
Abstract
We present an efficient method for synthesizing cationic poly(ethylene imine) derivatives using the multicomponent split-Ugi reaction to create a library of functional ionizable lipopolymers. Here we show 155 polymers, formulated into polyplexes, to establish structure-activity relationships essential for endosomal escape and transfection. A lead structure is identified, and lipopolymer-lipid hybrid nanoparticles are developed to deliver mRNA to lung endothelium and immune cells, including T cells, with low in vivo toxicity. These nanoparticles show significant improvements in mRNA delivery to the lung compared to in vivo-JetPEI® and demonstrate effective delivery of therapeutic mRNA(s) of various sizes. IL-12 mRNA-loaded nanoparticles delay Lewis Lung cancer progression, while human CFTR mRNA restores CFTR protein function in CFTR knockout mice. Additionally, we demonstrate in vivo CRISPR-Cas9 mRNA delivery, achieving gene editing in lung tissue and successful PD-1 knockout in T cells in mice. These results highlight the platform’s potential for systemic gene therapy delivery.
Similar content being viewed by others

Introduction
In the wake of the successful global deployment of lipid nanoparticle-delivered mRNA vaccines to stymie the COVID19 pandemic, there has been accelerated efforts to identify new non-viral delivery systems that are safe, more effective, systemically deliverable and tissue specific. Achieving these aims is essential to further broadening the clinical applications of mRNA delivery, including employment for replacement gene therapy and cancer immunotherapy. Regardless of components or applications non-viral delivery systems must contain chemical motifs which both protect the mRNA molecules from systemic degradation and facilitate nanoparticle entry into target cells1,2. In addition, these carriers must also facilitate mRNA escape from the endosome to allow efficient mRNA translation into functional proteins within the cells.
While there are a host of identified molecular species that effectively satisfy one or more of these requirements, the success of any given agent is also impacted by both the size of the mRNA cargo and complexity of cargo (multiple nucleic acid species). In general larger mRNA molecules are both more prone to degradation by nucleases and more inefficiently packaged into lipid nanoparticles than smaller mRNAs, reducing the efficiency of subsequent protein expression, while elevating the cost and effectiveness of manufacturing3,4. Thus far, to meet all of the above criteria, formulations in general have been uniquely tailored to excel at individual applications, resulting in the need for a broader regulatory oversight and manufacturing support system to enable eventual therapeutic deployment. Therefore, the design of a universal system that can efficiently encapsulate and deliver mRNA of various sizes or mixtures of mRNAs without requiring additional changes can have a significant impact in accelerating and extending the breadth of therapeutic mRNA applications.
Poly(ethylene imine) (PEI) has been intensively investigated as a cationic polymer for gene delivery applications for decades5,6. However, there is a tradeoff between effective transfection and cytotoxicity for higher molar mass PEIs, which limits its use in vivo7. The high charge density of PEI is believed to lead to strong interactions with cellular membranes, which contributes to its cytotoxicity8. However, the amine units of PEI are amenable to chemical modification. Indeed, chemical modifications, or partial hydrolysis and partial reduction9 of a poly(2-oxazoline) precursor10, can serve to lower the charge density thus reducing toxic effects6,11,12, while further modulating polyplex properties can increase transfection efficacy. For example, inclusion of hydrophobic groups onto polycations has been shown to strongly influence transfection13, presumably due to strengthening self-assembly behavior of the polyplex and modulation of polyplex-cell interactions14,15. Specifically, alkylation of primary amines in branched PEI with dodecyl chains was shown to improve transfection 5-fold while lowering toxicity16. Modification of a low molecular weight PEI (1.8 kg mol−1) with various methylcarboxytrimethylene carbonate derivatives showed improved transfection for ethyl and benzyl side chain substituents17. It was found that moderate degrees of modification of branched PEI with propionic acid improved transfection, demonstrating the importance of a balance of hydrophobic moieties18. A range of other promising modifications have been identified, including succination19, acetylation20, carbamoylation21, as well as conjugation with dexamethasone22, lipids23,24,25,26, fluoroalkanes27, and aromatic groups28, inter alia29,30,31,32,33.
While diverse libraries of cationic polymers are being synthesized to enable delivery across extrahepatic tissues, the therapeutic index remains limited for clinical development34,35,36,37. Moreover, the dispersity inherent to polymers presents further challenges for scale up and reproducibility. To this end, we hypothesized that a rapid high-throughput method of derivatization of PEI that results in structural diversity of PEI based ionizable lipopolymers and further formulation optimization can result in resolving these long-standing issues, enabling therapeutically relevant utilization of PEI based lipid nanoparticles that have hitherto been unrealized.
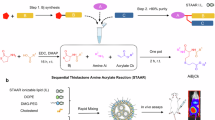
The Ugi multicomponent reaction38 is one of best known and versatile reactions and utilizes four different reagents - an amine, aldehyde, isocyanide and carboxylic acid -39 and has been utilized in polymer science as a step growth polymerization reaction40,41,42, a post-polymerization modification tool43 and as a polymer coupling reaction44. Here, we introduce the so-called split-Ugi45,46, to synthesize linear lipo-PEI derivatives optimized for RNA transfection. The secondary amine units of linear PEI require this variant of the Ugi reaction, effectively involving two equivalents of secondary amine as opposed to one equivalent of a primary amine in the standard Ugi reaction. The split-Ugi modification is expected to yield a product with two modified repeat units, one a tertiary amine with one substituent stemming from the isocyanide and the one from the aldehyde, and the other an amide group featuring the carboxylic acid moiety (Fig. 1). This introduces a convenient route to prepare PEI derivative libraries with a mixture of large variety of functional groups to explore the effect of polymeric structure on transfection efficiency both in vitro and in vivo.

The structures of molecules are in Supplementary Table 1.
In this study, a library of isocyanide/aldehyde/carboxylic acid reagents were selected to create a range of structures, and additionally molecular weights and modification densities of the PEIs samples were varied to further increase structural diversity (Fig. 1). Through initial screening we identified a lead lipo-PEI with enhanced endosomal escape, subsequent in vitro transfection and developed hybrid lipopolymer-lipid nanoparticle formulations for in-vivo mRNA-based gene delivery. We demonstrate a multiple order increase of in vivo mRNA delivery to the lungs via systemic administration compared to the traditional PEI formulation standard in vivo-JetPEI®. The produced system can deliver both small (IL-12 mRNA) and large (hCFTR) functional mRNA preserving expression of therapeutic proteins. Additionally, the delivery system showed efficient gene editing in lung endothelial and T cells by delivery of Cre mRNA and CRISPR-Cas9 mRNA mixed mRNA cargo system with minimal toxicity.
Results
Synthesis of modified PEI for mRNA delivery
The split-Ugi reaction47 is a sparingly employed version of the four component Ugi reaction48 that has not been utilized for polymer modification. We initially assessed the split-Ugi reaction to obtain linear PEI derivatives with a range of reagents and of different modifications to introduce lipid-like side chains featuring ionizable tertiary amines (Fig. 1). A linear PEI35 was reacted with formaldehyde (A1), acetic acid (C1) and four different isocyanides (B1, B5, B6 and B7) targeting a total modification of 50% of the secondary amine repeat units (4:1:1:1 molar ratio of amine:aldehyde:isocyanide:acid). 1H-NMR and 13C-NMR spectroscopic analysis showed successful integration of the isocyanide functionality (Supplementary Figs. 25 and 26), however, the degree of functionalization varied and was lower than expected, especially for the acetic acid component. In contrast to classic Ugi reactions (utilizing a 1:1 molar ratio of amine to carboxylic acid, or 2:1 for a split-Ugi), we only targeted partial conversion of the amine, requiring an excess of amine with respect to carboxylic acid. We hypothesize that the excess of amine may lower the reactivity of the carboxylic acid in the Ugi by formation of ion pairs. Accordingly, use of excess of the carboxylic acid up to an equimolar quantity with respect to the amine resulted in higher incorporation of all reagents into the polymer for a test system using A1/B1/C1 reagents (Supplementary Fig. 31). With this, degrees of functionalization close to the target was achieved for all aldehyde, isocyanide and carboxylic acid components, and therefore this procedure was selected for further derivative synthesis.
An initial library of 155 split-Ugi modified polymers was synthesized by this combinatorial approach, utilizing linear PEI of four different molecular weights (0.88, 1.6, 3.8 and 9.6 kg mol−1), and a 4 arm-star PEI, targeting various modification degrees (25%, 50% and 100%) with a small library of aldehydes, isocyanides and carboxylic acids (Fig. 1, Supplementary Table 1). Linear PEIs were chosen as the polymer backbone, as these can be readily synthesized with low dispersity and well-controlled molar masses using cationic ring opening polymerization of 2-oxazoline and subsequent hydrolysis. Additionally, they are easier to characterize without the complication of mixed primary/secondary/tertiary amine species found in hyperbranched PEIs. It should be noted that hyperbranched PEIs are typically preferred in polyplex formation as they display the highest in vitro transfection49.
Successful incorporation of the aldehyde/isocyanide functional groups with NMR spectroscopy, However, the carboxylic acid reagent showed variable incorporation across samples, in some cases we found as low as ~40% of the expected amide group formation (Supplementary Fig. 1), deviating from the results of the test system P1/A1/B1/C1 described above. This leads to a larger fraction of unreacted PEI units, although formation of other side products, unidentifiable in the NMR, cannot be completely ruled out. The incomplete incorporation of carboxylic acid reagent suggests that the imino-anhydride intermediate is not exclusively attacked by the secondary amine as expected in the modified Mumm rearrangement step but may react with another nucleophile in the system. This has been reported when using methanol as the Ugi reaction solvent47. Also the interception by water is known to occur in the 3 component-Ugi reaction, but this typically requires another catalyst and is considered unlikely to occur in the present system50. While a range of carboxylic acid reagents was explored, it was not considered to be of high relevance for structural variation due to the lowered incorporation of this reagent onto the polymer. Additionally, preliminary screening of acetic acid derivatives showed promising transfection, and thus acetic acid was used for most of the library. Size-exclusion chromatography (SEC) analysis indicated molecular weights in the expected range and monomodal distributions suggesting the modification otherwise proceeded smoothly (Supplementary Fig. 1b).
Degree of polymerization and length of carbon tail chain effects mRNA delivery
Polyplexes are widely investigated non-viral type of gene delivery system, which provide protection of mRNA and facilitate endosomal escape. Therefore, for an initial screening we formulated polyplexes from the synthesized PEI-derivative library via an ethanol injection method.
For in vitro studies a library of polyplexes with mRNA encoding Firefly luciferase (Fluc) was screened in HeLa cells to screen for mRNA transfection. Additionally, polyplexes were tested in Gal9-HEK293 cells to evaluate the endosomal escape efficacy as it was described previously51. In this system, endosomal rupture, enabling nanoparticle/mRNA escape, results in the redistribution of Gal9 which is visualized as GFP puncta in the Gal9-GFP reporter cells. Based on in vitro screening (Fig. 2a), several trends emerged from the library. First, the more hydrophobic samples appeared to perform better (e.g., U12, U15, U22, U46, U155) compared to less hydrophobic (U1-U8). Hydrophobicity of the polymers was varied with a combination of alkyl chain bearing isocyanide and aldehyde reagents. Such functionalities result in a polymer repeat unit with an ionizable tertiary amine unit in the backbone and two alkyl chain groups attached, reminiscent of cationic lipids, which are known as excellent transfection agents (e.g., DOTAP, DOTMA or DOGS)52. Notably, the position of hydrophobic chain impacted the transfection rates as well. Second, varying variations across carboxylic acid substitutions showed that the shortest alkyl chain (acetic acid) were the most efficient of the substitutions investigated on this platform backbone. Thirdly in general, the lower molar mass polymers and the higher modification density showed better performance as assessed by luciferase expression. In contrast, for PEI homopolymers, higher molar mass showed higher transfection, however, this trend does not apply for the hydrophobic derivatives in our library. The addition of hydrophobic interactions is predicted to strengthen interactions that lead to polyplex self-assembly, leading to more stable particles for even the low molar mass derivatives.

a In vitro library screening. HeLa cells were treated with Fluc mRNA-loaded lipopolyplexes. The relative luciferase expression (normalized to cell viability fluorescent signal) after 24 h incubation with lipopolyplexes is shown on the heat map (n = 6, 3 biological replicates). Gal9-HEK293 cells were treated with mCherry RNA- loaded lipopolyplexes. The percent of cells with endosomal escape (EndEsc) cases after 24 h incubation with polyplexes is presented on the heat map (n = 40–150 cells, one biological replicate). Data are presented as Means. Images of Gal9-HEK293 cells after treatment are shown; b Structures of in vitro high performing lipopolymers; c hydrodynamic diameter of lipopolyplexes loaded with Fluc mRNA or mCherry RNA. Red symbol - U155, blue symbol - U12, U25, U40, U46, U64, U100, black symbol - rest of lipopolyplexes. Source data are provided as a Source Data file.
Unexpectantly, our in vitro screening of the initial library revealed that most of the synthesized polymers robustly facilitated endosomal escape, while much fewer facilitated mRNA transfection/expression in the patterns described above (Fig. 2a). Out of the in vitro best performing polymers (Fig. 2b), U155 demonstrated preferable properties with regards to particle size and stability for in vivo studies compared to U12, U25, U40, U46, U64 and U100 (Fig. 2c).
The transfection efficiency of mRNA delivered by polymeric nanoparticles depends on factors such as complex uptake by cells, endosomal escape and cargo release. These processes can be controlled by balancing the molecular weight of the polycationic backbone and the incorporation of lipid-like moieties. Having selected U155 as the most ideal complex, we next performed in vitro screening (Fig. 3a, b) of U155 analogues with various molar masses of starting PEI and along with various targeting modifications to determine how these modifications impacted polyplex performance. These investigations revealed the following patterns: mRNA translation decreased as the molar mass (PEI length) of the polymer increased (U15-U16-U17, respectively). We presumed this was due to the increased stability of the complexes themselves along with tighter interactions with mRNA, which would be predicted to reducing rates of endosomal escape or cargo release. Conversely, when the molar mass of the polymer was significantly reduced (U154 compared to U15), transfection efficiency was also lower, likely due too far reduced particle stability. The preference of higher modification density (U155 compared to U154) may suggest a reduction in charge density is beneficial, due to the larger number of non-ionizable amide repeat units introduced. Additionally, the presence of sufficiently hydrophobic groups appears to be necessary for particle stability and optimal transfection. It should be noted that the split-Ugi not only transforms secondary amine to an amide, but it also transforms secondary amine into a tertiary amine.

a Representative images of Gal9-HEK293 cells after 24 h incubation with mCherry RNA- loaded lipopolyplexes from polymers of various chain length and modification; b HeLa cells were treated with Fluc mRNA-loaded lipopolyplexes. The relative luciferase expression (normalized to cell viability fluorescent signal) after 24 h incubation with polyplexes is shown. Data are presented as Mean ± SD (n = 6, 3 biological replicates with 2 technical replicates each); c In vivo bioluminescent images of BALB/c mice 5–6 h post i.v. injection of 5 μg Fluc mRNA per mouse. For In vivo JetPEI: shown summary of 2 batches of freshly prepared nanoparticles (n = 6 biological replicates); for U155 shown summary of 3 batches of lipopolyplexes from different batches of U155 polymer (n = 8 biological replicates). Data are presented as Mean ± SD, unpaired t test, two-tailed p = 0.0285; d ex vivo bioluminescent images of BALB/c mice 5–6 h post i.v. injection of 5 μg Fluc mRNA per mouse. K kidney, S spleen, H heart, LV liver, LN lungs. Source data are provided as a Source Data file.
Thus, a lead polymer with promising transfection performance was identified (U155), synthesized by modification of a PEI17 with decanal, dodecylisocyanide and acetic acid reagents via the split-Ugi (P5, A5, B4, C1) (Fig. 2b). The targeted 100 % modification should yield a polymer with 50% tertiary amine repeat units featuring a lipid-like tail, and by 1H NMR a functionalization of 47% (Supplementary Fig. 1c, d). Notably, the number of methyl amide repeat units formed was substantially lower at 19 % (target 50%), meaning a significant portion of secondary amine PEI units remain despite targeting full conversion.
We next explored the capability of the selected polymer for in vivo efficacy through Fluc mRNA delivery. Formulated by vortexing, polyplexes exhibited a diameter of 201 ± 41 nm (PDI 0.20 ± 0.05) and the ζ-potential of 57 ± 6 mV, as was characterized by dynamic light scattering (DLS). As a positive control, we used the standard in vivo PEI-based formulation in vivo JetPEI®, which demonstrated mRNA delivery and is established as an appropriate control in studies involving hybrid lipid-polymer nanoparticles for mRNA53. Injected (i.v.), Fluc mRNA encapsulated into U155 polyplexes showed strong bioluminescent signal and the preferential luciferase protein expression in lungs and spleen (Fig. 3c, d). Notably, bioluminescence signal outperformed in vivo JetPEI® at the same dose (5 μg mRNA per mouse) by 50-fold. To exclude other potent molecules from synthesized library in vivo, rather than in vitro testing, we formed polyplexes loaded with Fluc mRNA from a large number of PEI derivative in the library, combined them into distinct small groups, and injected them i.v. Surprisingly, in contrast to the in vitro results, none of the pooled groups exhibited significant luciferase production (Supplementary Fig. 2).
Accumulation of cationic gene delivery systems in the lungs is well documented and various pathways of nanoparticles fate after i.v. administration have been described54,55. One mechanism for lung accumulation of cationic nanoparticles involves opsonization of proteins like vitronectin55 It has also been observed that highly charged cationic particles interact with cellular blood components, especially erythrocytes, producing small aggregates, which first infiltrate and accumulate in the small lung capillaries vasculature after intravenous injection and then subsequently translocate to the spleen and liver54. Additionally, biodistribution studies have determine that delivery to the lungs and spleen is also influenced by particles size53. Due to the high hydrophobicity of the polymer, U155 polyplexes are prone to aggregation during the concentration process, resulting in the formation of particles exceeding 400 nm. Injection of these aggregates in vivo was associated with increased mRNA expression in the spleen at the expense of observing expression in the lung (Supplementary Fig. 3d). We hypothesized the reason for this was enhanced phagocytic clearance by splenic macrophages, known to occur more effectively than endocytose for particles ~500 nm in size56. Overall, the highly positively charged U155 polyplexes are rapidly eliminated from the circulation within 24 h (Supplementary Fig. 3a–c). To prevent the potential for the in vivo and formulation aggregation processes described above which could complicate in vivo utility, we pursued an approach to both reduce particle surface charge and enhance the size stability of U155 containing nanoparticles. To do so, we chose to employ a polyplex-lipid nanoparticle hybrid strategy and characterized the in vivo efficacy of this hybrid particle
Hybrid polymer-lipid U155@lipids nanoparticles outperform in vivo JetPEI® in mRNA delivery to the lung
Previously it has been shown that combining polymers with lipids in formulating for gene delivery systems can improve colloidal properties of such particles under physiological conditions, both enhancing target delivery and as well as increasing in vivo transfection efficiency53,57. To decrease the ζ -potential of resulting hybrid nanoparticles negatively charged phosphatidyl glycerol (PG) lipids can be added58. Pattipeiluhu et al. previously used 1,2-distearoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (DSPG) in LNPs to direct distribution of particles after i.v. injections59. Inclusion of such anionic lipid into LNPs reduces accumulation of particles in liver, while mean positive ζ -potential is associated with nanoparticle accumulation in the lungs60,61. However, it has also been noted that inclusion of anionic LNPs in single reaction nanoparticle assemblies results in lowered RNA loading efficiency, due to the charge repulsion against mRNA molecules62. To overcome this limitation while still capitalizing on the positive distribution effects of DSPG inclusions in nanoparticle formulations, we chose to form hybrid polymer lipid nanoparticle through a two-step approach63. Firstly, cationic polyplex formation occurs by separately mixing the cationic moiety (i.e. U155) with mRNA. In the second step a lipid mixture (i.e. DSPG) is used to encapsulate the cationic complexes with a lipid membrane. Others have demonstrated by employing such approaches improves, particle stability and is associated with reduce toxicity64. U155, hybrid polymer-lipid nanoparticles were formulated employing this approach (Fig. 4a). Briefly, U155/mRNA preformed polyplexes formed were mixed (2/1 wt/wt) with an ethanol solution containing DSPG, soy PC, cholesterol and DMG-PEG2000 (molar ratio 22/23/50/5) using a microfluidic mixing (Fig. 4b). The final formulation was then dialyzed against Tris-HCl buffer (25 mM, pH 7.4) for 3–4 h at room temperature.

a Schematic illustrating the formulation of a lipopolymer lipid hybrid nanoparticles U155@lipids (https://BioRender.com/c35l657); b Lipid composition of outer shell of U155@lipids; c Representative Cryo-TEM image of U155@lipids. Scale bar, 50 nm; d Hydrodynamic diameter (min 79.6 nm, max 195.3 nm) and PDI (min 0.084, max 0.241) of produced nanoparticles in 25 mM Tris-HCl buffer (pH 7.4); e ξ-potential of U155 lipopolyplexes and U155@lipids in 2.5 mM Tris-HCl buffer (pH 7.4); f Normalized TNS fluorescence of nanoparticles at various pH. TNS interacts with positively charged amines and produces fluorescence signal. Data are presented as Mean ± SD (n = 8); g Encapsulation efficiency (EE) and recovery of mRNA after loading into U155@lipids (n = 5); h Ex vivo images of bioluminescence in various organs 5–6 h post i.v. injection of 5 μg Fluc mRNA per mouse loaded in U155@lipids. K kidney, S spleen, H heart, LV liver, LN lungs. i Comparison of ex vivo bioluminescence in lungs revealed U155@lipids increased lungs specific of initial lipopolymer U155. Data are presented as Means ± SD (n = 5 for U155 and n = 6 for U155@lipids biological replicates), unpaired t test, two-tailed p = 0.0253; j Optimized U155@lipids formulation is orders of magnitude more potent than in vivo JetPEI across multiple Fluc mRNA doses. Data are presented as Means ± SD (n ≥ 3 biological replicates); k Non-linear dose response of Fluc mRNA transfection in lungs (BALB/c mice) 5–6 h post i.v. injection; l Representative images and quantification of tissue biodistribution of DiD-labeled U155@lipids nanoparticles. K kidney, S spleen, H heart, LV liver, LN lungs. Data are presented as Means ± SD; n = 2 for PBS and DiD controls and n = 3 for U155@lipids biological replicates. S spleen, LN lungs. Results are presented as Means ± SD. Source data are provided as a Source Data file.
The above-described molar ratios of LNP constituents were rationally selected as described below based upon previous literature as well as experimentally confirmed as follows. First, Cholesterol plays a crucial role in enhancing the in vivo stability of liposomes and lipid nanoparticles (LNPs), thus influencing nanoparticle clearance rates in the blood65,66. Additionally, cholesterol has been reported to enhance the transfection efficiency of RNA67. Studies suggest an optimal molar content of cholesterol typically in the range of 38.5–50%68,69, here, we chose 50% of cholesterol, aligned with literature70. With regards to PEG-Lipid, the optimal molar concentration of PEG-lipid in liposomes can vary depending on the specific application. To evade the body’s immune system and extend liposomes circulation time it was shown previously that 5% molar PEG-lipid is optimal71,72. Additionally, Kaczmarek et al. showed that inclusion of 2–5 mol % of C18-PEG2000 in hybrid polymer-lipid nanoparticles facilitates lung targeted expression53. Since the splenic and lung tropic (non-liver) expression of mRNA of our U155 polyplexes, we aimed to further retain this unique trait our hybrid polyplex formulation by aligning PEG2000-lipid usages at 5%.
Existing reports suggest optimal levels of DSPG range between 20 and 25 mol % of total lipids in liposomal membranes for in vivo studies73. Addition of DSPG into our formulation showed improved in vitro mRNA transfection (Supplementary Figs. 4a and 5). Further, supporting the literature recommendation, we found that increasing DSPG from 22 mol% to 31.5 mol% resulted in a dramatic reduction of mRNA transfection in the lung after i.v. injection (Supplementary Fig. 4b). We speculated that this reduction in mRNA transfection was due to lower nanoparticles stability, resulting from the increased density of negatively charged head groups of DSPG repelling one another and destabilizing the bilayer73. We also speculated that further reduction in overall positive charge of the particle may also alter in vivo nanoparticle molecular interactions, changing cellular uptake and systemic tropism.
We next utilized the above U155@lipids formulation described above to determine if the previously observed lung tropism of mRNA expression was retained in this hybrid polyplex formulation. Cryo-TEM images showed a rather irregular structure of hybrid nanoparticles (Fig. 4c). Mean hydrodynamic diameter was 120 nm with PDI < 0.2 (Fig. 4d). While Cryo-TEM showed some clear variability of particle morphology, DLS suggested a rather low dispersity. Covering the U155 polyplex with a lipid bilayer significantly decreased the total particle ζ-potential to 11.4 ± 7.7 mV (Fig. 4e). Titration with fluorescent dye TNS at various pH values showed a slowly falling trend line, consistent with multiple overlapping pKa values in the system (Fig. 4f). Encapsulation efficiency of produced nanoparticles was greater than 98% with displayed good mRNA recovery (>75%) (Fig. 4g). Once again, using i.v. delivery of Fluc mRNA, we observed that the hybrid-polyplex lipid nanoparticle not only retained lung tropism, but increased mRNA expression 5-fold compared to bare U155 polyplexes (Fig. 4h, i). Overall, the optimized particle demonstrated orders of magnitude greater effectiveness compared to the commercially available in vivo JetPEI® across multiple doses (Fig. 4j, Supplementary Fig. 6). We also determined the performance of our platform compared to previously reported lung-targeting SORT LNPs, offering an additional comparator for the potential of our polymer-based RNA delivery platform. DOTAP-LNPs were formulated according to the composition described previously60,74, with the molar ratio of 5A2-SC8/DOPE/cholesterol/DMG-PEG/DOTAP equal to 11.9/11.9/23.8/2.4/50 and were administered at two doses (5 µg and 10 µg per mouse, via i.v. injection). The results revealed that our particles performed at the same level (5 µg) or even exceeded (10 µg) in vivo transfection compared to SORT LNPs (Supplementary Fig. 7), and further optimization of both the lipopolymer structure and the lipid composition could dramatically improve performance. Interestingly, U155@lipids particles showed a non-linear dose response in lungs (Fig. 4k). Such effects have been previously reported in particular various non-liver targeted nanoparticles, an especially for polymeric particles75,76.
The highly efficient phagocytic and lipid scavenging properties of the liver has resulted in most lipid-based nanoparticle formulations demonstrating a dominant tropism for genetic expression in the liver. Consequently, there is presently a high research focus on identifying lipid formulations or nanoparticle delivery strategies that demonstrate non-liver tissue tropism. For example, it was previously shown tumor tissue uptake and expression of nanoparticle cargo could be increased by reducing nanoparticle uptake by phagocytic cells such as macrophages and dendritic cells in the liver and spleen76. Multiple strategies exist to deplete macrophages: pre-injection of clodronate or similar agents, or blank particles of the same composition as ones with the active molecule76,77,78,79. Given that we observed non-liver tropism without (1) intentionally inhibiting phagocytic uptake and (2) our polyplex-hybrid -formulation contains lipids which have been reported to demonstrate tropism to the liver, we sought to determine the biodistribution of U155@lipids by labeling them with fluorescent dye DiD. First, we demonstrated that dye labeled particles were stable in 50% serum at 37 °C for at least 4 h (Supplementary Fig. 8). Mice were then injected via tail vein with 5 µg Fluc mRNA nanoparticles with 0.1 mol % DiD. U155@lipids preferentially accumulated in the lungs, and to a lesser extent in the spleen and liver (Fig. 4l). We posited the low but noticeable liver accumulation may be due to phagocytosis of these lipids by monocyte/macrophages populations in the lungs that breakdown lipids and package them for removal by the liver or by phagocytic uptake of liver resident macrophages themselves (Kupffer cells)76. Liver uptake of nanoparticles could also be due to other non-immune resident hepatic cells known for their lipid and blood scavenging properties. Regardless of mechanism, we sought to determine whether pre-injection of non-mRNA bearing U155 hybrid-lipid polymeric nanoparticles (blank) would elevate subsequent non-liver tissue expression of genetic cargo as discussed above. To test this, we injected blank U155@lipids 12 h prior to administration of 5 µg Fluc mRNA nanoparticles (Supplementary Fig. 9). The blank particles were prepared using the same method as the mRNA-loaded particles, and their concentration was adjusted to match the 5 µg dose of loaded particles by volume. Indeed, the pretreatment increased the luciferase expression in the lungs ∼2-fold compared to standard scheme (Supplementary Fig. 9). In contrast, co-injection of a mixture of blank particles with Fluc mRNA loaded ones did not affect transfection in lungs. Interestingly, a similar effect was observed after multiple dosages of U155@lipids loaded with Fluc mRNA. Since luciferase expression is cleared from the body within 24–48 h after 5 µg Fluc mRNA administration (Supplementary Fig. 10), we injected a second dose of the same formulation 48 h after first administration. We observed 2-fold increase in protein expression after the second dose (Supplementary Fig. 11). Importantly, these data are consistent with a phagocytic clearance mechanism of these particles that can be saturated by pretreatment, enabling greater nanoparticle efficiency. Thus, using our particles we can perform multiple dosing of mRNA without compromising transfection efficiency.
For our nanoparticles to be useful in clinically translational applications they would need to be demonstrated to be safe and non-immunogenic. We next assessed the possibility of acute lung inflammation after first particle dose (pre-treatment). Such acute inflammation has been demonstrated previously to be one mechanism whereby secondary dose enhancement has been observed80. While increasing transfection efficiency, such inflammation dependent mechanisms would be predicted to limit therapeutic utility. Lung histological samples taken 24 h after 5ug nanoparticle injection, revealed no statistically significant difference in immune cells infiltration between U155@lipids and PBS injected animals and did not show signs of tissue damage (Fig. 5a, b). Additionally, no cytokines, involved in the acute inflammatory response such as IL-1, IL-6 or TNFα were detected in the serum samples taken before the lungs were removed (Fig. 5c–f). Considering the above results, we sought to first establish cellular level identification of mRNA expressing cells in the lung after which we could begin investigating the therapeutic utility of this hybrid-lipid polyplex nanoparticle through relevant animal models.

a Representative images of paraffin-embedded lung sections, which were stained with H&E. Red dashed loops indicate cells infiltration and occurrence of possible micro-abscesses; scale bar 200 μm (left panel) and 20 μm (right panel); b QuPath software quantification of the number of nuclei normalized to tissue area (n = 3 slides for PBS control, 3 biological replicates and n = 8 slides for U155@lipids, 3 biological replicates). Representative image of how QuPath’s tool selects nuclei can be found in Supplementary Fig. 12. c–f Plasma concentrations of cytokines. IL-1β, TNFα were below the detection threshold (<16 pg/mL), unpaired t test, two-tailed p = 0.28 and p = 0.401 (n = 5 biological replicates). Source data are provided as a Source Data file.
Hybrid U155@lipids enable mRNA delivery to the lungs and T cells
To identify the cell populations within the lungs that express the mRNA product mediated by U155@lipid particles delivery, we employed the Ai9 mouse strain. This mouse strain harbors a genetic construct in all of it cells which, upon administration and functional expression of Cre-recombinase, removes a stop codon from a tdTomato protein expression DNA construct present in the ROSA26 safe harbor locus (Fig. 6a) By identifying cells expressing tdTomato we can positively identify cells receiving U155@lipid delivered mRNA. We employed two independent approaches, multi-analyte IHC and flow cytometry, to determine cellular tdTomato expression in lungs, spleen and liver. For flow cytometric determination, we first, collected organs, performed single cell dissociation, then labeled the cells with a cocktail of antibodies that recognize cell specific proteins. Consistent with our whole mouse fluc mRNA experiments we observed. the lungs were the primary organ that showed significant transfection (Fig. 6b), while both the spleen and liver exhibited Cre mRNA transfection to a lower degree (Fig. 6a). Employing a flow gating strategy, we observed the majority of tdTomato+ cells in the lungs transfected by U155@lipids were identified as CD45−CD31+ (putative endothelial cells) as well as CD45+(immune) cells (Fig. 6b). While CD31+ is commonly used to identify endothelial cells both CD45+ and CD45− cells have been reported to express CD31+. We found that less than 30% of tdTomato+ cells are CD45−/CD31+, consistent with classical endothelial cells while more than 50% of the tdTomato positive cells were CD45+/CD31+ consistent with activated immune cells (T cells, B cells and myeloid cells), or more rare endothelial precursor cells (Fig. 6b). While immune cells are generally resistant to lipid nanoparticle transfection, especially in vivo, a small fraction of myeloid immune cells have regularly been observed to express lipid nanoparticle expressed genetic constructs, likely due to their highly phagocytic nature. Surprisingly, using flow cytometry we observed that a small but statistically significant range of immune cells expressed tdTomato after CRE administration including not only myeloid cells but T cells (TCRb+, ~5%) and B cells (B220+, ~1%) (Fig. 6b). T lymphocytes are critical regulators and effectors of adaptive immune response, and their transfection in vivo offers unique opportunities to advance cancer immunotherapy, autoimmune diseases treatment or vaccine development. Aside from observed transfection in the lung it was noted that a small portion of TCRb+/tdTomato+ cells (∼0.4%) was detected in spleen tissue as well (Supplementary Fig. 14). Overall, few endothelial and epithelial cells were recovered from the single cell digest as these protocols favor immune cell isolation and survival over others cell types81.

a Experimental scheme and representative tdTomato+ cells as a percentage of the total live cell population in each of several major organs: Ai9 mice were injected with U155@lipids encapsulating Cre mRNA, and tdTomato expression in single cell–level was quantified 3 days after injection using flow cytometry (https://BioRender.com/j34q758); b Quantification of tdTomato+ cells in the lungs in different cell types (tdTomato+ cells as a percentage of the overall population of each cell type). Data are presented as Means ± SD (n = 3 biological replicates), (unpaired t test, two-tailed p = 0.0219, multiple unpaired t test p = 0.005, p = 0.0003, p = 0.0008, p = 0.0002, p = 0.001). The gating strategy for flow cytometry analysis is shown in Supplementary Figs. 13 and 14; c Representative images of paraffin-embedded lung sections, which were stained with H&E and antibodies for multiplex IHC. Scale bar is 200 μm; d Multiplex IHC of lung sections; e Quantification of IHC in (c). Data are presented as Means ± SD (n = 3 biological replicates, unpaired t test, two-tailed p = 0.0392). Representative image of how QuPath’s tool select tdTomato+ stained areas is in Supplementary Fig. 15. Source data are provided as a Source Data file.
Given the low level of tdTomato expression in immune cells conflicted with the strong Fluc expression we observed previously in the lung, we sought additional confirmation of tdTomato expression through employment of multiplex IHC (mIHC) on formalin fixed paraffin embedded tissue samples stained with key markers (CD31, CD45 and E-cadherin). Processing of mIHC images of tdTomato+ stained tissue revealed higher transfection rates comparable to what would have been inferred from the Fluc mRNA experiments (Fig. 6d) and significantly higher compared to those observed by flow cytometry (Fig. 6b). Multiplex IHC images showed colocalization of tdTomato signal with both endothelial and immune cells (Fig. 6c), however, by IHC CD45-/CD31+ cells were chiefly responsible for the strong tdTomato signal. Notably, based on tissue morphology, U155@lipids nanoparticles not only reach blood vessels, but also Lyve1+ lymphatic vessels (Fig. 6c). Previously it has been reported that positively charged nanoparticles extravasate out of the blood vessel into lymphatics through fenestrations in the endothelium or through transcellular transport through increased interaction and adsorption to the negatively charged cell membranes of endothelial cells81,82. Some rare colocalizations with E-cadherin+ epithelial cells were also observed, while no transfection of large airways was found (Fig. 6c). Importantly H&E of lung (Fig. 6c) and liver (Supplementary Fig. 16) tissues 3 days post Cre mRNA injection did not reveal any signs of inflammation or tissue damage. Periodic Acid-Schiff (PAS) staining combined with Fast Green counterstaining, which is sensitive to liver function, also did not show evidence of liver toxicity (Supplementary Fig. 16).
Treatment with IL-12 mRNA loaded hybrid U155@lipids confers a significant survival advantage in Lewis Lung cancer model
As U155@lipids transfects mostly lung endothelial and immune cells we explored the therapeutic potential of our nanoparticles to facilitate cancer immunotherapy by delivering mRNA encoding for the secreted protein interleukin-12 (IL-12) in the Lewis Lung carcinoma mouse model of lung cancer. As a cytokine, IL-12 is secreted protein noted for its anti-tumor activity in a variety of preclinical models83. Previously it was reported that IL-12 mRNA delivery may be an effective immunotherapy against multiple cancer types84,85,86,87,88. Upon delivery, the host cells translate the IL-12 mRNA into IL-12 protein. IL-12 is a cytokine that plays a crucial role in polarizing the immune system towards Th1/cytotoxic immune milieu, associated with functional anti-tumor immune response. IL-12 is particularly necessary for polarizing T cells through which the tumor microenvironment can also be further changed to facilitate inhibition of tumor growth (Fig. 7a). To check the therapeutic potential of our platform we established an orthotropic lung cancer model by i.v. injection of Lewis lung carcinoma cells expressing luciferase (LLC-Lum) and evaluated treatment with IL-12 mRNA (Fig. 7b).

a Structure of IL-12 mRNA and protein; b Experimental scheme of treatment (https://BioRender.com/o56i111); c Serum IL-12p70 concentration 24 h post 1st and 2nd i.v. injections of IL-12 mRNA and mCherry RNA (5 ug dose per mouse), normal saline (NS). Data are presented as Means ± SD (n = 5 biological replicates), (unpaired t test, two-tailed, **p < 0.005, ***p < 0.0005); d Representative images of bioluminescence in lungs upon time; e Representative images of ex vivo lungs; f Kaplan–Meier survival analysis comparing overall survival (OS) between control (NS, n = 4 biological replicates), mCherry RNA (U155@lipids mCherry RNA, n = 4 biological replicates), and IL-12-mRNA treated (U155@lipids IL-12 mRNA, n = 5 biological replicates). P-values were calculated using log-rank test; g Days survival past in vivo bioluminescence signal reached 2 × 105 p/sec/cm2/sr. n = 4 for NS and mCherry RNA, n = 5 for IL-12 mRNA, unpaired t test, two-tailed, **p = 0.0072 and 0.0032. Source data are provided as a Source Data file.
We explored IL-12 protein expression (Fig. 7c) in serum 24 h post i.v. dose administrations. Notably, animals treated with IL-12 mRNA exhibited substantial protein levels, in contrast to the negligible cytokine presence with PBS or mCherry RNA treatments (Fig. 7c). Interestingly, IL-12 cytokine concentration was approx. the same after the first and second doses, once again confirming the effectiveness of our platform for multiple dosage administrations. Tumor growth was monitored by bioluminescence signal (Fig. 7d, e, Supplementary Fig. 17). Administration (i.v.) of IL-12 mRNA loaded U155@lipids significantly delayed tumor progression prolonged overall survival of mice compared to PBS and mCherry RNA loaded U155@lipids controls (Fig. 7f). While all mice eventually succumbed to tumor burden, mice receiving IL-12 survived significantly longer even once larger tumors were established (2 × 105 p/sec/cm2/sr) consistent with the presence of an active adaptive anti-tumor immune response (Fig. 7g). Despite transient weight loss for the first days after IL-12 mRNA treatment, animals recovered and, unlike the control groups, maintained weight for at least 10 days post the second dose (Supplementary Fig. 18). Notably two doses of mCherry RNA control also induced an initial delay in tumor progression but it did not significantly induce IL-12 production, nor extend overall survival, nor slow time to death post establishment of large tumors (Fig. 7c, d, f, g), establishing the essential importance of irrelevant cargo controls in such experiments to appropriately assess particle/cargo efficacy. Similar results utilizing similar approaches, but different delivery platforms have been previously reported, thus overall84, our results further support that IL-12 initiated immune stimulation provided by RNA delivery, could yield a significant yet transient therapeutic benefit.
Delivering of CFTR mRNA restores CFTR-mediated chloride efflux in CFTR KO mice
To assess our level of success in developing a broadly employable therapeutic platform across mRNA sizes, next, we sought to deliver a therapeutic mRNA larger than Fluc (1922 b) or IL-12 (1617 b) mRNA and subsequently check the functionality of expressed protein. Delivery of CFTR mRNA (6132 b) is a potential therapeutic approach for cystic fibrosis, a genetic disorder characterized by the non-functioning of the CFTR protein. CFTR mRNA delivery aims to introduce functional mRNA into cells, allowing them to transiently produce the CFTR protein (Fig. 8a). According to guidance from the cystic fibrosis foundation, targeting multiple cell types for gene therapies including respiratory, intestinal and nasal epithelial cells, immune cells and stem cells, with CFTR mRNA treatment is predicted to offer comprehensive therapeutic benefits and improve the overall quality of life for individuals with cystic fibrosis through restoration of a variety of functional activities in these varied cell types89,90. But our main goal is to use CFTR mRNA as a model large molecule to test if our platform can efficiently deliver it, preserving its structure for mRNA translation into functional protein.

a CFTR-WT is core-glycosylated (CG) in the endoplasmic reticulum, after which it moves through the trans-Golgi network to become complex-glycosylated (CxG) and reaches the plasma membrane, where it acts as a chloride channel(https://BioRender.com/z87d224); b RT-PCR analysis of BALB/c mice lungs 4 h after i.v. injection of U155@lipids hCFTR mRNA (n = 3 biological replicates). Negative control - untreated mouse; positive control - hCFTR mRNA; loading control, GAPDH; c); experimental scheme of treatment (https://BioRender.com/z87d224); d Schematic diagram illustrating the correlation between NPD traces and ion transport (https://BioRender.com/z87d224); e Representative NPD traces for a single CFTR KO mouse before and 48 h post treatment with U155@lipids hCFTR mRNA (2 days nasal installation with total 8 µg mRNA dose per mouse); f hCFTR response before and after treatment (n = 6 biological replicates). Data are presented as Mean ± SD; unpaired t test, two-tailed, **p = 0.0041. Source data are provided as a Source Data file.
To evaluate our platform for delivering large functional mRNA (human CFTR mRNA) to lungs, we first injected hCFTR mRNA (8 µg per mouse) loaded in our platform via tail vein and harvested lungs after 4 h followed by total mRNA isolation, cDNA synthesis, and amplification of exon 11 of hCFTR, as reported earlier91. Fig. 8b demonstrates a clear band of 150 bp, similar to the control human CFTR mRNA on 2% agarose gel, indicating the successful delivery of hCFTR to lung tissue. However, the CFTR protein undergoes post-translational modifications, becomes core-glycosylated (135 kDa) in the endoplasmic reticulum and is further modified to a functional complex through extensive glycosylation (180 kDa). To be functional it next needs to move through the trans-Golgi network before reaching its functional destination at the plasma membrane (Fig. 8a)92. This folding process is crucial for proper function of CFTR protein as a chloride ion channel and partly depends on integrity of delivered mRNA. Thus, we sought to rigorously assess the subcellular fraction containing the expressed CFTR protein to confirm membrane association. In these experiments we once again encapsulated human (h) hCFTR in our nanoparticle platform, this time administering i.v. in CFTR knockout (KO) mice. We again confirmed efficient expression of functional CFTR protein in the lungs, using western blot analysis (Supplementary Fig. 19a). Importantly, comparing the lysates from cytoplasm and membrane fractions we observed the presence of CFTR enriched strongly in the membrane fraction, indicating that exogenous hCFTR mRNA is translated and undergoes post-translational modification and subsequent membrane association (Supplementary Fig. 19a) analogous to endogenous CFTR mRNA.
Having previously observed that i.v. administration of CRE mRNA resulted in few epithelial cells expressing tdTomato, we altered our route of administration to intranasal introduction of nanoparticles anticipating that increasing both the frequency and diversity of lung cells targeted would yield greater therapeutic efficacy in lung functioning assays. Importantly we demonstrated that after altering route of administration we observed significant transfection of nasal epithelium postnasal instillation (Supplementary Fig. 19b). To evaluate the in vivo functionality of expressed CFTR protein we measured nasal potential difference (NPD) in CFTR KO mice before and after nasal instillation of hCFTR mRNA loaded platform (Fig. 8c). The difference in electrical potential across conductive epithelium arises from the movement of ions across the epithelial layer and is mainly influenced by the activity of three key channels: the epithelial sodium channel (ENaC), CFTR, and calcium-activated chloride channels (Fig. 8d)93. In CFTR KO mice, there is inadequate transport of chloride ions, leading to increased absorption of sodium and an exaggerated response to substances like amiloride, which inhibit ENaC. Notably, inhibition of CFTR with CFTRInh-172 does not produce any observable effects in CFTR KO mice due to the absence of the CFTR protein (Fig. 8e, f). Once we delivered hCFTR mRNA (total 8 µg per mouse) mice exhibited polarization in response to CFTR stimulation (Fig. 8f), followed by inhibition after addition of CFTRInh-172, confirming that the observed response was CFTR-dependent (Fig. 8f). Collectively, these data demonstrate our platform successfully delivered the large hCFTR mRNA to the lungs preserving functionality of the cargo and translated protein.
Hybrid U155@lipids enable CRISPR-Cas9 editing in the lungs
Demonstrating a high lungs tropism upon i.v. delivery and achieving successful transfection in endothelial and T cells with in vivo tolerability, we proceeded to investigate the potential of employing U155@lipids for efficient delivery of CRISPR-Cas9 systems to the lungs in the Ai9 mouse strain. To do this we delivered Cas9 and sgRNAs targeting the stop cassettes present in the ROSA26 locus, impeding expression of tdTomato expression. Introduction of insertions or deletions in the tdTomato STOP cassette (Fig. 9a) disrupts the transcriptional stop sequence, leading to tdTomato protein expression. The U155@lipids encapsulated Cas9 mRNA+sgRNA (1/1 wt/wt) at dose of 19 µg total RNA was injected via tail vein in Ai9 mice followed by gene editing quantification on day 9 post injection (Fig. 9a). The multiplex IHC of lung tissue revealed editing only in CD31+ endothelial cells (Fig. 6b). Notably Cre-recombinase editing was 15-fold more efficient than the CRISPR-Cas9 system (Figs. 6d and 9c), therefore the number of potentially edited CD45+ cells detected by IHC was expected to be substantially lower and difficult to quantify by IHC. To prove that tdTomato expression was a result of indels or frame shift near STOP codons, we used the next-generation sequencing (NGS) analysis of targeted DNA sequence, extracted from lungs. Editing was quantified as sum of the insertions and deletions, normalized to the number of reads, resulting in a % of reads containing indels. As per the NGS data, treated mice displayed 5.6 ± 2.4% of editing in lungs tissue (Fig. 9d).

a Experimental scheme: Ai9 mice were injected with U155@lipids encapsulating Cas9 mRNA/sgRNA (1/1 wt/wt ratio), and 9 days after injection lungs were collected and analyzed with next-generation sequencing (NGS) and IHC (https://BioRender.com/m23g662); b Representative images of paraffin-embedded lung sections, which were stained with antibodies for multiplex IHC. White arrows point out the tdTomato and CD31+ co-localization. Scale bar is 200 μm; c Quantification of IHC in (b). Data are presented as Means ± SD (n = 3 for control and n = 4 for U155@lipids biological replicates) unpaired t test, two-tailed, **p < 0.0202. Representative image of how QuPath’s tool select tdTomato+ stained areas is in Supplementary Fig. 12; d Quantification of editing events in lungs by NGS. Data are presented as Means ± SD (n = 5 for control and n = 4 for U155@lipids biological replicates), unpaired t test, two-tailed, *p = 0.0206; e Schematic of the Cas9 target site within the mouse PDCD−1 gene at the exon2 ___location; f Schematic of interaction of PD-1 positive and ΔPD-1 knockout T cells with tumor cells (https://BioRender.com/l54q041); g Quantification of editing events in CD4+/CD8+ T cells by NGS. Data are presented as Means ± SD (n = 4 for CD4+ and n = 5 for Cas9 mRNA and CD8+ biological replicates), unpaired t test, two-tailed, *p = 0.0354. Source data are provided as a Source Data file.
Encouraged by these results, we sought to extend the application of our particles to show functional editing in T cells. Administration of antibodies targeting PD-1 have significantly improved outcomes for cancer patients through removal of the “checkpoint” signal initiated through PD1 expressed by T cells that normally limits their anti-tumor activities (Fig. 9f). Ablation of PD1 signaling has been demonstrated to enhance not only the activity of resident T cells but also increase the persistence of transferred CAR-T cells94,95. Thus, we sought to design a guide which provides deletion and frame shift in mouse exon 2 of the PD1 locus, permanently blocking future expression of the PD-1 protein (Fig. 9e). I.v. administration of one dose (10 µg total RNA, Cas9 mRNA/sgRNA wt/wt ratio of 1/1) of U155@lipids nanoparticles after pre-activation with a single i.v. dose of IL-12 mRNA (5 µg) showed up to 0.29% of gene editing in both CD4+ and CD8+ T cells based on NGS analysis (Fig. 9g). While far from optimal frequencies, these results establish the in vivo feasibility of a wider spectrum of immunotherapeutic approaches enabled by a non-viral mRNA delivery platform.
Discussion
Non-viral systems for mRNA delivery to the lungs have gained significant interest due to their potential in treating various respiratory diseases, including cystic fibrosis, lung cancer, and infectious diseases like COVID-19. Presently, dozens of lipids and polymer-based platforms with lung tropism are designed for various routes of administration, however, most of them suffer from side effects such as immunogenicity and high frequencies of lung embolism due to nanoparticles being highly positive charged. Addressing these challenges is crucial for the successful development of safe and effective mRNA delivery systems for lung-targeted therapies.
We have successfully implemented a highly efficient method for synthesizing cationic PEI-based polymers utilizing the split-Ugi reaction. By formulating a structurally diverse library of these polymers into polyplexes and screening them in vitro and in vivo, we have resolved the problem of balancing particles stability with cargo release, which are shown to depend on the degree of polymerization and polymer hydrophobicity. In particular, the incorporation of lipid-like moieties at high modification degrees (~50% of polymer repeat units) was identified to yield lipopolymers with high transfection performance. Additionally, the introduced tertiary amine units of the polymer may be beneficial as they have lower proton buffering capacity at physiological pH compared to primary and secondary amines, resulting in reduced osmotic stress and decreased cellular toxicity96,97. In future work, synthesizing these split-Ugi derived polymers with biodegradable backbones would represent a promising advanced to our current approach.
It is worth noting, while clearly proven effective, both lipopolymer structure and lipid composition of the final formulation outlined here can likely be further improved. The main goal of the present work was to demonstrate the possible applications and perspectives of the offered approaches in mRNA delivery platform formulating.
Using a two-step approach, we produced lipopolymer-lipid hybrid nanoparticles. These nanoparticles selectively deliver and induce effective mRNA expression in lung endothelium and immune cells, including T and B cells, with minimal in vivo toxicity after i.v. administration. U155@lipids demonstrate orders of magnitude higher potency in systemic mRNA delivery to the lungs compared to in vivo-JetPEI®, which is considered as commercially available standard for PEI (polymeric) vehicle for in vivo mRNA delivery98,99. Similar to previous reports, we demonstrated that pre-injection of blank nanoparticles decreases the liver accumulation, improving U155@lipids mRNA delivery to the lungs. Notably, our platform demonstrated effective when provided by a multiple dosage administration schedule. This is important as mRNA is transient and depending upon cargo application, by require iterative administration, including utilization in gene editing gene therapy where edited cell frequency could be slowly, and perhaps more safely elevated over time to achieve therapeutic effect.
Another advantage of our platform and formulation approaches is the demonstrated successful delivery of therapeutic mRNAs of various sizes and complexities for effective expression of functional proteins and ablation of gene expression. With feasibility of in vivo edited T cells established, it is exciting to speculate how further optimization of particles, guides and particle administration routes and dosing may improve transfection efficiency. This approach could be extended beyond genetic ablation of T cell PD1 expression to include potentially integration or expression of chimeric antigen receptor cargo. Interestingly it remains to be seen with such approaches whether high transfection efficiency, gene introduction or gene ablation is favorable, as knocking out the PD-1 gene, or expressing CAR in many T cells may have significant consequences. While it might enhance the immune response against cancer, it could also lead to inflammatory syndromes or autoimmunity. Therefore, this approach and further application of our platform need to be carefully studied and potentially combined with other strategies to minimize potential side effects.
The capacity to deliver mRNA to the lungs through various routes of administration also increases the number of cell types that can be affected, enabling treatment of cystic fibrosis. Our platform successfully delivered large hCFTR mRNA to the lungs preserving functionality of the cargo and translated protein upon i.v. administration. Also, instead of i.v. injection, intratracheal installation could be implemented (Supplementary Fig. 20). However, for further improvement of therapeutic effect, the CFTR mRNA sequence should also be further optimized100. Furthermore, U155@lipids demonstrated the efficient delivery of CRISPR-Cas9 system to the lungs, resulting in significant gene editing within tissues. This could potentially enable an alternative strategy for cystic fibrosis treatment.
In considering the results above in sum, we speculate that the mechanism of lung tropism for our nanoparticle system fits the description of “passive’ organ tropism”. In our case the passive organ tropism of the lung in particular would be the preferential an accumulation of nanoparticles facilitated by the electrostatic interaction of positively charged nanoparticles and negatively charged blood components (proteins, cells)101. This effect is likely to favor the lung due to the high local blood flux, slower blood flow/pressure and the extensive surface area of pulmonary capillary endothelium. This would be predicted to enhanced delivery and uptake of nanoparticles in the lungs102 upon i.v. administration, with transfection efficiency in lung endothelial cells, as we have observed. This idea is further supported by the results our intranasal administration of CFTR. While not determined in the CFTR experiments, it is unlikely that improvement in lung function observed would be attributable to increased CFTR expression in lung endothelial cells, and thus likely that route of administration influenced cell targeting as predicted by a passive targeting mechanism. Additional experimentation is required to definitively determine the mechanisms and opportunities to modulate the lung/organ tropism of this hybrid-lipid polyplex formulation.
To conclude, our findings underscore the tremendous potential of both our synthetic and formulation strategies to identify constituents for lipid nanoparticle mediated mRNA delivery. In the present iteration we successfully identified a PEI-based polyplex demonstrating lung tropism, that when rationally formulated with a complementary lipid membrane enabled effective delivery of different sized mRNAs, permissive of repeat dosing and demonstrated compatible with gene editing approaches in the lungs, holding promise for a wide range of therapeutic applications.
Methods
Ethical statement
All experiments were conducted in accordance with the Institutional Animal Care and Use Committee (IACUC, protocol # IP00001707, #IP00002318, # IP00002286) of Oregon Health and Sciences University.
Animals
Female BALB/C, male C57BL/6 albino (Charles River Laboratory) and female CFTR-/-tm1Unc Tg(FABPCFTR)1 Jaw/J double-transgenic CFTR KO, male B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J (Ai9) (The Jackson Laboratory) mice, aged 8 weeks, were used in the study. All mice used in our experiments were maintained in the animal facility of Oregon Health and Sciences University with a 12 h light cycle and free access to water and food. All animal experiments applied to both sexes. Blinding was not applied in our studies, we used matched age and sex controls.
Reagents
Octylamine, decylamine, dodecylamine, cyclohexanecarboxaldehyde, octanal, decanal, benzylamine and trans,cis-2,6-nonadienal were obtained from TCI Chemicals and used as received. Ethyl formate, hexanal, dodecanal, formaldehyde (37 % in H2O), acetic acid, hexanoic acid, 3-(dimethylamino)propionic acid hydrochloride, cyclohexylisocyanide, ethyl isocyanoacetate and 2-morpholinoethyl isocyanide, cholesterol, 30% Hydrogen peroxide solution (7722-84-1), citric buffer (10x) antigen retriever were obtained from Sigma Aldrich and used as received. L-α-phosphatidylcholine (Soy-PC), 1,2-distearoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (sodium salt) (DSPG) were purchased from AvantiPolarLipids. 1,2-Dimyristoyl-rac-glycero-3-methylpolyoxyethylene (DMG-PEG2000) was obtained from NOF American Corporation. DiD’ solid (1,1’-Dioctadecyl-3,3,3’,3’-Tetramethylindodicarbocyanine, 4-Chlorobenzenesulfonate Salt) was purchased. From ThermoFisher Scientific. AMEC Red Substrate Kit, Peroxidase (HRP) (SK-4285) was obtained from Vector Laboratories. CleanCap Fluc mRNA, mCherry mRNA, Cre mRNA, customized IL-12 mRNA, human CFTR mRNA (NCBI: NM_000492.3, custom-made), Cas9 mRNA, each with fully substituted uridine by pseudouridine and cytidine by 5-methylcytidine, were purchased from TriLink Biotechnologies. Guide RNA for tdTomato and PD-1 gene editing, all primers for PCR were ordered from Integrated DNA Technologies.
Acetonitrile, benzonitrile, benzylbromide, methyl triflate and 2-ethyl-2-oxazoline were obtained from Sigma Aldrich and dried over CaH, then purified by distillation before use.
NMR spectra were recorded on a Bruker Ultrashield 500 MHz Plus system at 25 °C using deuterated solvents obtained from Sigma-Aldrich.
MALDI-ToF-MS was performed on a Shimadzu Axima Performance instrument in or positive-reflector mode. Trans-2-[3-(4-tert-Butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) (100 mg mL−1 in ACN) was used as the matrix without further purification (Sigma-Aldrich). NaTFA salt was used as the ionization agent (1 mg mL−1 in MeOH). Matrix, polymer, and salt solutions were mixed in a 1:1:0.5 volume ratio and then 1 μL of the mixture was deposited onto a ground steel target plate before insertion into the ion source chamber. The instrument was calibrated against a poly(ethylene glycol) methyl ether standard (Sigma Aldrich, Average Mn = 2000 g mol−1) prepared under the same conditions with DCTB matrix.
SEC-RI chromatography
Poly(ethyloxazoline) samples were analyzed with the Agilent 1260 Infinity II chromatography system with Stryagels HR2, HR4 and HT5 columns and Agilent 1260 infinity RI detector was used for the SEC studies. DMF + 0.1% LiBr was used as an eluent. The flow rate was 0.8 mL/min. The column was thermostated at 40 °C. The Ugi modified polymers were instead analyzed on a system using an eluent mixture of CHCl3, IPA and triethylamine (TEA) in ratios of 94:4:2. The system consisted of Waters 515 HPLC pump, Biotech DEGASi GPC Degasser, Waters 717 plus Autosampler and Waters 2410 Differential Refractometer together with Waters Styragel HR 2, HT 3, and HT 4 7,8 × 300 mm and guard column. The flow rate was set to 0.800 ml/min and the columns thermostated at 30 C PMMA standards obtained from Polymer Standards Service were used for calibration.
Synthesis of benzylisocyanide
Selected isocyanides were synthesized using a modified literature procedure103. Benzylamine (5 g, 46.7 mmol) was dissolved in ethyl formate (11.3 ml, 140 mmol) in a round bottom flask and refluxed for 3 h at an oil bath temperature of 65 °C. The reaction mixture was concentrated under reduced pressure.
The formate intermediate was dissolved in dichloromethane (DCM) (30 mL) under nitrogen atmosphere, triethylamine (32.5 mL, 233 mmol) was added, cooled with an ice water bath followed by dropwise addition of a solution of phosphorus oxychloride (4.56 mL, 49 mmol) in DCM (5 mL). The reaction mixture was stirred for 2 h and allowed to warm to room temperature and then purified directly by column chromatography over silica. Diethyl ether was initially used as the mobile phase, switching to a 50% DCM/Et2O mixture after elution of approximately one column volume. The product fractions were combined, concentrated and then purified by distillation under vacuum to yield a pale-yellow oil (yield = 2.3 g, 42 %). 1H NMR (500 MHz, CDCl3) δ 7.41 (m, 5H, C5H5), 4.67 (2H, s, CH2).
Synthesis of alkylisocyanides
The same protocol described above for benzylisocyanide was employed using 5 g of the alkylamine reagent, modifying quantities of the other reagents accordingly. For column chromatography a hexane/ethyl acetate (9:1 to 4:1) mixture was used. Only octylisocyanide was further purified by distillation.
Octylisocyanide
Yield = 3 g, 56%. Distilled at 60 °C, 0.2 mbar. 1H NMR (500 MHz, CDCl3) δ 3.40 (tt, 2H, J = 1.9, 6.7 Hz, CH2NC), 1.70 (2H, m, CH2CH2NC), 1.45 (2H, m, CH2(CH2)2NC) 1.32 (8H, m, CH3(CH2)4) 0.91 (3H, t, J = 7.0 Hz, CH3).
Decylisoycanide
Yield = 4.1 g, 77%. 1H NMR (500 MHz, CDCl3) δ 3.40 (tt, 2H, J = 1.9, 6.7 Hz, CH2NC), 1.70 (2H, m, CH2CH2NC), 1.45 (2H, m, CH2(CH2)2NC) 1.32 (8H, m, CH3(CH2)4) 0.91 (3H, t, J = 7.0 Hz, CH3).
Dodecylisocyanide
Yield = 3.8 g, 72%. 1H NMR (500 MHz, CDCl3) δ 3.40 (m, 2H, CH2NC), 1.70 (2H, m, CH2CH2NC), 1.46 (2H, m, CH2(CH2)2NC) 1.32 (16H, m, CH3(CH2)8) 0.91 (3H, t, J = 6.9 Hz, CH3).
1H NMR are presented in Supplementary Figs. 21–24.
Synthesis of poly(ethylene imine)s
Cationic ring opening polymerization of 2-ethyl-2-oxazoline
2-Ethyl-2-oxazoline (15 g, 151.3 mmol), benzonitrile (35.1 mL) and methyl triflate (0.69–4.32 mmol) were added to a dry Schlenk flask under inert atmosphere and then stirred at 80 °C for 4 h. Molar equivalents of methyl triflate (MeOTf) were altered to target degrees of polymerization 35, 85 and 220 accordingly. The reaction mixture was cooled to 40 °C, terminated by addition of benzylamine (10 eq. with respect to MeOTf) and left to stir overnight. The polymer was precipitated three times from diethyl ether and dried under vacuum to yield a colorless powder.
For the preparation of the lowest degree of polymerization PEtOx15, the initiator benzyl bromide was used in acetonitrile and the termination carried out with 1 M KOH solution instead. Yield = 4.9 g (88 %). SEC and MALD-ToF results are presented in Supplementary Fig. 25 and Table 2.
Hydrolysis of poly(2-ethyl-2-oxazoline)
Poly(2-ethly-2-oxazoline) was dissolved in 3 M HCl (75 mL) in a round bottom flask fitted with a stir bar and refluxed overnight for 18 h. The mixture was cooled to room temperature, then adjusted to pH 10 by addition of 4 M NaOH causing precipitation of the polymer. The solid was collected by centrifugation and washed five times with distilled water collecting again by centrifugation between washes. The solid was dried under vacuum to yield a colorless powder. 1H NMR is presented in Supplementary Fig. 26.
Ugi modification of poly(ethylenimine)
Reactions were run targeting total modification amounts of PEI units of 25-100%, assuming the reaction forms the split-Ugi product whereby 2 molar equivalents of PEI secondary amines are required with respect to the other reagents. The quantities of reagent were calculated as follows taking the repeat unit of PEI as 1 eq:
25% target modification: 0.125 eq. aldehyde, 0.125 eq. isocyanide, 1 eq. carboxylic acid.
50 % target modification: 0.25 eq. aldehyde, 0.25 eq. isocyanide, 1 eq. carboxylic acid.
100 % target modification: 0.5 eq. aldehyde, 0.5 eq. isocyanide, 1 eq. carboxylic acid.
PEI was dissolved in EtOH/H2O (9/1) at a concentration of 50 g/L polymer, and stirred with heating at 50 °C. When targeting the highest modification density, the concentration was lowered to 20 g/L polymer to ensure sufficient solubility. The aldehyde reagent was added to the PEI solution and stirred for 30 min, followed by addition of the isocyanide and then the acid. The mixture was stirred overnight with continued heating of 50 °C, then cooled to room temperature, diluted with ethanol, and transferred to a dialysis membrane (regenerated cellulose, MWCO = 1 kDa). Dialysis was performed first once against ethanol, and then twice against deionized water exchanging solvents every 24 h. The solution was freeze dried to yield the product, typically as a pale orange solid/oil. Alternatively, in early batches of samples (U1 – U11) the polymers were purified by precipitating three times into ice cold diethyl ether, resolubilizing in ethanol between steps. This procedure was unreliable, however, for the more hydrophobic derivatives which showed some degree of solubility in diethyl ether or hexane and thus dialysis was selected as the purification method of choice. NMR spectra are presented in Supplementary Figs. 26–32, Supplementary Table 3.
Polyplex preparation
For in vitro and in vivo screening polyplexes were formulated from the synthesized PEI-derivatives library via ethanol injection method. Briefly, a solution of the polymer in ethanol (2 g/L) was combined with an aqueous phase containing mRNA (28.6 g/L) in an acetate buffer (25 mM, pH 5, IS 154 mM) at the ratio water to ethanol 7/1 (v/v), resulting in a 10/1 (w/w) polymer to mRNA ratio. The mixture was then left to incubate for 30 min, allowing the polyplexes to form. Finally, the polyplexes were neutralized using Tris-HCl buffer (25 mM, pH 7.4, IS 154 mM). For in vivo studies polyplexes were dialyzed against Tris-HCl buffer (25 mM, pH 7.4) for 2 h at room temperature. Size distribution and polydispersity indexes (PDI) of polyplexes were determined with dynamic light scattering using Stunner (Unchained Labs, US) or Zetasizer Nano ZSP (Malvern Instruments, UK). Concentration of RNA in final formulation was assumed to be 100% yield.
Hybrid polymer-lipid particles preparation
Hybrid polymer-lipid nanoparticles were formulated in two steps. Before formulating, all lipids with specified molar ratios were dissolved and mixed in ethanol to form a complete lipid mix solution. Separately, polymer was dissolved in ethanol and mRNA was diluted in 25 mM acetate buffer (pH 5.0). Then, the ethanol polymer solution was rapidly mixed by vortexing with the aqueous buffer solution containing Fluc mRNA at a ratio of 20/1 (aqueous/ethanol, v/v) to achieve a final weight ratio of 10/1 (total polymer/mRNA, w/w). The resulting mixture was then left to incubate for 30 min, allowing the polyplexes to form. Next, the aqueous solution of polyplexes was combined with ethanol phase, containing DSPG, soy PC, cholesterol and DMG-PEG2000 lipids in molar ratio 22/23/50/5, by microfluidic mixing using NanoAssemblr Ignite+ (Precision Nanosystems). Polymer to lipids ratio was 2/1/(w/w). Final hybrid particles were dialyzed 3–4 h against Tris-HCl buffer (25 mM, pH 7.4) and concentrated with 10-kDa Amicon Ultra centrifuge filters (Millipore, Burlington, MA). Size distribution and PDI of polyplexes were determined with dynamic light scattering using Stunner (Unchained Labs, US) or Zetasizer Nano ZSP (Malvern Instruments, UK). RNA concentration in final formulation was measured with RiboGreen kit according to the manufacturer’s protocol with some modifications. Samples were diluted to 100 µg/L in 1× TE buffer with 2 g/L heparin (Sigma-Aldrich, U.K.). Standard solutions from the corresponding RNA stock were also prepared in a 1× TE buffer with 2 g/L heparin to account for any variation in fluorescence. Ribogreen reagent was diluted 2000-fold in 1× TE buffer. RNA encapsulation of samples was determined by comparing the signal of the RNA-binding fluorescent dye RiboGreen in the absence and presence of a detergent (2% Triton X-100). All samples were incubated 15 min at 37 °C before addition of reagent. In the absence of detergent, the signal comes only from unencapsulated RNA. In the presence of detergent, the particles were disrupted so that the measured signal comes from the total. Fluorescence was measured at λex = 485 nm and λem = 530 nm. Recovery of mRNA was evaluated as the ratio of total mRNA in final formulation to mRNA added for formulating.
To evaluate particles in vivo biodistribution, DiD dye was added to lipids mixture at 0.1% molar of total lipids concentration. For pretreatment studies, blank nanoparticles were prepared according to the protocol without mRNA in acetate buffer.
Cryo-transmission electron microscopy (TEM)
Cryo-TEM images were captured with Falcon III and K3 Summit cameras with DED at 300 kV. The Vitrobot Mark IV system (FEI) was used to plunge-freeze a copper lacey carbon film-coated grid (Quantifoil, R1.2/1.3 300 Cu mesh). U155@lipids (10 µL) was dispensed onto the glow discharged grids in the Vitrobot chamber maintained at a temperature of 23 °C and a relative humidity of 100% to freeze the samples. The sample was incubated for 30 s before being blotted with filter paper for 3 s before being submerged in liquid ethane cooled by liquid nitrogen. The frozen grids were clipped. The images were taken at an electron dose of 15–20 e−/Å2 using 45,000 nominal magnifications with 1.5 binning then processed and analyzed using ImageJ (Fiji ImageJ2 version: 2.9.0/1.53t).
TNS assay
TNS assay was performed as described previously104. The McIlvaine citric-phosphate buffer was used to prepare buffers at various pH values between about 3 and 9 for determining apparent pKa. Then a stock of 300 μM 6-(p-toluidino)-2-naphthalenesulfonic acid sodium salt (TNS reagent) in DMSO was prepared. Buffer solution (90 μL) of was added to wells. Then 3.3 μL of U155 polyplexes or U155@lipids sample and 2 μL of 300 μM TNS reagent solution were added. Each well was then carefully mixed, and fluorescence was measured. With the resulting fluorescence values, a sigmoidal plot of fluorescence versus buffer pH was created.
In vivo -JetPEI® formulation
Complexes of mRNA with In vivo-JetPEI® (Polyplus-transfection) were prepared according to the manufacturer’s instructions. Fluc mRNA (40 µg) and 6.4 µl In vivo-JetPEI® (N/P = 8) were each diluted in 200 µL of 5% sterile D-glucose. Both solutions were mixed, followed by a 10-min incubation at room temperature.
SORT LNPs with DOTAP
mRNA-loaded DOTAP LNP formulation was formed using the protocol described previously60,74. All lipids with specified molar ratios were dissolved in ethanol and Fluc mRNA was dissolved in 10 mM citrate buffer (pH 4.0). The two solutions were rapidly mixed at an aqueous to ethanol ratio of 3/1 by volume with a final weight ratio of 40/1 (total lipids/mRNA). Lipids were mixed at molar ratio of 5A2-SC8/DOPE/cholesterol/DMG-PEG/DOTAP equal to 11.9/11.9/23.8/2.4/50. The final mixture was dialyzed overnight against PBS (pH 7.4) and concentrated with 10-kDa Amicon Ultra centrifuge filters (Millipore, Burlington, MA). Size distribution and PDI of particles were determined with dynamic light scattering using Stunner (Unchained Labs, US) or Zetasizer Nano ZSP (Malvern Instruments, UK). RNA concentration in final formulation was measured with RiboGreen kit according to the manufacturer’s protocol with some modifications, described above.
In vitro transfection efficacy screening
HeLa cells were plated in white, clear-bottom 384-well plates (2000 cells per well in 50 µL of complete DMEM media) and allowed to adhere overnight. Then, cells were treated with polyplexes loaded with FLuc mRNA (100 ng per well). Cell viability results (CellTiter-Fluor, Promega), and luciferase expression data (ONE-Glo Luciferase Assay, Promega) were collected 24 h post-treatment using a microplate reader. Luminescent readout (in relative luminescence units) was normalized by cell counts commensurate with fluorescence (relative fluorescence units, RFU).
Endosomal escape studies
HEK293T/17 Gal9-GFP reporter cells were seeded (10,000 cells per well) in complete DMEM media in 96-wells black with clear-bottom plate. After overnight incubation, polyplexes loaded with mCherry RNA were added at a dose of 100 ng mRNA per well and incubated for 24 h. After incubation, media was gently aspirated, cells were then washed twice with PBS and fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. Once cells were fixed, wells were gently washed with PBS two more times and DAPI (Thermo Fisher, Federal Way, WA) was then added at 1:1000 in PBS for nuclear staining. Following DAPI staining, cells were washed again two times and left in PBS. Reporter cells were imaged for GFP-positive puncta with a Fluorescent EVOS microscope with objective at 20× to report maximum intensity projections. Images were processed using ImageJ (Fiji ImageJ2 version: 2.9.0/1.53t).
In vivo bioluminescence imaging
Fluc mRNA encapsulated in particles at a certain dose was injected via tail vein to female BALB/c mice. For bioluminescence imaging, mice received d-luciferin substrate (150 mg/kg) intraperitoneally and were imaged according to the manufacturer’s protocol. Image acquisition and analysis were performed using the IVIS Lumina XRMS and the manufacturer’s software (PerkinElmer). Mice were anesthetized by isoflurane inhalation during the procedure. For ex-vivo imaging, mice were sacrificed. Different organs were harvested, mocked in 1.5 g/L d-luciferin solution and analyzed.
In vivo particles biodistribution
DiD-labeled nanoparticles were injected via tail vein to female BALB/c mice at the dose 5 µg of Fluc mRNA per animal. Mice were sacrificed 4 h post-treatment, organs were harvested and imaged using the IVIS Lumina XRMS (λex/λem = 640 nm/670 nm). DiD concentration in formulation was measured in methanol and estimated using ε640 nm =2.45 × 105 cm−1M−1. Control solution was prepared using DiD dye in DMSO, which was then diluted with DPBS to achieve final concentration of 1 % (v/v) DMSO.
Lewis lung cancer model treatment
For the Lewis Lung cancer model, 2 × 106 LL/2-Luc2 cells (CRL-1642-LUC2, ATCC) in 100 µL PBS were tail vein injected into male C57BL/6 albino mice and randomly assigned to treatment and control groups. At days 2 and 6, IL-12 mRNA (5 µg), PBS (control) and mCherry mRNA (5 µg control) were i.v. injected. Tumor growth was monitored every 2–5 days by in vivo bioluminescence imaging. Animals were weighted every 2–5 days. For in vivo assessment of IL-12 mRNA transfection, mouse blood was collected 24 h post treatment and centrifuged at 10,000 × g for 15 min after 30 min of standing at room temperature. Concentration of IL-12 p70 in serum was determined using mouse ELISA Kit (Invitrogen) according to manufacturer’s instructions. On day of tumor instillation mice were measured for body weight and luciferase expression every other day. Five days post tumor instillation mice were then monitored daily for body weight. Mice were assessed for body condition (coat scruffiness, skin tenting), movement (lethargy) and euthanized upon either demonstrating low body condition scores, rapid loss of body weight (meeting or exceeding 10% body weight loss between measurements, or 20% overall body weight loss) or achieving an average luciferase expression value greater than or equal to 5 ×106 p/sec/cm2/sr) which was determined to represent a tumor burden no greater than 200 mm2 in compliance with approved institutional IACUC protocol (2318). At endpoint all mice and tumor burdens were in compliance with these established criteria.
NPD assay
For particle administration, mice were anesthetized with an intraperitoneal injection of a mixture of ketamine and xylazine (100 μg/10 μg/kg body weight). Particles were administered on two consecutive days to a single nostril (2 μL/application, 10 applications over 20 min, 4 µg /day).
NPD was measured using a modification of the previously described methods and using a previously described circuit93. Following sedation with IP ketamine/xylazine, the mouse is orally intubated to protect the airway. Mice were positioned at a 15° head-down tilt, and a high-impedance voltmeter (World Precision Instruments, Sarasota, FL) attached to chloride pellet electrodes was used to measure the potential difference between an exploring nasal bridge and subcutaneous reference probe. A syringe pump continuously perfused solution into the nostril through a polyethylene tube stretched to approximately 0.5 mm in diameter (PE10, 0.28 mm inner diameter [ID]; Clay-Adams, BD, Sparks, MD). Solutions were perfused into the nasal cavity sequentially (Ringer’s, Amiloride, Gluconate substituted Ringer’s with isoproterenol, CFTR inhibitor, and 172Adenosine Triphosphate solution). DMSO (0.5%) was added to the CFTRInh-172 solution to improve solubility; this has been shown previously not to affect NPD measurements. NPD assay was performed prior to nanoparticle administration to record a baseline and then repeated 48 h after dosing.
In vivo gene editing
For gene editing in mouse lungs with Cas9 mRNA/sgRNA in U155@lipids particles, Ai9 mice were injected with 100 μL of particles at dose total 19 μg per mouse (0.8 mg/kg; total RNA 1/1 mRNA/sgAi9, w/w). Animals were sacrificed on day 9 post-treatment, part of lungs was used for NGS analysis, another part of lungs was formalin-fixed, paraffin-embedded and analyzed with multiplex IHC.
In the sgRNA sequence, the asterisks (blue) and red font indicate the phosphorothioate bond and 2′-O-methyl ribonucleotides, respectively.
A*A*G*UAAAACCUCUACAAAUGGUUUUAGAGCUAGAAAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCU*U*U*U.
For PD-1 gene editing in mouse T cells with Cas9 mRNA/sgRNA, male C57BL/6 albino mice were treated (i.v.) with 5 μg dose of IL-12 mRNA loaded in U155@lipids and randomly assigned to treatment and control groups. At day 4, animals were treated (i.v.) with Cas9 mRNA/sgRNA system at dose total 10 μg per mouse (0.5 mg/kg; total RNA 1/1 mRNA/sgRNA, w/w) and Cas9 mRNA (control) at dose 5 μg per mouse. On day 10 post-treatment mice sacrificed, lungs and spleen were harvested, then dissociated and CD4+/CD8+ T cells were isolated using EasySepTM Mouse isolation kit (Stemcell Technologies) according to manufacturer’s instructions.
sgRNA sequence: TGGTTTGAGCCAACCCGTCC (exon 2, mouse).
Flow cytometry studies
To study the biodistribution and cell type transfection by mRNA, Ai9 mice were intravenously injected with 10 µg Cre mRNA loaded into U155@lipids particles. Untreated Ai9 mice were used as a control. On day 3 post-injection mice were sacrificed, lungs, spleen and liver were harvested and then dissociated. Single cells were generated with the organ dissociation protocol. Briefly, mouse organs were perfused with PBS and placed in a well of 12-wells plate with 0.5 ml of Click’s buffer on ice. Then 50 µl of collagenase IV and DNAse I mixture was added, and tissue was minced using tweezers and incubated for 30 min at 37 °C in 5% CO2 incubator and then treated with 50 mM EDTA in d-PBS for another 5 min at 37 °C in 5% CO2 incubator. Subsequently, the digested tissue was filtered through a 70 µm nylon mesh strainer to collect single cells. Cells were washed using flow wash buffer (PBS containing 2 mM EDTA and 0.5% BSA) and collected by centrifugation at 450 × g for 5 min at 4 °C. The pelleted cells were resuspended in 100–200 µl of ACK lysis buffer (BioWhittaker, Lonza) for 5 min at room temperature and quenched with the addition of equal volume of 5 mM EDTA in PBS. After washing, cells were stained with live dead cell staining dye NIR and then incubated with Fc block (BioLegend). Cells were further stained with the following fluorochrome-conjugated antibodies: CD45-BUV395 (BD Horizon, #565967, 1:400 dilution), B220-BUV805 (BD Horizon, #569199, 1:300 dilution), CD31-BV605 (BD Horizon, #740356, 1:200 dilution), TcrB-AF700 (BD Horizon, #560705, 1:300 dilution). For validation, please, see the above-mentioned vendors’ specification sheets. The stained cells were analyzed using Cytek Aurora.
Multiplex immunohistochemistry
For immunohistochemistry (IHC) studies, formalin-fixed, paraffin-embedded mouse lung samples were sectioned at 4 μm, deparaffinized and antigen retrieved with 10 mM citrate buffer for 20 min at 110 °C. Slides were then incubated 10 min in 3% peroxide in methanol, washed in TBS-tween buffer. Next, lungs sections were cyclically stained with antibodies in the following order: cycle 1 Rabbit RFP polyclonal antibodies (Biotium, 1:600 dilution, 1 h), cycle 2 Rabbit E-Cadherin (Cell Signaling, 24E10 #3195, dilution 1:200, 1 h), cycle 3 Rabbit CD31 (Abcam, ab182981, dilution 1:200, 1 h), cycle 4 Rabbit CD45 (Abcam, ab10558, dilution 1:200, 1 h), cycle 5 Rabbit Lyve1 (Abcam, ab33682, dilution 1:200, 1 h). Visualization was performed using the AMEC Red Substrate Kit, Peroxidase (HRP) (SK-4285) as instructed by the manufacturer. Digital scanning was performed using Aperio ImageScope AT2 (Leica Biosystems) at 20× magnification. Images were processed with ImageScopex64, QuPath (Version: 0.4.4) and ImageJ (Fiji ImageJ2 version: 2.9.0/1.53t). Between each cycle tissue sections were treated with 95% ethanol for 10 min to wash blue protein staining and then 1% SDS, 25 mM glycine solution (pH 2–3) at 70 °C for 1 h.
Hematoxylin and eosin staining
Formalin-fixed, paraffin-embedded mouse lung and liver samples were sectioned at 4 μm. Lungs and liver tissues were first stained with hematoxylin for 30 s at room temperature and then rinsed in tap water. Then, tissue sections were decolorized in acid alcohol (10% acetic acid and 85% ethanol in water), followed by water washing. Next, sections were immersed in a saturated sodium bicarbonate solution, washed in water, and then immersed in 95% alcohol. Finally, staining was performed with Eosin-Phloxine for 10 s, followed by a dehydration step and cover slipping. Digital scanning was performed using Aperio ImageScope AT2 (Leica Biosystems) at 20× magnification. Images were processed with QuPath (Version: 0.4.4) and ImageJ (Fiji ImageJ2 version: 2.9.0/1.53t).
PAS-fast green staining
Formalin-fixed, paraffin-embedded mouse liver samples were sectioned at 4 μm, deparaffinized and hydrated to water. Then sections were oxidized in 0.5% periodic acid solution for 10 min and washed with water. Next, sections were placed in Schiff’s reagent for 30 min and wash in warm tap water for 5 min. To stain PAS-negative background elements tissue samples were incubate in Fast green (0.02% solution) for 30 s, following washing with water, dehydration with 95% and absolute alcohols, and cover-slipping. Digital scanning was performed using Aperio ImageScope AT2 (Leica Biosystems) at 20× magnification. Images were processed with QuPath (Version: 0.4.4) and ImageJ (Fiji ImageJ2 version: 2.9.0/1.53t).
Serum cytokines screening
Blood serum from female BALB/c mice 24 h post injection with 5 µg Fluc mRNA, loaded in U155@lipids, via tail vein injection was collected and analyzed by IDEXX BioAnalytics. Samples were tested on the Milliplex MAP Mouse Cytokine/Chemokine Magnetic Bead Panel (Millipore, Cat No. MCYTOMAG-70K-PMX) according to the kit protocol as qualified. Data were collected by xPONENT® 4.3 (Luminex) and data analysis was completed using BELYSA® 1.1.0 software. The data collected by the instrument software are expressed as Median Fluorescence Intensity (MFI). MFI values for each analyte are collected per each individual sample well. Analyte standards, quality controls, and sample MFI values were adjusted for background.
PCR amplification and Western blot analysis
Lungs tissues were taken 4 h (for PCR analysis) and 48 h (for western blot analysis) post hCFTR mRNA loaded particles administration (i.v.). For PCR analysis, RNA was isolated from lysates using the RNEasy Plus Micro Kit (QIAGEN), and cDNA libraries were generated by RT-PCR using the ProtoScript II cDNA Synthesis Kit (BioLabs) according to the manufacturers’ protocols. To demonstrate the presence of LNP-delivered hCFTR, primers were designed to amplify exon 11 (forward: 5’-AAC TGG AGC CTT CAG AGG GT-3’; reverse: 5’ -TTG GCA TGC TTT GAT GAC GC-3’) using GAPDH as a loading control (forward: 5’- AAC AGC AAC TCC CAC TCT TC -3’; reverse: 5’ – AGC CGT ATT CAT TGT CAT ACC A -3’). PCR was carried out using KAPA kit (Invitrogen) in a 30-cycle reaction with a 10-min, 95 °C polymerase activation step. Each repeating cycle consisted of two steps: 15 s at 95 °C and 1 min at 60 °C. PCR products were analyzed using a 2% agarose gel. For western blot analysis, tissue was homogenized and then lysed using PierceTM RIPA buffer (from cytoplasm extraction) and Tm-PER-100 (FIVEphoton Biochemicals) (from membrane extraction) containing HALT protease inhibitor (Thermo Scientific) at 4 °C. Total protein concentration was quantified by Micro BCA Protein Assay Kit according to the manufacturer’s instructions (Thermo Scientific). Samples and migration standards were prepared under reducing conditions and incubated for 10 min at 37 °C prior to loading. Wells were loaded at 400 ng (standards) or 100 µg total protein/well into a Novex WedgeWell 8% Bis-Tris gel (Invitrogen) and separated in Tris-glycine SDS running buffer (Novex). Transfer to a nitrocellulose membrane (Novex) was performed in tris-glycine transfer buffer (12 mM Tris-base, 100 mM glycine, 20% methanol) at 20 V for 90 min (Mini Blot Module, Invitrogen). For blocking, we used Super Block Blocking buffer (ThermoScientific). For staining Ms-anti-CFTR Ab596 (University of North Carolina), 1:2000, and Ms-anti-b-actin (Abcam), 1:10000 antibodies were used. The secondary antibody was horseradish peroxidase (HRP)-Gt- anti-Ms, 1:5000 (Abcam). For detection and imaging, SuperSignal West Pico Chemiluminescent Substrate (ThermoScientific) and a myECL Imager (model 62236X, Thermo Scientific) were used.
Next generation sequencing (NGS)
NGS was performed via a two-step PCR as described previously105. Genomic DNA (gDNA) was harvested from lung and spleen tissues and CD4+/CD8+ T cells using the Qiagen DNeasy Blood & Tissue kit (Qiagen, #69506) according to the manufacturer’s instructions. Briefly, genomic regions of interest were amplified using two primer sets specific to the first or second/third stop codon of the Ai9 loxp-stop-loxp cassette or to the exon 2 of the PD-1 gene of T cells (Supplementary Table 4), and specific thermocycling conditions (Supplementary Table 5). A second PCR was performed to add the necessary sequences to bind the amplicon to the Illumina flow cell (Supplementary Table 5 S5). The samples were then run on a gel, excised, pooled together, and harvested. The pooled library DNA concentration was quantified via qPCR using the KAPA Quantification Kit (Roche, #07960140001). After quantification the library was prepared for NGS using Illumina’s standard denature and dilute protocol. The NGS library was run on an Illumina MiniSeq using a 300-cycle mid output kit (Illumina, FC-420-1004). Editing was quantified using CRISPResso2 in standard mode, using the indel count in the “CRISPResso_quantification_of_editing_frequency.txt” file. The insertions and deletions were added together before being normalized to the number of reads, resulting in a % of reads containing indels.
Statistical analysis
All statistical analyses and significance were performed by GraphPad Prism 10 software (La Jolla, CA) or Microsoft Excel. An unpaired t-test was used to compare between two groups. Statistical significance among different groups and the data were considered statistically significant if p < 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the results in this study are available within the paper and its Supplementary Information and Source data file. The raw NGS data have been deposited in the SRA database: PRJNA1130491. Source data are provided with this paper.
References
Yan, Y. et al. Non-viral vectors for RNA delivery. J. Controlled Rel. 342, 241–279 (2022).
Yang, W., Mixich, L., Boonstra, E. & Cabral, H. Polymer‐Based mRNA Delivery Strategies for Advanced Therapies. Adv. Healthc. Mater. 12, e2202688 (2023).
Saiding, Q. et al. Nano-bio interactions in mRNA nanomedicine: Challenges and opportunities for targeted mRNA delivery. Adv. Drug Deliv. Rev. 203, https://doi.org/10.1016/j.addr.2023.115116 (2023).
Sahin, U., Karikó, K. & Türeci, Ö. MRNA-based therapeutics-developing a new class of drugs. Nat. Rev. Drug Discov. 13, 759–780 (2014).
Casper, J. et al. Polyethylenimine (PEI) in gene therapy: Current status and clinical applications. J. Controlled Rel. 362, 667–691 (2023).
Jäger, M., Schubert, S., Ochrimenko, S., Fischer, D. & Schubert, U. S. Branched and linear poly(ethylene imine)-based conjugates: Synthetic modification, characterization, and application. Chem. Soc. Rev. 41, 4755–4767 (2012).
Fischer, D., Bieber, T., Li, Y., Elsässer, H. & Kissel, T. A Novel Non-Viral Vector for DNA Delivery Based on Low Molecular Weight, Branched Polyethylenimine: Effect of Molecular Weight on Transfection Efficiency and Cytotoxicity. Pharm. Res. 16, 1273–1279 (1999).
Moghimi, S. M. et al. A two-stage poly(ethylenimine)-mediated cytotoxicity: implications for gene transfer/therapy. Mol. Ther. 11, 990–995 (2005).
Halupczok, S. et al. Poly(2-ethyl-2-oxazoline-: Co-N-propylethylene imine)s by controlled partial reduction of poly(2-ethyl-2-oxazoline): Synthesis, characterization and cytotoxicity. Polym. Chem. 12, 680–688 (2021).
Shah, R. et al. In vitro study of partially hydrolyzed poly(2-ethyl-2-oxazolines) as materials for biomedical applications. J. Mater. Sci. Mater. Med. 26, 157 (2015).
Rezaee, M., Gholami, L., Gildeh, M. S., Ramezani, M. & Kazemi Oskuee, R. Charge reduction: an efficient strategy to reduce toxicity and increase the transfection efficiency of high molecular weight polyethylenimine. J. Pharm. Investig. 49, 105–114 (2019).
Wang, M. et al. High efficiency and low toxicity of polyethyleneimine modified Pluronics (PEI-Pluronic) as gene delivery carriers in cell culture and dystrophic mdx mice. J. Mater. Chem. 22, 6038–6046 (2012).
Liu, Z., Zhang, Z., Zhou, C. & Jiao, Y. Hydrophobic modifications of cationic polymers for gene delivery. Prog. Polym. Sci. 35, 1144–1162 (2010).
Sunshine, J. C., Akanda, M. I., Li, D., Kozielski, K. L. & Green, J. J. Effects of Base Polymer Hydrophobicity and End-Group Modification on Polymeric Gene Delivery. Biomacromolecules 12, 3592–3600 (2011).
Ong, Z. Y. et al. Biodegradable cationic poly(carbonates): Effect of varying side chain hydrophobicity on key aspects of gene transfection. Acta Biomater. 54, 201–211 (2017).
Thomas, M. & Klibanov, A. M. Enhancing polyethylenimine’s delivery of plasmid DNA into mammalian cells. Proc. Natl Acad. Sci. 99, 14640–14645 (2002).
Teo, P. Y. et al. Hydrophobic modification of low molecular weight polyethylenimine for improved gene transfection. Biomaterials 34, 7971–7979 (2013).
Meneksedag-Erol, D., KC, R. B., Tang, T. & Uludağ, H. A Delicate Balance When Substituting a Small Hydrophobe onto Low Molecular Weight Polyethylenimine to Improve Its Nucleic Acid Delivery Efficiency. ACS Appl. Mater. Interfaces 7, 24822–24832 (2015).
Zintchenko, A., Philipp, A., Dehshahri, A. & Wagner, E. Simple Modifications of Branched PEI Lead to Highly Efficient siRNA Carriers with Low Toxicity. Bioconjug Chem. 19, 1448–1455 (2008).
Forrest, M. L., Meister, G. E., Koerber, J. T. & Pack, D. W. Partial Acetylation of Polyethylenimine Enhances In Vitro Gene Delivery. Pharm. Res. 21, 365–371 (2004).
Yang, C. et al. Mitigated Cytotoxicity and Tremendously Enhanced Gene Transfection Efficiency of PEI through Facile One‐Step Carbamate Modification. Adv. Health Mater. 2, 1304–1308 (2013).
Kim, H., Kim, H. A., Bae, Y. M., Choi, J. S. & Lee, M. Dexamethasone‐conjugated polyethylenimine as an efficient gene carrier with an anti‐apoptotic effect to cardiomyocytes. J. Gene Med. 11, 515–522 (2009).
Hsu, C. Y. M., Hendzel, M. & Uludaǧ, H. Improved transfection efficiency of an aliphatic lipid substituted 2 kDa polyethylenimine is attributed to enhanced nuclear association and uptake in rat bone marrow stromal cell. J. Gene Med. 13, 46–59 (2011).
Neamnark, A. et al. Aliphatic Lipid Substitution on 2 kDa Polyethylenimine Improves Plasmid Delivery and Transgene Expression. Mol. Pharm. 7, 618–618 (2010).
Sawant, R. R. et al. Polyethyleneimine-lipid conjugate-based pH-sensitive micellar carrier for gene delivery. Biomaterials 33, 3942–3951 (2012).
Dahlman, J. E. et al. In vivo endothelial siRNA delivery using polymeric nanoparticles with low molecular weight. Nat. Nanotechnol. 9, 648–655 (2014).
Shen, W. et al. Screening of efficient polymers for siRNA delivery in a library of hydrophobically modified polyethyleneimines. J. Mater. Chem. B 4, 6468–6474 (2016).
Yu, Q.-Y. et al. Aromatic Modification of Low Molecular Weight PEI for Enhanced Gene Delivery. Polym 9, 362 (2017).
Wang, J., Meng, F., Kim, B.-K., Ke, X. & Yeo, Y. In-vitro and in-vivo difference in gene delivery by lithocholic acid-polyethyleneimine conjugate. Biomaterials 217, 119296 (2019).
Song, J. et al. Low molecular weight polyethyleneimine modified by 2-aminoimidazole achieving excellent gene transfection efficiency. Eur. Polym. J. 140, 110017 (2020).
Noske, S., Karimov, M., Aigner, A. & Ewe, A. Tyrosine-Modification of Polypropylenimine (PPI) and Polyethylenimine (PEI) Strongly Improves Efficacy of siRNA-Mediated Gene Knockdown. Nanomaterials 10, 1809 (2020).
Zheng, M., Zhong, Y., Meng, F., Peng, R. & Zhong, Z. Lipoic Acid Modified Low Molecular Weight Polyethylenimine Mediates Nontoxic and Highly Potent in Vitro Gene Transfection. Mol. Pharm. 8, 2434–2443 (2011).
Doody, A. M., Korley, J. N., Dang, K. P., Zawaneh, P. N. & Putnam, D. Characterizing the structure/function parameter space of hydrocarbon-conjugated branched polyethylenimine for DNA delivery in vitro. J. Controlled Rel. 116, 227–237 (2006).
Liu, Y., Li, Y., Keskin, D. & Shi, L. Poly(β‐Amino Esters): Synthesis, Formulations, and Their Biomedical Applications. Adv. Healthc. Mater. 8, 1801359 (2019).
Rotolo, L. et al. Species-agnostic polymeric formulations for inhalable messenger RNA delivery to the lung. Nat. Mater. 22, 369–379 (2023).
Zheng, N., Cudjoe, D. K. & Song, W. Multicomponent Polymerization toward Cationic Polymers for Efficient Gene Delivery. Macromol. Rapid Commun. 42, 2000464 (2021).
Miao, L. et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat. Biotechnol. 37, 1174–1185 (2019).
Ugi, I. Recent Progress in the Chemistry of Multicomponent Reactions. Pure Appl. Chem. 73, 187–191 (2001).
Rocha, R. O., Rodrigues, M. O. & Neto, B. A. D. Review on the Ugi Multicomponent Reaction Mechanism and the Use of Fluorescent Derivatives as Functional Chromophores. ACS Omega 5, 972–979 (2020).
Sehlinger, A., Dannecker, P.-K., Kreye, O. & Meier, M. A. R. Diversely Substituted Polyamides: Macromolecular Design Using the Ugi Four-Component Reaction. Macromolecules 47, 2774–2783 (2014).
Yang, B., Zhao, Y., Wei, Y., Fu, C. & Tao, L. The Ugi reaction in polymer chemistry: syntheses, applications and perspectives. Polym. Chem. 6, 8233–8239 (2015).
Gangloff, N., Nahm, D., Döring, L., Kuckling, D. & Luxenhofer, R. Polymerization via the Ugi-reaction using aromatic monomers. J. Polym. Sci. A Polym. Chem. 53, 1680–1686 (2015).
Sehlinger, A., Verbraeken, B., Meier, M. A. R. & Hoogenboom, R. Versatile side chain modification via isocyanide-based multicomponent reactions: tuning the LCST of poly(2-oxazoline)s. Polym. Chem. 6, 3828–3836 (2015).
Yang, B. et al. Introducing the Ugi reaction into polymer chemistry as a green click reaction to prepare middle-functional block copolymers. Polym. Chem. 5, 2704–2708 (2014).
Giovenzana, G. B., Tron, G. C., Di Paola, S., Menegotto, I. G. & Pirali, T. A Mimicry of Primary Amines by Bis‐Secondary Diamines as Components in the Ugi Four‐Component Reaction. Angew. Chem. Int. Ed. 45, 1099–1102 (2006).
Nichugovskiy, A. et al. Synthesis of Novel Lipophilic Polyamines via Ugi Reaction and Evaluation of Their Anticancer Activity. Molecules 27, 6218 (2022).
Tron, G. C. Off the beaten track: The use of secondary amines in the Ugi reaction. Eur. J. Organic Chem. 2013, 1849–1859 (2013).
Dömling, A. & Ugi, I. Multicomponent reactions with isocyanides. Angewandte Chem. Int. Ed. 39, 3168–3210 (2000).
Nakayama, Y. Hyperbranched Polymeric “Star Vectors” for Effective DNA or siRNA Delivery. Acc. Chem. Res. 45, 994–1004 (2012).
Flores-Reyes, J. C., Islas-Jácome, A. & González-Zamora, E. The Ugi three-component reaction and its variants. Organic Chem. Front. 8, 5460–5515 (2021).
Herrera, M., Kim, J., Eygeris, Y., Jozic, A. & Sahay, G. Illuminating endosomal escape of polymorphic lipid nanoparticles that boost mRNA delivery. Biomater. Sci. 9, 4289–4300 (2021).
Zhi, D. et al. A review on cationic lipids with different linkers for gene delivery. Adv. Colloid Interface Sci. 253, 117–140 (2018).
Kaczmarek, J. C. et al. Optimization of a Degradable Polymer-Lipid Nanoparticle for Potent Systemic Delivery of mRNA to the Lung Endothelium and Immune Cells. Nano Lett. 18, 6449–6454 (2018).
Merdan, T., Kopec̆ek, J. & Kissel, T. Prospects for cationic polymers in gene and oligonucleotide therapy against cancer. Adv. Drug Deliv. Rev. 54, 715–758 (2002).
Dilliard, S. A., Cheng, Q. & Siegwart, D. J. On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles. Proc. Natl Acad. Sci. 118, e2109256118 (2021).
Baranov, M. V., Kumar, M., Sacanna, S., Thutupalli, S. & van den Bogaart, G. Modulation of Immune Responses by Particle Size and Shape. Front. Immunol. 11, https://doi.org/10.3389/fimmu.2020.607945 (2021).
Kaczmarek, J. C. et al. Polymer–Lipid Nanoparticles for Systemic Delivery of mRNA to the Lungs. Angew. Chem. 128, 14012–14016 (2016).
Marra, J. Direct measurement of the interaction between phosphatidylglycerol bilayers in aqueous electrolyte solutions. Biophys. J. 50, 815–825 (1986).
Pattipeiluhu, R. et al. Anionic Lipid Nanoparticles Preferentially Deliver mRNA to the Hepatic Reticuloendothelial System. Adv. Mater. 34, e2201095 (2022).
Cheng, Q. et al. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nat. Nanotechnol. 15, 313–320 (2020).
LoPresti, S. T., Arral, M. L., Chaudhary, N. & Whitehead, K. A. The replacement of helper lipids with charged alternatives in lipid nanoparticles facilitates targeted mRNA delivery to the spleen and lungs. J. Controlled Rel. 345, 819–831 (2022).
Álvarez-Benedicto, E. et al. Optimization of phospholipid chemistry for improved lipid nanoparticle (LNP) delivery of messenger RNA (mRNA). Biomater. Sci. 10, 549–559 (2022).
Maeki, M. et al. Development of Polymer-Lipid Hybrid Nanoparticles for Large-Sized Plasmid DNA Transfection. ACS Appl. Mater. Interfaces 16, 2110–2119 (2024).
Nong, J. et al. Multi-stage-mixing to create a core-then-shell structure improves DNA-loaded lipid nanoparticles’ transfection by orders of magnitude. Preprint at https://doi.org/10.1101/2024.11.12.623321 (2024).
Hosta-Rigau, L., Zhang, Y., Teo, B. M., Postma, A. & Städler, B. Cholesterol – a biological compound as a building block in bionanotechnology. Nanoscale 5, 89–109 (2013).
Zhang, Y. & Anchordoquy, T. J. The role of lipid charge density in the serum stability of cationic lipid/DNA complexes. Biochim. Biophys. Acta Biomembranes 1663, 143–157 (2004).
Xu, L., Wempe, M. F. & Anchordoquy, T. J. The effect of cholesterol domains on PEGylated liposomal gene delivery in vitro. Ther. Deliv. 2, 451–460 (2011).
Semple, S. C. et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 28, 172–176 (2010).
Kauffman, K. J. et al. Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 15, 7300–7306 (2015).
Filipczak, N., Pan, J., Yalamarty, S. S. K. & Torchilin, V. P. Recent advancements in liposome technology. Adv. Drug Deliv. Rev. 156, 4–22 (2020).
Wei, X., Cohen, R. & Barenholz, Y. Insights into composition/structure/function relationships of Doxil® gained from “high-sensitivity” differential scanning calorimetry. Eur. J. Pharmaceutics Biopharmaceutics 104, 260–270 (2016).
Dos Santos, N. et al. Influence of poly(ethylene glycol) grafting density and polymer length on liposomes: Relating plasma circulation lifetimes to protein binding. Biochim. Biophys. Acta Biomembranes 1768, 1367–1377 (2007).
Mönkkönen, J., Liukkonen, J., Taskinen, M., Heath, T. D. & Urtti, A. Studies on liposome formulations for intra-articular delivery of clodronate. J. Controlled Rel. 35, 145–154 (1995).
Wang, X. et al. Preparation of selective organ-targeting (SORT) lipid nanoparticles (LNPs) using multiple technical methods for tissue-specific mRNA delivery. Nat. Protoc. 18, 265–291 (2023).
Stack, T. et al. Enhancing subcutaneous injection and target tissue accumulation of nanoparticles via co-administration with macropinocytosis inhibitory nanoparticles (MiNP). Nanoscale Horiz. 6, 393–400 (2021).
Ouyang, B. et al. The dose threshold for nanoparticle tumour delivery. Nat. Mater. 19, 1362–1371 (2020).
Sago, C. D. et al. Augmented lipid-nanoparticle-mediated in vivo genome editing in the lungs and spleen by disrupting Cas9 activity in the liver. Nat. Biomed. Eng. 6, 157–167 (2022).
Dutta, D. & Donaldson, J. G. Search for inhibitors of endocytosis. Cell Logist. 2, 203–208 (2012).
Zhang, Y.-N., Poon, W., Sefton, E. & Chan, W. C. W. Suppressing Subcapsular Sinus Macrophages Enhances Transport of Nanovaccines to Lymph Node Follicles for Robust Humoral Immunity. ACS Nano 14, 9478–9490 (2020).
Herber-Jonat, S. et al. Comparison of lung accumulation of cationic liposomes in normal rats and LPS-treated rats. Inflamm. Res. 60, 245–253 (2011).
Trac, N. & Chung, E. J. Overcoming physiological barriers by nanoparticles for intravenous drug delivery to the lymph nodes. Exp. Biol. Med. 246, 2358–2371 (2021).
Liu, J. et al. Enhanced Primary Tumor Penetration Facilitates Nanoparticle Draining into Lymph Nodes after Systemic Injection for Tumor Metastasis Inhibition. ACS Nano 13, 8648–8658 (2019).
Minnar, C. M., Lui, G., Gulley, J. L., Schlom, J. & Gameiro, S. R. Preclinical and clinical studies of a tumor targeting IL-12 immunocytokine. Front. Oncol. 13, 1321318 (2023).
Li, Y. et al. Multifunctional oncolytic nanoparticles deliver self-replicating IL-12 RNA to eliminate established tumors and prime systemic immunity. Nat. Cancer 1, 882–893 (2020).
Liu, M., Hu, S., Yan, N., Popowski, K. D. & Cheng, K. Inhalable extracellular vesicle delivery of IL-12 mRNA to treat lung cancer and promote systemic immunity. Nat. Nanotechnol. 19, 565–575 (2024).
Xu, S. et al. Tumor-Tailored Ionizable Lipid Nanoparticles Facilitate IL-12 Circular RNA Delivery for Enhanced Lung Cancer Immunotherapy. Adv. Mater. https://doi.org/10.1002/adma.202400307 (2024).
Lai, I. et al. Lipid nanoparticles that deliver IL−12 messenger RNA suppress tumorigenesis in MYC oncogene-driven hepatocellular carcinoma. J. Immunother Cancer 6, 125 (2018).
Di Trani, C. A. et al. Intratumoral injection of IL-12-encoding mRNA targeted to CSFR1 and PD-L1 exerts potent anti-tumor effects without substantial systemic exposure. Mol. Ther. Nucleic Acids 33, 599–616 (2023).
Turuvekere Vittala Murthy, N., Vlasova, K., Renner, J., Jozic, A. & Sahay, G. A new era of targeting cystic fibrosis with non-viral delivery of genomic medicines. Adv. Drug Deliv. Rev. 209, https://doi.org/10.1016/j.addr.2024.115305 (2024).
Allen, L. et al. Future therapies for cystic fibrosis. Nat Commun 14, 693 (2023).
Robinson, E. et al. Lipid Nanoparticle-Delivered Chemically Modified mRNA Restores Chloride Secretion in Cystic Fibrosis. Mol. Ther. 26, 2034–2046 (2018).
Cant, N., Pollock, N. & Ford, R. C. CFTR structure and cystic fibrosis. Int. J. Biochem. Cell Biol. 52, 15–25 (2014).
Macdonald, K. D. et al. Lubiprostone activates non-CFTR-dependent respiratory epithelial chloride secretion in cystic fibrosis mice. Am. J. Physiol. Lung Cell Mol. Physiol. 295, 933–940 (2008).
Chamberlain, C. A. et al. Highly efficient PD-1-targeted CRISPR-Cas9 for tumor-infiltrating lymphocyte-based adoptive T cell therapy. Mol. Ther. Oncolytics 24, 417–428 (2022).
Rupp, L. J. et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-Tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 7, 737 (2017).
Freeman, E. C., Weiland, L. M. & Meng, W. S. Modeling the proton sponge hypothesis: Examining proton sponge effectiveness for enhancing intracellular gene delivery through multiscale modeling. J. Biomater. Sci. Polym. Ed. 24, 398–416 (2013).
Neu, M., Fischer, D. & Kissel, T. Recent advances in rational gene transfer vector design based on poly(ethylene imine) and its derivatives. J. Gene Med. 7, 992–1009 (2005).
Peng, H. et al. Intracranial delivery of synthetic mRNA to suppress glioblastoma. Mol. Ther. Oncolytics 24, 160–170 (2022).
Kaczmarek, J. C. et al. Polymer–Lipid Nanoparticles for Systemic Delivery of mRNA to the Lungs. Angew. Chem. Int. Ed. 55, 13808–13812 (2016).
Zhang, H. et al. Algorithm for optimized mRNA design improves stability and immunogenicity. Nature 621, 396–403 (2023).
Fu, L. et al. Passive’ nanoparticles for organ-selective systemic delivery: design, mechanism and perspective. Chem. Soc. Rev. 52, 7579–7601 (2023).
Li, Y.-X. et al. Targeting pulmonary vascular endothelial cells for the treatment of respiratory diseases. Front. Pharmacol. 13, 983816 (2022).
Waibel, K. A., Nickisch, R., Möhl, N., Seim, R. & Meier, M. A. R. A more sustainable and highly practicable synthesis of aliphatic isocyanides. Green. Chem. 22, 933–941 (2020).
Sabnis, S. et al. A Novel Amino Lipid Series for mRNA Delivery: Improved Endosomal Escape and Sustained Pharmacology and Safety in Non-human Primates. Mol. Ther. 26, 1509–1519 (2018).
Gautam, M. et al. Lipid nanoparticles with PEG-variant surface modifications mediate genome editing in the mouse retina. Nat. Commun. 14, 6468 (2023).
Acknowledgements
Financial support from the Cystic Fibrosis Foundation (SAHAY19XX0), R01HL146736 from NHLBI (G.S), R01CA270783-01 (G.S). We thank PhD J. Kim, PhD Y. Eygeris, PhD A. Mukherjee for helpful advice and electron microscopy acquisition at Multiscale Microscopy Core (MMC), Oregon Health & Sciences University. We thank MS Elaine Huang for assisting with H&E staining, and PhD Shaposhnikov A.V. for help with data visualization. The images of nanoparticles, mice, syringes, organs (Figs. 4a, 6a, 7a, b, 8, 9, Supplementary Fig. 9) were created with BioRender.com.
Author information
Authors and Affiliations
Contributions
K.Y.V. and A.K. contributed equally to this work. G.S. and R.L. conceptualized the idea and directed the work. K.Y.V., A.K., N.D.P, G.S. and R.L. designed the experiments. A.K. and M.W.A. performed synthesis of molecules. K.Y.V., N.D.P., J.W. and K.D.M.D. established animal models. K.Y.V., N.D.P., A.J., D.K.S., M.G., N.T.V.M., A.R. and J.W. performed the experiments. K.Y.V., N.D.P., A.K., A.J., D.K.S., N.T.V.M., G.S, R.L. analyzed the data. K.Y.V., A.K, R.L., and G.S. wrote the article with contribution of all authors. All authors read and approved the final article.
Corresponding authors
Ethics declarations
Competing interests
K.Y.V., A.K., M.G., M.W.A., R.L., G.S. have filed a provisional patent based on this work. G.S. is a co-founder of EnterX Bio and has an advisory role to Rare Air Inc., Serina Tx and Mana Bio. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Vlasova, K.Y., Kerr, A., Pennock, N.D. et al. Synthesis of ionizable lipopolymers using split-Ugi reaction for pulmonary delivery of various size RNAs and gene editing. Nat Commun 16, 4021 (2025). https://doi.org/10.1038/s41467-025-59136-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59136-z
