
Abstract
The interplay between tumor cells and the microenvironment significantly influences cancer progression. Here, we report a significant role of the transcription factor FOXM1 in shaping the tumor immune landscape. Single-cell sequencing reveals that tumor-intrinsic FOXM1 creates an immune-suppressive tumor microenvironment by inhibiting expression of stress ligands (including ULBP1) on cancer cells, thereby blocking NKG2D-NKG2DL interactions critical for priming natural killer- and T cell-mediated cytotoxicity of cancer cells. FOXM1 suppresses ULBP1 expression by epigenetically silencing the DNA-sensing protein STING using a DNMT1-UHRF1 complex, which in turn inhibits the unfolded protein response protein CHOP from activating ULBP1. Importantly, cancer patients with higher levels of FOXM1 and DNMT1, and lower levels of STING and ULBP1, have worse survival and are less responsive to immunotherapy. Collectively, our findings provide key insight into how a tumor-intrinsic transcription factor epigenetically shapes the tumor immune microenvironment, with strong implications for refining existing and designing new cancer immunotherapies.
Similar content being viewed by others


Introduction
Tumor-intrinsic factors exert their influence on tumor initiation and progression through different mechanisms1,2. One crucial aspect of tumor development is the modulation of the tumor microenvironment, orchestrated by intrinsic factors3. Among the diverse changes induced by tumor-intrinsic factors, alterations in immune cell composition and function are pivotal in shaping the tumor-immune landscape4,5. For example, activation of the Wnt/β-catenin signaling pathway in cancer cells is associated with increased expression of immunosuppressive factors like programmed death-ligand 1 (PD-L1), which can inhibit the activity of cytotoxic T cells6. Oncogenic mutations in the genes encoding the Janus kinase (JAK) family of proteins also affect immune regulation7,8. Specifically, JAK mutations can lead to constitutive activation of downstream signaling pathways, such as the signal transducer and activator of transcription (STAT) pathway, resulting in immune evasion and tumor immune privilege8,9,10. These mutations also induce expression of immunosuppressive factors, such as indoleamine 2,3-dioxygenase- and T cell-inhibitory receptors, thereby suppressing anti-tumor immune responses and promoting tumor growth. Furthermore, dysregulation of the PI3K/AKT/mTOR pathway, which is common in various cancer types, can have profound effects on immune cell function within the tumor microenvironment11,12. Activation of this pathway in cancer cells can lead to increased production of immunosuppressive cytokines, such as interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β), which inhibit immune cell activation to create an immunosuppressive milieu13.
Despite the progress made in understanding the role of certain tumor-intrinsic factors in immune regulation, specific factors and the underlying mechanisms governing immune function within the tumor microenvironment remain incompletely understood. Notably, no reported intrinsic factors are known to epigenetically control NKG2D-dependent anti-tumor immunity and immune memory. In addition, the intricate interplay among distinct tumor-specific intrinsic and extrinsic factors and their collective impact on immune cell behavior and tumor evolution remain active areas of investigation.
The transcription factor FOXM1 is one such tumor-intrinsic factor that could potentially play a critical role in shaping the tumor immune microenvironment. FOXM1 belongs to the evolutionarily conserved Forkhead Box family of transcription factors implicated in promoting various aspects of cancer growth and progression, such as proliferation, survival, DNA repair, invasion, metastasis, and resistance to conventional therapies14,15,16,17,18. However, the mechanisms by which FOXM1 may promote tumor growth remain to be elucidated. In particular, the role of tumor-intrinsic FOXM1 in regulating anti-tumor immunity is unknown. Understanding the role of FOXM1 in shaping the immune landscape and its impact on tumor progression may provide a pathway for devising targeted therapies and immunotherapeutic interventions in cancers.
Here, we demonstrate that tumor-intrinsic FOXM1 suppresses anti-tumor immunity and immune memory by epigenetically silencing the cytosolic DNA-sensing protein STING. FOXM1-dependent silencing of STING inhibits the expression of stress ligands (NKG2DL), impairing the interaction between NKG2D and NKG2DL. This disruption leads to the evasion of natural killer- and T cell-mediated cytotoxicity of cancer cells, highlighting a crucial mechanism of immune escape facilitated by FOXM1.
Results
FOXM1 promotes tumor growth in immunocompetent mice
To investigate the importance of tumor-intrinsic FOXM1 in cancer growth, we used the CRISPR-Cas9 system to delete FOXM1 in two triple-negative breast cancer cell models, E0771 (mouse) and MDA-MB-231 (human) cells. We generated knockout clones using two different sgRNAs targeting FOXM1 (sg1 and sg3) (Fig. 1a, b). FOXM1 knockout had no effect on proliferation in these cell lines (Fig. 1c, d). Consistent with that finding, we saw no significant differences in cell cycle profiles upon FOXM1 knockout in cancer cells (Supplementary Fig. 1a, b).

a, b Representative Western blot of human FOXM1 levels in control and FOXM1 knockout MDA-MB-231 (a) or E0771 (b) cells using two different sgRNAs (sgRNA 1 and sgRNA 3). The blots are representative of three independent experiments. c Control and FOXM1 knockout MDA-MB-231 cells were plated at low density; then, cell growth/confluency measurements were performed using Incucyte assays. Results are shown as % cell confluency. d Control and Foxm1 knockout E0771 cells were plated at low density, and cell confluency measured using Incucyte assays. Results are shown as % cell confluence. Figures (c) and (d) are derived from three biological replicates, with each replicate containing two technical replicates. e Tumor volumes in NOD-scid-gamma (NSG) immunodeficient mice orthotopically implanted with scrambled control (Ctrl) or Foxm1 knockout (sgRNA 1 and sgRNA 3) E0771 cells (n = 7 mice/group). f Left: Tumor volumes in C57BL6/J immunocompetent mice orthotopically implanted with scrambled control (Ctrl) or Foxm1 knockout (sgRNA 1 and sgRNA 3) E0771 cells (n = 8 mice/group). Right: Tumor weights in scrambled control (Ctrl) and knockout (sg1 and Sg3) groups. g Left: Schema (Created in BioRender. Nirzhor, S. (2025) https://BioRender.com/s8dko5w) for rechallenge experiments to test immune memory in C57BL6/J mice implanted with scrambled control (n = 5) and Foxm1 knockout E0771 cells (n = 5). Right: Mice in the Foxm1 knockout sg3 group (n = 4) that did not form tumors were rechallenged with low passage scrambled control (Ctrl) E0771 cells at the original tumor inoculation site at day 65. Tumor volume for all experiments was calculated using V = [l x (w)2]/2. Control groups in all graphs are represented by dark blue circles, while FOXM1 knockout sg1 and sg3 groups are represented by red square or triangle, respectively. Results are displayed as mean +/− SEM. p-values for panels (c–f) (left side) were calculated using non-linear regression with Gompertz growth for panel (c, d) or Malthusian growth for panel (e, f). p-values for the panel f (right side) were calculated using Welch’s two-tailed t test. Source data are provided as a Source Data file.
To confirm these results in vivo, we performed xenograft assays in NOD-scid-gamma (NSG) mice, which lack adaptive immunity. FOXM1 knockout did not significantly impact tumor growth compared to the scrambled control (Fig. 1e). However, in immunocompetent mice, the Foxm1 sg3 clone was unable to form tumors in most mice or resulted in significantly smaller tumors in a few, whereas the Foxm1 sg1 clone produced notably smaller tumors in the majority of mice (Fig. 1f). To validate these findings, we used shRNA to silence Foxm1 in the 4T1 mouse tumor model. Consistent with the results in E0771 cells, Foxm1 knockdown in 4T1 cells led to a significant reduction in tumor growth in immunocompetent mice (Supplementary Fig. 1c). These results strongly suggest that FOXM1 plays a crucial role in promoting tumor growth in an immunocompetent microenvironment.
Immune memory is an important component of long-term tumor suppression and prevention of recurrence. Since knockout of Foxm1 led to tumor regression and long-term inhibition of tumor formation in immunocompetent mice, we hypothesized that these tumors were being suppressed through immune memory. To test this, we rechallenged mice in the knockout group that did not form tumors with wild-type cells. Mice rechallenged with wild-type cells again formed no tumors (Fig. 1g). These results indicate that Foxm1 depletion can induce immune memory.
FOXM1 shapes the tumor immune landscape
To more comprehensively understand how FOXM1 influences the tumor immune microenvironment, we conducted flow cytometry analysis on Foxm1 wild-type and knockout tumors from C57BL6/J mice (Supplementary Fig. 1d and Supplementary Fig. 2a, b). A significant surge in CD45+ immune cell infiltration occurred within the knockout group (Fig. 2a), and overall populations of T cells and CD8 + T cells increased (Fig. 2b, c). However, we detected no substantial differences in PD-1 expression between the control and knockout groups (Supplementary Fig. 3a). Regarding immunosuppressive cell populations, CD4 + FOXP3 + Treg cells were reduced in the knockout group (Fig. 2d). This reduction was primarily due to a decrease in CD4 + cells, as the proportion of FOXP3+ cells relative to CD4 + cells remained unchanged in knockout compared to control groups (Supplementary Fig. 3b, c). Given that over 70% of CD4 + cells are FOXP3+ cells, the CD8 + /CD4 + ratio and therefore CD8 + /FOXP3 + ratio is significantly higher in the knockout groups (Supplementary Fig. 3d). This is clinically relevant as cancer patients, including those with invasive breast cancer, who have a higher CD8 + /FOXP3 + ratio typically exhibit better prognosis and treatment response19.

a–d Representative results from flow cytometry analyses of infiltrated immune cells in tumors from scrambled control (Ctrl; n = 3) and Foxm1 knockout (Foxm1 KO; n = 3) groups. Data are presented as mean values +/- SEM. Representative gates for each group are shown directly below the graphs. e Left: Representative results from flow cytometry analyses of infiltrated myeloid/antigen presenting cell populations in scrambled control (Ctrl; n = 4) and Foxm1 KO (n = 4) tumors. Data are presented as mean values +/− SEM. % Total on the y axis represents % of total living single cells. Right: Representative gates for each myeloid population of cells. p-values for panels (a–d) were calculated using two-sided Welch’s t tests, while p- values for panel (e) was calculated using multiple t test with Benjamini, Krieger, and Yekutieli correction. Control and Foxm1 knockout groups in all bar graphs are represented by dark blue circles or red squares, respectively. Source data are provided as a Source Data file.
Because myeloid cells play a crucial role in initiating innate and adaptive immune responses, we explored whether FOXM1 knockout affected their recruitment and/or activation. Analyses of antigen-presenting cell markers, such as dendritic cells (DCs) and macrophages (CD11b, CD11c, and MHC-II), revealed a considerable increase in proportions of myeloid cells within tumors in the knockout group, including both CD11b + and CD11c + cells (Supplementary Fig. 2b and Fig. 2e). Moreover, we observed an increase in MHC-II-positive antigen-presenting cells (CD11b + CD11c + MHC-II +) in tumors in the knockout group. Consistent with these findings, 4T1 tumors showed increased infiltration of CD3+ cells on immunohistochemistry (Supplementary Fig. 3e, f). Collectively, these findings indicate that Foxm1 depletion leads to enhanced immune cell infiltration.
Altered immune cell composition and increased T/NK cell response pathways in a FOXM1-depleted tumor microenvironment
To gain a comprehensive understanding of the alterations in the tumor microenvironment following FOXM1 depletion, we employed single-cell RNA sequencing (scRNA-seq) on pooled tumor samples separately from control and Foxm1 knockout groups. The total number of cells sequenced from each pooled sample was 6732 for Control, 4673 for Knockout sg1, and 5147 for Knockout sg3. Careful annotation and clustering were performed based on copy number variations and established population markers to distinguish between different types of cells in the stroma and to differentiate cancer cells from normal epithelial cells (Supplementary Figs. 4, 5a, b, 6a, b, Supplementary Data File 1). We identified 30 distinct clusters of cells, including normal epithelial cells, cancer-associated fibroblasts, blood cells, various immune cell populations, and vasculature (Fig. 3a). The scRNA-seq data revealed significant changes in immune cell populations between the two groups, including T cells, natural killer (NK) cells, B cells, plasmablasts, myeloid progenitors, neutrophils, monocytes, dendritic cells (DCs), and macrophages (Fig. 3a). Knockout tumors exhibited a striking increase in the total number of immune cells, particularly in T and NK cell clusters (Fig. 3b, c). These results were further validated by immunofluorescence staining of CD8 and NK cells in tumors from control and Foxm1 knockout groups (Supplementary Fig. 6c).

a UMAP representation of 16,552 single cells from scrambled control (6732 cells) and Foxm1 KO (sg1: 4673 and sg3: 5147 cells) samples (n = 5) showing proportions of cell types in the tumor microenvironment. Tumor samples were pooled separately from control and Foxm1 knockout groups for single-cell sequencing. Cell type compositions are shown as cluster numbers (left), samples (middle), and cell type annotation assignments based on marker expression (also see Supp. Data File 1) (right). b Stacked bar plots showing the number of cells in each cell population of scrambled control (Ctrl) and Foxm1 KO (sg1 and sg3) tumors. c Pie chart showing the proportions of different cell populations in scrambled control (Ctrl) and Foxm1 KO (sg1 and sg3) tumors. d UMAP representation of 4483 T and NK cells from control (812 cells) and Foxm1 KO (sg1: 1560 and sg3: 2111) samples (n = 5) showing subpopulations of T and NK cells based on established markers (also see Supp. Data File 2). e T and NK cell subpopulations in scrambled control (Ctrl) and Foxm1 KO (sg1 and sg3) tumors. f Top enriched pathways (GSEA) in T and NK cells from control (Ctrl) and Foxm1 KO (sg1 and sg3) tumors. Normalized enrichment scores (NES) are shown below. g Inflammatory and activation markers in T and NK cells in (ctrl) and Foxm1 KO (sg1 and sg3) tumors. The percent of positive cells is indicated by the size of the dot, and the scaled expression level is indicated by the color. Source data are provided as a Source Data file.
We then examined the subtypes and activation states of T and NK cells in knockout tumors. De novo clustering identified 14 subclusters of T and NK cells assigned to 11 populations, including naïve T, exhausted CD8 + T, exhausted CD4 + T, proliferating T cells, effector CD4 + T cells, activated T cells, FOXP3 Treg, effector memory T cells, effector CD4 + T cells, NK cells, and doublets (Fig. 3d and Supplementary Data File 2). There was an overall increase in most subpopulations of T and NK cells in tumors from Foxm1 knockout groups (Fig. 3e). To determine the pathways activated in different immune cells, we performed gene set enrichment analysis (GSEA). Analyses revealed enhanced inflammatory signaling in multiple cell types, indicating potential recruitment or activation of immune cells in Foxm1 knockout tumors. Pathways involved in T and NK cell responses, such as T cell and NK cell-mediated cytotoxicity, were significantly upregulated in Foxm1 knockout tumors, consistent with their increased T/NK cell infiltration (Fig. 3f).
To assess the activation or proliferation state of immune cells, including T and NK cells, we analyzed the expression of T and NK cell markers in our scRNA-seq data. Effector molecules, such as interferon-γ (IFN-γ) and GZMB, were higher in T and NK cells within Foxm1 knockout tumors compared to controls (Fig. 3g). These observations collectively underscore the importance of FOXM1 in the tumor immune microenvironment, potentially influencing immune cell recruitment, activation, and anti-tumor response.
FOXM1 knockout leads to increased cell-cell communication and inflammatory signaling
To gain deeper insights into the communication dynamics between cancer cells and immune cells in Foxm1 knockout tumors, we conducted Cellchat analysis20. Foxm1 knockout tumors exhibited a distinctive cell communication pattern compared to those in controls (Fig. 4a and Supplementary Fig. 7a). While tumors in the control group showed prominent communication between cancer cells and the vasculature, Foxm1 knockout tumors displayed an increased communication between cancer and immune cells, particularly from T and NK cells to cancer cells or other immune cells (Fig. 4a and Supplementary Fig. 7a). In particular, interaction between molecules involved in cell death signaling, antigen presentation, and NK/T cell activation were increased. These included FAS, TRAIL, MHCI, and NKG2D, which activates CD8 + T and NK cells upon interaction with stress ligands (NKG2DL) expressed on cancer cells (Fig. 4b).

a Cellchat network showing the probability and patterns of communications between different cell populations in (ctrl) and Foxm1 KO (sg1 and sg3) tumors. The cellchat network was constructed using CellPhoneDB. Line thicknesses reflect the number of ligand-receptor interactions between different cell populations; circle sizes indicate which cells are contributing to cell-to-cell communication. b Interactions between ligands and receptors expressed on tumor and immune cells in scrambled (ctrl) and Foxm1 KO (sg1 and sg3) tumors. Line thicknesses indicate the intensity of interactions. Highly enriched pathways, including cell death signaling (FAS, TRAIL), antigen presentation (MHC-I), and NK cell activation (NKG2DL) in Foxm1 KO compared to control are shown. c Immunofluorescent staining of ULBP1 in E0771 Ctrl and Foxm1 KO xenograft sections. ULBP1:green; DAPI:blue. Scale bar = 20 μm. White arrows indicate ULBP1 localization. Images are representative of three independent biological replicates per group. d qRT-PCR analysis of ULBP1 in E0771 control and Foxm1 KO cells (n = 3 biological replicates /group). e Schematic (Created in BioRender. Nirzhor, S. (2025) https://BioRender.com/uws6f4d) showing injection schedule of antibodies to deplete CD8 or NK1.1 or block NKG2D. f Left: Tumor growth in Rag1 -/- mice and CD8 + T cell-depleted and IgG-control treated mice transplanted with Foxm1 KO (sg3) E0771 cells. Rag -/- (n = 3); IgG Isotype control and CD8 T depleted (n = 4). Right: Tumor growth in IgG isotype control, NK cell-depleted, and NKG2D cell-depleted mice transplanted with Foxm1 KO (sg3) E0771 cells(n = 4/group). g Differentially secreted factors in media collected from scrambled control (Ctrl) and Foxm1 knockout E0771 cells. Left: Red box indicates altered cytokines (CCL2/MCP-1 and CCL5/RANTES) in Foxm1 KO compared to Ctrl. Right: Quantification of bands shown in red box, normalized to negative control. Results are mean +/− SEM of three pooled biological replicates. p-value for panel (d and f) was calculated using two-sided Welch’s t test or non-linear regression with Malthusian growth, while for panel (g), multiple t test with Benjamini, Krieger, and Yekutieli correction was used. Control and Foxm1 KO groups in all bar graphs are represented by dark blue circles or red squares, respectively. Source data are provided as a Source Data file.
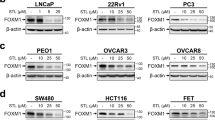
Single-cell analyses provided additional evidence for increased levels of these interacting molecules on corresponding cells (Supplementary Fig. 7b). We observed a significant increase in expression of the stress ligand ULBP1 in both mouse and human FOXM1 knockout tumors compared to controls (Fig. 4c, d and Supplementary Fig. 7c–f). ULBP1 belongs to the NKG2D ligand (NKG2DL) family, which includes MICA/B and ULBP1-6 in humans, and MULT1/ULBP1, HL60, and RAE1 in mice. To assess whether other NKG2DL family members may also be affected in FOXM1-depleted cells, we conducted qPCR analyses. In addition to ULBP1, FOXM1 depletion consistently induced expression of ULBP3, 4, and 5 in multiple cancer cell lines (Supplementary Fig. 7c–f). However, expression of other NKG2D ligands, such as MICA/B, was significantly altered in some but not all FOXM1-depleted cell lines (Supplementary Fig. 7c–f).
FOXM1 blocks activation of T and NK cells
Our results suggest that FOXM1 may support tumor growth by blocking the recruitment as well as activation of CD8 + T and NK cells in the tumor microenvironment. To further validate these data, we conducted tumor xenograft assays using sg3 Foxm1 knockout cells in several immune depletion models (Fig. 4e). Given the pivotal role of T cells, particularly CD8 + T cells, in the sustained control of cancer and their vital significance as prime targets in immunotherapy, we first asked whether increased CD8 + T cells in Foxm1 knockout tumors might be responsible for inhibiting tumor growth. We tested tumor growth of these cells in a Rag1 knockout mouse model, which lacks functional T and B cells, and in C57BL6/J mice treated with either IgG isotype control or CD8α antibody to deplete CD8 + T cells (Fig. 4e). Similar to the NSG mice, all mice injected with Foxm1 sg3 knockout cells exhibited tumor growth in both Rag1 knockout and CD8-depleted mice, displaying a rapid exponential trajectory that persisted until the experimental endpoint, unlike wild-type C57BL6/J mice (Fig. 4f, left). Tumor growth rate resembled that of the control cells, with no distinction between Rag1 knockout and CD8-depleted mice (Fig. 4f, left).
In addition to CD8 + T cells, our single-cell results indicated increased numbers of NK cells in Foxm1 knockout tumors, suggesting an equally important role for NK cells in mediating tumor suppression in these tumors. To test that concept, we depleted NK cells using NK1.1 antibody in C57BL6/J mice implanted with Foxm1 KO cells (Fig. 4e). Similar to CD8 + T cells, depletion of NK cells also led to marked tumor growth in the Foxm1 knockout group, while IgG-treated mice had no tumor growth (Fig. 4f, right).
Our single-cell analyses identified increased expression and cell-cell communication of stress ligands, suggesting a critical role for NKG2D in activating NK and T cells in Foxm1 knockout tumors. To directly test that idea, we blocked NKG2D signaling using an antibody against NKG2D in C57BL6/J mice transplanted with Foxm1 knockout cancer cells (Fig. 4e). Interestingly, blockade of NKG2D promoted tumor growth of Foxm1 knockout cells comparable to CD8 + T- and NK cell-depleted groups, indicating a direct link between FOXM1 and tumor-specific expression of stress ligands and their interaction with the NKG2D receptor on NK and T cells (Fig. 4f, right).
FOXM1 inhibits cGas-STING and NF-κb signaling to block T/NK-cell recruitment and activation
Our results indicated that loss of FOXM1 in cancer cells leads to increased recruitment and activation of immune cells in the tumor microenvironment. To understand the mechanism by which FOXM1 may regulate this process, we first used a cytokine array to test cell culture supernatant from FOXM1 knockout human and mouse breast cancer cells (Supplementary Fig. 7j, k). RANTES (CCL5) and MCP-1 (CCL2) were the two chemokines that showed significantly increased secretion in mouse and human FOXM1 knockout cells compared to their wild-type counterparts (Fig. 4g and Supplementary Fig. 7g). Tumor-intrinsic CCL5 promotes T/NK cell trafficking to tumor sites by interacting with receptor CCR5 expressed on T and NK cells21. Our single-cell seq results further confirmed the increase in CCL5 levels and the predominance of CCR5+ cells in Foxm1 knockout tumors (Supplementary Fig. 7h, i). To further validate the increase in immunogenic signaling in FOXM1 knockout cells, we determined the levels of cytokines (TNFA, IFNB, CXCL10), antigen presentation molecules (MHC-I, β2M), and the immunoproteasome in our control and FOXM1 knockout cells. Many were significantly upregulated in both mouse and human cells (Supplementary Fig. 8a–c).
To gain a deeper understanding of the mechanisms by which tumor-intrinsic FOXM1 may block the recruitment and activation of immune cells in the tumor microenvironment, we performed pathway analysis in cancer cells using scRNA-seq. Cytosolic DNA sensing cGas-STING and its associated signaling, as well as TNFα/NF-kB pathways, were consistently highly upregulated in FOXM1 knockout cancer cells (Fig. 5a). Based on our results, which show increased immune cell infiltration and activation, the activation of the cGAS/STING pathway in FOXM1 knockout cells is logical, given that cGAS-STING and NF-κB signaling are crucial for eliciting an effective anti-tumor immune response17,22. Moreover, the cGAS-STING pathway activates both type I and type II interferon signaling proteins, as well as NF-κB p65, which in turn induces expression of CCL2 and CCL517,18,23,24. There is extensive crosstalk between these pathways, enhancing their mutual activation. Consistent with that finding, Western blot analysis showed markedly increased activation of cGas-STING and NF-κb signaling proteins in both mouse and human FOXM1 knockout/knockdown cancer cells from diverse lineages compared with controls (Fig. 5b and Supplementary Fig. 8d–f). In addition to protein, we observed STING mRNA expression was significantly induced in FOXM1 knockout cancer cells (Fig. 5c and Supplementary Fig. 8g, h). Further supporting this, single-cell analyses showed markedly increased RNA levels of STING and downstream signaling molecules in FOXM1 knockout tumor cells (Supplementary Fig. 8i).

a Heatmap showing the log-normalized expression of top genes from GSEA pathway enrichment analysis. Genes representing pathways with the highest enrichment in E0771 Foxm1 knockout tumors compared to scrambled control are shown. b Western blots of cGas-STING pathway proteins in scrambled (Ctrl) and FOXM1 knockout E0771 (left) and MDA-MB-231 cells (right). β-actin or GAPDH was used as a loading control. The arrow indicates the band for total STING. c, d qRT-PCR analysis of STING (c), and DNMT1 and UHRF1 mRNA (d) in MDA-MB-231 Ctrl and FOXM1 KO cells. e Western blot analysis of DNMT1 and UHRF1 in MDA-MB-231 and E0771 Ctrl and FOXM1 KO cells. f ChIP-qPCR in MDA-MB-231 cells showing FOXM1 enrichment on the DNMT1 promoter. g Western blot analysis of MDA-MB-231 and BT549 cells treated with DNMT1 inhibitor (GSK-3484862) using antibody against STING. h Western blot analysis of MDA-MB-231 transfected with scrambled (siCtrl) or siDNMT1 cells using antibodies against cGas-STING pathway proteins. i ChIP-qPCR in MDA-MB-231 cells showing DNMT1 enrichment on STING promoter. j ChIP-qPCR in MDA-MB-231 Ctrl and FOXM1 KO cells showing enrichment of H3K4me3 on STING promoter. k MethyLight qRT-PCR assay using primers spanning STING promoter in Ctrl and FOXM1 KO MDA-MB-231 cells. The bar graph shows the relative log2 of percentage STING promoter methylation in FOXM1 wild-type and FOXM1 knockout groups. l Western blot analyses in scrambled and Foxm1 KO E0771 cells using antibodies against the indicated UPR proteins. m qRT-PCR on Foxm1 KO cells transfected with scrambled or Sting siRNA using primers against MHC-I, IFNβ, and ULBP1. All bar graphs represent log2 of fold change in gene expression or fold enrichment on promoters. All qPCR data represent the mean +/− SEM of three biological replicates with two technical replicates per group. Western blots are representative of three independent experiments. p-values for panels (c, f, i–k) were calculated using a two-sided Welch’s t test. p-values for (d and m) were calculated using multiple t test with Benjamini, Krieger, and Yekutieli correction. Control and FOXM1 knockout groups in all bar graphs are represented by dark blue circles or red squares, respectively. Source data are provided as a Source Data file.
We next sought to further address the mechanism by which FOXM1 may regulate STING signaling. Increased expression of the STING transcript in FOXM1 knockout cancer cells suggested that FOXM1 may block the transcription of STING. Such a premise is supported by findings that transcription factors can nucleate the assembly of epigenetic repressor complexes to silence target gene promoters. For example, Myc recruits DNA methyltransferase DNMT1 on STING gene promoter25. Moreover, UHRF1, an E3 ubiquitin ligase protein, recruits DNMT1 to maintain DNA methylation status at specific gene promoters26. To test whether FOXM1 may be involved with assembling an epigenetic repressor complex on the STING promoter, we examined levels of DNMT1 and UHRF transcripts and proteins in FOXM1 knockout cells. FOXM1-depleted cancer cells showed that both were reduced, suggesting that FOXM1 transactivates their expression (Fig. 5d, e).
To address how DNMT1 is regulated by FOXM1, we asked whether FOXM1 may bind to the DNMT1 promoter and establish a permissive chromatin state. ChIP-qPCR showed enrichment of FOXM1 and H3K4me3 on the DNMT1 promoter (Fig. 5f and Supplementary Fig. 8j). To further confirm this, we performed ChIP-qPCR on control and FOXM1 knockout cells using the antibody against H3K4me3 and primers spanning the DNMT1 and UHRF1 promoter region. FOXM1 depletion resulted in a marked reduction in H3K4me3 enrichment on both DNMT1 and UHRF1 promoters, compared to control (Supplementary Fig. 8k,m). To substantiate our results, we performed a meta-analysis of ENCODE datasets for H3K4me3 ChIP-seq, focusing on the promoter regions of DNMT1 and UHRF1. This analysis confirmed significant H3K4me3 enrichment at the promoters of both DNMT1 and UHRF1 (Supplementary Fig. 8l,n). To determine whether DNMT1 could regulate expression of STING in FOXM1 knockout cancer cells, we first determined the levels of STING in cancer cells treated with a DNMT1 inhibitor (GSK-3484862); STING expression was significantly increased in those cells (Fig. 5g). In addition, downstream effectors of STING signaling pathway were highly induced in cancer cells depleted for DNMT1 (Fig. 5h). To understand how FOXM1-DNMT1 signaling may regulate STING expression, we conducted ChIP-qPCR assays using antibodies against DNMT1, FOXM1, and H3K4me3 and a set of primers spanning the promoter region of STING. We found robust enrichment of DNMT1 at the STING promoter in cancer cells with intact FOXM1 expression (Fig. 5i). Further supporting these data, H3K4Me3 at the STING promoter was notably enriched in cancer cells with depleted FOXM1 versus intact FOXM1 (Fig. 5j). However, we did not observe enrichment of FOXM1 on STING promoter (Supplementary Fig. 8o). To identify potential factors that may facilitate the recruitment of DNMT1 on STING promoter, we utilized JASPAR and analyzed the STING promoter region where DNMT1 binds. Meta-analysis showed potential binding sites for multiple transcription factors on the STING promoter. Notably, our meta-analysis of the ENCODE dataset revealed significant enrichment of MYC at the STING promoter, which is particularly relevant given MYC’s known role in recruiting DNMT1 to gene promoters (Supplementary Fig. 8p).
Given DNMT1’s role in gene silencing via DNA methylation, we investigated the methylation status of the STING promoter in FOXM1 wild-type cancer cells. We conducted a methyLite assay on bisulfite-treated DNA using primers specific to the STING promoter. FOXM1 knockout cells showed significantly decreased methylation levels compared to wild-type cells (Fig. 5k). To determine whether this regulation was specific to STING or applicable to other DNMT1 target genes, we analyzed the expression of genes reported to be regulated by DNMT1 in multiple cancer cell lines. Notably, while some DNMT1 targets, including immunity-related genes such as ICAM1, RAPGEF3, were induced in FOXM1 knockout tumor cells, others were not, suggesting that FOXM1 selectively establishes epigenetic repressor complexes at certain gene promoters (Supplementary Fig. 8q, r). To corroborate these findings, we conducted a meta-analysis using ENCODE datasets for DNMT1 ChIP-seq on liver cancer and leukemia cell lines. The meta-analysis revealed 31,000 and 15,000 DNMT1 binding peaks in ~ 8000 genes across the genome in HepG2 and K562 cells, respectively (Supplementary Fig. 9a, b). Out of those 8000 genes, only 2000 genes exhibited enrichment in both cancers, indicating a specificity of DNMT1 target genes in different cancers (Supplementary Fig. 9a, b). Further analysis revealed a significant DNMT1 enrichment at the promoter regions of approximately 40–45% of the roughly 8000 genes examined across both cancer types (Supplementary Fig. 9c). Common genes showing DNMT1 enrichment included STING and several other immunity-related genes, which were induced in FOXM1 knockout cells (Supplementary Fig. 9d, 8l, m). Conversely, other reported tumor-associated genes did not show enrichment in the ENCODE dataset, suggesting the specificity of DNMT1 binding to certain but not all genes (Supplementary Fig. 9e). Collectively, these findings suggest that FOXM1 may help establish a chromatin state conducive to repressing STING expression in cancer cells through DNMT1-mediated DNA methylation. Another mechanism of STING activation is through the accumulation of cytosolic DNA. Since FOXM1 regulates several DNA repair pathways, we surmised that FOXM1 depletion may induce DNA damage and result in the accumulation of cytosolic DNA. In support of this premise, immunofluorescence analyses showed the presence of micronuclei, indicating the formation of acentric chromosomal fragments in response to DNA damage in FOXM1 knockout cancer cells (Supplementary Fig. 10a).
Our results showed that FOXM1 blocks NKG2D-dependent activation of NK/T cells by inhibiting stress ligands in cancer cells. Expression of stress ligands is induced by external and internal stimuli, including DNA damage, suggesting that the FOXM1-STING signaling axis may regulate expression of stress ligands in general and ULBP1 in particular in cancer cells. To further elucidate how FOXM1-STING may regulate ULBP1 expression, we determined the status of unfolded protein response (UPR) proteins in FOXM1 KO cancer cells, since endoplasmic reticulum stress induces expression of stress ligands. Western blot analyses showed increased expression of several UPR proteins in FOXM1 KO cells (Fig. 5l and Supplementary Fig. 10b). Of all the UPR proteins, CHOP is of particular importance, as it can directly transactivate ULBP127. To further substantiate that the induction of stress ligands and other immunogenic molecules in FOXM1 knockout cancer cells depends on STING activation, we conducted rescue experiments. Silencing STING in FOXM1 knockout cells resulted in decreased activation of cGAS-STING pathway genes, along with reduced levels of ULBP1 and other immunogenic molecules (Fig. 5m and Supplementary Fig. 10c). Furthermore, STING depletion resulted in strong suppression of CHOP levels in both mouse and human cells, indicating crosstalk among FOXM1, DNA sensing, UPR, and stress ligand pathways in cancer cells (Supplementary Fig. 10d, e).
FOXM1 expression is correlated with immunosuppressive signatures in cancer patients
To establish the clinical relevance of our findings, we measured infiltration of immune cells in tumors from breast cancer patients with high and low FOXM1 expression. Our analysis revealed an increased infiltration of CD3 + CD8 + and CD56 + immune cells in tumors with low FOXM1 expression, in contrast to those with high FOXM1 expression (Fig. 6a). Importantly, like CD8+ and NK cells, tumors from breast cancer patients with low FOXM1 showed higher expression of ULBP1, while those with high FOXM1 showed lower expression of ULBP1 (Fig. 6a). To further substantiate these observations, we performed a correlation analysis between FOXM1 expression and immune signatures in cancer patients. Employing a meta-analysis approach utilizing TIMER2.0, our results demonstrated an inverse relationship emerged between FOXM1 levels and tumor-suppressive immune cells, encompassing CD8, NK cells (including activated NK cells), and central memory CD4 + T cells (Fig. 6b, c and Supplementary Fig. 11a, b). Concurrently, there was a positive correlation between FOXM1 expression and immunosuppressive cell types, such as Tregs and MDSC, within the tumor microenvironment (Fig. 6d). Notably, we also found negative correlations between FOXM1 and STING expression in samples from breast cancer and lung cancer patients (Fig. 7a and Supplementary Fig. 11c). Furthermore, FOXM1, DNMT1, and UHRF1 expression were significantly positively correlated in breast cancer patients (Fig. 7b, c).

a (top) Immunofluorescent staining of FOXM1, CD3 + CD8 + T cells, and CD56 + NK cells in tumor tissues from breast cancer patients based on levels of high versus low FOXM1 expression. Top images: FOXM1 indicated by red; CD56 indicated by green. Bottom images: CD3 is indicated by red; CD8 is green (bottom image). DAPI is stained as blue. The circle indicates a cluster of NK cells in a FOXM1 low patient. (bottom) ULBP1 staining in tissues from breast cancer patients based on levels of high versus low FOXM1 expression. Scale bar indicates 50μm. The experiment was performed with n = 3 patients for each group (FOXM1 high and FOXM1 low). Representative pictures from two patients per group are shown. The number shown on the top of the figure indicates de-identified breast cancer patients. b–d Spearman correlations of FOXM1 RNA levels with tumor purity and immune cell infiltration in breast cancer patients from the TIMER2.0 database. The populations of cells examined include cytotoxic cells (NK cells, CD8 + T cells) and immunosuppressive cells (regulatory T cells [Tregs] and myeloid-derived suppressive cells [MDSC]. p-values for panels b-d were calculated by the TIMER2.0 database using the Spearman correlation coefficient.

a–c Pearson correlation analyses between FOXM1 and various genes, including STING, DNMT1, and UHRF1, in breast cancer patients (n = 1085) from the GEPIA2 database. d–f Kaplan-Meier survival analysis showing associations between FOXM1, DNMT1, and STING (TMEM173) expression and overall survival (FOXM1 and DNMT1) or relapse-free survival (STING). (d) n = 942 patients for FOXM1 low and n = 937 patients for FOXM1 high; (e) n = 236 patients for STING low and n = 234 patients for STING high; (f) n = 139 patients for DNMT1 low and n = 75 patients for DNMT1 high;values for panels (a–c) were calculated by GEPIA2 using Pearson correlation coefficient and a two-tailed t distribution.
To further understand the significance of FOXM1, DNMT1, STING, and ULBP1 on patient survival, we calculated Kaplan-Meier survival curves in breast cancer patients for changes in overall survival or relapse-free survival based on high or low expression of these genes. Tumors with high FOXM1 or DNMT1 levels were associated with worse overall survival compared with those with low expression (Fig. 7d, e). Inversely, patients whose tumors had high STING (TMEM173) or ULBP1 levels showed better disease-free survival compared to low tumors (Fig. 7f and Supplementary Fig. 11d). To understand the relevance of these findings to clinical outcomes in cancer patients undergoing immunotherapy, we performed expression analyses of FOXM1 in patients who were and were not responsive to immunotherapy in clinical trials, using ClinicalOmicsDB. FOXM1 expression was higher in patients who did not respond to either anti-PD-L1(atezolizumab) or anti-PD-L1(pembrolizumab) immunotherapy compared to those who did respond (Supplementary Fig. 11e, f). Collectively, these observations illuminate a critical facet of FOXM1’s influence on the tumor immune landscape and responsiveness to standard immunotherapies by modulating DNA sensing, UPR, and stress ligand pathways (Fig. 8).

We propose that high levels of FOXM1 expression in cancer cells create an immunosuppressive microenvironment by promoting epigenetic silencing of the DNA-sensing protein STING, leading to reduced expression of stress ligands, MHC-1 class genes, and pro-inflammatory molecules. FOXM1 depletion leads to STING activation through several mechanisms. First, FOXM1 depletion leads to marked reductions in DNMT1 and UHRF1, constituting an epigenetic repressor complex targeted to the STING promoter. Second, FOXM1 depletion leads to an increase in micronuclei, likely through increased DNA damage or lapses in cell cycle, and consequently activation of the cytosolic DNA sensing cGas-STING-NFkB and unfolded protein response (UPR) pathways. Activation of STING-NFκB signaling induces expression of chemokines/cytokines such as CCL5, which in turn promotes recruitment of immune cells to the tumor. In addition, activation of STING-UPR stimulates expression of MHC1 and the stress ligand ULBP1, which facilitates interaction and activation of NKG2D-expressing CD8 + T and NK cells and subsequent killing of cancer cells. The model was created in BioRender. Nirzhor, S. (2025) https://BioRender.com/v49t420.
Discussion
The intricate interplay between tumor cells and the immune microenvironment helps determine the trajectory of cancer progression and therapeutic responses1,2. In this study, we present extensive evidence to elucidate the importance of tumor-intrinsic FOXM1 in shaping the immune landscape of tumors. Our findings provide insights into the oncogenic role of FOXM1 by revealing its multifaceted engagement in key epigenetically reprogramming events associated with immune modulation within the tumor microenvironment. Our studies demonstrate that FOXM1 in tumor cells suppresses infiltration, recruitment, and activity of myeloid cells, CD8 + T cells, and NK cells. Further mechanistic investigations show that FOXM1 epigenetically suppresses the DNA-sensing protein STING, leading to reduced expression of stress ligands that act through the cell surface receptor NKG2D to prime NK- and T cell-mediated cytotoxicity of cancer cells.
The tumor microenvironment is shaped by stressors such as DNA damage, cellular stress, and inflammation28,29. These stressors help prime the DNA sensing pathway by activating STING and other sensors to activate immune effectors through the induction of interferons and proinflammatory cytokines30,31,32,33,34,35,36. In our study, we have shown how FOXM1 epigenetically regulates the STING DNA sensing pathway. While a previous study showed that FOXM1 silences GATA-3 expression in the mammary gland and luminal breast cancers by binding to its promoter and recruiting the DNMT3B-Rb complex37,38,39, our findings indicate that FOXM1 does not repress GATA-3 in triple negative breast cancer cells. This suggests that FOXM1 modulates GATA-3 through a distinct mechanism in a cell-type-specific manner. Consistent with this, we demonstrate that FOXM1 does not directly bind to the STING promoter. Instead, we show that tumor-intrinsic FOXM1 epigenetically silences STING by activating transcription and recruiting a DNMT1-UHRF repressor complex to the STING promoter. Since UHRF binds to hemi-methylated DNA and interacts with DNMT1 and HDAC1, our study indicates that FOXM1 may play a key role in ensuring a robust silencing of STING1 by facilitating DNMT1-HDAC-dependent methylation of the second strand. Recruitment of the FOXM1-UHRF-DNMT1 epigenetic repressor complex to specific gene loci such as STING must be context-dependent, as not all DNMT1 target genes are silenced. The specific binding sequence or base pair modifications could be one plausible context for assembly of the FOXM1-dependent DNMT1-UHRF1 repressor complex on the STING promoter. For example, in addition to hemi-methylated CpG sites, UHRF binds to carboxylated or hybrid methylated/carboxylated CpG dyads40. Furthermore, DNMT1’s catalytic activity is strongly influenced by flanking sequences outside the core CpG site41. Therefore, it is possible that the STING gene promoter region possesses such base pair modifications/specific flanking sequences conducive for UHRF1-DNMT1-HDAC recruitment and activity. Future genome-wide studies aimed at identifying sequence arrangements may unveil deeper insights into sequence specificity at STING and other gene loci that are targeted by FOXM1-dependent epigenetic silencing.
FOXM1’s role in negatively regulating STING levels and consequently inhibiting IRF3 activation sheds light on how FOXM1 may impair the initiation of anti-tumor immune responses. One way that FOXM1-STING-IRF3 signaling could affect anti-tumor immunity is by transcriptionally regulating expression of stress ligands42,43,44. In addition to STING, FOXM1 may regulate the expression of a stress ligand via UPR. FOXM1 depletion results in the induction of UPR proteins, including CHOP, which transactivates ULBP127. This interaction posits that FOXM1 is a potential regulator of the UPR pathway, amplifying the intricate interplay between FOXM1, UPR, and stress ligand expression in modulating immune recognition and response. Recent studies showing UPR activation of the STING pathway and STING activation leading to upregulation of endoplasmic reticulum stress response genes45,46, suggest that the dual activation of UPR and STING, induced by FOXM1 depletion, could exert a concerted response where tumor cells, stressed by DNA damage and protein misfolding, release signals that attract immune cells to the tumor site. Furthermore, FOXM1’s influence on the NF-κB signaling pathway introduces another layer of complexity to its immune regulatory roles. NF-κB-mediated cytokine induction, exemplified by CCL5, contributes to immune cell recruitment. FOXM1 depletion enhances NF-κB signaling, potentially leading to augmented CCL5 expression and subsequent immune cell infiltration. This nexus, where FOXM1 intricately intertwines with NF-κB-mediated immune cell recruitment and stress ligand expression, unveils its significance as a master regulator of immune response dynamics.
Our findings suggest that FOXM1 might also exert a hitherto unexplored influence on immune memory. Cancer cells with intact DNA repair mechanisms might be more efficient at repairing DNA damage induced by the immune response itself, such as immune cell-mediated attack or inflammation. This enhanced ability to repair DNA might allow cancer cells to evade immune recognition and destruction, contributing to tumor progression and immune escape47. It is conceivable that cancer cells with proficient DNA repair, supported by FOXM1, could potentially impede the formation of an effective immune memory response. This could occur if cancer cells manage to survive immune attacks and escape immune surveillance. Consequently, the immune system might fail to generate a strong memory against these cancer cells, impairing the long-term effectiveness of immunotherapeutic approaches aimed at targeting those cells.
The precise reasons for impairment of STING expression and signaling in cancers remain elusive. However, the high activity of FOXM1 in proliferating cancer cells indicates that FOXM1-mediated epigenetic silencing of STING may provide a survival advantage to cancer cells. This advantage could extend beyond immune evasion, as FOXM1 supports various DNA repair events counteracting STING’s role in promoting DNA damage through modulation of reactive oxygen species homeostasis48. It is likely that FOXM1’s repression of STING could prevent activation of a DNA damage response that is unfavorable for rapidly dividing cancer cells. Thus, by silencing STING, FOXM1 may tip the balance towards cellular proliferation over apoptosis, promoting tumorigenesis and cancer cell survival. In addition, the STING pathway plays a role in the activation of autophagy, which can limit cancer growth49. Therefore, FOXM1’s multi-faceted role in facilitating DNA repair by blocking autophagy and silencing anti-tumor immunity signaling via STING could be pivotal in promoting tumorigenesis and cancer cell survival.
In conclusion, our study unveils an important role of FOXM1 in shaping the tumor immune microenvironment. The intricate interplay among FOXM1, STING, UPR, NF-kB signaling, and stress ligand expression introduces layers of complexity to the immune evasion strategies employed by tumor cells. Through its regulation of these pathways, FOXM1 appears to bridge the gap between DNA repair, cellular stress responses, and immune activation, presenting a future avenue for therapeutic exploration. In the broader context of cancer immunotherapy, the ability to sensitize tumors to immune recognition through stress ligand upregulation is of paramount importance. Strategies that exploit this facet of tumor-immune interaction, in conjunction with approaches targeting FOXM1 and/or immune checkpoint inhibitor/s, might hold the key to potent therapeutic interventions that unleash the full potential of the immune system against cancer. In particular, our findings suggest that targeting FOXM1 expression in cancer cells may facilitate the development of immune cell memory to prevent tumor recurrence. This approach thus holds promise as a therapeutic vaccine against cancer.
Methods
Ethics statement
This study complies with all relevant ethical regulations, including the approval from the UTHCSA IACUC committee. Mice were regularly monitored for signs of morbidity. Mice were euthanized using C02 inhalation either upon completion of the experiment, when tumors reached the maximum permitted size (<2 cm3), or when they experienced more than 20% body weight loss.
Breast cancer cell lines
MDA-MB-231, BT-549, BT-20, MDA-MB-468, 143B, 4T1, and E0771 cells were purchased from ATCC and maintained at 37 °C in ATCC recommended growth medium in a humidified incubator containing 5% CO2. Cell lines were authenticated using STR analysis and tested for mycoplasma using the Universal Mycoplasma Detection Kit (ATCC # 30-1013 K). MDA-MB-231-pCW-Cas9 control and FOXM1 knockout cells were cultured in DMEM media containing 10% tetracycline-free FBS (Sigma) and 1% antibiotic/antimycotic (Gibco). E0771-pCW-Cas9-expressing control and FOXM1 knockout cells were cultured in RPMI media containing 10% tetracycline-free FBS and 1% antibiotic/antimycotic.
Generation of Cas9-inducible knockout cells
pCW-Cas9 and lenti-sgRNA-Blast vectors were purchased from addgene. pCW-Cas9 vector (addgene, Plasmid # 50661) encodes a doxycycline inducible Cas9, while lentisgRNA Blast (addgene, Plasmid # 104993) encodes a vector that can constitutively express an sgRNA of choice. Antibiotic kill curves were determined for both vectors (puromycin for pCW-Cas9 and blasticidin HCL for lentisgRNA-Blast) before viral transduction. E0771 cells were transduced with lentivirus encoding pCW-Cas9 and then selected with puromycin to generate cells with stable expression of Cas9. Stable expression of Cas9 was verified by Western blots after induction with doxycycline and subsequently maintained in media without tetracycline.
Scrambled sgRNA and 2 different sgRNAs against human or mouse FOXM1 were cloned into lenti-sgRNA-Blast plasmids, and successful cloning was verified using Sanger sequencing. Sequences for scrambled and FOXM1 sgRNAs are listed in Supplementary Table 1. Stable Cas9- expressing cells were transduced with lentivirus containing scrambled or FOXM1 sgRNAs. The stable cells were plated at low density in 96-well plates to select for single clones. The clones were maintained in media containing doxycycline and blasticidin for a week until screening for FOXM1 knockout using Western blots. Once generated, all knockout cells were cultured in tetracycline-free media.
Incucyte assays
MDA-MB-231 and E0771 control and FOXM1 knockout cells were plated at low density in 96- well plates. Cells were incubated in Incucyte (Sartorius) and monitored for cell confluency using Incucyte Zoom 2016B software. The results were visualized using GraphPad Prism software. p-values were calculated using non-linear regression analysis with Gompertz growth.
In vivo tumor xenograft studies
All the in vivo experiments were done in female mice (no sex or gender analysis was performed) because breast cancer predominantly occurs in females (99%). All mice were housed in ventilated cages with 5 mice per cage. The animal housing facility was maintained at 68–72 °F with humidity at 30% to 70% and 12 hr:12 hr light to dark cycles. 4–6-week-old female C57BL6/J, Balb/c, and NOD-scid-gamma (NSG) mice were obtained from Jackson Laboratories. On the day of injection, sub-confluent E0771 control and/or knockout cells were harvested and mixed with growth factor-reduced Matrigel (Corning, Cat # 354248). Approximately 250,000 cells were orthotopically injected into the fourth mammary gland of 5- to 6-week-old female C57BL6/J or NSG mice. 8– to 10-week-old female Rag1 knockout mice were obtained from Dr. Nu Zhang. Orthotopic xenograft assays were performed by injecting cells into the 4th mammary gland using 250,000 Foxm1 knockout cells. For the 4T1 xenograft model in Balb/c mice, approximately 100,000 4T1 cells were injected into the mammary gland of 4–6 week old female mice. Once tumors formed, tumors were measured every few days using a digital caliper. The volume of the tumor was calculated using the formula- V = ½ (Length × Width2).
For immune depletion and blockade studies, IgG Isotype control (Cat # BE0089) or depletion antibodies against CD8 (Cat # BE0004-1, Clone 53-6.7), NK1.1(Cat # BE0036, Clone PK136), or a blocking antibody against NKG2D (BE0111, Clone HMG2D) were purchased from BioXCell. C57BL6/J mice were injected with 200 µg per mouse of IgG or depletion antibodies 1 day before injection of E0771 Foxm1 knockout sg3 cells. Following inoculation of cells, mice were treated twice a week with the respective antibodies for the remainder of the experiment. Once tumors formed, tumors were measured every few days using a digital caliper. Volumes of tumors were calculated using the formula- V = ½ (Length × Width2). For rechallenge experiments, mice were injected with control and knockout cells then monitored for tumor growth. Mice in the knockout group that did not form tumors were rechallenged with low passage control cells at day 65 after initial inoculation. Tumor growth was monitored. All experiments were approved by IACUC at UT Health San Antonio and performed according to the protocol, following ethical guidelines. The results were visualized using GraphPad Prism. The p-value was calculated using non-linear regression analysis with Malthusian growth.
RNA and protein analyses
Human and mouse scrambled control and FOXM1 knockout samples were collected and lysed with qiazol (Qiagen, Cat#79306) for RNA and RIPA buffer containing halt protease and phosphatase inhibitor for protein. RNA samples were isolated using RNeasy kits (Qiagen, Cat#74004) and then reverse transcribed into cDNA using iscript rt-pcr kits (Bio-Rad, Cat#1708890). qRT-PCR was performed on samples using iTaq sybr green supermix (Bio-Rad, Cat#1725124) or PowerUp SYBR Green Mastermix (Applied Biosystems, Cat # A25776), using primers of interest. Protein samples were quantified using Bradford assays and then run on 10% or 4–20% gradient SDS-PAGE protein gels (Bio-Rad, Cat#4561094). Western blots were performed for proteins involved in cGAS-STING and other pathways. β-actin or GAPDH were used as a loading control. SiRNAs against FOXM1 or DNMT1 were obtained from Sigma. For FOXM1 or DNMT1 knockdown experiments, wild-type cells were plated then transfected with either FOXM1 or DNMT1 siRNA for 72 h before harvesting for RNA and protein. For rescue experiments, E0771 FOXM1 knockout cells were plated then transfected with siRNA against STING. After 72 hrs, cells were harvested for RNA and protein. After validation of STING knockdown, qRT-PCR was performed for target genes of interest. Fold change was calculated using the ΔΔCt method, after normalization to GAPDH. Log2 of fold change was calculated and used for all statistical tests and visualization of the data. All primers, siRNAs, and antibodies used in the study are listed in Supplementary Tables 2 and 3.
Tumor processing for flow cytometry
Mice were euthanized using CO2 inhalation and cervical dislocation. Tumors and spleen were harvested and collected in media containing 2% FBS. Tumors were weighed, dissected into small pieces, and pieces with similar weights were taken for flow cytometry. Tumors were cut into small pieces and then digested with RPMI media containing 2% FBS, 1 mg/ml Collagenase IV(Worthington, Cat #LS004188), and 20 µg/ml DNAse I(Sigma, Cat #4536282001) at 37 degrees for 1 hr with shaking. Samples were mashed on top of a 70 µM strainer in RPMI media and then filtered into a 50 ml tube. RBC lysis was performed before cells were counted, and 1 million cells were taken per panel for each sample. Cells were centrifuged and then washed once more before incubation with CD16/32 Fc block (1:200, BD Biosciences, Cat #553142) and Zombie Violet fixable viability dye (Biolegend, Cat # 423114, 1:500) for 30 min at 4 °C. Cells were washed then stained with surface antibodies for 30 min, based on the panel. The samples were washed once before fixation and staining for intracellular proteins. Once staining was finished, the cells were fixed with 2% PFA at 37 °C for 10 min before washing with FACS buffer and storage at 4 °C. Spleen samples were used as single-color antibody controls. The data were acquired on an LSR II Fortessa X-20 cytometer. The panels used for the study are as follows: Myeloid panels (CD45-APC-Cy7, CD11b-PE, CD11c-APC, and MHC-II-BV711), CD4 T cell panels (CD45-APC-Cy7, CD4-PE, FOXP3- FITC), and CD8 T cell panels (CD45-APC-Cy7, CD3-APC, CD8-V500, PD-1-PE). Compensation was performed using spleen samples stained with single-color anti-CD4 antibodies conjugated to various fluorophores. Gating strategy used in the study are listed in Supplementary Figs. 1d, 2a, b. Flow cytometry antibodies used in the study are listed in Supplementary Table 3. Compensation and data analyses were performed on Flowjo v10 software.
Immunofluorescence and immunohistochemistry staining for immune infiltration
For immunofluorescence analyses of mouse tumors, samples were harvested and frozen on dry ice then stored at −80 °C for future use. The frozen tumors were thawed and embedded in OCT solution before processing them into 5 µm sections. The sections were stored at −80 °C until further use.
Frozen de-identified breast cancer patient slides of samples from breast cancer patients were obtained from Mays Cancer Center. Both mouse and human slides were stained using the same protocol. Briefly, sections were thawed and incubated with acetone/methanol buffer (60:40 mixture), followed by washes with PBS. The samples were blocked for 15 minutes at room temperature with TBST containing 2% goat serum and 0.25% BSA, and then incubated overnight with the primary antibody. Slides were incubated with either FOXM1(1:150, Cell Signaling Technologies, Cat #5436) and CD56(1:100, Bio-Rad, Cat #MCA2693GA) or CD3(1:100, Bio-Rad), CD8 (1:150, Biolegend), and ULBP1 antibodies (1:100, Booster Biological Technologies, Cat # A04323-1). Slides were washed with 0.25% BSA in TBST several times and then incubated with a combination of secondary antibodies (Invitrogen, anti-Rabbit Alexa Fluor 594, or anti-mouse Alexa Fluor 488, 1:500). Slides were washed and mounted with ProLong™ Gold Antifade Mountant with DNA Stain DAPI (Thermo Fisher Scientific, Cat # P36935) before imaging on Nikon Eclipse Ni fluorescent microscope. Initially, the patient tumors were categorized as FOXM1 high or FOXM1 low based on the intensity of staining or positivity. Subsequently, we compared the number of infiltrating CD3 + CD8 + T cells, CD56 + NK cells, as well as ULBP1 staining between tumors from breast cancer patients with high and low FOXM1 expression.
For immunohistochemical staining of CD3, 4T1 controls and Foxm1 knockdown mouse tumor samples were harvested and then fixed in 10% formalin overnight. The samples were removed from formalin and then kept in 70% ethanol until paraffin embedding and processing into 5 µm sections. Staining was performed using ImmPRESS Goat anti-Rat IgG kits (Vector Labs, Cat # MP-444). Sections were deparaffinized and stained using standard protocols, including antigen retrieval, endogenous peroxidase inactivation using hydrogen peroxide, blocking with normal goat serum, and staining with CD3(1:250, Cat# MCA1477, Bio-Rad). Sections were then incubated with ImmPRESS Polymer Reagent containing an HRP-conjugated secondary antibody before washing and developing with DAB substrate. A total of 5 images were taken per sample, and total CD3-positive cells were counted. The investigators were blinded to the group during analysis.
Cytokine arrays
E0771 and MDA-MB-231 cells were plated in 6-well plates using complete tetracycline-free media. After 72 h, the media was collected and stored at −80 °C for future analysis. Human C5 arrays (Cat # AAH-CYT-5-4) and mouse C3 arrays (Cat # AAM-CYT-3-4) were obtained from Ray Biotech. The membranes were blocked according to the manufacturer’s protocol. Following this, the media from 2 different biological replicates (either control or knockout samples) was combined and incubated overnight at 4 °C with the respective membranes. The next day, the membranes were washed, incubated with primary and secondary antibodies, then developed using x-ray film. The film was scanned and the signal quantified using ImageJ software. The signal was normalized, and the fold change was calculated using the manufacturer-provided sheet in Excel software. The figures were created using GraphPad Prism software. Cytokine array plate layout for Mouse and Human samples are shown in Supplementary Fig. 7j, k.
Micronuclei analysis using immunofluorescence
Cells were plated at high density in 35 mm glass bottom dishes wiath 14 mm micro-wells (Cellvis, Cat # D35141.5 N). After 48 h, the media was removed, and cells were fixed with 4% paraformaldehyde for 15 min. Then cells were washed twice with PBS. The PBS was removed, and 10 µl of DAPI solution containing mounting media was added before coverslipping. Cells were incubated in DAPI for 10 min before imaging using an Olympus FV3000 confocal microscope. We imaged 9-10 fields containing 500–1000 cells at 20x magnification. Micronuclei were counted and normalized to the total number of nuclei present in each image and graphed as the number of micronuclei per 1000 cells. The images and box and whisker plots are derived from three independent experiments. The investigators were blinded to the group during analysis.
Chromatin immunoprecipitation (ChIP)-qPCR and ENCODE ChIP-seq analysis
Chromatin immunoprecipitation was performed using SimpleChIP Plus Sonication Chromatin IP kits (Cell Signaling Technologies, Cat # 56383). Sub-confluent MDA-MB-231 control and FOXM1 knockout cells were grown and then harvested following the manufacturer’s protocol. Chromatin immunoprecipitation was performed using DNMT1, FOXM1, H3K4me3, and IgG antibodies. Following ChIP pulldown and washing, samples were reverse-crosslinked at 65 degrees for 2 h in the presence of proteinase K, followed by column-based purification and elution of DNA. The eluted DNA was analyzed by qRT-PCR using primers targeting the STING, UHRF1, and DNMT1 promoters. The fold change over IgG was calculated for each sample. Log2 of fold change values were calculated and subsequently used for statistical analysis and visualization of data. Statistical significance was determined using an unpaired two-way Welch’s t test.
For the FOXM1, H3K4me3, and MYC ChIP-seq analysis, ChIP-seq data of FOXM1, H3K4me3, and MYC from MCF7 cell line was downloaded from ENCODE website (https://www.encodeproject.org/). For the DNMT1 ChIP-seq analysis, ChIP-seq data of DNMT1 from HepG2 and K562 cell lines were downloaded from the ENCODE website (https://www.encodeproject.org/). We selected p-value (bigwig file) and IDR ranked peaks (bed file) from combined 2 repeats for each cell line was extracted. The peaks were further filtered with scores less than 200 removed.
DNA Methylation analysis
MethyLight qPCR assay for measuring methylation status of STING promoter region was developed using the method described by Wu et al.50. Genomic DNA was extracted from cultured cells using QIAamp DNA mini kit (Qiagen) following the manufacturer’s protocol. The concentration and purity of the extracted DNA were measured using Eppendorf Biophotometer Implen NanoPhotometer N60. Bisulfite conversion was performed by treating genomic DNA with EZ DNA Methylation Gold Kit (Zymo Research Corp) following the manufacturer’s protocol. Equal amounts of modified DNA was used as template for MethyLight. The sequences of these primers are as follows: methylated forward primer: 5’-TTAGGTTGGAGTGTAATGGTACG-3’, methylated reverse primer: 5’-AAATTAACTAAACGTAATAACGCA-3’, unmethylated forward primer: 5’TAGGTTGGAGTGTAATGGTATGA-3’, and unmethylated reverse primer: 5’AAAAATTAACTAAACATAATAACACAT-3’. Amplification was performed in a 20-mL reaction mixture containing an equal amount of bisulfite-modified DNA, 1 μM of each primer, 10 μL PowerUp SYBR Green Master Mix (Thermo Fisher Scientific), and nuclease-free water. PCR was performed using the following conditions: initial denaturation at 95 °C for 10 min; 40 cycles of denaturation at 94 °C for 30 s, annealing at 60.5 °C (for methylated STING primers) or 58 °C (for unmethylated STING primers). Data are shown as log2 of fold change in % methylation, relative to control. Statistical significance was determined using an unpaired two-sided Welch’s t test.
Single cell RNA-seq sample preparation and data collection
Freshly dissected tissue was micro-dissected into small chunks and processed as above for flow cytometry. Following tissue dissection, single cells were filtered using a 40 µm filter to remove residual clumps. 10 million cells were taken, and dead cells were removed using the dead cell removal kit (Miltenyi Biotec; #130-090-101). Cells were pooled from 2 tumor samples for control and Foxm1 KO 1 tumors; we used one sample for Foxm1 KO 3 tumors due to limited numbers of samples. Cells were then counted and loaded onto the 10X Chromium controller, targeting 10,000 cells for capture per well. Single-cell RNA sequencing libraries were generated using the Chromium Single Cell 3’ Library & Gel Bead Kit V3.1 kit and Chromium Single Cell 3’ Chips according to the manufacturer’s instructions (10X Genomics, Pleasanton, CA). In brief, all single-cell samples and required reagents were loaded on a 10X Chromium controller for droplet generation, followed by reverse transcription in the droplets, cDNA amplification, fragmentation, adapter, and index addition following the manufacturer’s instructions. Barcoded single-cell transcriptome libraries were sequenced with 100 bp paired-end reads on a NovaSeq S4 platform.
Single-cell RNA sequencing analysis
Raw sequencing reads were aligned to the GRCm38 genome (mouse) using Cell Ranger (v5; RRID:SCR_017344) software from 10X Genomics with default parameters. Subsequently, genes were quantified as UMI counts using Cell Ranger (v5;RRID:SCR_017344). Doublets were removed using DoubletFinder (v2.0.3; RRID:SCR_018771) with an expectation of 7.5% doublet rate assuming Poisson statistics. Downstream analysis was performed on filtered feature counts generated by Cell Ranger, and low-quality single cells containing <200 expressed genes or >15% mitochondrial transcripts were removed. Single cells were then normalized and clustered using Seurat51(v4.9.9.9060; RRID:SCR_016341). Single-cell gene expression counts were normalized to the library size and log2-transformed. Seurat51 was used to perform PCA reduction using the top 2000 most variable genes in the dataset. Computed PCAs were batch-corrected for variations between samples using Harmony (v1.1.0; RRID:SCR_018809). Batch-corrected PCs (1:40 for all clusters and 1:50 for T cell de novo analyses) were used as input for Louvain-based graphing with a resolution of 1 for all clusters and 0.5 for T cells de novo clustering. Cluster-specific marker genes were identified using the fast Wilcoxon rank-sum test wilcoxauc () function from the presto R package (V1.0.0 “github.com/immunogenomics/presto”) and used to identify the cell types. Two complementary methods were used to differentiate between normal epithelial cells and cancer cells. First, the inferCNV package (v3.9; https://github.com/broadinstitute/infercnv) was utilized to differentiate between malignant and normal cells, based on Copy Number Variations. Tumor cells show significantly higher proportion CNV in most chromosomes compared to normal epithelial cells. Second, tumor specific genes from bulk RNAseq data performed on control tissue cultured E0771 cells (#GSE139239). The top marker genes from this sample were utilized as E0771-specific markers. We used ggplot2 (v3.3.3; RRID:SCR_014601) and ComplexHeatmap (v2.16.0; RRID:SCR_017270) packages in R to produce boxplots and heatmaps.
Data and Code availability for scRNA-seq
The raw data for single-cell sequencing are publicly available without restriction through the GEO series GSE266617. Source data are provided with this paper. All codes used in the single-cell data analyses is publicly available through GitFront: https://gitfront.io/r/nourhanemad/nkZwBQVsds3y/Timilsina-FOXM1-Manuscript-Figs/.
Pathway analysis
Gene Set Enrichment Analysis (GSEA): We used the fGSEA (v1.26.0 -RRID:SCR_020938) R package to test for enrichment of hallmark gene sets downloaded from MsigDB (RRID:SCR_016863, msigdbr R package v7.5.1). For input, we used pre-ranked gene lists generated using a fast Wilcoxon rank-sum test (scRNAseq) (presto R package v1.0.0 “github.com/immunogenomics/presto”).
Cell-cell communication analysis
The CellChat R package (v1.6.1;RRID:SCR_021946) was used to assess cell-cell interaction between the cell types in each sample. The Seurat object was subsetted by sample to provide separate inputs for the createCellChat() function. CellChat’s internal literature validated ligand-receptor databases containing secreted, extracellular rmatrix-receptor, and cell-cell contact signaling were then used to construct cell-cell communication networks using standard scripts. CellChat’s suite of visualization tools were used to plot significant interactions through chord plots, heatmaps, and bubble plots.
Meta-analysis of patient datasets for gene correlation, immune infiltration, and response to immunotherapy
No sex/gender analysis was performed in this study for meta-analysis of patient samples because the majority of breast cancer(>99%) occurs in females. The purity and immune infiltration scores and the figures, along with p values and Rho scores, for FOXM1 were obtained directly from TIMER2.0 web portal52,53. Correlation of gene expression between FOXM1 and STING for breast and lung cancers were obtained from the GEPIA2 web portal54. p-values and R-scores were obtained directly from the web portal. The Kaplan-Meier survival analysis plot was obtained for overall survival or disease-free survival based on median expression of FOXM1, DNMT1, STING, and ULBP1 in breast cancer samples using KMplotter or GEPIA2. The p value and hazard ratio displayed in the graph are directly from GEPIA2. The ClinicalOmicsDB1 dataset was used to correlate FOXM1 gene expression with patient response to immune checkpoint inhibitors (Anti-CTLA-4 Mab, Anti-PD-L1 MAb, Anti-PD1 MAb). We observed significant differences (p < 0.05) between responders and non-responders in two studies, Prins_GBM_2019 for pembrolizumab and IMmotion150 atezolizumab. The data are available through ClinicalOmicsDB1 (https://trials.linkedomics.org/study_info/?study=Prins_GBM_2019.csv) and (https://trials.linkedomics.org/study_info/?study=IMmotion150_atezo.csv). Figures were generated using the ggplot2 package in R version 4.2.3 using the data directly downloaded from the web portal, and p values were calculated using two-sided Wilcoxon rank-sum test.
Statistics and reproducibility
All micrographs and data are derived from three biological replicates. GraphPad Prism software was used for data visualization and statistical analyses, while BioRender was used to create the schematics and the model used in the paper. For in vivo mouse studies, using a Chi-squared test with 1 degree of freedom from a Generalized Estimating Equations (GEE) analysis, we estimated that at least four mice (each for Ctrl and KO groups) were required to achieve a statistical power of ~ 90% with significance at 0.05. The mice were randomized before tumor implantation and treatment. All data are represented as mean ± SEM. All qRT-PCR data points, including ChIP-qPCR and Sting promoter methylation, are represented as mean +/− SEM of the log2 of fold change relative to Ctrl or IgG. The data include three biological replicates, each derived from two technical replicates. The p-values for all the experiments except scRNA-seq were determined using either an unpaired one-sided or two-sided Welch’s t test, multiple t tests with Benjamini, Krieger, and Yekutieli correction, or non-linear regression with Malthusian or Gompertz growth, depending on the experiment. The exact statistical methodology is written in the figure legend. The exact p-values are displayed on the graphs. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the conclusions in this paper are present in the paper and/or the Supplementary Materials. The raw data for single-cell sequencing are publicly available without restrictions through the GEO series GSE266617. The ClinicalOmicsDB1 database was used to correlate FOXM1 gene expression with patient response to immune checkpoint inhibitors (Anti-CTLA-4 Mab, Anti-PD-L1 MAb, Anti-PD1 MAb) using the Prins_GBM_2019 (https://trials.linkedomics.org/study_info/?study=Prins_GBM_2019.csv) and Immotion150 study (https://trials.linkedomics.org/study_info/?study=IMmotion150_atezo.csv). Source data are provided in this paper.
Code availability
All codes used in the single-cell data analyses are publicly available through GitFront: https://gitfront.io/r/nourhanemad/nkZwBQVsds3y/Timilsina-FOXM1-Manuscript-Figs/.
References
Wellenstein, M. D. & Visser, K. E. Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity 48, 399–416 (2018).
Gonzalez, H., Hagerling, C. & Werb, Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 32, 1267–1284 (2018).
Spranger, S. & Gajewski, T. F. Mechanisms of tumor cell–intrinsic immune evasion. Annu. Rev. Cancer Biol. 2, 213–228 (2018).
Li, J. et al. Tumor cell-intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity 49, 178–193.e7 (2018).
Nguyen, K. B. & Spranger, S. Modulation of the immune microenvironment by tumor-intrinsic oncogenic signaling. J. Cell Biol. 219, e201908224 (2020).
Du, L. et al. β-Catenin induces transcriptional expression of PD-L1 to promote glioblastoma immune evasion. J. Exp. Med. 217, e20191115 (2020).
Sansone, P. & Bromberg, J. Targeting the interleukin-6/Jak/Stat pathway in human malignancies. JCO 30, 1005–1014 (2012).
Owen, K. L., Brockwell, N. K. & Parker, B. S. JAK-STAT Signaling: A double-edged sword of immune regulation and cancer progression. Cancers 11, 2002 (2019).
Caetano, M. S. et al. IL6 Blockade reprograms the lung tumor microenvironment to limit the development and progression of K-ras–mutant lung cancer. Cancer Res. 76, 3189–3199 (2016).
Albacker, L. A. et al. Loss of function JAK1 mutations occur at high frequency in cancers with microsatellite instability and are suggestive of immune evasion. PLOS ONE 12, e0176181 (2017).
Fruman, D. A. et al. The PI3K pathway in human disease. Cell 170, 605–635 (2017).
Mafi, S. et al. mTOR-Mediated regulation of immune responses in cancer and tumor microenvironment. Front. Immunol. 12, 774103 (2022).
Antoniv, T. T. & Ivashkiv, L. B. Interleukin-10-induced gene expression and suppressive function are selectively modulated by the PI3K-Akt-GSK3 pathway. Immunology 132, 567–577 (2011).
Koo, C.-Y., Muir, K. W. & Lam, E. W.-F. FOXM1: From cancer initiation to progression and treatment. Biochim. Biophys. Acta 1819, 28–37 (2012).
Kalathil, D., John, S. & Nair, A. S. FOXM1 and Cancer: Faulty cellular signaling Derails homeostasis. Front. Oncol. 10, 626836 (2021).
Zhang, Y.-L. et al. A narrative review of research progress on FoxM1 in breast cancer carcinogenesis and therapeutics. Ann. Transl. Med. 9, 1704 (2021).
Hopewell, E. L. et al. Lung tumor NF-κB signaling promotes T cell–mediated immune surveillance. J. Clin. Invest. 123, 2509–2522 (2013).
Yeo, H., Lee, Y. H., Koh, D., Lim, Y. & Shin, S. Y. Chrysin inhibits NF-κB-dependent CCL5 transcription by targeting IκB kinase in the atopic dermatitis-like inflammatory microenvironment. Int. J. Mol. Sci. 21, 7348 (2020).
Goda, N. et al. The ratio of CD8 + lymphocytes to tumor-infiltrating suppressive FOXP3 + effector regulatory T cells is associated with treatment response in invasive breast cancer. Discov. Onc. 13, 27 (2022).
Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Dangaj, D. et al. Cooperation between constitutive and inducible chemokines enables T cell engraftment and immune attack in solid tumors. Cancer Cell 35, 885–900 (2019).
Gan, Y. et al. The cGAS/STING pathway: A novel target for cancer therapy. Front. Immunol. 12, 795401 (2022).
Lin, R., Heylbroeck, C., Genin, P., Pitha, P. M. & Hiscott, J. Essential role of interferon regulatory factor 3 in direct activation of RANTES chemokine transcription. Mol. Cell. Biol. 19, 959–966 (1999).
Deng, X. et al. Transcriptional regulation of increased CCL2 expression in pulmonary fibrosis involves nuclear factor-κB and activator protein-1. Int. J. Biochem. Cell Biol. 45, 1366–1376 (2013).
Wu, S.-Y. et al. MYC suppresses STING-dependent innate immunity by transcriptionally upregulating DNMT1 in triple-negative breast cancer. J. Immunother. Cancer 9, e002528 (2021).
Liu, X. et al. UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nat. Commun. 4, 1563 (2013).
Hosomi, S. et al. Intestinal epithelial cell endoplasmic reticulum stress promotes MULT1 up-regulation and NKG2D-mediated inflammation. J. Exp. Med. 214, 2985–2997 (2017).
Greten, F. R. & Grivennikov, S. I. Inflammation and cancer: Triggers, mechanisms, and consequences. Immunity 51, 27–41 (2019).
Aboelella, N. S., Brandle, C., Kim, T., Ding, Z.-C. & Zhou, G. Oxidative stress in the tumor microenvironment and its relevance to cancer immunotherapy. Cancers 13, 986 (2021).
Ding, L. et al. PARP Inhibition elicits STING-dependent antitumor immunity in Brca1-deficient ovarian cancer. Cell Rep. 25, 2972–2980 (2018).
Woo, S.-R. et al. STING-Dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842 (2014).
Bhattacharya, S. et al. RAD51 interconnects between DNA replication, DNA repair and immunity. Nucleic Acids Res. 45, 4590–4605 (2017).
Reisländer, T., Groelly, F. J. & Tarsounas, M. DNA Damage and cancer immunotherapy: A STING in the Tale. Mol. Cell 80, 21–28 (2020).
Reisländer, T. et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat. Commun. 10, 3143 (2019).
Chabanon, R. M. et al. PARP inhibition enhances tumor cell–intrinsic immunity in ERCC1-deficient non–small cell lung cancer. J. Clin. Invest. 129, 1211–1228 (2019).
Heijink, A. M. et al. BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat. Commun. 10, 100 (2019).
Carr, J. R. et al. FoxM1 Regulates Mammary Luminal Cell Fate. Cell Rep. 1, 715–729 (2012).
Kopanja, D. et al. Transcriptional repression by FoxM1 suppresses tumor differentiation and promotes metastasis of breast cancer. Cancer Res. 82, 2458–2471 (2022).
Mukhopadhyay, N. K. et al. Plk1 Regulates the Repressor Function of FoxM1b by inhibiting its Interaction with the Retinoblastoma Protein. Sci. Rep. 7, 46017 (2017).
Schneider, M. et al. Systematic analysis of the binding behaviour of UHRF1 towards different methyl- and carboxylcytosine modification patterns at CpG dyads. PLOS ONE 15, e0229144 (2020).
Adam, S. et al. DNA sequence-dependent activity and base flipping mechanisms of DNMT1 regulate genome-wide DNA methylation. Nat. Commun. 11, 3723 (2020).
Xu, Y. et al. Decreased expression of the NKG2D ligand ULBP4 may be an indicator of poor prognosis in patients with nasopharyngeal carcinoma. Oncotarget 8, 42007–42019 (2017).
de Kruijf, E. M. et al. NKG2D ligand tumor expression and association with clinical outcome in early breast cancer patients: an observational study. BMC Cancer 12, 24 (2012).
Lam, A. R. et al. RAE1 Ligands for the NKG2D Receptor Are Regulated by STING-Dependent DNA Sensor Pathways in Lymphoma. Cancer Res. 74, 2193–2203 (2014).
Wu, J. et al. STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J. Exp. Med. 216, 867–883 (2019).
Smith, J. A. STING, the Endoplasmic reticulum, and mitochondria: Is three a crowd or a conversation? Front. Immunol. 11, 611347 (2021).
Xu, Y., Nowsheen, S. & Deng, M. DNA Repair deficiency regulates immunity response in cancers: Molecular mechanism and approaches for combining immunotherapy. Cancers 15, 1619 (2023).
Hayman, T. J. et al. STING enhances cell death through regulation of reactive oxygen species and DNA damage. Nat. Commun. 12, 2327 (2021).
Liu, D. et al. STING directly activates autophagy to tune the innate immune response. Cell Death Differ. 26, 1735–1749 (2019).
Wu, C.-S. et al. Methylation status of the stimulator of interferon genes promoter in patients with chronic hepatitis B. Medicine 97, e13904 (2018).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Li, T. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 48, W509–W514 (2020).
Li, T. et al. TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 77, e108–e110 (2017).
Tang, Z., Kang, B., Li, C., Chen, T. & Zhang, Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 47, W556–W560 (2019).
Acknowledgements
Single-cell RNA sequencing data were generated in the Genome Sequencing Facility, which is supported by UT Health San Antonio, P30 CA054174, and NIH Shared Instrument grant S10OD030311 and CPRIT Core Facility Award RP220662. We acknowledge the histology core at Greehey Children’s Cancer Research Institute for their help with immunohistochemical processing and staining of tissues. M.K.R. is supported by R01CA179120, R01CA239227, P30CA054174, and CPRIT RP200110. RV is supported by R01CA179120, R01CA239227, VA1 I01 BX006280-0 1, and P30CA054174. N.Z. is supported by VA Merit Award 1I01BX005955, American Cancer Society grant RSG-18-222-01-LIB, and a W.M. Keck Foundation award. Y.C. is supported by RP160732 and P30 CA054174. S.T. was supported by T32CA148724. J.Y.H. is supported by T32CA279363), ST-MSTP T32GM113896, and T32GM145432. T.P.D. is supported by T32CA279363), ST-MSTP T32GM113896, and T32CA148724.
Author information
Authors and Affiliations
Contributions
S.T. and M.K.R. conceived the study. S.T., M.K.R., N.Z., and N.A. designed experiments. S.T. and M.K.R. wrote the manuscript. S.T., D.M., D.S., S.A., J.Y.H., S.P., T.P.D., S.N., P.V., and J.P. performed the experiments. S.T., S.P., N.A., S.A., J.Y.H., T.P.D., M.B., Y.C., S.Z., V.K., P.S., R.H., N.M., G.H., R.V., N.Z., P.V., and M.K.R. analyzed the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Pradip Raychaudhuri and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Timilsina, S., Huang, J.Y., Abdelfattah, N. et al. Epigenetic silencing of DNA sensing pathway by FOXM1 blocks stress ligand-dependent antitumor immunity and immune memory. Nat Commun 16, 3967 (2025). https://doi.org/10.1038/s41467-025-59186-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59186-3
