
Abstract
Prime editing is a genome engineering tool that allows installation of various small edits with high precision. However, prime editing efficiency and purity can vary widely across different edits, genomic targets, and cell types. Prime editing typically offers more precise editing outcomes compared to other genome editing methods such as homology-directed repair. However, it can still result in significant rates of unintended editing outcomes, such as indels or imprecise prime edits. This issue is particularly notable in systems utilizing a second nicking gRNA, such as PE3 and PE5, as well as in dual pegRNA systems and fully active nuclease systems such as PEn, which increase efficiency but compromise precision. In this work, we show that pharmacological inhibition of DNA-PK and Polϴ, two major mediators of mutagenic DNA repair pathways, improves precision of PEn, PE3, PE5, PE7, and dual pegRNA editing systems, including TwinPE, HOPE, and Bi-PE, across multiple genomic loci and edit types. We show that co-inhibition of DNA-PK and Polϴ mitigates both prime editing-unrelated indels and prime editing by-products such as template duplications. Moreover, in the case of PEn, this strategy also substantially improved the off-target editing profile. Together, our data establish small molecule modulation of DNA repair pathways as a general strategy to maximize the precision of diverse prime editing systems.
Similar content being viewed by others

Introduction
Prime editing is a next generation genome engineering approach that allows DNA donor-free installation of various types of small genomic edits including insertions, deletions, and substitutions1. Compared to more traditional approaches for precise genome editing such as homology-directed repair (HDR), prime editing induces lower levels of undesired editing outcomes and off-targets2,3. A typical prime editor (PE) comprises a Cas9 nickase fused to a reverse transcriptase (RT) via a flexible linker2. PE operates by reverse-transcribing an RT template (RTT) which is part of a prime editing guide RNA (pegRNA). As a result, PE introduces a 3’ genomic flap containing a desired edit and a homology region that anneals and integrates into the targeted genomic locus. Prime editing is a highly modular method and numerous prime editing systems were developed for various genome editing applications1. Currently, broad adaptation of PE is impeded by several limitations, such as the need for extensive pegRNA optimizations and the widely varying editing efficiency observed across different types of edits, target sites, and cell types2,4,5,6.
Multiple strategies were developed to increase efficiency of the prototypical PE2 system. Some of these centers around modulating the mismatch repair pathway (MMR) that was shown to impede prime editing, likely by shifting resolution of prime editing heteroduplex intermediates back to the original unedited state2,4,7. MMR impairment via different means has proven to be an effective, albeit target- and cell type-specific, strategy to improve prime editing efficiency. The PE3 system introduces an additional nicking gRNA (ngRNA) to induce a nick in the unedited strand opposite to the 3’ flap, and shifts the heteroduplex repair outcome in favor of the desired edit2. More recently, the PE4 system was developed, where co-expression of a dominant-negative form of MLH1 (MLH1dn), one of the essential mediators of MMR in human cells, improved efficiency and purity of prime editing outcomes. To increase prime editing efficiency further, PE3 and PE4 systems were combined into the PE5 system (PE2 + MLH1dn + ngRNA). While both PE3 and PE5 can be more efficient than PE2, the presence of a second nick in proximity (<100 bp) to the pegRNA nick might result in a DNA double strand break (DSB) and often leads to high levels of non-RT templated insertions or deletions (indels), generally caused by larger deletions that extend beyond the two DNA nicks8.
In addition, a dual pegRNA system (TwinPE) was developed to enable installation of larger edits as well as replacements of genomic fragments9. TwinPE does not involve formation of heteroduplex intermediates and thus avoids being negatively impacted by MMR activity. However, like PE3 and PE5, because of two separate DNA nicks, TwinPE can also lead to high rates of indels. Similar dual pegRNA systems like HOPE and Bi-PE, with variations to the design of the homology arms on the RTT template, can also introduce unwanted mutations10,11.
As a different approach to increase efficiency of prime editing, several studies reported prime editing systems based on the wild-type, fully active SpCas9 nuclease (Prime Editing nuclease – PEn)12,13,14,15,16,17. PEn can rescue editing at target sites resistant to nickase-based PE, likely due to a more robust DNA repair mechanism promoted by the presence of DSBs. However, the presence of DSBs activates mutagenic DNA end-joining mechanisms such as non-homologous end joining (NHEJ) and alternative end-joining (alt-EJ) pathways, therefore severely impeding the utility of PEn when high precision is required.
Thus, while increasing the efficiency of intended editing, the prime editing systems that employ either a second ngRNA (PE3, PE5), two pegRNAs (TwinPE) or a wild-type Cas9 nuclease (PEn) often lead to high frequencies of editing by-products due to mutagenic DSB repair.
Here we present a method to improve prime editing by substantially reducing these undesired by-products. We introduce ‘2-inhibitor’ PE (2iPE), a small molecule combination approach that uses inhibitors of DNA repair pathways, modulating the choice of DSB repair during PE editing. We and others have previously demonstrated the advantages of employing dual NHEJ/alt-EJ inhibition in HDR-based methods to enhance editing efficiency and precision while simultaneously reducing off-target editing18,19,20,21. The small molecule inhibitors target the core NHEJ and alt-EJ factors DNA-dependent protein-kinase (DNA-PK) and DNA polymerase ϴ (Polϴ), respectively, thereby minimizing undesired by-products formed by NHEJ and alt-EJ. With 2iPE, we apply this principle across a wide range of prime editing systems to substantially improve the purity of editing outcomes without negatively impacting on-target efficiency.
Results
Co-inhibition of DNA-PK and Polϴ improves precision of PEn editing
Based on recent work from our and other laboratories that utilized inhibitors of DNA-PK and Polϴ to improve HDR knock-ins18,19, we hypothesized that simultaneous inhibition of DNA-PK and Polϴ with small molecules could similarly improve the precision of prime editing. For initial testing of this concept, we opted for PEn editing, as it generally displays the highest levels of indels due to its fully active Cas9 nuclease, and the mechanisms leading to various by-products of PEn editing are well understood (Fig. 1a). We previously demonstrated that treatment with the highly selective and potent DNA-PK inhibitor AZD764822 (referred to as DNA-PKi) increased the precision (proportion of precise prime edits compared to overall editing) of PEn editing (Peterka et al. 2022). In a subsequent study, Li et al. co-expressed PEn with a ubiquitin variant that inhibits tumor suppressor p53-binding protein 1 (53BP1) (uPEn)15. Both systems suppress NHEJ-mediated indels and imprecise prime edits, resulting in an increase in PEn editing precision and efficiency. Nevertheless, in both cases, the levels of by-products remained high. In the case of PEn editing with DNA-PKi, disabled NHEJ was compensated for by alt-EJ activity, and depletion of Polϴ decreased indels exacerbated by DNA-PKi16,20. To rigorously test the effects of chemical co-inhibition of DNA-PK and Polϴ on the precision of PEn editing, we installed a point mutation in the KCNA1 locus in HEK293T cells, using a pegRNA previously shown to be highly efficient but imprecise when used with PEn and inefficient when used with PE515. We tested PEn with different combinations of DNA-PKi, and Polϴ inhibitors PolQi1 and PolQi2 (Fig. 1b). PolQi1 targets the polymerase ___domain whereas PolQi2 inhibits the helicase ___domain of Polϴ18, both of which are required for DSB repair23. We used targeted amplicon sequencing (amplicon-seq) to compare editing outcomes for each of these conditions to uPEn (PEn-P2A-i53 configuration)15 as well as to nickase-based PE54. As reported previously, PEn allowed efficient editing of KCNA1 but suffered from increased levels of indels and imprecise prime edits (Fig. 1b). As expected, PEn editing in the presence of DNA-PKi increased the precision compared to PEn alone but did not mitigate indels unrelated to prime editing, which were shifted from insertions towards deletions, suggesting a shift from NHEJ to alt-EJ-mediated DNA repair (Supplementary Fig. 1a). Co-administration of DNA-PKi with PolQi1 or PolQi2 further reduced indels and improved editing precision compared to uPEn, PEn, and PEn in combination with DNA-PKi alone. Finally, to inhibit DNA-PK and both the polymerase and helicase domains of Polϴ, we combined DNA-PKi with PolQi1 and PolQi2. This combination led to a near-complete editing purity while maintaining over 5-fold higher efficiency compared to PE5 (Fig. 1b). The effect of DNA-PK and Polϴ inhibition on PEn editing is evident in the allele types generated post-editing. DNA-PK inhibition notably reduces the insertion of the homology flap from the pegRNA RTT, while Polϴ inhibition decreases small deletions surrounding the cut site (Supplementary Fig. 1b). Treatments with PolQi1 and PolQi2 alone had no impact on DNA repair outcome (Supplementary Fig. 1c) and cell proliferation was unaffected by inhibitor treatments (Supplementary Fig. 1d).

a Schematic representation of PEn editing depicting molecular mechanisms of four possible PEn editing outcomes and their modulation by DNA repair inhibitors. b Installation of a point mutation in KCNA1 in HEK293T cells. Cells treated with indicated combinations of DNA-PK and Polϴ inhibitors were co-transfected with a pegRNA and indicated gene editing systems. Editing outcomes were analyzed by amplicon-seq and quantified using CRISPResso2 in prime editing mode. Plots show mean ± SD of n = 3 biological replicates. Source data are provided as a Source Data file. PBS Primer Binding Site, RTT Reverse-Transcriptase Template, alt-EJ alternative End-Joining pathway, NHEJ Non-Homologous End-Joining pathway, indels non-templated insertions and deletions.
To test the consistency of this effect on PEn editing, we installed point mutations, insertions, and short deletions in HEK293T and HeLa cells using a panel of pegRNAs that were previously shown to be highly active but imprecise with PEn or uPEn15,16 (Fig. 2a–c). Based on our initial results at the KCNA1 locus, we focused on the two most promising combinations of DNA-PK and Polϴ inhibitors: DNA-PKi+ PolQi1 (referred to as “2iPEn”) and a triple combination of DNA-PKi+ PolQi1+ PolQi2 (referred to as “2+iPEn”). For comparison, we also performed editing with PE5, uPEn, and PEn in combination with DNA-PKi only (“1iPEn”). Both 2iPEn and 2+iPEn consistently reduced indel outcomes of PEn editing across multiple targets and edits and for both HEK293T and HeLa cells (Fig. 2a–c). Although for some individual targets we observed both increased editing efficiency (Fig. 2a, b) and one case of decreased efficiency (Fig. 2b – CDKL5), the efficiency of precise editing across all the tested loci was generally unaffected by inhibitor treatments (Supplementary Fig. 2). 2+iPEn consistently led to near-complete precise editing purity, matching the precision levels of PE5 (Fig. 2d), while maintaining the high efficiency of PEn. The precision increase of 2+iPEn compared to PEn and uPEn was 4.3-fold and 2.1-fold, respectively, in HEK293T cells and 9.8-fold and 7.1-fold, in HeLa cells (Fig. 2d). In summary, we demonstrate that combining small molecule inhibitors to target both NHEJ and alt-EJ significantly improves the editing outcome of PEn by reducing by-products to the level of nickase-based PEs while maintaining the efficiency of PEn editing.

Genome editing with six different prime editing systems to install indicated point mutations or insertions (ins.) using plasmid co-transfection in (a) HEK293T and b HeLa cells. c PEn editing to delete 3 bp at HEK3 and RUNX1 sites in indicated cell line. d Calculated precision score of the six different prime editing systems across all loci tested in (a and b). “Precision score” was calculated as the number of amplicon-seq reads with precise prime edit per the number of amplicon-seq reads with any edit. Plots represent mean ± SD of n = 8 precision scores calculated at target sites in (a and b). P-values were determined using one-way ANOVA (two-tailed, paired). Multiple comparisons were corrected using Tukey’s test. Displayed pairwise comparisons are relative to 2+iPEn. Calculated P-values: HEK293T: PE5 vs 2i+PEn =0.2371 (ns), uPEn vs 2i+PEn =<0.0001 (****), PEn vs 2i+PEn = <0.0001 (****), 1iPEn vs 2i+PEn = <0.0001 (**), 2iPEn vs 2i+PEn = 0.2794 (ns). HeLa: PE5 vs 2i+PEn = 0.3556 (ns), uPEn vs 2i+PEn = <0.0001 (****), PEn vs 2i+PEn = <0.0001 (****), 1iPEn vs 2i+PEn = <0.0001 (****), 2iPEn vs 2i+PEn = 0.0008 (**). ns not significant. All amplicon sequencing data described above was analyzed using CRISPResso2 in prime editing mode. All bar plots show mean ± SD of n = 3 biological replicates. Drug treatments: 1i = DNA-PKi; 2i = DNA-PKi + PolQi1; 2+i = DNA-PKi + PolQi1 + PolQi2. Source data are provided as a Source Data file.
Co-inhibition of DNA-PK and Polϴ reduces PEn off-target editing
PEn increases off-target editing when used with promiscuous pegRNAs compared to editing with Cas9 alone16, similar to what is observed for uPEn15. This is at least in part due to the promiscuous priming by RT at the off-target cuts leading to their inability to be seamlessly repaired back to the original wild-type state16. As we have shown earlier that co-inhibition of DNA-PK and Polϴ substantially mitigates off-target editing in Cas9-treated samples18, we hypothesized that editing with 1iPEn, 2iPEn or 2+iPEn will result in a similar alleviating effect on PEn off-target editing. To explore the impact of small molecule inhibitors of DNA repair on PEn off-target editing, we systematically investigated four promiscuous pegRNAs targeting the EMX1, FANCF, HEK3, and HEK4 loci together with 13 corresponding off-target sites in HEK293T cells2,24 using amplicon-seq (Fig. 3a). Consistent with our prior findings, PEn editing showed cleavage activity across most interrogated off-target sites. uPEn or 1iPEn equally reduced off-target editing at all tested sites by 2.9-fold on average. 2iPEn further diminished off-target editing by 10.2-fold on average compared to PEn editing alone. Importantly, 2+iPEn not only improved editing precision at the on-target sites compared to PEn and uPEn, but also reduced off-target editing the most by an average of 27.9-fold. A complete overview of all on- and off-target editing events is provided in Supplementary Fig. 3. To account for variable on-target editing levels across different PEs, we computed specificity scores to normalize off-target editing to on-target editing levels. The specificity score is calculated as 1 minus the ratio of editing frequency at the off-target site to that at the on-target site resulting in a range between 0 and 1. A specificity score of 1 denotes the absence of off-target editing25. This analysis substantiated the off-target editing results, confirming a significant decrease in off-target editing for 2iPEn and 2+iPEn compared to PEn (Fig. 3b). In summary, our findings demonstrate a substantial reduction of PEn off-target editing through the concurrent inhibition of DNA-PK and Polϴ.

a Amplicon-seq and CRISPResso2 analysis at four on-target and 13 off-target sites for the indicated PEn configurations using plasmid co-transfections in HEK293T cells. Editing is shown as percentages of modified reads in each sample. Mismatches to the on-target gRNA sequence are highlighted in red lower-case letters, the PBS region is underlined. b Computed specificity scores for all off-target data shown in (a). Specificity scores are calculated as 1 minus the ratio of editing frequency at the off-target site to that at the on-target site resulting in a range between 0 and 1. Each dot in the graph represents one individual off-target, horizontal lines represent median and 95% confidence intervals of n = 3 biological replicates. Significance levels of specificity scores were calculated with the non-parametric Friedman test (paired, two-tailed). Multiple comparisons were corrected using Dunn’s test. Calculated P-values: PEn vs. uPEn >0.9999 (ns), PEn vs. 1iPEn = 0.7544 (ns), PEn vs. 2iPEn = 0.0008 (***), PEn vs. 2+iPEn <0.0001 (****). ns not significant. Drug treatments: 1i = DNA-PKi; 2i = DNA-PKi + PolQi1; 2+i = DNA-PKi + PolQi1 + PolQi2. Source data are provided as a Source Data file. PBS Primer Binding Site (PBS), PAM Protospacer Adjacent Motif.
Co-inhibition of DNA-PK and Polϴ improves precision of nickase-based prime editing systems in different cell types
PE3 and PE5 have been shown to induce higher levels of indels than PE2 and PE4, due to a second DNA nick proximal to the target locus2,4,8. This leads to the generation of indels that are possibly mediated by NHEJ and/or alt-EJ, analogous to PEn-induced indels. We thus speculated that both PE3 and PE5 could benefit from DNA-PK and Polϴ co-inhibition similar to PEn. To test this hypothesis, we used a pegRNA to install an 11 bp insertion into the PCSK9 locus16 in HEK293T and HeLa cells. To gain a comprehensive view of how different drug treatments impact different prime editing systems, we used PE2, PE3, PE4 and PE5 in combination with DNA-PKi alone (“1iPE”), DNA-PKi+ PolQi1 (“2iPE”) and DNA-PKi+ PolQi1+ PolQi2 (“2+iPE”). As expected, editing with PE2 and PE4 led to lowest editing levels but high precision with no detectable indels or other by-products, as compared to PE3 and PE5. Consequently, DNA-PK and Polϴ co-inhibition had no impact on PE2 and PE4 editing precision (Fig. 4a). On the other hand, both PE3 and PE5 were more efficient but less precise, showing both imprecise prime editing by-products as well as indels that were both reduced, in ascending order, with 1iPE, 2iPE, and 2+iPE (Fig. 4a). PE3-installed precise prime edits represented only 60% and 36% of overall editing in HEK293T and HeLa cells, respectively. The precision of both PE3 and PE5 were improved with each drug combination, reaching up to 98% and 96% with 2+iPE3 in HEK293T and HeLa cells, while maintaining high efficiency. Corresponding allele frequency plots show that PE3-induced by-products consisted mostly of deletions spanning the two nicked sites and that these by-products disappeared upon 2i treatment (Supplementary Fig. 4).

a 11 bp insertion in PCSK9 locus in HEK239T and HeLa cells using PE2, PE3, PE4, PE5. b Installation of indicated edits in HEK293T and HeLa cells with PE3 and PE5. c Calculated precision score across all tested loci in (a and b) and Supplementary Fig. 5a). Plots represent mean ± SD of n = 12 precision scores calculated at target sites in Fig. 2a, b. P-values were determined using one-way ANOVA (two-tailed, paired) with Tukey’s multiple comparisons test. Displayed pairwise comparisons are relative to PE3 or PE5. Calculated P-values: HEK293T: PE3 vs 1iPE3 = 0.9860 (ns); PE3 vs 2iPE3 = 0.2791 (ns); PE3 vs 2+iPE3 = 0.0106 (*); PE5 vs 1iPE5 = 0.9946 (ns); PE5 vs 2iPE5 = 0.4350 (ns); PE5 vs 2+iPE5 = 0.0277 (*). HeLa: PE3 vs 1iPE3 = 0.0655 (ns); PE3 vs 2iPE3 < 0.0001 (****); PE3 vs 2+iPE3 < 0.0001 (****); PE5 vs 1iPE5 = 0.2166 (ns); PE5 vs 2iPE5 < 0.0001 (****); PE5 vs 2+iPE5 < 0.0001 (****). d 11 bp insertion of PCSK9 locus in HEK239T using PE2, PE7, and PE7 with ngRNA. e +4T>G substitution of FANCF locus in iPSCs, PHH, HepG2, and Huh7 cell lines using PE3 (mRNA) and synthetic pegRNA/ngRNA. f +5G>A substitution of HBB locus in K562 cells using PEn (mRNA) and synthetic pegRNA/ngRNA. Editing outcomes and precision scores were analyzed as described above. P-values were determined using one-way ANOVA (two-tailed, paired) with Tukey’s multiple comparisons test. Displayed pairwise comparisons are relative to 2i+PEn or PE5. Calculated P-values = PEn vs. 2+iPEn = 0.0109 (*), 1iPEn vs. 2+iPEn = 0.0215 (*). All amplicon sequencing data described above was analyzed using CRISPResso2 in prime editing mode. All bar plots show mean ± SD of n = 3 biological replicates. Drug treatments: 1i = DNA-PKi; 2i = DNA-PKi + PolQi1; 2+i = DNA-PKi + PolQi1 + PolQi2. Source data are provided as a Source Data file.
To further test the beneficial effect of 2+iPE on prime editing precision we used 12 pegRNAs previously shown to induce high levels of indels4,26,27 to target 4 loci (FANCF, HEK3, RUNX1 and EMX1), with various edit types (substitutions, insertions, and deletions) in combination with PE3 and PE5 in HEK293T and HeLa cells. In the cases where indels were generated under untreated conditions, we observed a similar beneficial effect with inhibitor treatment across the different target sites and types of edit for both PE3 and PE5 (Fig. 4b, Supplementary Fig. 5a, b). The purity of prime edits was significantly increased across all target sites and edits when combining the DNA-PK and Polϴ inhibitors with a nearly complete elimination of imprecise prime edits and indels (Fig. 4b). Precision between PE3/PE5 and 2+iPE3/2+iPE5 was improved from 59.5%/62.25% to 93.4%/93.1% in HEK293T cells, and from 48.6%/54.0% to 91.9%/93.9% in HeLa cells, respectively (Fig. 4c). Variation in the prime editing purity between different target sites with PE3/PE5 was also reduced with the combination of inhibitors, giving a more consistent editing outcome regardless of the target site or edit (Fig. 4c). Next, we tested if the beneficial effects of 2iPE are also applicable to the recently developed PE7. This system co-expresses the La protein, which binds to and stabilizes poly-U tailed pegRNAs, thereby enhancing editing efficiency28. We compared the editing outcomes of PE2, PE7, and PE7 with an additional ngRNA for an 11 bp insertion at the PCSK9 locus in HEK293T cells. As expected, PE7 showed improved efficiency over PE2, with the highest efficiency observed when a ngRNA was included (Fig. 4d). However, the ngRNA also increased indels, which were substantially reduced by co-inhibition of DNA-PK and Polϴ. These findings demonstrate that dual inhibition approach enhances the precision of PE3, PE5, and PE7 systems.
To evaluate the broader applicability of the 2iPE and 2iPEn strategies, we tested them across multiple cell types, such as HepG2, Huh7 and K562 cell lines and also included human induced pluripotent stem cells (iPSCs) and primary human hepatocytes (PHHs). We transfected synthetic pegRNAs and ngRNAs alongside mRNA-encoded PE2 editors to introduce point mutations in the FANCF locus. The 2i strategy generally reduced indel formation across all tested cell types (Fig. 4e), except in PHHs, where baseline indel frequency was already low, and inhibitor treatment had no additional effect. Furthermore, using PEn mRNA and synthetic pegRNA in K562 cells, we assessed editing efficiency at an additional HBB locus29. In this experiment, the inclusion of the two inhibitors substantially reduced indels without compromising precise editing efficiency, significantly increasing precision of editing compared with editing with only DNA-PKi or without inhibitors (Fig. 4f). These data highlight the broad utility of the 2iPE and 2iPEn strategies across diverse cell types.
Co-inhibition of DNA-PK and Polϴ improves precision of dual-pegRNA prime editing systems
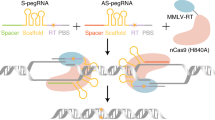
Lastly, we tested if DNA-PK and Polϴ co-inhibition can improve the precision of dual pegRNA editing systems. Nickase and nuclease versions of dual pegRNA editing systems have been developed9,13. This strategy is mostly employed for replacing the genomic sequence between the two spacers with a long heterologous sequence but can also be used to generate large deletions or to introduce multiple substitutions across a wide genomic segment. Due to their double-cutting nature, dual pegRNA approaches often lead to unwanted indels resulting from imprecise repair (Fig. 5a). We first applied DNA-PK and Polϴ co-inhibition on the TwinPE system applied to remove genomic sequence between each spacer and replace it with the serine integrase Bxb1 attachment sites (50 bp attP and 38 bp attB) within the AAVS1, ACTB, and CCR5 loci30. TwinPE installed these replacements with high efficiency (up to 70%), but also introduced indels (up to 9%) (Fig. 5b). Addition of both DNA-PKi and PolQi reduced indels to 1% and less, while maintaining similar rates of precise replacement (Fig. 5b). Sequence alignment of variants generated by TwinPE revealed integration of the genomic flaps, including end-to-end ligation of the two genomic flaps as evidenced by insertion of the homology arms (Supplementary Fig. 6). The beneficial effect of 2+i-TwinPE motivated us to apply the inhibitors to other dual pegRNA prime editing systems, such as HOPE and Bi-PE10,11. Both these strategies use a nickase version of prime editors with a pair of pegRNAs with variable designs, such as extra homology arms to the genomic site, and have different editing applications, including the installation of multiple substitutions (Fig. 5c). Similar to TwinPE, application of DNA-PK and Polϴ co-inhibition on HOPE and Bi-PE substantially reduced indels at all target sites but maintained the same levels of precise editing efficiency achieved without inhibitors (Fig. 5d). Thus, the 2i-strategy can be employed for improving the precision of dual pegRNA editing strategies.

a Schematic representation of TwinPE-mediated replacement with possible editing outcomes. b TwinPE mediated attP (50 bp) or attB site (38 bp) installation at three different sites (AAVS1, ACTB, CCR5) with indicated drug treatments in HEK293T cells. c Schematic representation of HOPE and Bi-PE editing. d Editing outcomes of HOPE (insertion) and Bi-PE2 and Bi-PE3 (five individual substitutions) at indicated target site subjected to indicated drug treatments in HEK293T cells. e Schematic representation of TwinPEn-mediated replacement and possible editing outcomes. f TwinPEn mediated attP (50 bp) or attB site (38 bp) installation at three different sites (AAVS1, ACTB, CCR5) with indicated drug treatments in HEK293T cells. All amplicon sequencing data described above was analyzed using CRISPResso2 in HDR mode. All bar plots show mean ± SD of n = 3 biological replicates. Drug treatments: 1i = DNA-PKi; 2+i = DNA-PKi + PolQi1 + PolQi2. Source data are provided as a Source Data file. RTT Reverse-Transcriptase Template, bp base pairs, del. deletion, ins. insertion.
We also tested dual pegRNA approach with nuclease-based TwinPE (referred here as TwinPEn) (Fig. 5e). For replacement editing, TwinPEn reached lower levels of intended editing compared to nickase-based TwinPE and, as expected, generated much higher amounts of indels (Fig. 5f). However, addition of the DNA-PK and Polϴ inhibitors significantly reduced these indels and increased precise editing outcomes compared to the untreated samples. The primary types of indels generated with nuclease-based TwinPEn are direct end-joining of one of the genomic flaps, a repair profile similar to the previously published homology-independent prime editing strategy (PRINS)16 or end-joining of one flap with the DSB end of the second spacer (Supplementary Fig. 7). In conclusion, we demonstrated the versability of our 2i-strategy for diverse types of dual pegRNA editing. Our data demonstrate that by-products of dual pegRNA prime editing systems are mediated by NHEJ and alt-EJ, and DNA-PK and Polϴ co-inhibition is a universal method to improve precision of these systems.
Discussion
Prime editing has emerged as a powerful technique for precise repair of small genomic mutations and indels, making it a valuable tool in disease modeling as well as a potential therapeutic modality. Unlike HDR, which is constrained by the cell cycle, and base editing, which is limited to specific point mutations, prime editing offers an alternative to address a broader range of genetic alterations. While prime editing exhibits various advantages over other precise genome editing tools, it generally suffers from low efficiency and high target-to-target variability. The low efficiency of the original PE2 system can be circumvented by various means, such as PE3 and PE5 systems, where a second ngRNA is used to bias DNA repair towards the desired editing outcome. Another approach that showed potential to increase PE efficiency is application of PEn, a prime editor with a fully active SpCas9 nuclease. However, the increased efficiency of these systems comes at the cost of decreased precision due to induction of DSBs that lead to introduction of indels and imprecise prime editing by-products. Here, we used small molecule inhibitors of DNA-PK and Polϴ to show that co-inhibition of NHEJ and alt-EJ pathways that give rise to undesired editing by-products, can be used to improve precision of prime editing. To this end, we adapted a small molecule cocktail previously reported to improve the outcomes of HDR editing. The NHEJ-inhibiting component of this cocktail is AZD7648 – a highly potent and selective DNA-PK inhibitor that targets the ATP-binding pocket of DNA-PK22. For alt-EJ inhibition, we target Polϴ with two selective inhibitors – PolQi1 and PolQi2, which target the polymerase and the helicase ___domain of Polϴ, respectively18. We demonstrate that this “2iPE” approach can be broadly applied across different prime editing systems such as PEn, PE3, PE5, PE7, and several dual pegRNA prime editing modalities, including TwinPE, HOPE, and Bi-PE. Using the 2iPE strategy, we achieved near-complete purity of precise editing outcomes while retaining high efficiency of these prime editing systems. Importantly, in addition to positive effects on on-target precision, we demonstrate that 2iPE shows beneficial effect on prime editing off-targets.
DNA-PK and Polϴ inhibition applied to HDR in our previous study reduced by-products as well as increased DNA template integration efficiency18. Mechanistically, the inhibition of DNA-PK and Polϴ likely increases the accessibility of DSB ends to resection factors that initiate HDR. Consequently, HDR capacity is enhanced under inhibitory conditions. In contrast, our current work shows that while 2iPE reduces by-products, it generally does not increase the efficiency of precise prime editing outcomes. This is expected for canonical nickase-based prime editing where the factors involved in DSB repair do not compete with the prime editing mechanism and HDR factors are not involved4,7. Interestingly, we observe a similar trend with PEn, which also typically reaches its maximal efficiency before DNA-PK and Polϴ inhibition. This suggests different mechanisms of PEn and HDR, despite both of them being dependent on homology-based DSB repair. This can be possibly explained by different characteristics of the repair templates; HDR uses long flanking homology arms, while PEn employs a single short homologous flap.
An alternative, non-exclusive hypothesis for the observed differences in precise editing outcomes between 2iHDR and 2iPE is related to cell cycle phases. HDR is mainly restricted to the G2/S phases, meaning only a fraction of an unsynchronized cell population can perform HDR repair. Consequently, DNA-PK and Polϴ inhibition typically enhances HDR rates by favoring HDR-capable cells over those performing NHEJ or alt-EJ. Conversely, prime editing seems independent of the cell cycle—although this (in)dependency has not been fully established. Therefore, even without DNA-PK and Polϴ treatment, the prime editing pathway can be effectively engaged across most unsynchronized cells. While DNA-PK and Polϴ inhibition prevents indel and by-product formation during prime editing, it does not generally enhance prime editing efficiency.
Although our work already demonstrates the utility of combined inhibition of DNA-PK and PolƟ on numerous types of prime editing, we believe the 2iPE strategy can be employed even more broadly for additional types of prime editing modalities to reduce the generation of imprecise editing when they occur26,31,32. Given the similar DNA repair pathways involved in all prime editing modalities, they should equally benefit from the co-inhibition of the NHEJ and alt-EJ pathway to achieve purer editing outcomes.
Methods
Ethical statement
AstraZeneca has a governance framework and processes in place to ensure that commercial sources have appropriate patient consent and ethical approval in place for collection of the samples for research purposes including use by for-profit companies.
DNA and RNA constructs
PE2 and PE4 (pCMV-PEmax and pCMV-PEmax-P2A-hMLH1dn)4, uPEn (uPEn3)15, PEn16, and a C-terminal fusion of PEmax with truncated La protein (1–194), referred in the text as PE728, were generated by gene synthesis (Genscript). The sequence of PEn corresponds to PEmax with the H840A mutation reversed to the original histidine, restoring nuclease activity. All pegRNAs and ngRNAs were generated by gene synthesis and cloned in pMA vector together with an upstream U6 promoter (Invitrogen). All pegRNA sequences used in this study are listed in Supplementary Data 1. Capped PEmax and PEnmax mRNA was synthesized through T7-directed in vitro transcription using a linearized PEnmax DNA template. This process yielded fully modified mRNA, in which uridines were replaced with N1-Methyl-pseudouridine and the mRNA was capped using TriLink’s CleanCap-AG Cap1 analog. The mRNA was then purified using the MEGAClear transcription clean-up kit (ThermoFisher), and its purity was analyzed with a fragment analyzer (Agilent).
Small molecule compounds and drug treatments
DNA-PK inhibitor AZD7648 was purchased from MedChemExpress (HY-111783). PolQi1 (WO2021028643) and PolQi2 (WO2020243459) were provided by AstraZeneca (Gothenburg, SE). All compounds were dissolved in dimethyl sulfoxide (DMSO) at a stock concentration of 10 mM.
Cell culture
HEK293T cells (ATCC, CRL-3216) were maintained in DMEM-GlutaMAX with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. HeLa cells (ATCC, CCL-2) were cultured in MEM-HEPES-GlutaMAX with 10% FBS and 1% penicillin/streptomycin. All reagents were purchased from Gibco. HepG2 cells were maintained in MEM-HEPES-GlutaMAX with 10% FBS, 1X MEM NEAA, 1 mM sodium pyruvate, and 1% penicillin/streptomycin. Huh7 cells (Riken BRC, RCB1366) were maintained in DMEM-GlutaMAX with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Primary human hepatocytes (PHHs) from a deceased female donor (LifeNet Health, LOT: 2018274-01) were first thawed in pre-warmed Human Hepatocytes Thawing Medium (MED-HHTM4C-50ML, LifeNet Health), washed once with PBS, then re-suspended in 20 mL of Human Hepatocytes Plating Medium with supplement (MED-HHPM-250ML, MED-HHPMS-15ML, LifeNet Health). The following day, the medium was replaced with maintenance medium (William’s E medium, no phenol red (Gibco), 5% FBS, 0.1 µM Dexamethasone, 0.5% penicillin/streptomycin, 1X Insulin-Transferrin-Selenium (ITS, 41400-045 (Gibco), 2 mM L-Glutamine, and 15 mM HEPES pH 7.4) for the remainder of the experiment. iPSCs (AstraZeneca-generated cell line, R-iPSC Clone J, LineID: iPS.1) were maintained in Cellartis DEF-CS Culture System (Cat. No. Y30010). K562 cells (ATCC CRL-243) were maintained in RPMI 1640-GlutaMAX supplemented with 10% heat inactivated FBS, 20 mM HEPES, 1 mM sodium pyruvate and 1% penicillin/streptomycin. Cells were maintained at 37 °C in a 5% CO2 atmosphere and regularly tested for mycoplasma contamination. Cell line identity was authenticated through STR profiling (IDEXX BioAnalytics). Deoxynucleosides (SIGMA- dG (D0901) dT (T1895), dC (D0776), and dA (D8668)) were resuspended at 25 mM in water, filter-sterilized, and stored at −20 °C.
Transfection protocols
Based on pilot experiments with different transfection reagents and plasmid concentrations, the optimal conditions for highest transfection efficiency were used for each cell lines. One day prior to transfections 12,500 HEK293T or 15,000 HeLa cells were seeded per well of a 96-well plate. If applied, treatment with small molecule compounds or DMSO control was initiated 1 h before transfections and continued for 72 h until harvest. AZD7648, PolQi1 and PolQi2 were used at a final concentration of 1 µM, 3 µM and 3 µM, respectively. HEK293T cells were transfected with FuGENE HD (Promega) using a 6:1 Fugene-to-DNA ratio and 100 ng of total DNA per well of 96-well plate (75 ng of editor and 25 ng of pegRNA). HeLa cells were transfected with FuGENE HD (Promega) using a 3:1 Fugene-to-DNA ratio and 200 ng of total DNA per well of a 96-well plate (150 ng of editor and 50 ng of pegRNA). For PE5, an additional 8.3 ng (HEK293T) or 16.7 ng (HeLa) of ngRNA was added to the transfection mix, which is the same ratio of editor/pegRNA/ngRNA used previously for comparing editing efficiency between PEn and PE515. After transfection, plates were incubated for 72 h before harvesting the cells.
For PE3 experiments in HepG2, Huh7, iPSCs, and PHHs, synthetic pegRNA to mutate FANCF with +4T>G substitution and ngRNA were ordered from Genscript at 2 nmol scale where both ends were modified with phosphorothioated 2’ O-methyl RNA bases. HepG2 and Huh7 cells were seeded at 10,000 cells per well of a 96-well plate in their respective growth medium one day prior to transfection. PHH vials from donors were thawed, washed, and seeded on a 96-well plate (80,000 cells/well) with respective growth medium one day prior to the transfection. PHH medium was replaced the next day and supplemented with 50 µM of each deoxynucleosides (dA, dT, dG, dC), to improve prime editing efficiency33,34 before treatment and transfection. iPSCs were passaged one day before transfection. On the day of transfection, medium was changed with DEF-CS GF-3 (1:1000) approximately 4 h prior. Cells were washed with PBS, counted (14,000 cells/well, 96-well plate), and reverse transfected during seeding. The medium was replaced every 24 h post-transfection. All cells were transfected with Lipofectamine Messenger Max (Invitrogen) using 150 ng of mRNA, 2 pmol of pegRNA, and 0.67 pmol of ngRNA together with 0.3 µL of Messenger Max reagent diluted in 10 µL of OptiMEM (Gibco) per well. After transfection, plates were incubated for 72 h before harvesting the cells. For iPSCs medium was small molecule compounds or DMSO were added to the medium fresh every 24 h post transfection.
For nucleofection of K562 cells, 54 pmol PEn mRNA, 100 pmol pegRNA and 66 pmol ngRNA were combined in buffer SF and incubated at room temperature for 10 min to allow RNP formation. 5 µL of complexes were added to 15 µL of cells suspension (4 × 105 cells) in buffer SF, yielding a total volume of 20 µL. The suspension was electroporated using Lonza 4D (program FF-120). After nucleofection 200.000 cells/ well were plated in 200 µL of prewarmed growth medium in a 96 well plate. Treatment with small molecule compounds or DMSO control was initiated 1 h before transfections and continued for 72 h until harvest. AZD7648, PolQi1 and PolQi2 were used at a final concentration of 1 µM, 1.5 µM and 1.5 µM, respectively.
For TwinPE, HOPE, Bi-PE, and TwinPEn experiments, each pair of pegRNA were ordered as plasmid expression constructs. One day prior to transfection, 12,500 HEK293T cells were seeded per well of a 96-well plate. Treatment with small molecule compounds or DMSO control was initiated 1 h before transfections and continued for 72 h until harvest. AZD7648, PolQi1 and PolQi2 were used at a final concentration of 1 µM, 3 µM and 3 µM, respectively. HEK293T cells were transfected with FuGENE HD (Promega) using a 6:1 Fugene-to-DNA ratio and 100 ng of total DNA, 50 ng of editor and 25 ng of each pegRNAs per well (96-well plate). After transfection, plates were incubated for 72 h before harvesting the cells.
Genomic DNA extraction and amplicon sequencing
Cells were harvested using Quick Extract solution (Lucigen) or Fast Extract solution (VWR). Amplicons were generated with Phusion Flash High-Fidelity 2x Mastermix (F548, Thermo Scientific) or Q5 Hot Start High-Fidelity 2x Mastermix (M0492, NEB) in a 15 μL reaction, containing 1.5–2 μL of genomic DNA extract and 0.2 μM of target-specific primers with barcodes and adapters for next generation sequencing (NGS). All primer sequences are listed in Supplementary Data 2. PCR cycling conditions for Phusion Flash High-Fidelity 2x Mastermix were: 98 °C for 3 min, followed by 30 cycles of 98 °C for 10 s, 60 °C for 20 s, and 72 °C for 30 s. For Q5 Hot Start High-Fidelity 2x Mastermix the following PCR protocol was applied: 98 °C for 30 s, followed by 30 cycles of 98 °C for 10 s, 60 °C for 20 s, and 72 °C for 30 s, and final elongation at 72 °C for 2 min. For off-target analysis, amplicons were generated using Q5 Hot Start High-Fidelity 2x Mastermix with the following cycling conditions: 98 °C for 3 min, followed by 30 cycles of 98 °C for 10 s, 65 °C for 15 s, and 72 °C for 30 s, and final extension at 72 °C for 2 min. All amplicons were purified using HighPrep PCR Clean-up System (MagBio Genomics). The size, purity, and concentration of amplicons were determined using a fragment analyzer (Agilent). To add Illumina indexes to the amplicons, samples were subjected to a second round of PCR. Indexing PCR was performed using KAPA HiFi HotStart Ready Mix (Roche), 0.067 ng of PCR template and 0.5 µM of indexed primers in the total reaction volume of 25 µL. PCR cycling conditions were 72 °C for 3 min, 98 °C for 30 s, followed by 10 cycles of 98 °C for 10 s, 63 °C for 30 s, and 72 °C for 3 min, with a final extension at 72 °C for 5 min. Samples were purified with the HighPrep PCR Clean-up System (MagBio Genomics) and analyzed using a fragment analyzer (Agilent). Samples were quantified using a Qubit 4 Fluorometer (Life Technologies) and subjected to sequencing using Illumina NextSeq system according to the manufacturer’s instructions.
Bioinformatic analysis
Demultiplexing of the amplicon-seq data was performed using bcl2fastq software. The fastq files were analyzed using CRISPResso2 V2.2.12 in the prime editing mode35. Detailed parameters are listed in the Supplementary Data 3. Histograms in Supplementary Fig. 1 and allele frequency plots in Supplementary Figs. 1 and 4 were generated using CRISPResso2.
Statistics & reproducibility
Data visualization and statistical analysis were conducted using GraphPad Prism 9 (GraphPad Software, Inc.). Figure legends contain information on statistical tests, sample sizes, and P-values. No data were excluded from the analyses. No statistical method was used to predetermine sample size. The experiments were not randomized. The investigators were not blinded during experiments and outcome assessment. Detailed statistical analyses are listed in the Supplementary Data 4.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All details and data to support the findings of this study are part of the manuscript or available in a publicly accessible repository. Amplicon sequencing samples are described in Supplementary Data 5 and source sequencing data are available in the NCBI Sequence Read Archive database, BioProject accession code PRJNA1247113. Source data are provided with this paper.
References
Chen, P. J. & Liu, D. R. Prime editing for precise and highly versatile genome manipulation. Nat. Rev. Genet 24, 161–177 (2023).
Anzalone, A. V. et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019).
Liang, S. Q. et al. Genome-wide profiling of prime editor off-target sites in vitro and in vivo using PE-tag. Nat. Methods 20, 898–907 (2023).
Chen, P. J. et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell 184, 5635–5652.e5629 (2021).
Koeppel, J. et al. Prediction of prime editing insertion efficiencies using sequence features and DNA repair determinants. Nat. Biotechnol. 41, 1446–1456 (2023).
Yu, G. et al. Prediction of efficiencies for diverse prime editing systems in multiple cell types. Cell 186, 2256–2272.e2223 (2023).
Ferreira da Silva, J. et al. Prime editing efficiency and fidelity are enhanced in the absence of mismatch repair. Nat. Commun. 13, 760 (2022).
Habib, O., Habib, G., Hwang, G.-H. & Bae, S. Comprehensive analysis of prime editing outcomes in human embryonic stem cells. Nucleic Acids Res. 50, 1187–1197 (2022).
Anzalone, A. V. et al. Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing. Nat. Biotechnol. 40, 731–740 (2022).
Zhuang, Y. et al. Increasing the efficiency and precision of prime editing with guide RNA pairs. Nat. Chem. Biol. 18, 29–37 (2022).
Tao, R. et al. Bi-PE: bi-directional priming improves CRISPR/Cas9 prime editing in mammalian cells. Nucleic Acids Res. 50, 6423–6434 (2022).
Adikusuma, F. et al. Optimized nickase- and nuclease-based prime editing in human and mouse cells. Nucleic Acids Res. 49, 10785–10795 (2021).
Jiang, T., Zhang, X.-O., Weng, Z. & Xue, W. Deletion and replacement of long genomic sequences using prime editing. Nat. Biotechnol. https://doi.org/10.1038/s41587-021-01026-y (2021).
Kweon, J. et al. Targeted genomic translocations and inversions generated using a paired prime editing strategy. Mol. Ther. 31, 249–259 (2023).
Li, X. et al. Development of a versatile nuclease prime editor with upgraded precision. Nat. Commun. 14, 305 (2023).
Peterka, M. et al. Harnessing DSB repair to promote efficient homology-dependent and -independent prime editing. Nat. Commun. 13, 1240 (2022).
Tao, R. et al. WT-PE: prime editing with nuclease wild-type Cas9 enables versatile large-scale genome editing. Sig. Transduct. Target Ther. 7, 108 (2022).
Wimberger, S. et al. Simultaneous inhibition of DNA-PK and Polϴ improves integration efficiency and precision of genome editing. Nat. Commun. 14, 4761 (2023).
Schimmel, J. et al. Modulating mutational outcomes and improving precise gene editing at CRISPR-Cas9-induced breaks by chemical inhibition of end-joining pathways. Cell Rep. 42, 112019 (2023).
Riesenberg, S. et al. Efficient high-precision homology-directed repair-dependent genome editing by HDRobust. Nat. Methods 20, 1388–1399 (2023).
Arai, D. & Nakao, Y. Efficient biallelic knock-in in mouse embryonic stem cells by in vivo-linearization of donor and transient inhibition of DNA polymerase θ/DNA-PK. Sci. Rep. 11, 18132 (2021).
Fok, J. H. L. et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 10, 5065 (2019).
Wood, R. D. & Doublié, S. Genome protection by DNA polymerase θ. Annu. Rev. Genet. 56, 207–228 (2022).
Tsai, S. Q. et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197 (2015).
Kim, Y. H. et al. Sniper2L is a high-fidelity Cas9 variant with high activity. Nat. Chem. Biol. 19, 972–980 (2023).
Truong, D.-J. J. et al. Exonuclease-enhanced prime editors. Nat. Methods 21, 455–464 (2024).
Doman, J. L. et al. Phage-assisted evolution and protein engineering yield compact, efficient prime editors. Cell 186, 3983–4002.e3926 (2023).
Yan, J. et al. Improving prime editing with an endogenous small RNA-binding protein. Nature 628, 639–647 (2024).
Everette, K. A. et al. Ex vivo prime editing of patient haematopoietic stem cells rescues sickle-cell disease phenotypes after engraftment in mice. Nat. Biomed. Eng. 7, 616–628 (2023).
Pandey, S. et al. Efficient site-specific integration of large genes in mammalian cells via continuously evolved recombinases and prime editing. Nat. Biomed. Eng. 9, 22–39 (2025).
Xiong, Y. et al. EXPERT expands prime editing efficiency and range of large fragment edits. Nat. Commun. 16, 1592 (2025).
Liang, R. et al. Prime editing using CRISPR-Cas12a and circular RNAs in human cells. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-02095-x (2024).
Levesque, S., Cosentino, A., Verma, A., Genovese, P. & Bauer, D. E. Enhancing prime editing in hematopoietic stem and progenitor cells by modulating nucleotide metabolism. Nat. Biotechnol. https://doi.org/10.1038/s41587-024-02266-4 (2024).
Liu, P. et al. Increasing intracellular dNTP levels improves prime editing efficiency. Nat. Biotechnol. https://doi.org/10.1038/s41587-024-02405-x (2024).
Clement, K. et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226 (2019).
Acknowledgements
We thank the AstraZeneca Discovery Sciences Genome Engineering team for support and input on this work. We thank Shalini Andersson and Steve Rees for supporting this project. We are grateful to the AstraZeneca NGS & Transcriptomic team for support with targeted amplicon sequencing. P.A. is funded by the European Union’s Horizon Europe research and innovation programme under grant agreement No 101057659 (EDITSCD). L.D. is funded by Promega Corporation. This Project has received funding by the European Union. Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or the European Research Executive Agency. Neither the European Union nor the granting authority can be held responsible for them.
Author information
Authors and Affiliations
Contributions
L.D., P.A., N.A., M.M. and M.P. conceptualized the study and prepared the manuscript. L.D., P.A., N.A. and M.P. performed most of the experimental work and analysis with support from A.M., N.S., H.M., P.P.H., S.M., G.T., S.W. and M.F.
Corresponding authors
Ethics declarations
Competing interests
All authors are current or former employees of AstraZeneca and may be AstraZeneca shareholders. L.D. is an employee of Promega corporation. AstraZeneca filed patents related to this work (WO2021204877A2 and WO2023052508A2).
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dacquay, L.C., Antoniou, P., Mentani, A. et al. Dual inhibition of DNA-PK and Polϴ boosts precision of diverse prime editing systems. Nat Commun 16, 4290 (2025). https://doi.org/10.1038/s41467-025-59708-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59708-z
