
Abstract
Benzamides constitute an important class of bulk and fine chemicals as well as essential parts of many life science molecules. Currently, all these compounds are majorly produced from petrochemical-based feedstocks. Notably the selective aerobic oxidative conversion of lignin and lignin-derived compounds to primary, secondary, and tertiary amides and phenols offers the potential for a more sustainable synthesis of valuable building blocks for fine chemicals, monomers for polymers, biologically active molecules, and diverse consumer products. Here we present the concept of “lignin to amides” which is demonstrated by a one-pot, multi-step oxidation process utilizing molecular oxygen and a 3d-metal catalyst with highly dispersed and stable cobalt species (Co-SACs) supported on nitrogen-doped carbon in water as solvent. Moreover, our cobalt-based methodology allows for the cost-efficient transformation of a lignin and its variety of derivates simply using O2 and organic amines. Mechanistic investigations and control experiments suggest that the process involves an initial dehydrogenation of Cα-OH, cleavage of the Cβ-O as well as C(O)-C bond and condensation of the resulting carboxylic acids with amines. Spectroscopic studies indicate that the formation of superoxide species (O2●−) and specific Co-nitrogen sites anchored on mesoporous carbon sheets are key for the success of this transformation.
Similar content being viewed by others
Introduction
Over the past two centuries, the growing world population and the excessive consumption of fossil resources have caused significant environmental problems. Among other problematic issues, billions of tonnes of CO2 have been released into the atmosphere, far exceeding the capacity of the Earth’s ecological cycle and leading to the global climate problems of today1,2,3,4. In this context, “carbon peaking”4 and “achieving carbon neutrality”5 have become important topics of concern for the global scientific community including researchers from social, economic, natural, and other sciences. In addition to the development of climate-neutral and carbon dioxide-free energy technologies that are in line with the Paris Agreement, circular chemistry can make a significant contribution to achieve sustainability6,7,8,9. The United Nations Intergovernmental Panel on Climate Change (IPCC) found that the chemical and petrochemical industry, which manufactures important products from fossil raw materials, is responsible for over seven percent of greenhouse gas emissions10. According to a report from the International Energy Agency (IEA) on the future of petrochemicals, this feedstock accounts for 12% of global oil demand and continues to grow. Explicitly, it was stated: “Our economies are heavily dependent on petrochemicals, but the sector receives far less attention than it deserves”11. To move away from the use of oil and gas as feedstocks in the chemical industry, it is necessary to develop a toolbox of technologies which allows the production of all kinds of valuable chemicals without concomitant formation of carbon dioxide. Specifically, the increased use of waste as well as abundant, non-eatable biomass as feedstocks can be part of the solution to meet the ever-growing demand and to reduce the dependence on fossil resources in this area12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28.
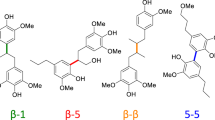
One of the most abundant biomass resources is lignocellulose, which is available from agricultural residues, forestry wastes, and energy crops4,20,21,22,23,24,25,26,27,28. It has an annual production of around 170 billion tons via plant photosynthesis and consists of three main components: cellulose (40–50 wt%), hemicellulose (20–30 wt%), and lignin (10–35 wt%). While cellulose and hemicellulose are polymers of C5 and C6 sugars, lignin is a heterogeneous, highly crosslinked polymer akin to phenol-formaldehyde resins, which is made up of various building blocks with certain bond linkages, e.g., β-O-4, β-5, α-O-4, β-β, 4-O-5, β-1 and others (see Fig. 1a)4,20,21,22,23,24,25,26,27,28. Due to the large content of aromatic structures, it is regarded as a potential alternative resource to replace fossil-based aromatic chemicals. Hence, research on lignin depolymerization and its downstream products have attracted much attention from both academic and industrial researchers in the past years. The majority of the investigated lignin transformations include hydrogenolysis29,30,31,32,33, hydrolysis34,35,36, and nucleus exchange processes18,37,38,39,40,41,42,43,44, which provide a variety of interesting building blocks. Compared to these works, selective oxidations, especially using air or molecular oxygen, to provide carboxylic acid derivatives are scarce12,40,41,42,44,45,46,47. Among these derivatives, amides are highly valuable compounds, which are widely used in organic synthesis. Meanwhile, these products are used in numerous agrochemicals and pharmaceuticals48,49,50,51. In fact, 73 of the top 200 selling drugs of the year 2022 were based on amides and its derivatives, mainly aromatic one (Fig. 1b)52.

a Representative lignin structure and typical linkages, (b) selected benzamide containing drugs, (c) classical and current methods for the preparation of amides, (d) this work on the renewable synthesis of amides with Co-SACs.
Conventional strategies for amide bond formation typically involve the condensation of a carboxylic acid or its derivatives with an amine, leading to the elimination of one equivalent of water (Fig. 1c)53,54,55. Functionalized amides are generally synthesized through the reaction of activated carboxylic acid derivatives, such as acyl chlorides and anhydrides, with amines. Alternatively, direct amidation of carboxylic acids with amines can be achieved using stoichiometric amounts of coupling reagents. However, these methods often generate considerable waste, raising environmental and sustainability concerns. Additionally, other methods, such as nitrile hydration56, Ritter reaction57, Passerini reaction58, Schmidt reaction59, and Beckmann rearrangement60 are applied for amide synthesis (Fig. 1c).
With respect to lignin model compounds, Loh and Chiba reported the oxidative synthesis of α-ketoamides by Cβ-H oxidation and Cβ-O bond cleavage of β-O-4 models in the presence of CuI and secondary amines40. More recently, the groups of Wang and Liu reported the synthesis of amides by oxidation of 2-phenoxyacetophenones applying homogeneous copper complexes41,42. In 2022, Xu and co-workers reported the preparation of benzanilides by oxidation of 2-phenoxyacetophenones with H2O243. In the same year, Dai and co-workers reported the heterogeneous manganese oxide-catalyzed cleavage of C-C bonds in alcohols using oxygen as the environmentally benign oxidant and ammonia as the nitrogen source44. Despite these advancements, all reported methods make only use of a limited number of lignin model compounds and have not been used for “real” lignin.
With the aim of developing a general methodology for the synthesis of various aromatic amides from renewable raw materials, we became interested in the oxidative amidation of lignin, its derivatives, and model compounds. Due to stability, ease of separation, and potential recycling, heterogeneous materials are generally preferred over homogeneous catalysts, and they might be readily used in flow reactors, facilitating production using continuous processes. Unfortunately, to the best of our knowledge, there are rare examples of heterogeneous materials for amides synthesis starting from lignin derivates. To ensure sufficient reactivity, we aimed to use highly dispersed redox-active 3d-metal centers merging the advantages (superior reactivity, high selectivity, and stability) of both homogeneous and heterogeneous catalysts61,62,63,64,65,66. Herein, we describe a generally applicable protocol which allows for highly selective oxidative amidation of lignin derivates in the presence of molecular O2 and H2O. The identified single cobalt atom catalyst (Co-L1@NC-800) shows high stability and broad functional group tolerance.
Results
Catalyst synthesis and model investigations
At the start of this project, several highly dispersed cobalt materials were generated on nitrogen-doped carbon via a so-called template-sacrificial method. Typically, Co(NO3)2.6H2O was mixed with ammonium chloride (NH4Cl) and 1,10-phenanthroline (L1, Phen) in ethanol to form a solution of the corresponding homogeneous metal-N complexes, which are subsequently supported on a specific SiO2 template (LUDOX®HS-40; 40 wt.% suspension in H2O). Subsequently, the obtained samples were pyrolyzed at 700–1000 °C under an N2 flow for 2 h. Finally, the isolated Co-based material is obtained by etching the pyrolyzed residue with ammonium bifluoride (4 M NH4HF2) and washing with 1 M H2SO4 to remove the silica template and unactive species (Fig. 2a). The resulting materials are represented as Co-L@NC-T, where L and T are the ligands and pyrolysis temperature, respectively. Similar materials based on Fe, Mn, and Cu were prepared and other cobalt catalysts were also prepared by using different ligands, such as o-phenylenediamine (L2, OPDA), p-phenylenediamine (L3, PPDA), 2,6-diaminopyridine (L4, DAP), aniline (L5, AN), hexamethylenediamine (L6, HMDA).

a Schematic representation of catalyst preparation; (b) investigating the effect of different ligands; (c) testing the activity of different metal-based materials; (d) investigating the effect of different solvents. e Stability and recycling tests of Co-L1@NC-800. Reaction conditions: 0.3 mmol 2-phenoxy-1-phenylethanol, 0.6 mmol aniline, 0.15 mmol K3PO4, 40 mg catalyst (4.5–5 mol% metal), 5 bar O2, 3 mL H2O, 140 °C, 20 h, the conversion and yields were calculated by GC based on 1 with mesitylene as internal standard. Reaction conditions for (e): 0.6 mmol 2-phenoxy-1-phenylethanol, 1.2 mmol aniline, 0.3 mmol K3PO4, 80 mg Co-L1@NC-800 (4.52 mol% Co), 5 bar O2, 5 mL solvent H2O, 140 °C, 7 h.
Next, all these prepared materials (M-L@NC-T) were tested for the oxidative amidation of 2-phenoxy-1-phenylethanol 1 with aniline 2 to obtain benzanilide 3 in presence of molecular O2 and H2O solvent as benchmark reaction. 2-Phenoxy-1-phenylethanol 1 is a commonly used model compound for lignin and the inherent β-O-4 structure is the most abundant lignin linkage. Furthermore, the desired product 3 has been used as an important precursor for synthesis of pharmaceuticals, dyes, and flavors42. As shown in Fig. 2b, among the tested catalysts the one prepared from L1 displayed the best performance (99% conversion of 1; 84% yield of benzanilide 3; Fig. 2b, column 1). Apart from the desired product 3, small amounts of α-ketoamide 5 (14%) and N-phenylformamide (20%) were formed. The latter side-product is formed by condensation of aniline and formic acid. Other materials obtained from the pyrolysis of Co(NO3)2.6H2O in the presence of L2-L6 showed lower activities and chemoselectivities towards the formation desired product 3. As expected, the non-pyrolyzed material (Co-L1@SiO2) displayed very poor activity (Supplementary Table 2, entry 5). Other materials with remaining SiO2 and the one prepared without nitrogen ligands (Co@SiO2-800) as well as the homogeneous cobalt complexes all showed poorer performance (Supplementary Table 2, entries 6-8).
In addition to the cobalt materials, related Fe-, Mn-, and Cu-L1@NC-800 samples were also synthesized using the same procedure. In catalytic tests, however, they showed a significantly lower conversion in the benchmark reaction (Fig. 2c). Recently it was shown that so-called dual atomic catalysts, e.g., Co/Fe, Co/Cu, have a higher activity compared to monometallic SACs due to synergistic interatomic interactions67,68,69. Hence, Co/Fe-, Co/Mn-, and Co/Cu-L1@NC-800 were also prepared and tested for the benchmark reaction. Unfortunately, the activity and selectivity of these catalysts were not better than that of Co-L1@NC-800 (Fig. 2c, column 3 and Supplementary Table 3, entries 1-3).
In general, solvents can affect the dispersion of solid materials, which has an impact on their activity and selectivity. The effect of different solvents on the product distribution is shown in Fig. 2d and Supplementary Table 4. The best chemoselectivity of 3 is achieved when water is used as the only solvent (Fig. 2d, column 1). In other organic solvents as well as mixed solvent systems increased formation of α-ketoamide 5 is observed (Fig. 2d and Supplementary Table 4). Furthermore, the influence of other reaction parameters, such as the reaction time, oxygen pressure and temperature were also examined, and the details are shown in Supplementary Table 5.
In addition to the high activity and selectivity, the stability, recyclability and scale-up aspects are crucial for practical applications. Thus, we performed reactions at a controlled conversion of 50% and reused the catalyst. As shown in Fig. 2e, Co-L1@NC-800 could be reused six times without significant decrease in activity. To exclude the possibility of Co leaching, a hot filtration experiment was also performed (Supplementary Fig. 10). After filtration of the catalyst material after 6 h, no further conversion was observed, indicating that the reaction was exclusively heterogeneous. In addition, the model experiment on a gram scale proceeded smoothly under standard conditions with 80% yield (Supplementary Fig. 14). All these results clearly demonstrate the reusability, stability and scalability of Co-L1@NC-800 catalytic system.
Characterization of Co-based materials
To understand the structural features of the prepared materials, the optimal catalyst (Co-L1@NC-800) and a recycled sample (Co-L1@NC-800R) were characterized using X-ray powder diffraction (XRD), transmission electron microscopy (TEM), high-angle annular dark-field scanning TEM (HAADF-STEM), X-ray spectroscopy (EDS), X-ray photoelectron spectroscopy (XPS), and X-ray absorption spectroscopy (XAS). The content of Co was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis and found to be approximately 2.0 wt%. The XRD patterns of Co-L1@NC-800 indicate the presence of two distinct diffraction peaks around 25° and 44°, which can be assigned to the (002) and (101) crystal planes of graphitic support, respectively (Supplementary Fig. 1)67,70. Notably, there are no diffraction peaks related to crystalline metallic cobalt or cobalt oxides in this material, thus excluding the presence of any large cobalt-containing crystalline particles. XRD data of the recycled catalyst sample revealed similar patterns compared to the fresh catalyst (Supplementary Fig. 1). In accordance with XRD findings and the absence of nanoparticles in TEM image (Fig. 3a and Supplementary Fig. 3), we propose the presence of highly dispersed Co species on the carbon matrix. Meanwhile, the porosity of the optimal material is clearly observed in TEM (Fig. 3a), which is explained by the removal of SiO2. Indeed, adsorption desorption isotherms confirm the highly mesoporous nature of the sample exhibiting a large surface area (584.4 m2/g) and pore volume (1.46 cm3/g) (Supplementary Fig. 2a). The pore size distribution corresponds well with the size distribution of original silica showing the maximum at around 5.3 nm (Supplementary Fig. 2b). These properties should support the absorption and transport of oxygen and substrates during the oxidation reaction and thus contribute to the excellent catalytic activity71. Interestingly, no cobalt species were observed in the HR-TEM images (Fig. 3a and Supplementary Fig. 3), which rules out large cobalt nanoparticles in this material. The highly dispersed Co-containing species were then analyzed using HAADF-STEM. The representative images (Fig. 3b) clearly show that the Co species (yellow cycles) are atomically dispersed on the N-doped carbon. EDS analyzes of the catalyst also confirm the presence of cobalt, even if the content is very low due to the highly dispersed form (Supplementary Fig. 4). Similarly, energy-dispersive X-ray spectroscopy (EDX) images reveal the presence of Co, N, and C elements in a homogeneous distribution over the entire N-doped carbon matrix in this sample (Fig. 3c–e). The elemental composition of the surface and the electronic state of Co-L1@NC-800 and Co-L1@NC-800-R were then determined using XPS. As illustrated in Supplementary Fig. 7, the survey spectra of the composites exhibited distinctive peaks of C, N, O and Co, respectively. The N 1 s spectra (Fig. 3f) displayed four peaks fitted with binding energies at 398.3, 399.7, 400.4 and 401.8 eV corresponding to pyridinic, Co-N, pyrrolic, and graphitic-N atoms, respectively. In addition, two peaks with Raman shifts at around 1351 cm−1 (D bond) and 1586 cm−1 (G bond, ID/IG 1.2) are observed in the Raman spectra of Co-L1@NC-800 (Supplementary Fig. 6), which are attributed to defect carbon and graphitic carbon, respectively.

a HR-TEM images, (b) HAADF-STEM images (Co single atoms are marked by yellow cycles), (c–e) EDS mapping C (red), N (green), Co (yellow), (f) N 1 s XPS of Co-L1@NC-800, (g) XANES spectra at the Co K-edge (inset is the magnified image), (h) FT-EXAFS at R-space, (i) EXAFS fitting shown in R-space and (j) Wavelet transform of Co-L1@NC-800, CoPC, Co foil and Co3O4.
To further elucidate the bonding state and local coordination structure of Co atoms of Co-L1@NC-800, Co K-edge synchrotron radiation-based X-ray absorption near-edge structure (XANES) measurements were performed with Co foil, CoO, Co3O4 and cobalt phthalocyanine (CoPc) as references. The near-edge feature of Co-L1@NC-800 is located between CoO and Co3O4, indicating an electronic structure of Coδ+ (2 <δ < 3) (Fig. 3g). The Fourier-transformed k3-weighted extended X-ray absorption fine structure (FT-EXAFS) spectrum in the R space (Fig. 3h) displays a main peak at 1.40 Å, which can be assigned to Co-N first coordination shell, and no typical Co-Co coordination peak at 2.16 Å can be detected, confirming the sole presence of isolated metal sites in Co-L1@NC-800 and excluding the possibility of the aggregation of anchored Co atoms. Furthermore, wavelet transform (WT) plot was used further identified the absence of Co-Co scattering (Fig. 3j). Here, WT maximum at 1.4 Å and 4.0 Å−1, assigned to the Co-N bond by comparing Co foil, Co3O4 and CoPc, and no corresponding Co-O and Co-Co intensity was detected. The results of the EXAFS analysis show that each Co atom has a statistical mean coordination number of 5.2 and the Co-N bond distance is 1.89 Å (Supplementary Table 1). Again, these results confirm the proposed Co-N5 configurations in Co-L1@NC-80069.
Selective synthesis of amides: scope and limitations
In order to use lignin or lignin-derived compounds as a raw material for the synthesis of fine chemicals or specialties, it is important to know the general range of applications in relation to the substrates. Once suitable reaction conditions and an optimal catalyst (Co-L1@NC-800) were in hand for the benchmark reaction, the oxidative amidation of lignin model compounds and substances obtainable by the degradation of lignin was therefore carried out. Initially, a series of amides were produced from 2-phenoxy-1-phenylethanol and various amines. The resulting products can be easily further functionalized and can therefore be used as central building blocks in organic chemistry. As shown in Fig. 4, the oxidative amination of aromatic, heterocyclic and aliphatic amines is easily achieved under standard reaction conditions and the corresponding secondary amides were produced in good to excellent yields. Simple anilines as well as substituted ones bearing aromatic or alkyl groups gave the corresponding secondary amides in yields of up to 88% (Fig. 4; products 6–8, 21–22). In a similar way, halogenated amides were prepared in high yields, which are versatile intermediates in organic synthesis as well as for pharmaceuticals and agrochemicals (Fig. 4; products 9–15, 25–26). In addition, functionalized amines containing methoxy-, benzyloxy-, hydroxy-, nitro-, cyano-, as well as amide groups gave the corresponding products in up to 90% yield (Fig. 4; products 16–20, 24, 27–28). Di- and tri-substituted amides were conveniently prepared, too (Fig. 4; products 25–29). Next, several heterocyclic amines containing pyridine, pyrimidine, imidazole, thiazole, indole, quinoline, benzodioxole, and benzodioxane units are well tolerated and the corresponding heterocyclic amides were smoothly obtained (Fig. 4; products 30-41). Performing the oxidative amidation of 2-phenoxy-1-phenylethanol with secondary amines, provided poorer chemoselectivity due to C-N bond cleavage (Fig. 4; products 42–45, 51). For example, when diphenylamine was reacted with 2-phenoxy-1-phenylethanol, apart from the desired product 43, 40% of 3 was obtained. On the other hand, applying aliphatic amines bearing different lengths of carbon chains provided the corresponding amides smoothly (Fig. 4; products 47-50). Interestingly, an adenine-typed amide was successfully prepared in 61% yield, too (Fig. 4; product 52).

Reaction conditionsa: 0.3 mmol 2-phenoxy-1-phenylethanol, 0.6 mmol amine, 40 mg Co-L1@NC-800 (4.52 mol% Co), 0.15 mmol K3PO4, 5 bar O2, 3 mL H2O, 140 °C, 20 h, isolated yield; b: same as “a” with 0.3 mmol K3PO4; c: GC yield based on the lignin model compounds.
Encouraged by the above results, different lignin derivatives were tested. To demonstrate the activity of the Co-L1@NC-800 catalyst for such transformations, three types of amides were selectively prepared starting from different lignin model compounds in the presence of aniline (product a), aqueous dimethylamine (product b), and aqueous ammonia (product c), respectively. Firstly, several β-O-4 compounds bearing different methoxy and γ-OH group were reacted with different kind of amines and smoothly produced corresponding amides and phenols (Fig. 5; products 53–57). In the case of substrate 57, in addition to Cβ-O cleavage, a further C-C cleavage reaction is necessary to generate the desired product. Nevertheless, our cobalt catalyst system showed the desired activity for this substrate. Subsequently, α-O-4 (Fig. 5; products 58–60) and β-1 (product 65) compounds were tested and the corresponding primary, secondary, and tertiary amides are delivered in yields up to 95%. Furthermore, more sensitive lignin derivatives can be applied in this transformation under ambient pressure (Fig. 5; products 61–63). To demonstrate the potential of this Co-SACs for biorefinery applications, we next tested the one-pot conversion of different mixtures into the corresponding amides. For example, a mixture of substrates 53, 55, 57, 58, 59, 60 and 65 was effectively converted to the desired product 53c in the presence of aqueous ammonia, obtaining 83% yield. It should be noted that the boiling point and polarity of the amide are much higher than that of substrates and minor byproducts. Therefore, the obtained amides can be easily isolated from the reaction mixture. Similarly, other compound mixtures were also converted to the corresponding amides (Fig. 5; products 68 and 69). In general, primary amines, especially anilines as well as ammonia reacted well with 2-phenoxy-1-phenylethanol to produce corresponding secondary and primary amides. However, secondary amines are less reactive towards amidation and gave <55% yields of corresponding amides (Fig. 4, products 42–45). In case of morpholine C-N bond cleavage occurred and the formation of desired product did not observe. Encouraged by these results from model compound conversion, pine wood chips were subjected to a one-pot amidation process in MeCN solvent, with ammonia acetate (NH4OAc) was used as nitrogen source. Notably, through this lignin-first strategy, amide 62c can be produced at a yield of 13 wt.% (relative to lignin content) in presence of the Co-L1@NC-800. Given that the theoretical monomer yield of 23 wt.% through nitrobenzene oxidation assays (Supplementary Tables 7–8), the Co-SACs mediated amidation process is highly efficient. As far as we know, this is the first example of the conversion of authentic lignin or raw biomass to amides using a heterogeneous catalyst.

a: Reaction conditions for products a: 0.3 mmol lignin model, 0.6 mmol amine, 40 mg Co-L1@NC-800 (4.52 mol% Co), 0.15 mmol K3PO4, 5 bar O2, 3 mL H2O, 140 °C, 20 h, GC yield based on the lignin model compounds; reaction conditions for products b: same as “products a” with 200 μL 1 M dimethylamine (aq.); reaction conditions for products c: same as “products a” with 100 μL aq. NH3 (25–28%). b: same as “a” without K3PO4; c: same as “a” at 70 °C with 1 bar air and performed in 20 mL pressure tube; d: 0.2 mmol for each lignin models, 200 mg Co-L1@NC-800 (4.83 mol% Co), 500 μL aq. NH3 (25–28%), 0.75 mmol K3PO4 5 bar O2, 20 mL H2O, 150 °C, 24 h; e: same as “a” with 40 mg Co-L1@NC-800 (4.52 mol% Co), 150 μL aq. NH3 (25–28%), 0.15 mmol K3PO4, 3 mL H2O; f: same as “a” with 150 mg Co-L1@NC-800 (5.1 mol% Co), 300 μL aq. NH3 (25–28%), 0.5 mmol K3PO4, 10 mL H2O; g: reaction conditions for pine lignin: 100 mg pine lignin, 20 mg Co-L1@NC-800, 3.0 mmol NH4OAC, 0.5 mmol H2SO4, 20 bar air, 10 mL MeCN, 180 °C, 20 h, HPLC yield with 1,4-dimethoxybenzene as internal standard. The yields of phenols were determined by GC, 4b: guaiacol, 4c: 4-methoxyphenol, 4 d: hydroquinone.
Mechanistic investigations
Several control experiments, a kinetic profile of the model reaction and spectroscopic in-situ measurements were carried out in order to understand the catalytic processes and reaction pathways during oxidative amination and to gain an insight into possible reaction pathways. Firstly, Co-L1@NC-800 catalyzed conversion of 2-phenoxy-1-phenylethanol was converted to phenoxyacetophenone (1b), acetophenone (1c), benzoylformic acid (1d), benzaldehyde (1e), and benzoic acid (1f) in the absence of amines over 12 h (Figs. 6a, 1). Subsequently, the control experiments of these intermediates with aniline were carried out and benzanilide was smoothly produced (Figs. 6a, 2) and Supplementary Fig. 12. As can be seen in Fig. 6b, the overall reaction using Co-L1@NC-800 succeeds with >90% conversion of 2-phenoxy-1-phenylethanol after 12 h, with the intermediates phenoxyacetophenone (1b), acetophenone (1c), benzoylformic acid (1d), benzaldehyde (1e), and benzoic acid (1f) building up and decomposing. This clearly indicates these products as intermediates in this oxidative amidation reaction. To further explore the reaction mechanism, next we conducted an isotope labeling experiment with H218O and O2 as oxygen sources. However, after the reaction there is no 18O labeled amide detected, which reveals that oxygen atoms of the amide groups originate from O2 rather than H2O (Figs. 6a, 3).

a Control experiments; (b) course of reaction time on the oxidative amidation of 2-phenoxy-1-phenylethanol; (c) radical trapping experiments; (d) DMPO spin-trapping EPR spectrum of O2 with Co-L1@NC-800, aniline and 2-phenoxy-1-phenylethanol in MeCN; (e) suggested reaction pathway.
Further, Experiments were carried out in the presence of radical scavengers in order to elucidate the reactive oxygen species (ROS) that occur during these transformations. As shown in Fig. 6c (columns 2 and 4), the addition of t-BuOH or NaN3, which are known to quench singlet oxygen (1O2) and hydroxyl (●OH) radicals, respectively, basically did not affect the reaction efficiency. Thus, the involvement of these types of radicals in the reaction system is ruled out. Even the addition of 2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO) to the reaction system to scavenge the free radicals involved did not result in a significant reduction of 3 (Fig. 6c, column 3). However, after the addition of 1,4-benzoquinone (PBQ), the reaction is significantly inhibited (Fig. 6c, column 6). The latter experiment clearly indicates that O2●− is formed during the reaction, as PBQ is known to react with superoxide (O2●−) species. This assumption is further supported by in-situ electron paramagnetic resonance (EPR) experiments in the presence of 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a spin trap agent. Here, a spin signal with the intensity ratio of 1:1:1:1:1:1 was observed (Fig. 6d), which is assigned to the signal of a DMPO-O2●− adduct44. Meanwhile, nitrogen radicals were also detected by in-situ EPR (Fig. 6d). Next, we also performed poisoning experiment of SAC-Co with potassium thiocyanate (KSCN), as shown in Fig. 6c (column 5), the catalyst’s activity showed significant decrease under the standard condition when KSCN was applied in the catalytic system, suggesting the involvement of atomic Co-N sites in the present catalytic reaction.
Based on all these results, we propose the following general reaction pathway: Initially, molecular oxygen interacts with Co-L1@NC-800 to produce O2●− on the catalyst surface, which then reacts with 2-phenoxy-1-phenylethanol to generate 2-phenoxyacetophenone via Cα-OH dehydrogenation. Next, oxidative cleavage of 2-phenoxyacetophenone forms phenol and acetophenone undergoes β-C-O scission. The latter one is further converted to benzoic acid via C(O)-C bond cleavage45,47. Ultimately, the desired amide formation proceeds through further reaction of benzoic acid with amine (Fig. 6e).
In this work, we presented the renewable and sustainable synthesis of a variety of functionalized amides via cobalt-catalyzed aerobic oxidative amidation of lignin and its derivates in the presence of a highly dispersed mesoporous cobalt-based catalyst. Apart from the specific catalyst, this process makes use of a biomass-based feedstock, molecular oxygen, amines, H2O and does not need any toxic solvents or other reagents. The developed catalytic material is easily prepared by a simple template-sacrificial method and contains isolated Co-N centers with Co-N5 configuration. This structural motif and the high surface area seem crucial for generating reactive oxygen species (O2●−) and to allow for efficient C-O/C-C bond cleavage reactions to synthesize different kinds of amides. The developed protocol is a first step towards a general, practical, and green synthesis of aromatic amides from renewable substrates utilizing earth abundant metal based single atoms.
Methods
Synthesis of secondary and tertiary amides from lignin model structure and amines
The magnetic stirring bar, 0.3 mmol lignin model compound, 0.6 mmol amine, 0.15 mmol K3PO4, 40 mg Co-L1@NC-800 were transferred to an 8 mL glass vial and then 3 mL H2O was added. Next, the vial was fitted with septum, cap and needle and the reaction vials (8 reactions vials at a time containing different substrates) were placed into a 300 mL autoclave. The autoclave was sealed and flushed with 4–5 bar O2 and then it was pressurized with 5 bar O2. Subsequently, the autoclave was placed into an aluminum block preheated at 140 °C and the reactions were stirred for the required time. After the completion of the reactions, the autoclave was cooled to room temperature, the remaining oxygen was discharged, and the samples were removed from the autoclave. The solid catalyst was filtered and washed thoroughly with ethanol and ethyl acetate. The filtrate containing product was extracted thoroughly with ethyl acetate and then analyzed by GC-MS. The corresponding amides were purified by column chromatography (silica; pentane-ethyl acetate mixture) and characterized by NMR analysis. The yields were determined by GC for the selected amides: After completion of the reaction, mesitylene (50 µL) as standard was added to the reaction vials and the reaction products were diluted with ethyl acetate followed by filtration using plug of silica and then analyzed by GC.
Data availability
The experimental and analytical data generated in this study are provided in the Supplementary Information. All data are available from the corresponding author upon request.
References
Annual global emissions of carbon dioxide 1940–2023: https://www.statista.com/statistics/276629/global-CO2-emissions/.
Zhong, J. W. et al. State of the art and perspectives in heterogeneous catalysis of CO2 hydrogenation to methanol. Chem. Soc. Rev. 49, 1385–1413 (2020).
Liu, Z., Deng, Z., Davis, S. J. & Ciais, P. Global carbon emissions in 2023. Nat. Rev. Earth Environ. 5, 253–254 (2024).
Zhang, C. F. et al. Catalytic strategies and mechanism analysis orbiting the center of critical intermediates in lignin depolymerization. Chem. Rev. 123, 4510–4601 (2023).
Kohse-Höinghaus, K. Combustion, chemistry, and carbon neutrality. Chem. Rev. 123, 5139–5219 (2023).
Fankhauser, S. et al. The meaning of net zero and how to get it right. Nat. Clim. Chang. 12, 15–21 (2022).
van Soest, H. L. et al. Net-zero emission targets for major emitting countries consistent with the Paris agreement. Nat. Commun. 12, 2140 (2021).
The Paris Agreement. https://unfccc.int/process-and-meetings/the-paris-agreement.
Rogelj, J. et al. Net-zero emissions targets are vague: three ways to fix. Nature 591, 365–368 (2021).
Climate change widespread, rapid, and intensifying-IPCC. https://www.ipcc.ch/2021/08/09/ar6-wg1-20210809-pr/.
The Future of Petrochemicals. https://www.iea.org/reports/the-future-of-petrochemicals.
Luo, H. et al. Oxidative catalytic fractionation of lignocellulosic biomass under non-alkaline conditions. J. Am. Chem. Soc. 143, 15462–15470 (2021).
Sullivan, K. P. et al. Mixed plastics waste valorization through tandem chemical oxidation and biological funneling. Science 378, 207–211 (2022).
Cen, Z. et al. Upcycling of polyethylene to gasoline through a self-supplied hydrogen strategy in a layered self-pillared zeolite. Nat. Chem. 16, 871–880 (2024).
Liu, M. Y., Wu, X. B. & Dyson, P. J. Tandem catalysis enables chlorine-containing waste as chlorination reagents. Nat. Chem. 16, 700–708 (2024).
Liu, M. Y., Han, B. X. & Dyson, P. J. Simultaneous generation of methyl esters and CO in lignin transformation. Angew. Chem. Int. Ed. 61, e202209093 (2022).
Zhang, P. et al. Streamlined hydrogen production from biomass. Nat. Catal. 1, 332–338 (2018).
Zhang, B. et al. Sustainable production of benzylamines from lignin. Angew. Chem. Int. Ed. 60, 20834–20839 (2021).
Li, N. et al. Selective lignin arylation for biomass fractionation and benign bisphenols. Nature 630, 381–386 (2024).
Li, C. Z., Zhao, X. C., Wang, A. Q., Huber, G. W. & Zhang, T. Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 115, 11559–11624 (2015).
Zhang, C. F. & Wang, F. Catalytic lignin depolymerization to aromatic chemicals. Acc. Chem. Res. 53, 470–484 (2020).
Zakzeski, J., Bruijnincx, P. C. A., Jongerius, A. L. & Weckhuysen, B. M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 110, 3552–3599 (2010).
Shen, X. J., Zhang, C. F., Han, B. X. & Wang, F. Catalytic self-transfer hydrogenolysis of lignin with endogenous hydrogen: road to the carbon-neutral future. Chem. Soc. Rev. 51, 1608–1628 (2022).
Wong, S. S., Shu, R. Y., Zhang, J. G., Liu, H. C. & Yan, N. Downstream processing of lignin derived feedstock into end products. Chem. Soc. Rev. 49, 5510–5560 (2020).
Sun, Z. H., Fridrich, B., de Santi, A., Elangovan, S. & Barta, K. Bright side of lignin depolymerization: toward new platform chemicals. Chem. Rev. 118, 614–678 (2018).
Wu, X. et al. Advancements and perspectives toward lignin valorization via O-demethylation. Angew. Chem. Int. Ed. 136, e202317257 (2024).
Battin-Leclerc, F. et al. Possible use as biofuels of monoaromatic oxygenates produced by lignin catalytic conversion: a review. Catal. Today 408, 150–167 (2023).
Sang, Y. et al. Catalysis and chemistry of lignin depolymerization in alcohol solvents - a review. Catal. Today 408, 168–181 (2023).
Meng, Q. L. et al. Sustainable production of benzene from lignin. Nat. Commun. 12, 4534–4546 (2021).
Wang, W. Y. et al. Catalytic refining lignin‐derived monomers: seesaw effect between nanoparticle and single-atom Pt. Angew. Chem. Int. Ed. 63, e202404683 (2024).
Shuai, L. et al. Formaldehyde stabilization facilitates lignin monomer production during biomass depolymerization. Science 354, 329–333 (2016).
Van den Bosch, S. et al. Reductive lignocellulose fractionation into soluble lignin-derived phenolic monomers and dimers and processable carbohydrate pulps. Energy Environ. Sci. 8, 1748–1763 (2015).
Gao, F. et al. Fragmentation of lignin samples with commercial Pd/C under ambient pressure of hydrogen. ACS Catal. 6, 7385–7392 (2016).
Deuss, P. J. et al. Aromatic monomers by in situ conversion of reactive intermediates in the acid-catalyzed depolymerization of lignin. J. Am. Chem. Soc. 137, 7456–7467 (2015).
Rahimi, A., Ulbrich, A., Coon, J. J. & Stahl, S. S. Formic-acid-induced depolymerization of oxidized lignin to aromatics. Nature 515, 249–252 (2014).
Lan, W., de Bueren, J. B. & Luterbacher, J. S. Highly selective oxidation and depolymerization of α,γ-Diol-protected lignin. Angew. Chem. Int. Ed. 58, 2649–2654 (2019).
Chen, Z. W. et al. Formal direct cross-coupling of phenols with amines. Angew. Chem. Int. Ed. 54, 14487–14491 (2015).
Elangovan, S. et al. From wood to tetrahydro-2-benzazepines in three waste-free steps: modular synthesis of biologically active lignin-derived scaffolds. ACS Cent. Sci. 5, 1707–1716 (2019).
Liu, M. Y. & Dyson, P. J. Direct conversion of lignin to functionalized diaryl ethers via oxidative cross-coupling. Nat. Commun. 14, 2830–2837 (2023).
Zhang, J., Liu, Y., Chiba, S. & Loh, T. P. Chemical conversion of β-O-4 lignin linkage models through Cu-catalyzed aerobic amide bond formation. Chem. Commun. 49, 11439–11441 (2013).
Li, H. J. et al. Amine-mediated bond cleavage in oxidized lignin models. ChemSusChem 13, 4660–4665 (2020).
Liu, X. W. et al. Copper-catalyzed synthesis of benzanilides from lignin model substrates 2-phenoxyacetophenones under an air atmosphere. N. J. Chem. 42, 1223–1227 (2018).
Liu, X. W. et al. H2O2-promoted C-C bond oxidative cleavage of β-O-4 lignin models to benzanilides using water as a solvent under metal-free conditions. Green. Chem. 24, 4395–4398 (2022).
He, P. P. et al. Heterogeneous manganese-oxide-catalyzed successive cleavage and functionalization of alcohols to access amides and nitriles. Chem 8, 1906–1927 (2022).
Luo, H. H. et al. Cobalt nanoparticles-catalyzed widely applicable successive C-C bond cleavage in alcohols to access esters. Angew. Chem. Int. Ed. 59, 19268–19274 (2020).
Gu, N. X. et al. Autoxidation catalysis for carbon-carbon bond cleavage in lignin. Acs. Cent. Sci. 9, 2277–2285 (2023).
Liu, S. J. et al. Oxidative cleavage of β-O-4 bonds in lignin model compounds with a single-atom Co catalyst. Green. Chem. 21, 1974–1981 (2019).
Gao, J. et al. Streamlining the synthesis of amides using Nickel-based nanocatalysts. Nat. Commun. 14, 5013–5025 (2023).
Wang, X. et al. Challenges and outlook for catalytic direct amidation reactions. Nat. Catal. 2, 98–102 (2019).
Lundberg, H., Tinnis, F., Selander, N. & Adolfsson, H. Catalytic amide formation from non-activated carboxylic acids and amines. Chem. Soc. Rev. 43, 2714–2742 (2014).
Zhang, D. W., Zhao, X., Hou, J. L. & Li, Z. T. Aromatic amide foldamers: structures, properties, and functions. Chem. Rev. 112, 5271–5316 (2012).
Williams, R. E. & Marshall, C. M. Top 200 brand name drugs by retail sales in 2023. https://njardarson.lab.arizona.edu/sites/njardarson.lab.arizona.edu/files/NjardarsonGroup2023Top200PosterV5.pdf.
Wang, S. P. et al. Direct amidation of carboxylic acids with nitroarenes. J. Org. Chem. 84, 13922–13934 (2019).
Kumar, V. et al. Highly selective direct reductive amidation of nitroarenes with carboxylic acids using cobalt(II) phthalocyanine/PMHS. RSC Adv. 4, 11826–11830 (2014).
Krause, T., Baader, S., Erb, B. & Goossen, L. J. Atom-economic catalytic amide synthesis from amines and carboxylic acids activated in situ with acetylenes. Nat. Commun. 7, 11732–11739 (2016).
Zou, Q. Q. et al. Manganese-pincer-catalyzed nitrile hydration, α-deuteration, and α-deuterated amide formation via metal ligand cooperation. ACS Catal. 11, 10239–10245 (2021).
Shen, T. & Lambert, T. H. Electrophotocatalytic diamination of vicinal C-H bonds. Science 371, 620–626 (2021).
Baker, R. H. & Stanonis, D. The passerini reaction. III. stereochemistry and mechanism, Robert H. Baker and David Stanonis. J. Am. Chem. Soc. 73, 699–702 (1951).
Liu, J. Z. et al. Nitromethane as a nitrogen donor in Schmidt-type formation of amides and nitriles. Science 367, 281–285 (2019).
Blatt, A. H. The beckmann rearrangement. Chem. Rev. 12, 215–260 (1933).
Wang, A. Q., Li, J. & Zhang, T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2, 65–81 (2018).
Cui, X. J. et al. Bridging homogeneous and heterogeneous catalysis by heterogeneous single metal-site catalysts. Nat. Catal. 1, 385–397 (2018).
Li, Z. et al. Well-defined materials for heterogeneous catalysis: from nanoparticles to isolated single-atom sites. Chem. Rev. 120, 623–682 (2019).
Kaiser, S. K. et al. Single-atom catalysts across the periodic table. Chem. Rev. 120, 11703–11809 (2020).
Bates, J. S. et al. Heterogeneous M-N-C catalysts for aerobic oxidation reactions: lessons from oxygen reduction electrocatalysts. Chem. Rev. 123, 6233–6256 (2023).
Singh, B. et al. Single-atom (iron-based) catalysts: synthesis and applications. Chem. Rev. 121, 13620–13697 (2021).
Liu, C. et al. Catalytic activity enhancement on alcohol dehydrogenation via directing reaction pathways from single- to double-atom catalysis. J. Am. Chem. Soc. 144, 4913–4924 (2022).
Shi, Y. Z. et al. Atomically dispersed cobalt/copper dual‐metal catalysts for synergistically boosting hydrogen generation from formic acid. Angew. Chem. Int. Ed. 62, e202313099 (2023).
Zhao, X. et al. Dual-metal hetero-single-atoms with different coordination for efficient synergistic catalysis. J. Am. Chem. Soc. 143, 16068–16077 (2021).
Liu, W. G. et al. Discriminating catalytically active FeNx species of atomically dispersed Fe-N-C catalyst for selective oxidation of the C-H bond. J. Am. Chem. Soc. 139, 10790–10798 (2017).
Sun, K. K. et al. Efficient iron single-atom catalysts for selective ammoxidation of alcohols to nitriles. Nat. Commun. 13, 1848–1856 (2022).
Acknowledgements
We gratefully acknowledge the German Research Foundation (DFG project number 447724917), European Union’s Horizon2020 research and innovation program under grant agreement No 101006744, and the State of Mecklenburg-Vorpommern for financial and general support. R.V.J. thanks the support of European Union under the REFRESH-Research Excellence for Region Sustainability and High-tech Industries project number CZ.10.03.01/00/22_003/0000048 via the Operational Program Just Transition. Z.M. and X.L. thank China Scholarship Council for research fellowship. X.L. and Z.Y. thank the National Natural Science Foundation of China (Grant No. 22267025, 22065042), Science and Technology Innovation Team of Higher Education of Guizhou Provincial Education Department (Qianjiaoji [2023]073). We thank the analytical team of the Leibniz-Institut für Katalyse e.V. for their excellent service.
Author information
Authors and Affiliations
Contributions
M.B., R.V.J. and X.L. supervised the project. Z.M., X.L., R.V.J. and M.B. planned and developed the project, and designed the experiments. Z.M. and X.L. prepared catalysts and performed all catalytic experiments. Y.C., Z.Y. and C.R. involved in performing catalytic experiments. Z.M., X.L., B.Z. and Z.C. carried out the catalyst characterization and analysis. Z.M., X.L., R.V.J. and M.B. wrote the paper with the input and contribution of all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Na Ji, Zhuohua Sun, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, Z., Chen, Z., Yuan, Z. et al. Synthesis of aromatic amides from lignin and its derivatives. Nat Commun 16, 3476 (2025). https://doi.org/10.1038/s41467-025-58559-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58559-y
