
Abstract
Alcohols are one of the most abundant functional groups in commercially available materials and biologically active compounds. Herein, we report a metal-free photocatalytic method for the deoxygenative Z-selective olefination of aliphatic alcohols. Key to this methodology is the radical olefination and isomerization of unstabilized open-shell species generated in situ by a catalytic reductive scission of benzoate esters. These processes are simultaneously orchestrated by a single phenothiazine photocatalyst via singlet and triplet excited states. Our protocol is distinguished by its wide substrate scope and broad applicability, even in the context of pharmaceuticals and saccharides. Given the mild and water-compatible conditions, our chemistry can also be utilized to functionalize DNA headpieces for DELs applications.
Similar content being viewed by others
Introduction
Over the past few decades, metal-catalysed cross-coupling reactions have revolutionized the field of synthetic chemistry, enabling the rapid construction of complex molecular architectures from simple organic materials, as well as the late-stage functionalization of bioactive compounds and advanced synthetic intermediates1,2,3,4. While a wide range of cross-couplings protocols are available for the synthesis of unsaturated compounds from sp2-hybridized coupling partners5, significant limitations remain with respect to the formation of C(sp2)–C(sp3) and C(sp3)–C(sp3) bonds6, with alkyl halides7 and alkyl-organometallics8 being the most commonly employed sp3-hybridized starting materials.
The advent of metallaphotoredox catalysis9,10,11, cross-electrophile coupling reactions12 and synthetic organic electrochemistry13 has enabled the use of carboxylic acids and alcohols as sp3-hybridized coupling partners. Especially attractive are the deoxygenative C(sp2)–C(sp3) cross-coupling reactions of alcohols, the latter being the most abundant functional groups in commercially available materials and biologically active compounds14. While several outstanding methods have been developed for the coupling of aliphatic alcohols and heteroarenes15,16,17,18,19,20,21, only few examples of deoxygenative olefination have been reported22,23,24. Notably, all the methodologies provide the thermodynamically favoured E isomer as the sole product. Given the ubiquity of Z-alkenes in synthetic and biologically relevant molecules, and the sporadic examples of Z-selective olefinations from native and abundant functional groups25,26,27, a cross-coupling reaction that can directly provide Z-olefins using aliphatic alcohols as sp3-hybridized starting materials is highly desirable yet elusive.
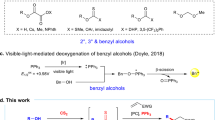
Herein, we present a mild photocatalytic method for the deoxygenative Z-selective olefination of benzoate esters derived from aliphatic alcohols (Fig. 1B). Feedstock and complex aliphatic alcohols can be readily converted into value-added olefin products in high yields, even in the case of pharmaceuticals and saccharides. Given the mild reaction conditions and compatibility with dilute aqueous media, our chemistry can also be utilized to functionalize DNA headpieces for applications in DNA-encoded libraries synthesis.

A Deoxygenative C(sp2)–C(sp3) cross-coupling reactions of alcohols. B This work: deoxygenative Z-selective olefination of aliphatic alcohols. C Neutral benzoates as precursors for radical functionalization reactions. D Design plan for the deoxygenative Z-selective olefination of alcohols. PC photocatalysis, FG functional group, PHT phenothiazine, SET single electron transfer, EnT energy transfer.
Results
Design plan
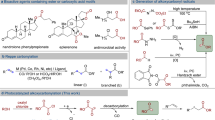
At the outset of our investigation, we envisioned a photocatalytic system that would simultaneously orchestrate: i) the activation of aliphatic alcohols to liberate unstabilized radicals, ii) the radical functionalization of alkenyl species and iii) the in-situ E to Z isomerization of the final olefin product. We recognized that the main challenge in our design plan is the merger of a general catalytic method that allows the generation of unstabilized radicals from aliphatic alcohols and the alkenylation/isomerization processes. We identified the reductive fragmentation of readily accessible benzoates as ideal compatible activation mode (Fig. 1C). Indeed, previous works have established that neutral benzoylated alcohols can undergo C(sp3)–O bond cleavage upon single electron reduction, releasing unstabilized alkyl radicals28. However, stoichiometric reductants29,30, UV photosensitizers31, or deeply reducing electrodes32, in combination with elevated temperatures, were necessary to enable the reductive scission, due to the very low reduction potential of benzoate substrates (E1/2(MeOBz/MeOBz•−) = −2.2 V vs SCE)33 and slow mesolytic fragmentation. Recently, a mild procedure for the deoxygenation of benzoates was reported34. Nevertheless, the reaction still requires the use of a stoichiometric reductant (CO2•−), which was generated in situ from formate by photoredox catalysis. Moreover, to the best of our knowledge, all reported methods for the reductive activation of neutral benzoates have been used only for the deoxygenation reaction, without further functionalization of the resulting alkyl radicals. Here, we proposed a photocatalytic system (Fig. 1D) where a phenothiazine (5) can directly perform the required electron reduction via the singlet excited state (6, E1/2(PHT•+/PHT*) = −2.5 V vs SCE)35,36. We reasoned that an alkenyl boron species (3) would be the perfect coupling partner for our strategy: not only is it a source of the sp2-hybridized fragment37,38,39,40, but can also form a Lewis acid-base complex (4) with the alcohol benzoates, increasing the kinetics of the challenging reductive β-scission (8)34. Subsequent radical addition (9) provides the corresponding benzyl radical (10), which is converted to carbocation 11 via a radical-polar crossover process by the oxidized phenothiazine (7), concomitantly restoring the ground state of the photocatalyst (5). Elimination of B(OR)3 provides the thermodynamically favoured E-alkene 12. Given the high triplet state energy of phenothiazine (~62 kcal/mol)41, we anticipated that the photocatalyst would also promote in situ E to Z isomerization of the product (E = 60.5 kcal/mol for E-propenylbenzene)42 via an energy transfer mechanism, providing the desired Z-olefin 13 as a final product43,44. It is noteworthy that both the singlet and the triplet excited states of the phenothiazine would be active in our deoxygenative olefination, representing a rare example where a single photocatalyst promotes both SET and energy transfer events through different excited states45.
Optimization studies
We began our investigation by evaluating the efficiency of alkyl radical formation from alcohol benzoate 14 with an N-phenylphenothiazine photocatalyst 16, in the presence of γ-terpinene (as a H atom source) and DMSO under 390 nm purple light irradiation (Fig. 2A).

A Screening of phenothiazine derivatives for the photocatalytic activation of neutral benzoates. Reaction conditions: 14 (0.1 mmol), γ-terpinene (0.2 mmol), photocatalyst (10 mol%) in DMSO (0.1 M) at rt for 16 h, 390 nm LED. B Optimization and control experiments for the deoxygenative Z-selective olefination of alcohols. Reaction conditions: 14 (0.1 mmol), styryl boron species (0.3 mmol), catechol (0.1 mmol), photocatalyst 19 (10 mol%) in DMSO (0.33 M) at rt for 16 h, 390 nm LED. a 1H NMR yields using methyl 3,5-dinitrobenzoate as internal standard. b Z:E ratio determined by 1H NMR. DMSO: dimethyl sulfoxide.
Pleasingly, deoxygenated product 15 was obtained in 25% yield, confirming that phenothiazine 16 is a matched reductant to reduce benzoate 14. A fast screening of substituted phenothiazine derivatives (17-20) identified thioether 19 as the best photocatalyst, providing product 15 in a promising 41% yield, along with 58% of unreacted 14. As expected, the slow mesolytic fragmentation for the generation of unstabilized radicals hampered the efficient activation of benzoate 14, confirming the necessity of using a Lewis acid as an additive34.
Encouraged by these preliminary results, we next evaluated our proposed deoxygenative Z-selective olefination of 14 using various styryl boronic acid derivatives (22-24) in the presence of catechol, phenothiazine 19 and DMSO under 390 nm purple light irradiation (Fig. 2B). Surprisingly, no conversion was observed when styryl boronic acid 22 was used (entry 1), while styryl trifluoroborate salt 24 provided the desired olefination product 21 in 74% yield with a Z:E ratio of 84:16 (entry 3). Variation of the catechol electronics showed a moderate effect on the outcome of the deoxygenative coupling (entries 4-7). The best result was obtained with t-butyl catechol, affording olefin 21 in 81% yield and Z:E ratio = 84:16 (entry 6). The molecular structure of the photocatalyst is strictly correlated with the energy of its triplet state and therefore the photostationary state composition of the E to Z isomerization process. A final screening of phenothiazine derivatives confirmed that thioether 19 is the best choice in terms of yield and Z:E ratio (Table S4). As expected, other types of photocatalysts such as Ru(bpy)3, Ir(ppy)3 and 4-CzIPN failed to activate benzoate 14. Lastly, additional control reactions demonstrated that purple light, photocatalyst and catechol are fundamental for the observed reactivity (entries 8 − 10), consistent with the mechanistic blueprint outlined in Fig. 1D.
Scope of the reaction
With optimal conditions in hand (entry 6, Fig. 2B), we next evaluated the alcohol scope for this transformation. As evident from the results compiled in Fig. 3, our photocatalytic deoxygenative Z-selective olefination could be conducted with a wide variety of benzoate esters derived from primary and secondary aliphatic alcohols. Cyclic (25-42) and acyclic (43-53) secondary alcohols were readily converted to the coupling products in good yields and Z:E ratio. Viable motifs in this transformation include amides (25, 38), sulfonamides (26), terminal alkynes (28), furans (52), thiophenes (55), indoles (50), triazoles (53), caffeine (51) and N-Boc protected amines (29, 39, 41 and 42).

Reaction conditions as Fig. 2B (entry 6), 0.2 mmol scale. Yield and Z:E ratio determined by 1H NMR using methyl 3,5-dinitrobenzoate as internal standard. In brackets isolated yields of Z-olefin, unless otherwise noted. All yields refer to the photocatalytic process only. ‡2 mmol scale. †Catechol is used instead of 4-t-Bu catechol, 16 h. *Isolated as mixture of isomers. #2 lamps. §dr = 1.6:1. ‖dr > 20:1. DMSO: dimethyl sulfoxide.
Bicyclic and spirocyclic structures (40-42), acetate (46), p-OMe-benzoate (48) and unprotected tertiary alcohol (49) were well tolerated, delivering the corresponding Z-olefins in moderate yields to good yields. When a substrate containing both primary and secondary benzoates was exposed to our olefination protocol (47), we observed the selective formation of the product derived from the activation of the secondary OBz. Notably, we could apply our methodology for the activation of the simple primary alcohols such as ethanol (57) and methanol (58); the latter being a rare example of a methylation cross-coupling reaction. While the Z:E ratio of these last examples was not optimal, the product mixtures were readily converted in situ to the corresponding E-olefins (please see section 4: “Photocatalytic Deoxygenative Z-selective olefination of aliphatic alcohols” in the SI). Remarkably, phenothiazine 19 was recovered in 98% yield from a scale-up experiment (25), demonstrating the robustness of our photocatalytic system.
Finally, we were delighted to find that this method is also amenable to a variety of biologically relevant molecules (59-65).
Substrates containing pharmaceutical cores such as etodolac (59) and oxaprozin (60) were converted into the desired Z-olefins with moderate to good yields. Notably, late-stage deoxygenative olefination of atorvastatin (61) proceeded smoothly, providing the coupled product in 84% yield. The hemiacetal at the anomeric carbon of commercially available furanose (63) could be activated to provide the coupled product with excellent diastereoselectivity. Similarly, a chiral secondary alcohol on the glucose core (64) and the primary alcohol on the galactose (65) were activated to generate the corresponding Z-olefins.
Pleasingly, our deoxygenative Z-selective olefination was found to be applicable with a wide array of alkenyl trifluoroborate salts (Fig. 4). Electron-rich and electron-poor substituents on the styryl moiety in the para (66-70) and meta (71-73) positions posed no problems, including the methyl group in ortho position (74). Heteroaromatics such as thiophene (76) and indole (77) were well tolerated, delivering the corresponding coupling products in moderate yields. Finally, a substrate containing the pharmaceutical core of gemfibrozil (78) was successfully coupled to obtain the desired Z-olefin.

Reaction conditions as Fig. 2B (entry 6), 0.2 mmol scale. Yield and Z:E ratio determined by 1H NMR using methyl 3,5-dinitrobenzoate as internal standard. In brackets isolated yields of Z-olefin, unless otherwise noted. All yields refer to the photocatalytic process only. †Catechol is used instead of 4-t-Bu catechol, 16 h. *Isolated as mixture of isomers. #2 lamps, 6 h. DMSO: dimethyl sulfoxide.
Preliminary mechanistic studies
To gain mechanistic insight into the proposal detailed in Fig. 1D, we conducted a series of preliminary experiments summarized in Fig. 5. Stern–Volmer quenching studies confirmed the activation of benzoate 14 by phenothiazine photocatalyst 18, showing a linear correlation between the amount of benzoate and the ratio I0/I (Fig. 5A). When the standard reaction was conducted in the presence of 1,4-dinitrobenzene (Ered = −0.64 V vs. SCE)46,47, benzoate 14 was completely recovered, suggesting the single electron reduction of the ester in standard conditions. A radical-clock experiment using primary benzoate 79 furnished the coupled product 80, demonstrating the open-shell nature of the deoxygenated intermediate (Fig. 5B). When E isomer 81 was exposed to phenothiazine 19 and purple light, a mixture of isomers with Z:E ratio of 70:30 was recovered after 16 hours, confirming the ability of the triplet excited state of phenothiazine 19 to promote in-situ photoisomerization of the coupled product (Fig. 5C). Finally, we sought to better understand the role of catechol in our olefination protocol. Several studies have shown that phenoxides can be used as photocatalysts and photoreductants48,49,50,51. While we have never observed the formation of catecholates, we performed UV-Vis studies and control experiments to exclude the possible photoactivity of these species (please see section 5: “Preliminary mechanistic studies” in the SI). The results, together with the control experiment without photocatalyst (entry 9, Fig. 2B), strongly suggest that catechol and catecholates are not productive photoactive molecules in our reaction conditions. However, we have found a possible non-covalent interaction between the styryl-BF3K 24 and catechol (please see section 5: “Preliminary mechanistic studies” in the SI), which may be responsible for a unique activation mode of the styryl-BF3K and the overall reactivity observed.

A Stern-Volmer quenching studies. B Radical-clock experiment. C Photoisomerization of the couped product E isomer. D On-DNA reactions for DELs applications. DMSO dimethyl sulfoxide, PHT phenothiazine.
On-DNA reactions for DELs applications
Given the mild reaction conditions, we aimed to investigate the aqueous compatibility of our system for its use in synthesizing DNA-encoded libraries (DELs). DELs are a powerful technology that has gained extensive use in medicinal chemistry as an efficient and cost-effective platform for discovering new pharmaceutical candidates52,53,54. In our previous studies, we reported a straightforward method for the deoxygenative cross-coupling of alcohols and DNA-pyridine conjugates21. However, only benzylic alcohols could be activated and incorporated onto DNA headpieces. Given the importance of DELs in medicinal chemistry, and the abundance of alcohols building blocks available, we focused our efforts on expanding the scope towards aliphatic alcohols and saccharides.
Preliminary results are shown in Fig. 5D. We were delighted to find that our deoxygenative olefination could be used to functionalize DNA–styryl trifluoroborate conjugate 82 with mannofuranose 83 in 60% yield. Despite the dilute aqueous conditions, the key coupling process took place in just 20 minutes. Remarkably, the DNA remained intact during the photochemical process and no oxidation was observed by mass spectrometry. Nevertheless, when other aliphatic alcohols were tested, the desired coupling products were obtained in low yields (10-20%, please see section 6: “On-DNA Deoxygenative Z-selective olefination of aliphatic alcohols” in the SI). Further investigations into the reaction mechanism under aqueous conditions and optimization of the on-DNA reaction are currently ongoing in our laboratory.
Discussion
In summary, we have developed a catalytic protocol for the deoxygenative Z-selective olefination of aliphatic alcohols. Key to this methodology is the use of a phenothiazine photocatalyst, which concurrently activates the aliphatic alcohols, promotes the cross-coupling of bench-stable alkenyl trifluoroborate salts and enables the isomerization of the final product to deliver Z-olefins. This method is distinguished by its wide substrate scope and broad applicability, even in the context of pharmaceuticals and saccharides. Given the mild and water-compatible reaction conditions, our chemistry can also be used to functionalize DNA headpieces for DELs applications. Overall, we believe that the flexibility and simplicity of our deoxygenative Z-selective olefination will make this procedure appealing to chemists in both industrial and academic settings, particularly those in medicinal chemistry.
Methods
General procedure for the photocatalytic Z-selective olefination of aliphatic alcohols
In a 10 mL microwave vial equipped with stir bar, the appropriate alcohol derivative (0.2 mmol, 1 eq.), (E)−2-arylvinyl-BF3K (0.6 mmol, 3.0 eq.), appropriate phenothiazine photocatalyst (0.02 mmol, 0.1 eq.) and 4-tert-butyl catechol or catechol (0.2 mmol, 1 eq.) were dissolved in DMSO (0.6 mL) under air. The vial was then flushed under Argon for 15 seconds and capped. The reaction was then degassed via Argon purging for 10 minutes and irradiated with 390 nm light. Note: The M.W. vial was placed at 5.0 cm distance from the Kessil 390 nm LED lamp and the reaction temperature was kept constant using a fan placed on top of the reaction at 33 cm distance. After 16 or 24 hours, the vial was decapped and (5 mL) of 1 M NaOH was added and stirred for 5 minutes. The reaction mixture was then transferred using EtOAc (sonication to ensure homogenization) to a separating funnel and the organic phase was extracted. The organic phase was washed once again with (2 mL) of 1 M NaOH and (10 mL) of saturated brine solution, dried over sodium sulfate and concentrated under reduced pressure. The reaction crude was purified by flash column chromatography (silica gel) under the stated conditions to provide the pure Z-olefin product.
General procedure for the photocatalytic Z-selective olefination of primary alcohols
In a 10 mL microwave vial equipped with stir bar, the appropriate alcohol derivative (0.2 mmol, 1 eq.), (E)−2-arylvinyl-BF3K (0.6 mmol, 3.0 eq.), appropriate phenothiazine photocatalyst (0.02 mmol, 0.1 eq.) and 4-tertbutyl catechol or catechol (0.2 mmol, 1 eq.) were dissolved in DMSO (0.6 mL) under air. The vial was then flushed under Argon for 15 seconds and capped. The reaction was then degassed via Argon purging for 10 minutes and irradiated with 390 nm light. Note: The M.W. vial was placed between two Kessil 390 nm LED lamps at 3 cm distance from each lamp and the reaction temperature was kept constant using a fan placed on top of the reaction at 33 cm distance. After 16 or 24 hours, the vial was decapped and (5 mL) of 1 M NaOH was added and stirred for 5 minutes. The reaction mixture was then transferred using EtOAc (sonication to ensure homogenization) to a separating funnel and the organic phase was extracted. The organic phase was washed once again with (2 mL) of 1 M NaOH and (10 mL) of saturated brine solution, dried over sodium sulfate and concentrated under reduced pressure. The reaction crude was purified by flash column chromatography (silica gel) under the stated conditions to provide the pure Z-olefin product.
General procedure for on-DNA Z-selective olefination of protected saccharides
A 2 mL microwave vial was charged with the appropriate alcohol derivative (5 μL of 50 mM stock solution in DMSO, 250 nmol, 100 eq), catechol (5 μL of 50 mM stock solution in DMSO, 250 nmol, 100 eq), PHT 84 (5 μL of 20 mM stock solution in DMSO, 100 nmol, 40 eq), DMSO (22.5 μL), H2O (11.5 μL), and DNA derivative 82 (1.0 μL of 1 mM stock solution in H2O, 1.0 nmol, 1 eq). The vial was flushed 15 seconds with Argon and sealed. The reaction was irradiated with 390 nm light for 20 or 30 minutes. Note: the vial was placed at 8 cm distance from the Kessil LED lamp and the reaction temperature was kept constant using a fan placed on top of the reaction at 33 cm distance. After irradiation, the reaction was transfer to a 1,5 mL Eppendorf tube and 5 µL of 3.0 M acetate buffer (pH=7) and 1 mL of cold absolute (EtOH) were added. The mixture was vortexed and allowed to stand at –65 °C for 30 min. The resulting suspension was centrifuged at 12446 x g for 5 min. The supernatant was discarded, and the sample was washed with 1 mL of cold absolute ethanol and centrifuged again at 12446 x g for 5 min. After discarding the supernatant, the pellets were dried under a flux of compressed air and redissolved in H2O to provide a stock solution of the desired concentration.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. Should any raw data files be needed in another format they are available from the corresponding author upon request.
References
Corbet, J.-P. & Mignani, G. Selected Patented Cross-Coupling Reaction Technologies. Chem. Rev. 106, 2651–2710 (2006).
Ruiz-Castillo, P. & Buchwald, S. L. Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev. 116, 12564–12649 (2016).
Campeau, L. C. & Hazari, N. Cross-Coupling and Related Reactions: Connecting Past Success to the Development of New Reactions for the Future. Organometallics 38, 3–35 (2019).
Buskes, M. J. & Blanco, M.-J. Impact of Cross-Coupling Reactions in Drug Discovery and Development. Molecules 25, 3493–3514 (2020).
Walters, W. P., Green, J., Weiss, J. R. & Murcko, M. A. What Do Medicinal Chemists Actually Make? A 50-Year Retrospective. J. Med. Chem. 54, 6405–6416 (2011).
Choi, J. & Fu, G. C. Transition metal–catalyzed alkyl-alkyl bond formation: Another dimension in cross-coupling chemistry. Science 356, eaaf7230 (2017).
Kambe, N., Iwasaki, T. & Terao, J. Pd-catalyzed cross-coupling reactions of alkyl halides. Chem. Soc. Rev. 40, 4937–4947 (2011).
Jana, R., Pathak, T. P. & Sigman, M. S. Advances in Transition Metal (Pd,Ni,Fe)-Catalyzed Cross-Coupling Reactions Using Alkyl-organometallics as Reaction Partners. Chem. Rev. 111, 1417–1492 (2011).
Zuo, Z. et al. Merging photoredox with nickel catalysis: Coupling of α-carboxyl sp3-carbons with aryl halides. Science 345, 437–440 (2014).
Chan, A. Y. et al. Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev. 122, 1485–1542 (2022).
Zhang, J. & Rueping, M. Metallaphotoredox catalysis for sp3 C–H functionalizations through hydrogen atom transfer (HAT). Chem. Soc. Rev. 52, 4099–4120 (2023).
Everson, D. A. & Weix, D. J. Cross-Electrophile Coupling: Principles of Reactivity and Selectivity. J. Org. Chem. 79, 4793–4798 (2014).
Horn, E. J., Rosen, B. R. & Baran, P. S. Synthetic Organic Electrochemistry: An Enabling and Innately Sustainable Method. ACS Cent. Sci. 2, 302–308 (2016).
Ertl, P. & Schuhmann, T. A Systematic cheminformatics analysis of functional groups occurring in natural products. J. Nat. Prod. 82, 1258–1263 (2019).
Anwar, K., Merkens, K., Troyano, F. J. A. & Gómez-Suárez, A. Radical Deoxyfunctionalisation Strategies. Eur. J. Org. Chem. 26, e202200330 (2022).
Jin, J. & MacMillan, D. W. C. Alcohols as alkylating agents in heteroarene C-H functionalization. Nature 525, 87–90 (2015).
Zhang, X. & MacMillan, D. W. C. Alcohols as latent coupling fragments for metallaphotoredox catalysis: sp3-sp2 cross-coupling of oxalates with aryl halides. J. Am. Chem. Soc. 138, 13862–13865 (2016).
Dong, Z. & MacMillan, D. W. C. Metallaphotoredox-enabled deoxygenative arylation of alcohols. Nature 598, 451–456 (2021).
Guo, P. et al. Dynamic kinetic cross-electrophile arylation of benzyl alcohols by nickel catalysis. J. Am. Chem. Soc. 143, 513–523 (2021).
Li, Z. et al. Electrochemically enabled, nickel-catalyzed dehydroxylative cross-coupling of alcohols with aryl halides. J. Am. Chem. Soc. 143, 3536–3543 (2021).
Kolusu, S. R. N. & Nappi, M. Metal-free Deoxygenative Coupling of Alcohol-Derived Benzoates and Pyridines for Small Molecules and DNA-Encoded Libraries Synthesis. Chem. Sci. 13, 6982–6989 (2022).
Xie, H., Wang, S., Wang, Y., Guo, P. & Shu, X.-Z. Ti-catalyzed reductive dehydroxylative vinylation of tertiary alcohols. ACS Catal. 12, 1018–1023 (2022).
Guo, H.-M., He, B.-Q. & Wu, X. Direct Photoexcitation of Xanthate Anions for Deoxygenative Alkenylation of Alcohols. Org. Lett. 24, 3199–3204 (2022).
Chi, B. K. et al. In-situ bromination enables formal cross-electrophile coupling of alcohols with aryl and alkenyl halides. ACS Catal. 12, 580–586 (2022).
Filippini, D. & Silvi, M. Visible Light-Driven Conjunctive Olefination. Nat. Chem. 14, 66–70 (2022).
Mastandrea, M. M., Cañellas, S., Caldentey, X. & Pericàs, M. A. Decarboxylative Hydroalkylation of Alkynes via Dual Copper-Photoredox Catalysis. ACS Catal. 10, 6402–6408 (2020).
Zheng, C., Cheng, W.-M., Li, H.-L., Na, R.-S. & Shang, R. Cis-Selective Decarboxylative Alkenylation of Aliphatic Carboxylic Acids with Vinyl Arenes Enabled by Photoredox/Palladium/Uphill Triple Catalysis. Org. Lett. 20, 2559–2563 (2018).
Masnovi, J. Radical anions of esters of carboxylic acids. Effects of structure and solvent on unimolecular fragmentations. J. Am. Chem. Soc. 111, 9081–9089 (1989).
Lam, K. & Markó, I. E. Using Toluates as Simple and Versatile Radical Precursors. Org. Lett. 10, 2773–2776 (2008).
Paquette, L. A. & Geng, F. A Highly Abbreviated Synthesis of Pentalenene by Means of the Squarate Ester Cascade. Org. Lett. 4, 4547–4549 (2002).
Saito, I., Ikehira, H., Kasatani, R., Watanabe, M. & Matsuura, T. Photoinduced reactions. 167. Selective deoxygenation of secondary alcohols by photosensitized electron-transfer reaction. A general procedure for deoxygenation of ribonucleosides. J. Am. Chem. Soc. 108, 3115–3117 (1986).
Lam, K. & Markó, I. E. Organic electrosynthesis using toluates as simple and versatile radical precursors. Chem. Commun. 95-97 https://doi.org/10.1039/B813545B (2009).
Gennaro, A., Isse, A. A., Savéant, J.-M., Severin, M.-G. & Vianello, E. Homogeneous Electron Transfer Catalysis of the Electrochemical Reduction of Carbon Dioxide. Do Aromatic Anion Radicals React in an Outer-Sphere Manner? J. Am. Chem. Soc. 118, 7190–7196 (1996).
Williams, O. P. et al. Practical and General Alcohol Deoxygenation Protocol. Angew. Chem. Int. Ed. 62, e202300178 (2023).
Speck, F., Rombach, D. & Wagenknecht, H.-A. N-Arylphenothiazines as strong donors for photoredox catalysis – pushing the frontiers of nucleophilic addition of alcohols to alkenes. Beilstein J. Org. Chem. 15, 52–59 (2019).
Jockusch, S. & Yagci, Y. The active role of excited states of phenothiazines in photoinduced metal free atom transfer radical polymerization: singlet or triplet excited states? Polym. Chem. 7, 6039–4043 (2016).
An, X.-D., Zhang, H., Xu, Q., Yu, L. & Yu, S. Stereodivergent Synthesis of α-Aminomethyl Cinnamyl Ethers via Photoredox-Catalyzed Radical Relay Reaction. Chin. J. Chem. 36, 1147–1150 (2018).
Shen, X., Huang, C., Yuan, X.-A. & Yu, S. Diastereoselective and Stereodivergent Synthesis of 2-Cinnamylpyrrolines Enabled by Photoredox-Catalyzed Iminoalkenylation of Alkenes. Angew. Chem. Int. Ed. 60, 9672–9679 (2021).
Brals, J., McGuire, T. M. & Watson, A. J. B. A Chemoselective Polarity-Mismatched Photocatalytic C(sp3)−C(sp2) Cross-Coupling Enabled by Synergistic Boron Activation. Angew. Chem. Int. Ed. 62, e202310642 (2023).
Lu, J. et al. Photoinduced Electron Donor Acceptor Complex-Enabled α-C(sp3)−H Alkenylation of Amines. Angew. Chem. Int. Ed. 63, e202409310 (2024).
Borowicz, P. et al. Nature of the lowest triplet states of 4′-substituted N-phenylphenothiazine derivatives. Phys. Chem. Chem. Phys. 2, 4275–4280 (2000).
Ni, T., Caldwell, R. A. & Melton, L. A. The relaxed and spectroscopic energies of olefin triplets. J. Am. Chem. Soc. 111, 457–464 (1989).
Singh, K., Staig, S. J. & Weaver, J. D. Facile Synthesis of Z-Alkenes via Uphill Catalysis. J. Am. Chem. Soc. 136, 5275–5278 (2014).
Neveselý, T., Wienhold, M., Molloy, J. J. & Gilmour, R. Advances in the E → Z Isomerization of Alkenes Using Small Molecule Photocatalysts. Chem. Rev. 122, 2650–2694 (2022).
Martínez-Gualda, A. M. et al. Chromoselective access to Z- or E- allylated amines and heterocycles by a photocatalytic allylation reaction. Nat. Commun. 10, 2634 (2019).
Arceo, E., Jurberg, I., Álvarez-Fernández, A. & Melchiorre, P. Photochemical activity of a key donor–acceptor complex can drive stereoselective catalytic α-alkylation of aldehydes. Nat. Chem. 5, 750–756 (2013).
Mendkovich, A. S. et al. Integrated Study of the Dinitrobenzene Electroreduction Mechanism by Electroanalytical and Computational Methods. Int. J. Electrochem. 5, 346043 (2011).
Liu, C., Shen, N. & Shang, R. Photocatalytic defluoroalkylation and hydrodefluorination of trifluoromethyls using o-phosphinophenolate. Nat. Commun. 13, 354 (2022).
Liu, C., Zhang, Y. & Shang, R. BINOLates as potent reducing photocatalysts for inert bond activation and reduction of unsaturated systems. Chem 11, 102359 (2025).
Liang, K. et al. Deprotection of benzyl-derived groups via photochemically mesolytic cleavage of C–N and C–O bonds. Chem 9, 511–522 (2023).
Filippini, G., Dosso, J. & Prato, M. Phenols as Novel Photocatalytic Platforms for Organic Synthesis. Helv. Chim. Acta 106, e202300059 (2023).
Gironda-Martínez, A., Donckele, E. J., Samain, F. & Neri, D. DNA-Encoded Chemical Libraries: A Comprehensive Review with Succesful Stories and Future Challenges. ACS Pharmacol. Transl. Sci. 4, 1265–1279 (2021).
Song, M. & Hwang, G. T. DNA-Encoded Library Screening as Core Platform Technology in Drug Discovery: Its Synthetic Method Development and Applications in DEL Synthesis. J. Med. Chem. 63, 6578–6599 (2020).
Flood, D. T., Kingston, C., Vantourout, J. C., Dawson, P. E. & Baran, P. S. DNA Encoded Libraries: A Visitor’s Guide. Isr. J. Chem. 60, 268–280 (2020).
Acknowledgements
The authors thank CIQUS, MICIU (PID2020-113067GA-I00, TED2021-129833A-I00, RYC2022-035515-I), the Xunta de Galicia (ED431F 2024/027, Centro de investigación do Sistema universitario de Galicia accreditation 2023-2027, ED431G 2023/03, and for C. A.-R. predoctoral fellowship, ED481A-2023-178) and the European Union (European Regional Development Fund - ERDF) for the financial support of this work. We sincerely thank Dr Aaron Trowbridge for proofreading this manuscript. We are also thankful for the use of CIQUS and RIAIDT-USC analytical facilities.
Author information
Authors and Affiliations
Contributions
M.N. and S.R.N.K conceived the project. M.N., S.R.N.K., I.S.-S., C.A.-R. and E.A. designed the experiments. S.R.N.K., I.S.-S., C.A.-R. and E.A. performed the experiments. M.N. wrote the manuscript, secured the fundings and supervised the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer?review
Peer review information
Nature Communications thanks Gang Chen, Rui Shang and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kolusu, S.R.N., Sánchez-Sordo, I., Aira-Rodríguez, C. et al. Photocatalytic deoxygenative Z-selective olefination of aliphatic alcohols. Nat Commun 16, 3155 (2025). https://doi.org/10.1038/s41467-025-58469-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58469-z
