “hicPlotMatrix” function of HiCExplorer (v3.5.1) (Fig. S5a). These matrices indicated that numerous interactions between PCDH-β and PCDH-γ genes occurred in non-human brains, thus affirming the absence of the boundary in these species. Subsequently, we assessed the intensity of CTCF-mediated interactions that crossed the human-specific boundary and the syntenic locus in human and macaque primary neuronal cells, respectively.
To further investigate the occurrence of the human-specific boundary, we embarked on an exploration of the genome sequence bases within the CTCF motifs in humans, as well as within the syntenic loci of chimpanzees, gorillas, and macaques. However, these alignments revealed no discernible base variants. Given that changes in CTCF binding efficacy are known to anticorrelate with DNA methylation levels within the CTCF DNA binding sequences118,119,120, we proceeded to assess the DNA methylation levels within the PCDH-β and PCDH-γ gene clusters in both human and macaque neuronal cells. To undertake this, we downloaded whole-genome bisulfite sequencing data of neuronal cells sorted from the dorsolateral prefrontal cortex tissues of 25 human individuals (GSE108066) and 15 macaques (GSE151768). To quantify DNA methylation levels within the PCDH gene cluster, we partitioned the genomic region into 1-kb bins and subsequently calculated the frequency of methylated cytosines within each bin to generate methylation profiles (Fig. S5b). Additionally, we meticulously computed the frequency of methylated cytosines across 19 bases within each CTCF binding site and subsequently compared the methylation levels of these sites between human and macaque neuronal cells (Fig. S5c). We discerned CTCF sites exhibiting significantly higher methylation levels in human neuronal cells at the PCDH-β locus compared to macaques (Fig. S5d).
Quantification of the expression combinatory of PCDH genes in individual neuronal cells using scRNA-seq data
Possessed scRNA-seq data from primary neuron samples, we obtained 8060 and 11,480 cells in humans and macaques, respectively. These cells were meticulously annotated based on their expression profiles of well-established marker genes. The annotation procedure facilitated the classification of these cells into distinct categories, namely, neuron progenitor cells (NPC), intermediate progenitor cells (IPC), and excitatory neurons (EN) (Fig. S1c, d). There are 1648/1534 NPC, 1253/2764 IPC, and 5159/7182 EN in the human/macaque primary neuron samples, respectively (Fig. S1e). It is noteworthy that the majority of cells in the primary neuron samples represented excitatory neurons.
To evaluate the differences in the expression of the PCDH gene cluster between human and macaque primary neuronal cells, we counted the number of expressed PCDH genes in each individual excitatory neuronal cell. A bubble plot was employed to visualize the expression levels and proportions of individual PCDH genes (Fig. S6g). Furthermore, Fig. S6e illustrates the distribution of cells with different numbers of expressed PCDH genes. To delve deeper into the combination of expressed PCDH genes between human and macaque neurons, we utilized the TF-IDF (term frequency-inverse document frequency) normalization method on the gene count matrices of human and macaque excitatory neurons. After combining the normalized matrices, hierarchical clustering was applied to sub-matrices of the PCDH cluster genes. Consequently, all excitatory neurons were divided into 9 clusters, each expressing unique combinations of PCDH genes (Fig. 3h). We then calculated the proportion of human and macaque excitatory neurons in each cluster. Notably, the majority of cells in clusters 1 and 2 were human excitatory neurons, while Clusters 8 and 9 primarily consisted of macaque excitatory neurons (Fig. S6f).
Identification of human-specific CTCF loops
Processing CTCF ChIA-PET data from human, chimpanzee, gorilla, and macaque B-lymphoblastoid cells, we yielded a substantial number of CTCF loops. However, due to inherent disparities in the coordinates of these CTCF loops across species and the inconsistency in the genomic span between anchors of each CTCF loop, direct identification of differences using the DESeq2 (1.30.1) R package proved challenging. To address this issue, we performed a coordinate transformation, employing the liftOver tool, to lift the coordinates of CTCF loop anchors from non-human primates to the human genome, and reciprocally from humans to non-human primates. This enabled the establishment of a set of syntenic anchors, representing those anchors amenable to alignment across the four species. The “minMatch” parameter was set to 0.9 for chimpanzees or gorillas and 0.85 for macaques during the conversion from humans to non-human primates, with a uniform 0.9 for all non-human primates in the reversed conversions. It is worth noting that a few CTCF loop anchors were inevitably lost due to genome assembly limitations and genuine genomic structural variations. Impressively, ~97.8% of the CTCF loop anchors in human B-lymphoblastoid cells could be successfully lifted to the genome of chimpanzees, with 92.1% and 82.5% for gorillas and macaques, respectively. Reciprocally, 98.4% of CTCF loop anchors from chimpanzee B-lymphoblastoid cells, 97.3% from gorillas, and 87.8% from macaques were aligned with the human genome, underscoring the efficacy of this anchoring approach. While it is acknowledged that non-human primate genome assembly quality posed certain limitations, these did not significantly impede the anchor alignment process. Consequently, a set of 30,743 syntenic anchors was successfully identified across the genomes of humans, chimpanzees, gorillas, and macaques.
To identify human-specific CTCF loops, we initially combined syntenic anchor pairs to construct putative CTCF loops, and quantified CTCF iPETs for each loop across all replicates of CTCF ChIA-PET datasets in B-lymphoblastoid cells. A meticulous selection process ensued, considering only CTCF loops with a cumulative iPET count exceeding 10 across all replicates of the four species. This comprehensive approach culminated in the identification of a total of 78,146 CTCF loops. The critical task of discerning human-specific CTCF loops was achieved by employing the DESeq2 (v1.30.1) R package to contrast the intensity of CTCF loops in GM12878 with those in EB176, EBJC, and LCL8664, respectively. The criterion for classifying a CTCF loop as species-specific was established as “|log2(fold change)| > 1, FDR < 0.05”. This rigorous assessment yielded a total of 3579 human-specific gained and 2567 lost CTCF loops between humans and chimpanzees, 4674 and 3773 between humans and gorillas, and 6289 and 7333 between humans and macaques in B-lymphoblastoid cells (Fig. S7a). Notably, the results corroborated the evolutionary relatedness among the species, elucidating that humans share a closer phylogenetic kinship with chimpanzees and gorillas compared to macaques. The outcomes of these three comparative analyzes were amalgamated, resulting in a refined selection of 20,479 CTCF loops, which were subsequently classified using a self-organizing map model based on fold changes in normalized loop intensity. A final total of 9670 human-gained and 8913 -lost CTCF loops were obtained in B-lymphoblastoid cells, further categorized into 2133 human-specific (HS) gained, 2418 HS lost, 2322 great apes-specific (APE) gained, 2623 APE lost, and 9147 miscellaneous (Misc) types of CTCF loops in B-lymphoblastoid cells (Fig. S7c).
Expanding the scope of comparison to human and macaque primary neuronal cells, 80,678 syntenic CTCF loops were identified. Employing DESeq2 (1.30.1), 5873 human gained and 6708 lost CTCF loops were identified using the parameters “| log2(fold change) | > 1, FDR < 0.05” (Fig. S7b). This comprehensive analysis facilitated the identification and categorization of human-specific CTCF loops across diverse cell types, shedding light on the intricate landscape of CTCF interactions among different primate species.
Aggregate peak analysis for human-specific CTCF loops
The process of visualizing human-specific gained and lost CTCF loops involved the utilization of Aggregate peak analysis (APA) through GENOVA (v0.9.98) package. In the initial step, all intra-chromosomal iPETs were transformed into contact maps with a 1-kb resolution by leveraging the juicer_tool (v0.7.5)121. Notably, to ensure the coordinate concordance with the human reference genome, the intra-chromosomal iPET from non-human libraries were systematically lifted to the human reference genome. Notably, iPETs with genomic spans surpassing 2 Mb were excluded. Subsequently, the contact maps of human, chimpanzee, gorilla, and macaque B-lymphoblastoid cells were translated into the requisite input format of GENOVA (v0.9.98), featuring bin1, bin2, and interaction counts, facilitated by the “hicConvertFormat” function within the HiCExplorer tool (v3.5.1). To mitigate the influence of background signals in proximity to the diagonal and to rectify discrepancies arising from differences in sequencing depth among the samples, a series of data preprocessing steps was executed. Specifically, interactions within intra-bins were systematically filtered out, and the interaction counts were normalized by the total number of appropriately aligned interactions. Immediately following this, APA heatmaps of HS gained, lost, and Consv CTCF loops were generated using the “APA” function in GENOVA (v0.9.98) package with the parameter “size = 40 + 1”. The resultant APA outcomes were then visualized using the “visualise.APA.ggplot” function in GENOVA (v0.9.98) package (Fig. 4b, c). To illustrate the impact of CTCF-mediated modifications in chromatin architecture on the transcriptional regulatory dynamics driven by RNAPII, identical computational procedures were executed to visualize the spatial distribution of RNAPII iPETs in the proximity of the CTCF loop (Fig. 4e, f).
Characteristics at the anchors of human-specific CTCF loops
To provide a comprehensive characterization of human-specific CTCF loops, we first characterized the attributes of the anchors of these loops. The anchor sequences were meticulously filtered based on the presence of nucleotide variations. This was accomplished by aligning the sequences of the CTCF binding sites in humans with their syntenic loci located on the loop anchors in non-human primates. For human-specific gained and lost CTCF loops, loop anchors that lacked any nucleotide variation among the four species were systematically eliminated, and only those with discernible base variations were retained for subsequent analysis. Conversely, in the case of conserved CTCF loops, only the loop anchors that displayed no base variation at the CTCF binding site were retained. In the context of B-lymphoblastoid cells, the research identified a total of 1129 anchors associated with human-specific gained CTCF loops, 1254 anchors linked to human-specific lost CTCF loops, and 8044 loop anchors classified as conserved. On the other hand, in primary neuronal cells, there are 2184 anchors for human-specific gained CTCF loops, 2039 anchors associated with human-specific lost CTCF loops, and 9260 anchors designated to conserved loops. To further elucidate the distinctiveness of these loop anchors, we conducted CTCF binding profiling on these anchors using the “computMatrix” function of deep tools (v3.4.3). Fig. S7e showed that in human B-lymphoblastoid cells, the strongest and weakest CTCF binding was predominantly observed at the anchors of human-specific gained and lost CTCF loops, respectively. Additionally, the GERP and the distribution of human-specific base sequences around the CTCF binding sites within the human-specific and conserved CTCF loop anchors were assessed using the “computMatrix” function. As Fig. S7f illustrated, the sequences of the CTCF binding sites within human-specific loop anchors exhibited lower levels of conservation compared to those in conserved CTCF loop anchors. As Fig. S7g illustrated, the human-specific CTCF loop anchors displayed a distinct enrichment of human-specific nucleotide substitutions at their CTCF binding sites.
To investigate the distribution of transcriptional regulatory elements within the human-specific CTCF loop anchors, we employed the “ame” function of the MEME suite122 to determine the enrichment of 443 transcription factor (TF) motifs obtained from the JASPAR database (https://jaspar.genereg.net/) at the human-specific CTCF loop anchors. Notably, we redefined the 30-kb region downstream of the CTCF binding site on the anchor as the CTCF loop anchor region (Fig. 4d). The TF enrichment was quantified using q values computed from the result of “ame” function. The result revealed a notable enrichment of TFs associated with specific cell types at anchors in both B-lymphoblastoid cells and primary neuronal cells (Fig. 4g, h). Furthermore, we investigated the distribution of HSEs within a defined genomic range of the anchors of the human-specific CTCF loops. Specifically, the range extended from 40 kb upstream to 100 kb downstream of the anchors. To ensure the robustness of this analysis, random anchors were also selected from the conserved CTCF loop anchors. In total, 1000 random samples were generated to serve as a control group. The results demonstrated that gained and lost HSEs were most densely concentrated within human-specific gained and lost CTCF loop anchor regions, respectively, with their frequency diminishing as the distance from the CTCF sites increased (Fig. 4i, j). These HSEs, found within the human-specific CTCF loop anchor regions, were denoted as human-specific loop-associated HSEs.
To gain further insights into the functional implications of these human-specific loop-associated HSEs, we calculated the intensity of RNAPII-mediated chromatin interactions involving these human-specific loop-associated HSEs. The results conclusively showed that these human-specific gained and lost loop-associated HSEs were significantly more and less involved in RNAPII-mediated chromatin interactions, respectively, in humans compared to other species, in both B-lymphoblastoid cells and primary neuronal cells (Fig. 4k, l). Moreover, a comparison of the expression levels of target genes regulated by these human-specific CTCF loop-associated HSEs through RNAPII-mediated chromatin interactions was undertaken. As presented in Fig. 4m, n, this analysis revealed that these target genes displayed significantly differential expression levels between humans and other species. These differences were consistently observed in both B-lymphoblastoid cells and neuronal cells, underscoring the potential functional significance of human-specific CTCF loops and HSEs in gene expression regulation across these cell types.
GWAS traits enrichment of human-specific gained CTCF loops
To explore potential associations between human-specific gained CTCF loops and diseases, we specifically focused on the GWAS traits enriched on the enhancers situated in the vicinity of the anchor regions of these loops. In the initial step, we identified all enhancers situated within 50 kb of the anchors within the human-specific CTCF loops, encompassing both HSEs and conserved enhancers. Subsequently, we conducted a comprehensive trait enrichment analysis utilizing genetic data from the European population in conjunction with trait annotations derived from the GWAS catalog (https://www.ebi.ac.uk/gwas/). Specifically, we curated single nucleotide polymorphisms (SNPs) of the European population and retrieved trait annotations for these SNPs from the GWAS catalog. For each trait, we quantified the number of SNPs that coincided with these delineated enhancers and evaluated the statistical significance of enrichment through chi-square tests. As illustrated in Figs. S7n and 7o, the SNPs located within B-lymphoblastoid cells exhibited noteworthy enrichment in entries associated with immune system disorders, including SLE and rheumatism. On the other hand, SNPs in primary neuronal cells displayed significant enrichment for traits linked to neurological conditions, such as schizophrenia and Alzheimer’s disease.
The InDel rs558245864 disrupts the human-specific CTCF site and results in the alteration of the BLK expression
To validate the impact of rs558245864 on CTCF binding to DNA, CTCF ChIP-seq data from six B-lymphoblastoid cell lines (ERP002168) were obtained. The six cell lines display different genotypes, comprising two homozygotes for the rs558245864 allele (GM12006 and GM12287), two heterozygotes (GM12003 and GM12815) for the rs558245864 allele, and two homozygotes for the reference allele (GM12814 and GM12004). Subsequent processing and analysis of these CTCF ChIP-seq data, as demonstrated in Fig. S8a, confirmed that rs558245864 indeed affected CTCF binding strength to DNA.
Furthermore, additional data, including ChIP-seq data for YY1 (GSM803406), NIPBL (GSM2443454), MED1 (GSM2443457), RNAPII (GSM935386), BRD4 (GSM1536175), and H3K27ac (GSM1233894) in GM12878, were processed, which revealed the enrichment of H3K27ac modifications and transcription factor downstream of the human-specific CTCF site, constrained within the CTCF loop. Notably, the analysis identified a super-enhancers located just downstream of the CTCF binding site (Fig. S8c). The presence of rs558245864 further demonstrated its influence on CTCF binding to DNA, which potentially consequently affected the regulation of nearby enhancers on target genes.
To assess the effect of rs558245864 on BLK expression across different genotypes, RNA-seq data from HG00514 (GSM2431197) and GM12878 (GSE92521) were analyzed. This analysis indicated that BLK expression was notably lower in HG00514 compared to GM12878. To extend this analysis to a population level, a dataset comprising 465 RNA-seq profiles (ERP001942) from human B-lymphoblastoid cell lines was employed. Based on the genotype information available from the 1,000 Genomes Project, the analysis revealed that BLK expression was significantly lower in homozygotes and heterozygotes for the rs558245864 allele compared to homozygotes for the wild-type allele (Fig. 5f).
Furthermore, data from the Vindija Neanderthal, Altai Neanderthal, and Denisova VCF files were obtained from the Max Planck Institute for Evolutionary Anthropology website (http://cdna.eva.mpg.de/neandertal/Vindija/VCF/), to gain insights into the genotypes of archaic humans at the loci of rs558245864 (Fig. S8e).
Linkage disequilibrium analysis
To elucidate the potential implications associated with rs558245864, an in-depth exploration was undertaken. Initially, we extracted the genetic information of all SNPs within the European population residing at the BLK locus from the International HapMap Project (1000 Genomes Project) database, facilitating subsequent linkage disequilibrium analysis. Remarkably, we observed that rs558245864 was within the same linkage disequilibrium region as other variants, including rs2736337, rs2736340, and rs13277113 (Fig. 5c). These SNPs have been previously identified through GWAS as SLE48,49,123.
Processing of 4C-seq data
To further substantiate the role of rs558245864 in modulating the transcriptional regulation and expression of the BLK gene in an SLE patient, we conducted a rigorous analysis of 4C-seq data from a control normal individual and an SLE patient using 4 Cseqpipe124. Notably, the SLE patient is a homozygote for the rs558245864 allele, while the control is a homozygote for the reference allele. Custom configurations were applied to the “4cseqpipe.conf” file, followed by the execution of the “4cseqpipe.pl” script. This 4Cseqpipe tool systematically performed a series of essential tasks, encompassing sequence extraction, alignment, data normalization, and generation of near-cis domainograms. To facilitate a visually informative representation of the 4C-seq data, the tool employed a color-coded multi-scaled heatmap approach, with sliding window sizes spanning the range from 2 to 50 kb. Additionally, the core trends in contact intensities were computed using a 5-kb window size and subsequently depicted in the form of trend curves. The visualizations also incorporated gray bands to signify the 20–80% quantiles within the windows, yielding an illustrative representation (Fig. S8f).
To ascertain the impact of rs558245864 on interactions involving the VP with the BLK and the upstream region in the patient, we conducted an in-depth analysis. Notably, the VP is within the human-specific enhancer upstream of the BLK promoter. Specifically, we extracted three successive interaction values on the core trends in the proximity of the promoter of BLK and the upstream region, which represent the strength of interactions between these loci and the VP. Then, we performed a Student’s t-test to assess the statistical significance of the observed differences between the control and patient. Remarkably, the analysis revealed a substantial reduction in the interaction of the VP with the BLK promoter in the patient relative to control (P = 0.0043). Conversely, a significantly increased interaction with the upstream region was observed in the patient (P = 0.0356), underscoring the potential functional implications of the rs558245864 allele in altering these specific chromatin interactions.
Processing of scISO-seq data
To assess the influence of human-specific CTCF loops on DLGAP1 isoform expression, a comprehensive analysis of scISO-seq was conducted to quantify the expression abundance of DLGAP1-201 and DLGAP1-206 in both human and macaque primary neuronal cells. The analysis of PacBio sequencing reads from the scISO-seq libraries was carried out using the dedicated scISO-seq analysis pipeline (https://isoseq.how). The initial step involved the processing of sequencing reads to generate circular consensus sequences (CCS) from the raw subread data using the “ccs” tool (v6.2.0) with a parameter setting of “--min-rq 0.99”, ensuring high data quality and accuracy. Subsequently, the obtained CCS reads underwent further refinement, which encompassed the removal of cDNA primers and poly(A) tails and the extraction of UMIs and cell barcodes. Specifically, for cDNA primer removal, the “Lima” tool (v2.4.0) was employed with parameters “--same --split”, effectively eliminating the primers and improving data quality. Cell barcodes (16-bp) and UMIs (10-bp) located at the 5′ end of the processed CCS reads were extracted using the “isoseq3 tag” function of “isoseq3” (v3.4.0) with the parameter “--design 16B-10U-T”. These extracted tags were subsequently associated with their respective reads, facilitating later deduplication. To ensure high-quality data, the removal of poly(A) tails was performed using the “isoseq3 refine” function with the parameter “--require-polya”. The removal of PCR duplicates was executed based on UMIs and cell barcodes, utilizing the “isoseq3 dedup” tool. The resulting deduplicated reads were then aligned to the human reference genome (hg38) or the macaque reference genome (rheMac10) using “minimap2” (v2.2.1)125 with parameters “-ax splice -uf-secondary = no -C5 -O6,24 -B4”. Following alignment, the alignment files were sorted based on the coordinates of the reads using the “samtools sort” function within “samtools” (v1.3.1). In total, 3,260,361 and 3,129,918 long reads were identified and sorted for humans and macaques, respectively.
Focusing on the DLGAP1 genes, 3109 and 990 long reads were extracted from the entirety of long reads in humans and macaques, respectively. The sorted long reads sharing identical splice site tags were subsequently collapsed into consensus transcript isoforms, while redundant isoforms were meticulously removed using the “collapse_isoforms_by_sam.py” script from Cupcake (https://github.com/Magdoll/cDNA_Cupcake) with specific parameters “-c 0.99 -i 0.95 --gen_mol_count”. This process yielded 134 non-redundant isoforms for DLGAP1 in humans and 109 non-redundant isoforms for DLGAP1 in macaques. The resulting non-redundant isoforms were visualized using the Integrative Genomics Viewer (IGV, v2.9.4)126, and the numbers of DLGAP1-201 and -206 isoforms were manually quantified. It is important to note that isoforms inconsistent with the exonic structure of DLGAP1-201 or -206 were excluded, with only those retaining less than four exons missing from the 3′ end considered. In the final analysis, 40 DLGAP1-201 and 26 DLGAP1-206 isoforms were identified in human primary neuronal cells, while 41 DLGAP1-201 and 9 DLGAP1-206 isoforms were identified in macaques (Fig. 6f). A chi-square test indicated that the proportion of DLGAP1-206 isoforms was significantly higher in humans compared to in macaques (P < 0.01).
Processing of scTSS-seq and TSS-seq data
To elucidate disparities in the expression of DLGAP1 isoforms, we subjected scTSS-seq data derived from primary neuronal cells and TSS-seq data obtained from dorsal forebrain organoids to comprehensive analysis. The scTSS-seq data had a distinct library structure in the R1 reads, characterized by a 16-bp cell barcode, a 10-bp UMI, and the TSO sequences. Conversely, the TSS-seq data lacked the cell barcode but retained the 10-bp UMI and the TSO sequences in the R1 reads. During the initial data preprocessing stage, guided by the specific library structures of scTSS-seq and TSS-seq, we extracted the cell barcodes and UMI sequences from the R1 reads of scTSS-seq data, and the UMI sequences from the R1 reads of TSS-seq data for subsequent analytical procedures. Subsequently, we employed Cutadapt (v1.13)127 to meticulously excise the TSO sequence and the contiguous sequence preceding it from the reads, incorporating the parameter “-g TTTCTTATATGGG”. The R1 reads that remained after this process were then aligned to the reference genomes of either human (hg38) or macaques (rheMac8) employing HISAT2 (v2.1.0) with the default settings. In order to ensure the highest data quality and precision, only the R1 reads that exhibited unique mapping, signified by MAPQ > 20, were retained for further analysis. In the following step, the removal of PCR duplicates was executed. For the scTSS-seq data, PCR duplicates were effectively eradicated through the application of a custom script that leveraged the UMI and cell barcode information associated with each read. In the case of the TSS-seq data, the removal of PCR duplicates was solely based on the UMI information. Following the successful removal of duplicates, the remaining R1 reads, especially those aligned with the promoters of DLGAP1-201 and DLGAP1-206, were counted using the “samtools view” function within Samtools (v1.3.1).
Chromatin accessibility of the human-specific CTCF-binding site at DLGAP1 locus during brain development
To dissect the dynamics of chromatin accessibility at the human-specific CTCF-binding site and enhancers and DLGAP1-201/206 promoters during human brain development, we processed single-cell assay for transposase-accessible chromatin with sequencing (scATAC-seq) data from four primary neuron samples at PCW16, 20, 21, and 24 (GSE162170). The scATAC-seq data underwent a reanalysis, resulting in a comprehensive count matrix that characterizes the chromatin accessibility status of each cell at every peak throughout the developmental stages of the brain. To ensure data quality, this count matrix was subjected to normalization through TF-IDF, following which a Seurat object was constructed using the “CreateSeuratObject” function within the Seurat (v3.2.3). The subsequent step involved the selection of the most variable 2000 genes through the “FindVariableFeatures” function, employing the “vst” method. The matrix was further subjected to scaling and centering procedures, achieved via the “ScaleData” function. PCA was carried out on these 2000 variable genes to extract the top 30 PCs with the “RunPCA” function. The construction of a k-nearest neighbor network was performed to facilitate cell visualization, realized through the “FindNeighbors” function, followed by cell clustering employing the “FindClusters” function with a resolution parameter of “resolution = 0.8”. Visualization was rendered using Uniform Manifold Approximation and Projection (UMAP), and the cell types were annotated based on the chromatin accessibility of marker genes as in the original publication128. It”s noteworthy that multiple cell types were annotated within this UMAP representation, encompassing categories such as glutamatergic neurons (GluN), inhibitory neurons (IN), neuronal intermediate progenitor cells (nIPC), and others.
The subsequent examination focused on scrutinizing the chromatin accessibility of the human-specific CTCF-binding site at the DLGAP1 locus throughout the various stages of brain development. DLGAP1 is prominently expressed in neurons and plays a pivotal role in neuronal signal transmission in the postsynaptic density region56. Therefore, a subset of cells was isolated for in-depth analysis, constituting 17,341 GluN and 1,896 nIPC cells. To elucidate the cellular trajectories, Monocle3 (v1.0.0)129 was employed, with the nIPC cells designated as the “root” cells. The TF-IDF-normalized matrix was subsequently sorted along the trajectory, enabling the construction of a genome-wide chromatin accessibility matrix spanning from nIPC to GluN. In the final phase, a submatrix was extracted from the sorted accessibility matrix, housing the accessibility peaks corresponding to the human-specific CTCF site, HSEs, DLGAP1 promoters, and weak enhancers (Fig. 6g). These weak enhancers were chromatin accessibility regions recorded in scATAC-seq data that do not overlap with the H3K27ac peaks from the in-house CUT&Tag data of primary neuronal cells.
Integration of scRNA-seq data from WT and KO dorsal forebrain organoids
For the scRNA-seq data from human dorsal forebrain organoids, we procured a comprehensive dataset, comprising 6436 cells from day-14 WT organoids, 9368 cells from day-14 KO organoids, 6789 cells from day-60 WT organoids, and 9502 cells from day-60 KO organoids. The primary objective of this analysis was to unveil the developmental distinctions between the WT and KO organoids. To ensure the harmonious integration of data from these diverse sources and to eliminate potential batch effects, we harnessed the Harmony (v1.0)130, a technique that can integrate multiple datasets into a shared space for unsupervised clustering. The Harmony embeddings, after integration, were subsequently subjected to dimensionality reduction using the “RunUMAP” function within the Seurat (v3.2.3) package. The accurate classification and annotation of individual cells were of paramount importance in this analysis. This was achieved using the “FindAllMarkers” function in Seurat (v3.2.3), with a minimum percentage threshold “min.pct = 0.25” and a minimum log-fold change threshold “min.logFC = 0.25” to determine the marker genes of clusters. These determined markers and known marker genes were applied in annotating cell types, and the subsequent examination revealed the presence of six distinct cell populations within the merged UMAP. These cell types were identified as follows: 13,197 cells in the G2, M, and S phases of the cell cycle (G2M/S), 5477 neuroepithelial cells (NEC), 3665 NPC, 2501 IPC, 4803 excitatory neurons, and 2452 inhibitory neurons. The cell types along the neuron differentiation were visualized using SPRING131. Briefly, the Euclidean distance between the top 30 PCs for each pair of cells was meticulously calculated. Subsequently, a k-nearest neighbor network was constructed, employing a parameter setting of “k = 20”. This network, which encapsulated the proximity between cells, was effectively visualized using SPRING. Within the SPRING plot, each individual cell is represented as a dot, and the spatial arrangement of these dots conveys the degree of similarity in their transcriptional profiles (Figs. 7e and S10f).
Constructing organoid developmental trajectory and identifying abnormal developmental processes in KO dorsal forebrain organoids
To elucidate the intricate developmental trajectory emerging from the amalgamated scRNA-seq data of KO and WT organoids, an in-depth analysis was conducted leveraging Monocle3 (v1.0.0). The initial step involved a transformation of the Seurat object into a Monocle3 object, accomplished through the use of the “as.cell_data_set” function. Thereafter, the cell population was subjected to clustering through “cluster_cells” function, incorporating the Leiden community detection algorithm. The trajectory graph was subsequently unveiled by employing the “learn_graph” function, which harnessed reverse graph embedding techniques, effectively mapping the developmental trajectory in a dimension-reduced space. Subsequently, each individual cell was assigned a pseudotime value, achieved through the “order_cells” function with the selection of a specific root. Notably, the root designation was based on a random selection of 500 cells derived from the G2M/S cell cycle stage.
To dissect the discrepancies in the developmental processes of excitatory neurons within WT and KO organoids, cellAlign (v 0.1.0)72 was employed to compare the pseudotemporal trajectories of “G2M/S-NEC-NPC-IPC-EN” between WT and KO organoids (Fig. 7f). the tool applies dynamic time warping to compare the dynamics of two trajectories by analyzing a normalized expression matrix derived from a shared gene set. The algorithm partitions the Monocle trajectory into 200 segments, estimating the expression profiles for each pseudopoint as the mean profile of all assigned cells. It subsequently calculates pairwise distances between ordered points along the trajectories of WT and KO organoids, ultimately discerning an optimal alignment that conserves the pseudotemporal order and minimizes the overall distance between matched cells. Together, the result unveiled a more advanced maturation state in KO organoids at the G2M/S-NEC-NPC stage in comparison to the WT counterparts.
To uncover the alterations in gene expression levels beneath the phenotypic variations between KO and WT organoids, an investigation was carried out to identify differentially expressed genes during NEC and NPC stages. We employed the “FindMarkers” function within the Seurat (v3.2.3) to find the differential expressed genes, incorporating parameters such as “min.pct = 0.1” and “logfc.threshold = 0.5”. This rigorous analysis led to the identification of 375 significant differentially expressed genes, which were further categorized into 196 upregulated and 179 downregulated genes using the self-organizing maps (Fig. S10j). Subsequently, these significant differentially upregulated and downregulated genes were subjected to gene ontology (GO) enrichment analysis, achieved through the DAVID database (DAVID, https://david.ncifcrf.gov.). The significantly enriched GO terms were plotted in Fig. S10k.
Statistics and reproducibility
Statistical methods and P values were indicated in the corresponding figure legends and the toolboxes are publicly available as described in Supplementary Data 3. Difference significance levels were defined as follows: P < 0.05 (*, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001), and no significance (ns) as P > 0.05. The number of biological replicates for each experiment is specified in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The ChIA-PET, ChIP-seq, CUT&Tag, scRNA-seq, scISO-seq, scTSS-seq, and RNA-seq data generated from human primary neuronal cells, B-lymphoblastoid cell lines (GM12878 and HG00514), and dorsal forebrain organoids, macaque primary neuronal cells and B-lymphoblastoid cell line (LCL8664), and B-lymphoblastoid cell lines from chimpanzees (EB176) and gorillas (EB(JC)) have been deposited with the GSA (Genome Sequence Archive in BIG Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences): PRJCA020156. The human data were under the accession number HRA007191 and the non-human data were under the accession number CRA015949. The human datasets generated during the current study are not publicly available due to privacy restrictions. Access to the data is subject to approval by the Data Access Committee to ensure compliance with data protection regulations. Researchers who meet the criteria for access to confidential data may request the data by contacting the Correspondence author at [email protected]. Requests will be reviewed within 14 working days, and approved applicants will be required to sign a data access agreement. This paper additionally included analyzes of publicly available datasets. The accession numbers for all these datasets are listed in the Supplementary Data 3. Source data are provided with this paper.
Code availability
The custom scripts used in this manuscript are available on Zenodo [https://doi.org/10.5281/zenodo.15017787]132.
References
Chimpanzee sequencing analysis consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69–87 (2005).
Varki, A. & Altheide, T. K. Comparing the human and chimpanzee genomes: searching for needles in a haystack. Genome Res. 15, 1746–1758 (2005).
Pollard, K. S. et al. An RNA gene expressed during cortical development evolved rapidly in humans. Nature 443, 167–172 (2006).
Franchini, L. F. & Pollard, K. S. Human evolution: the non-coding revolution. BMC Biol. 15, 89 (2017).
Mack, K. L., Campbell, P. & Nachman, M. W. Gene regulation and speciation in house mice. Genome Res. 26, 451–461 (2016).
Gorkin, D. U., Leung, D. & Ren, B. The 3D genome in transcriptional regulation and pluripotency. Cell Stem Cell 14, 762–775 (2014).
Dowen, J. M. et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 159, 374–387 (2014).
Lupianez, D. G. et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012–1025 (2015).
Ji, X. et al. 3D chromosome regulatory landscape of human pluripotent cells. Cell Stem Cell 18, 262–275 (2016).
Rowley, M. J. & Corces, V. G. Organizational principles of 3D genome architecture. Nat. Rev. Genet. 19, 789–800 (2018).
Kim, S. & Shendure, J. Mechanisms of Interplay between Transcription Factors and the 3D Genome. Mol. Cell 76, 306–319 (2019).
Misteli, T. The self-organizing genome: principles of genome architecture and function. Cell 183, 28–45 (2020).
Tang, Z. H. et al. CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell 163, 1611–1627 (2015).
Dixon, J. R. et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012).
Hnisz, D. et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 351, 1454–1458 (2016).
Narendra, V. et al. CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 347, 1017–1021 (2015).
Choudhary, M. N. K., Quaid, K., Xing, X., Schmidt, H. & Wang, T. Widespread contribution of transposable elements to the rewiring of mammalian 3D genomes. Nat. Commun. 14, 634 (2023).
Cournac, A., Koszul, R. & Mozziconacci, J. The 3D folding of metazoan genomes correlates with the association of similar repetitive elements. Nucleic Acids Res. 44, 245–255 (2016).
Kaaij, L. J. T., Mohn, F., van der Weide, R. H., de Wit, E. & Buhler, M. The ChAHP complex counteracts chromatin looping at CTCF sites that emerged from sINE expansions in mouse. Cell 178, 1437–1451 e1414 (2019).
Schmidt, D. et al. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell 148, 335–348 (2012).
Trizzino, M. et al. Transposable elements are the primary source of novelty in primate gene regulation. Genome Res. 27, 1623–1633 (2017).
Acemel, R. D. & Lupianez, D. G. Evolution of 3D chromatin organization at different scales. Curr. Opin. Genet. Dev. 78, 102019 (2023).
Real, F. M. et al. The mole genome reveals regulatory rearrangements associated with adaptive intersexuality. Science 370, 208–214 (2020).
Fudenberg, G. & Pollard, K. S. Chromatin features constrain structural variation across evolutionary timescales. Proc. Natl. Acad. Sci. USA 116, 2175–2180 (2019).
Sadowski, M. et al. Spatial chromatin architecture alteration by structural variations in human genomes at the population scale. Genome Biol. 20, 148 (2019).
Luo, X. et al. 3D Genome of macaque fetal brain reveals evolutionary innovations during primate corticogenesis. Cell 184, 723–740.e721 (2021).
Rekaik, H. et al. Sequential and directional insulation by conserved CTCF sites underlies the Hox timer in stembryos. Nat. Genet. 55, 1164–1175 (2023).
Pollen, A. A., Kilik, U., Lowe, C. B. & Camp, J. G. Human-specific genetics: new tools to explore the molecular and cellular basis of human evolution. Nat. Rev. Genet. 24, 687–711 (2023).
Quintana-Murci, L. Human immunology through the lens of evolutionary genetics. Cell 177, 184–199 (2019).
Vietri Rudan, M. et al. Comparative Hi-C reveals that CTCF underlies evolution of chromosomal ___domain architecture. Cell Rep. 10, 1297–1309 (2015).
Galupa, R. & Heard, E. Topologically associating domains in chromosome architecture and gene regulatory landscapes during development, disease, and evolution. Cold Spring Harb. Symp. Quant. Biol. 82, 267–278 (2017).
McArthur, E. & Capra, J. A. Topologically associating ___domain boundaries that are stable across diverse cell types are evolutionarily constrained and enriched for heritability. Am. J. Hum. Genet. 108, 269–283 (2021).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015).
Gazal, S. et al. Linkage disequilibrium-dependent architecture of human complex traits shows action of negative selection. Nat. Genet. 49, 1421–1427 (2017).
Hujoel, M. L. A., Gazal, S., Hormozdiari, F., van de Geijn, B. & Price, A. L. Disease heritability enrichment of regulatory elements is concentrated in elements with ancient sequence age and conserved function across species. Am. J. Hum. Genet. 104, 611–624 (2019).
Hirano, S. & Takeichi, M. Cadherins in brain morphogenesis and wiring. Physiol. Rev. 92, 597–634 (2012).
Wu, Q. & Maniatis, T. A striking organization of a large family of human neural cadherin-like cell adhesion genes. Cell 97, 779–790 (1999).
Esumi, S. et al. Monoallelic yet combinatorial expression of variable exons of the protocadherin-alpha gene cluster in single neurons. Nat. Genet. 37, 171–176 (2005).
Ribich, S., Tasic, B. & Maniatis, T. Identification of long-range regulatory elements in the protocadherin-alpha gene cluster. Proc. Natl. Acad. Sci. USA 103, 19719–19724 (2006).
Rubinstein, R. et al. Molecular logic of neuronal self-recognition through protocadherin ___domain interactions. Cell 163, 629–642 (2015).
Tasic, B. et al. Promoter choice determines splice site selection in protocadherin alpha and gamma pre-mRNA splicing. Mol. Cell 10, 21–33 (2002).
Guo, Y. et al. CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell 162, 900–910 (2015).
Hirayama, T., Tarusawa, E., Yoshimura, Y., Galjart, N. & Yagi, T. CTCF is required for neural development and stochastic expression of clustered Pcdh genes in neurons. Cell Rep. 2, 345–357 (2012).
Monahan, K. et al. Role of CCCTC binding factor (CTCF) and cohesin in the generation of single-cell diversity of protocadherin-alpha gene expression. Proc. Natl. Acad. Sci. USA 109, 9125–9130 (2012).
Nakahashi, H. et al. A genome-wide map of CTCF multivalency redefines the CTCF code. Cell Rep. 3, 1678–1689 (2013).
Jeong, H. et al. Evolution of DNA methylation in the human brain. Nat. Commun. 12, 2021 (2021).
Mendizabal, I. et al. Cell type-specific epigenetic links to schizophrenia risk in the brain. Genome Biol. 20, 135 (2019).
Castillejo-Lopez, C. et al. Genetic and physical interaction of the B-cell systemic lupus erythematosus-associated genes BANK1 and BLK. Ann. Rheum. Dis. 71, 136–142 (2012).
Hom, G. et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N. Engl. J. Med. 358, 900–909 (2008).
Dang, J. et al. Gene-gene interaction of ATG5, ATG7, BLK and BANK1 in systemic lupus erythematosus. Int. J. Rheum. Dis. 19, 1284–1293 (2016).
Okada, Y. et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381 (2014).
Furlanis, E., Traunmuller, L., Fucile, G. & Scheiffele, P. Landscape of ribosome-engaged transcript isoforms reveals extensive neuronal-cell-class-specific alternative splicing programs. Nat. Neurosci. 22, 1709–1717 (2019).
Hardwick, S. A. et al. Single-nuclei isoform RNA sequencing unlocks barcoded exon connectivity in frozen brain tissue. Nat. Biotechnol. 40, 1082–1092 (2022).
Traunmuller, L., Gomez, A. M., Nguyen, T. M. & Scheiffele, P. Control of neuronal synapse specification by a highly dedicated alternative splicing program. Science 352, 982–986 (2016).
Husi, H., Ward, M. A., Choudhary, J. S., Blackstock, W. P. & Grant, S. G. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat. Neurosci. 3, 661–669 (2000).
Kim, E. et al. GKAP, a novel synaptic protein that interacts with the guanylate kinase-like ___domain of the PSD-95/SAP90 family of channel clustering molecules. J. Biol. Chem. 136, 669–678 (1997).
Shin, S. M. et al. GKAP orchestrates activity-dependent postsynaptic protein remodeling and homeostatic scaling. Nat. Neurosci. 15, 1655–1666 (2012).
Kirov, G. et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 17, 142–153 (2012).
Li, J. et al. Integrated systems analysis reveals a molecular network underlying autism spectrum disorders. Mol. Syst. Biol. 10, 774 (2014).
Rasmussen, A. H., Rasmussen, H. B. & Silahtaroglu, A. The DLGAP family: neuronal expression, function and role in brain disorders. Mol. Brain 10, 43 (2017).
Stessman, H. A. et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 49, 515–526 (2017).
Birey, F. et al. Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59 (2017).
Pasca, A. M. et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 12, 671–678 (2015).
Taniguchi, K. et al. Genetic and molecular analyses indicate independent effects of TGIFs on Nodal and Gli3 in neural tube patterning. Eur. J. Hum. Genet. 25, 208–215 (2017).
Wotton, D. & Taniguchi, K. Functions of TGIF homeodomain proteins and their roles in normal brain development and holoprosencephaly. Am. J. Med. Genet. C. Semin. Med. Genet. 178, 128–139 (2018).
El-Jaick, K. B. et al. Functional analysis of mutations in TGIF associated with holoprosencephaly. Mol. Genet. Metab. 90, 97–111 (2007).
Gripp, K. W. et al. Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nat. Genet. 25, 205–208 (2000).
Kuang, C. et al. Intragenic deletion of Tgif causes defectsin brain development. Hum. Mol. Genet. 15, 3508–3519 (2006).
Melhuish, T. A. & Wotton, D. The interaction of the carboxyl terminus-binding protein with the Smad corepressor TGIF is disrupted by a holoprosencephaly mutation in TGIF. J. Biol. Chem. 275, 39762–39766 (2000).
Taniguchi, K., Anderson, A. E., Sutherland, A. E. & Wotton, D. Loss of Tgif function causes holoprosencephaly by disrupting the SHH signaling pathway. PLoS Genet 8, e1002524 (2012).
Kanton, S. et al. Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 574, 418–422 (2019).
Alpert, A., Moore, L. S., Dubovik, T. & Shen-Orr, S. S. Alignment of single-cell trajectories to compare cellular expression dynamics. Nat. Methods 15, 267–270 (2018).
Chang, I. & Parrilla, M. Expression patterns of homeobox genes in the mouse vomeronasal organ at postnatal stages. Gene Expr. Patterns 21, 69–80 (2016).
Lee, B. K. et al. Tgif1 counterbalances the activity of core pluripotency factors in mouse embryonic stem cells. Cell Rep. 13, 52–60 (2015).
Thai, C. W. et al Comparative chromatin dynamics of stem cell differentiation in human and rat. bioRxiv, Preprint at https://doi.org/10.1101/2021.1102.1111.430819 (2021).
van de Leemput, J. et al. CORTECON: a temporal transcriptome analysis of in vitro human cerebral cortex development from human embryonic stem cells. Neuron 83, 51–68 (2014).
Graham, V., Khudyakov, J., Ellis, P. & Pevny, L. SOX2 functions to maintain neural progenitor identity. Neuron 39, 749–765 (2003).
Knepper, J. L., James, A. C. & Ming, J. E. TGIF, a gene associated with human brain defects, regulates neuronal development. Dev. Dyn. 235, 1482–1490 (2006).
Zhang, X. et al. Pax6 is a human neuroectoderm cell fate determinant. Cell Stem Cell 7, 90–100 (2010).
Marletaz, F. et al. The little skate genome and the evolutionary emergence of wing-like fins. Nature 616, 495–503 (2023).
Grove, J. et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 51, 431–444 (2019).
Caglayan, E. et al. Molecular features driving cellular complexity of human brain evolution. Nature 620, 145–153 (2023).
Marasco, L. E. & Kornblihtt, A. R. The physiology of alternative splicing. Nat. Rev. Mol. Cell Biol. 24, 242–254 (2023).
Verta, J. P. & Jacobs, A. The role of alternative splicing in adaptation and evolution. Trends Ecol. Evol. 37, 299–308 (2022).
Wright, C. J., Smith, C. W. J. & Jiggins, C. D. Alternative splicing as a source of phenotypic diversity. Nat. Rev. Genet. 23, 697–710 (2022).
Barrio-Hernandez, I. et al. Network expansion of genetic associations defines a pleiotropy map of human cell biology. Nat. Genet. 55, 389–398 (2023).
Stearns, F. W. One hundred years of pleiotropy: a retrospective. Genetics 186, 767–773 (2010).
Tarantal A. F. Ch. 20 - Ultrasound imaging in rhesus (macaca mulatta) and long-tailed (macaca fascicularis) macaques: reproductive and research applications. In: The Laboratory Primate (ed. Wolfe-Coote, S) (Academic Press, 2005).
Workman, A. D., Charvet, C. J., Clancy, B., Darlington, R. B. & Finlay, B. L. Modeling transformations of neurodevelopmental sequences across mammalian species. J. Neurosci. 33, 7368–7383 (2013).
Brewer, G. J. & Torricelli, J. R. Isolation and culture of adult neurons and neurospheres. Nat. Protoc. 2, 1490–1498 (2007).
Glynn, M. W. & McAllister, A. K. Immunocytochemistry and quantification of protein colocalization in cultured neurons. Nat. Protoc. 1, 1287–1296 (2006).
Shi, Y., Kirwan, P., Smith, J., Robinson, H. P. & Livesey, F. J. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat. Neurosci. 15, 477–486 (2012).
Wen, Z. et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 515, 414–418 (2014).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Kaya-Okur, H. S., Janssens, D. H., Henikoff, J. G. & Ahmad, K. Henikoff S. Efficient low-cost chromatin profiling with CUT&Tag. Nat. Protoc. 15, 3264–3283 (2020).
Kaya-Okur, H. S. et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10, 1930 (2019).
Krijger, P. H. L., Geeven, G., Bianchi, V., Hilvering, C. R. E. & de Laat, W. 4C-seq from beginning to end: a detailed protocol for sample preparation and data analysis. Methods 170, 17–32 (2020).
Wang, P. et al. In situ chromatin interaction analysis using paired-end tag sequencing. Curr. Protoc. 1, e174 (2021).
Heigwer, F., Kerr, G. & Boutros, M. E-CRISP: fast CRISPR target site identification. Nat. Methods 11, 122–123 (2014).
Trevino, A. E. et al. Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution. Cell 184, 5053–5069.e5023 (2021).
Velasco, S. et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 570, 523–527 (2019).
Zhao, H. et al. CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics 30, 1006–1007 (2014).
Zhu, Y., Li, M., Sousa, A. M. & Sestan, N. XSAnno: a framework for building ortholog models in cross-species transcriptome comparisons. BMC Genom. 15, 343 (2014).
Massingham T., Goldman N. simNGS and simLibrary–software for simulating next-gen sequencing data. http://www.ebi.ac.uk/goldman-srv/simNGS/ (2012).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Ramirez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Zheng, G. X. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e1821 (2019).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Putri, G. H., Anders, S., Pyl, P. T., Pimanda, J. E. & Zanini, F. Analysing high-throughput sequencing data in Python with HTSeq 2.0. Bioinformatics 38, 2943–2945 (2022).
Salameh, T. J. et al. A supervised learning framework for chromatin loop detection in genome-wide contact maps. Nat. Commun. 11, 3428 (2020).
Ramirez, F. et al. High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat. Commun. 9, 189 (2018).
van der Weide, R. H. et al. Hi-C analyses with GENOVA: a case study with cohesin variants. NAR Genom. Bioinform. 3, lqab040 (2021).
Habegger, L. et al. RSEQtools: a modular framework to analyze RNA-Seq data using compact, anonymized data summaries. Bioinformatics 27, 281–283 (2011).
Lyu, X., Rowley, M. J., Kulik, M. J., Dalton, S. & Corces, V. G. Regulation of CTCF loop formation during pancreatic cell differentiation. Nat. Commun. 14, 6314 (2023).
Phillips, J. E. & Corces, V. G. CTCF: master weaver of the genome. Cell 137, 1194–1211 (2009).
Wang, H. et al. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 22, 1680–1688 (2012).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Bailey, T. L. & Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2, 28–36 (1994).
Acosta-Herrera, M. et al. Genome-wide meta-analysis reveals shared new loci in systemic seropositive rheumatic diseases. Ann. Rheum. Dis. 78, 311–319 (2019).
van de Werken, H. J. et al. Robust 4C-seq data analysis to screen for regulatory DNA interactions. Nat. Methods 9, 969–972 (2012).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011).
Trevino, A. E. et al. Chromatin accessibility dynamics in a model of human forebrain development. Science 367, eaay1645 (2020).
Cao, J. Y. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Weinreb, C., Wolock, S. & Klein, A. M. SPRING: a kinetic interface for visualizing high dimensional single-cell expression data. Bioinformatics 34, 1246–1248 (2018).
Wu X., et al. Evolutionary divergence in CTCF-mediated chromatin topology drives transcriptional innovation in humans. Zenodo, https://doi.org/10.5281/zenodo.15017787 (2025).
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (No. 31970562 for Z.T., No. 32100415 for D.X., and No. 82201314 for T.W.), the Science and Technology Planning Project of Guangdong Province (No. 2023B1212060018 for Z.T.), the Key-Area Research and Development Program of Guangdong Province (No. 2023B1111020007 for Z.T.), and the Fundamental Research Funds for the Central Universities starting fund (No. BMU2022RCZX038 for T.W.). We thank Dr. Yijun Ruan of Zhejiang University and Dr. Evan Eichler of the University of Washington for helpful discussions during the preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Z.T. conceived, designed, and supervised the study. X.W., R.L., Z.X., J.D., L.H., and X.Z. performed the experiments. D.X., Y.T., X.G., and W.D. analyzed the data. X.L. and A.X. performed the CRISPR/Cas9-mediated genome editing in human induced pluripotent stem cells. L.C., T.X., and W.X. performed the electrophysiological experiments on human dorsal forebrain organoids. T.W. analyzed the genomic mutations at the CTCF anchors among autism families. F.F. and X.Y. provided the human fetal brain samples, and X.C. provided the non-human primate B-lymphoblastoid cell lines. Z.T., X.W., D.X., and R.L. wrote the manuscript with input from D.P. and M.K.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Andrea Chiariello, Hongjie Yao, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, X., Xiong, D., Liu, R. et al. Evolutionary divergence in CTCF-mediated chromatin topology drives transcriptional innovation in humans. Nat Commun 16, 2941 (2025). https://doi.org/10.1038/s41467-025-58275-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58275-7
