
Abstract
Electrochemically mediated carbon capture presents an energy-efficient and cost-effective strategy to combat climate change due to its ability to utilize renewable energy and operate at ambient conditions. However, many current approaches suffer from operational instability and limited scalability potential due to a lack of reliable, low-cost redox-active absorbent materials. Here, we introduce a class of chemically robust and economical redox-tunable Brønsted acids to electrify amine carbon capture. The redox-tunable acids exhibit a reversible tunability in pKa spanning over 20 units in organic solvents in response to electrochemical potential, thereby enabling the regeneration of classic amines for CO2 separation via proton-coupled electron transfer. Remarkably, the RAs maintain their chemical integrity for over 400 h of operation in a symmetric carbon capture flow cell under 10% CO2 and 21% O2 at ambient temperature and pressure. By harnessing electrification, our approach can effectively mitigate shortcomings inherent to thermochemical carbon capture processes, facilitating a more sustainable drop-in replacement for incumbent amine scrubbing.
Similar content being viewed by others


Introduction
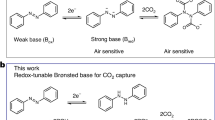
The urgent need to address climate change has sparked a growing interest in carbon capture technologies, which play a crucial role in reducing greenhouse gas emissions. Currently, the most mature method for point source carbon capture is amine scrubbing, which employs aqueous amines as absorbents and thermal swing processes to regulate CO2 separation (Fig. 1a)1,2,3,4. In this approach, amines are regenerated by stripping with water vapor at 100–120 °C, leading to a multitude of challenges, including low energy efficiency, fast absorbent loss via evaporation or degradation, equipment corrosion, and complexities in integrating this technology with existing infrastructures. Recent efforts in developing water-lean CO2 absorbents are attractive as they can enhance CO2 capacity through different binding mechanisms from aqueous systems5 and mitigate the evaporative and sensible heat losses by utilizing non-volatile organic solvents6,7,8. Nevertheless, these nonaqueous absorbents still require thermal regeneration.

a Conventional amine scrubbing via thermal-swing processes and previous aqueous/nonaqueous electrochemical amine regeneration based on heterogeneous electrochemistry involving heavy metal species. b Chemical versatility of redox-tunable Brønsted acids (RAs) and schematic showing the mechanism of redox-tunable Brønsted acids mediated amine regeneration (RAMAR).
The increasing availability of renewable electricity has created a compelling opportunity to enhance the efficiency and sustainability of carbon capture through electrification9,10, as highlighted in recent reviews11,12,13,14,15,16,17,18,19. Electrochemically meditated carbon capture (EMCC) can be achieved via various mechanisms, such as redox-driven reversible CO2 complexation using organic sorbents20,21,22,23,24 or pH swing25,26,27,28,29. Unlike conventional thermochemical methods, EMCC is compatible with intermittent renewable energy and can operate under mild, isothermal conditions. Moreover, the modular nature of EMCC allows the development of plug-and-play systems, enhancing their adaptability to the diverse and multiscale demands of carbon capture. However, current redox-active materials used in EMCC face fundamental limitations. Specifically, existing redox-active CO2 sorbents and pH mediators are often sensitive to molecular oxygen (O2) in their activated (reduced) form22,30,31, and parasitic reactions during repeated capture-release cycles lead to the accumulation of unstable species and rapid sorbent degradation21,23. Additionally, costly synthetic procedures limit the scalability of current EMCC materials for scalable implementations.
To capitalize on the exceptional CO2 removal efficiencies and optimized process equipment of amine absorption while reducing parasitic energy and capital costs, we present a scale-transferrable approach to electrify amine scrubbing, termed redox-tunable (Brønsted) acid mediated amine regeneration (RAMAR). RAMAR leverages Wurster-type compounds as redox-tunable Brønsted acids (RAs) to modulate CO2 capture and release across a broad spectrum of classic amines (Fig. 1b). In their oxidized state, RAs function as strong Brønsted acids (protonated iminium), facilitating CO2 release by protonating amine-CO2 adducts. Conversely, in the reduced state, RAs transition into potent Brønsted bases (deprotonated azanide), effectively regenerating amines for CO2 capture by abstracting protons from ammoniums. At the molecular level, RAs exhibit highly reversible pKa modulation spanning over 20 units in nonaqueous solvents through a redox cycle, which is the largest tunability reported among known stimuli-responsive acids/bases to our best knowledge32,33,34. This wide dynamic pKa range is the key enabler for electrochemical amine regeneration. Moreover, RAs demonstrate exceptional redox stability and molecular designability and are synthesized and isolated in pure form by facile chemical methods. At the process level, unlike previous attempts at electrifying amine carbon capture, such as those using copper redox35,36,37,38,39 and dual-salt cation swing40 (Fig. 1a), RAMAR eliminates the need for heavy metals and poorly reversible electroplating/intercalation chemistries. Importantly, all species involved in the RAMAR process are thermodynamically stable in ambient conditions (especially O2), thereby offering opportunities for practical implementation in complex gas environments.
Herein, RAMAR is evaluated in symmetric, flow-based carbon capture prototypes, where the electrochemical cell design can enhance the energy efficiency and cycling stability compared to many previous EMCC configurations29. Using the readily available N1,N4-diphenylbenzene-1,4-diamine (RA 1), we showcase continuous CO2 capture-release operation over 80 cycles (400 h) under realistic aerobic conditions (10% CO2 and 21% O2) with near-unity electrochemical efficiency and nearly half the energy consumption compared to previous EMCC processes. The RAMAR mechanism lays the foundation for developing efficient EMCC systems and can potentially serve as a drop-in replacement for amine scrubbing to accelerate the widespread adoption of carbon capture technologies.
Results
Validating and understanding redox-tunable Brønsted acids
Governed by the hard-soft acid-base theory, alkaline CO2 sorbents, such as sp3-N amines, form strong adducts with CO2 via acid-base interaction, thus necessitating substantial energy input to break the N–C bond for CO2 release. In principle, chemical acidification and subsequent basification could liberate CO2 and regenerate amines for reversible CO2 separation at ambient temperatures. To avoid introducing non-recyclable chemical reagents and additional separation steps, pH swing strategies employing redox-active mediators, which can reversibly bind and release protons at distinct electrochemical potentials, present a promising avenue for electrified amine scrubbing. Specifically, the mediators should have much lower pKa values than amines in their oxidized state while exhibiting much higher pKa values than amines in their reduced state. Nonetheless, reported redox-active pH mediators do not have the required dynamic pKa range28,29,41,42. To bridge this knowledge gap, we judiciously explore a class of Wurster-type compounds to investigate their potential as redox-tunable proton acceptors/donors in the presence of amines. These compounds are secondary amines/azanides (proton acceptors) in the reduced state and iminiums/imines (proton donors) in the oxidized state, which are considered strong bases and acids, respectively. Moreover, RAs are stable and isolatable in both reduced and oxidized forms via straightforward synthetic steps (see Synthetic Procedures in Methods and Supplementary Figs. 1–8). We postulate that oxidizing RAs in the presence of CO2-loaded amines can result in the donation of protons to amines for CO2 release, whereas subsequent reduction of oxidized RAs can attract protons from ammonium to regenerate amines for CO2 capture (Fig. 2a).

a Reversible proton-coupled oxidation/reduction reactions between 1 and 1-ox in the presence of MEA. b Crude 1H NMR of 1 and 1-ox after bulk electrolysis in the presence of MEA under 10% CO2 atmosphere. Bulk electrochemical oxidation of 1 affords 1-ox identical to chemical oxidation methods with negligible impurities. c CV curves of 1 and 1-ox under various conditions. 1 or 1-ox (5 mM) was measured in DMSO saturated with N2 and with tetrabutylammonium hexafluorophosphate (NBu4PF6, 100 mM) as the supporting salt. 1 and 1-ox were scanned anodically and cathodically at a rate of 50 mV s−1, respectively. Source data for Fig. 2 are provided as a Source Data file.
To validate the proposed chemistry, we conducted bulk electrolysis of compound 1 in deuterated dimethyl sulfoxide (DMSO-d6) in the presence of two molar equivalents of monoethanolamine (MEA) under 10% CO2 (Fig. 2b and Supplementary Figs. 9 and 10). The crude proton nuclear magnetic resonance (1H NMR) spectrum after bulk electrochemical oxidation of 1 suggests the full conversion to N1,N4-diphenylcyclohexa-2,5-diene-1,4-diimine (1-ox) with negligible impurities, as compared to the 1H NMR of 1-ox derived from the chemical oxidation method43. Besides, the formation of monoethanolammonium indicates successful proton transfer from 1 to MEA after oxidation (see Supplementary Figs. 11–13 for the NMR spectra of pristine MEA, its protonated form, and carbamic acid/carbamate form). Furthermore, the reaction exhibits high chemical reversibility, as suggested by the complete regeneration of 1 and carbamic acid/carbamate in the crude 1H NMR after subsequent bulk electrochemical reduction of 1-ox.
Cyclic voltammetry (CV) experiments were also conducted on 1 and 1-ox under various conditions (Fig. 2c, see Supplementary Fig. 14 for the corresponding chemical reaction under each condition). CV reveals an irreversible electrochemical reduction process of the imine compound 1-ox in the absence of free protons (curve i). The rather negative reduction potential of 1-ox indicates the strong basicity of 1’s conjugate base (deprotonated form). By adding two molar equivalents of superacidic bistriflimidic acid (HTFSI) to the 1-ox solution, the CV of 1-ox (cathodic scan, curve ii) becomes electrochemically reversible and mirrors that of 1 (anodic scan, curve iii). The results strongly evidence the reversible electrochemical interconversion between 1 and protonated 1-ox (iminium form). In the presence of a stoichiometric amount of MEA, the CV of 1 (anodic scan, curve iv) becomes electrochemically irreversible again, accompanied by cathodically shifted reduction and oxidation potentials compared to curve iii. This implies a proton-coupled electron transfer (PCET) process, where the basic MEA attracts protons from 1 via hydrogen bonding and facilitates its oxidation but impedes the proton transfer to 1-ox and thus negatively shifts its reduction potential. Similarly, the cathodic scan of 1-ox conducted with a stoichiometric amount of MEA and HTFSI (curve v) exhibits an identical CV behaviour to that observed in curve iv, further confirming the reversibility of the reaction shown in Fig. 2a.
We further compared the CV of 1 in the absence and presence of MEA under N2 and CO2 atmospheres (Supplementary Fig. 15a). As mentioned earlier, the addition of MEA breaks the redox reversibility of 1 through the introduction of a PCET process, with both cathodically shifted onset potentials for reduction (−0.9 V vs. Fc+/0) and oxidation (−0.4 V vs. Fc+/0). Interestingly, when the solution is saturated with pure CO2, the oxidation onset potential returns to −0.08 V vs. Fc+/0, identical to that in the absence of MEA. This indicates the consumption of free MEA by forming CO2 adducts, which eliminates the hydrogen bonding that promotes oxidation. Alternatively, the presence of CO2 anodically shifts the reduction potential of 1 with MEA compared to that under N2, which we attribute to the amine-CO2 binding that facilitates proton transfer from ammonium to 1-ox. The CV of 1 was also tested and analyzed at lower CO2 partial pressures (10–20%) (Supplementary Figs. 15a and 16 and Supplementary Note 1).
To gain fundamental insights into the PCET process of 1 in the absence/presence of MEA in DMSO under N2 and CO2, we proposed the nine-state equilibrium chart and calculated the potential of each electron transfer step and the Gibbs free energy change of each proton transfer step using density functional theory (DFT, Fig. 3a). We consider three scenarios for the proton transfer steps (Supplementary Fig. 17), including proton transfer between different forms of 1 and the DMSO solvent molecule (reaction i), MEA (reaction ii), and MEA-CO2 adduct (reaction iii), which represents PCET without amine, with amine under N2, and with amine under CO2, respectively. The blue-shaded region in Fig. 3a shows the most energetically favourable reaction pathway in the presence of MEA, where the two electron transfer steps occur at the same potential, corroborating our experimental CV observation. Besides, the proton transfer steps show the lowest free energy in the presence of MEA under N2 (reaction ii), which also agrees well with the most negatively shifted CV oxidation potential. Furthermore, we calculated the pKa of each state of 1, revealing a wide swing between −7.3 and 30.1 (Fig. 3a).

a PCET pathways under conditions without amine (i), with MEA and N2 (ii), and with MEA and CO2 (iii). The potentials (vs. Fc+/0) for the electron transfer steps (E), the changes in Gibbs free energy for the proton transfer steps (∆G), and the pKa values of each state are calculated by DFT and compared with experimental results if available. The most favourable pathway in the presence of proton donors with varying pKa is shaded in pink (1), blue (2), green (3), and yellow (4), corresponding to the four different regions in the pKa-potential diagram in (b). b Reduction potentials of 1-ox in the presence of proton donors with varying pKa (−14.9 to 32 in DMSO). The data was obtained by the average of three independent CV measurements in the presence of proton donors under N2. The diagram is divided into four regions, which undergo different reaction pathways highlighted in (a). Source data for Fig. 3 are provided as a Source Data file.
To experimentally estimate the pKa values of 1 and the conjugate acid of 1-ox, we measured the CV of 1-ox in DMSO under N2 in the presence of two molar equivalents of acids (proton donors) with various pKa (−14.9 to 31.3 in DMSO)44,45. The pKa value of each proton donor used is shown in Supplementary Fig. 18. The reduction potential-pKa diagram reveals two pKa-independent regions and two pKa-dependent regions in between (Fig. 3b). The independent regions indicate the upper and lower bound pKa of the system as 1.6 (pK”a2) and 21.1 (pKa2), respectively (see Fig. 3a for the pKa labels of the different forms of 1). The pKa-dependent regions exhibit two different slopes of ~113.2 mV pKa−1 (1.6 to 7.47) and ~45.4 mV pKa−1 (7.47 to 21.1), corresponding to two protons and one proton transfer per electron transfer, respectively. The deviation from the idealized Nernstian slope is likely caused by homoconjugation of the acids in proton-accepting DMSO44,45. The pKa tunability range of RA 1 is, to the best of our knowledge, the widest among known stimuli-responsive acids and bases. In comparison, typical photoacids exhibit pKa shifts of only 1–3 units33, while photobases show shifts in the range of 1–9 units32.
Based on the reduction potential-pKa diagram, the PCET pathways of 1/1-ox under different pKa environments are proposed in Supplementary Fig. 18. For proton donors with pKa below 1.6 or above 21.1, no proton transfer is involved in the redox process. This is because protons cannot dissociate from 1-ox ([1-ox + 2H]2+, iminium form) under strongly acidic conditions, while protons cannot bind to 1 ([1 − 2H]2−, azanide form) under strongly basic conditions. In the region of 1.6 <pKa < 7.5, one electron reduction of 1-ox can result in doubly protonated [1’ + H]+ due to the relatively strong acidic environment and the strong basicity of 1’. In the region of 7.5 <pKa < 21.1, an overall two-proton-two-electron-transfer process dominates the redox reactions between 1 and 1-ox.
With an onset reduction potential of −0.9 V (vs. Fc+/0), MEA falls into the pKa region between 7.5 and 21.1, implying that CO2 separation using 1 and MEA operates under a two-proton-two-electron mechanism. Furthermore, Fig. 3b provides a guideline for pairing suitable amines with RAs by matching their pKa values. For energy efficiency considerations, it is ideal to drive amine regeneration under a one-electron-two-proton mechanism. This requires selecting an amine with lower Brønsted basicity (low pKa for its conjugate acid) but sufficient nucleophilicity for CO2 absorption. However, such amines rarely exist, as strong nucleophiles are usually strong bases. To circumvent this challenge, molecular engineering of RAs may potentially shift the one-electron-two-proton region to larger pKa values to encompass the common amine absorbents.
Expanding the chemical space of RAs and candidate amines
With the RAMAR mechanism validated using model compound 1 and MEA, we further expanded the chemical space of RAs to tailor their physicochemical properties. As shown in Fig. 1b, the Wurster-type structural motif serves as a versatile platform for molecular engineering. Specifically, we demonstrate three additional structures with the ease of encoding functionality at N-substitutes and side chains, which are N1,N4-di(naphthalen-2-yl)benzene-1,4-diamine (2), N1,N4-di-sec-butylbenzene-1,4-diamine (3), and diethyl 2,5-bis(phenylamino)terephthalate (4). These compounds are either commercially available or can be obtained via facile synthetic steps from simple precursors (see Synthetic Procedures in Methods).
The redox potential and the reversibility of RAs in the presence of amine can be tuned by induction and resonance effects through structural modification, respectively (Supplementary Fig. 15 and Supplementary Table 1). Specifically, electron-donating or electron-withdrawing groups (EDGs or EWGs) can be introduced into the Wurster-type motif, shifting the redox potential cathodically or anodically governed by the free-energy relationship in the Hammett equation46. Interestingly, the redox reversibility (the voltage gap between oxidation and reduction) can be enhanced by adding EDGs or elongating RA conjugation, thereby lowering the theoretical work of CO2 separation. In a practical RAMAR process, RAs are oxidized under a CO2-rich atmosphere to desorb concentrated CO2 and reduced under a CO2-lean atmosphere to regenerate amines. In the presence of MEA, the CV peak difference between oxidation under 100% CO2 and reduction under N2 are 0.76, 0.68, 0.57, and 0.96 V for 1, 2, 3, and 4, respectively (Supplementary Fig. 15 and Supplementary Table 1). A smaller voltage gap via molecular design corresponds to a lower theoretical energy consumption.
For practical applications, RAs need to be stable against O2, which is a common contaminant in the gas streams of interest for carbon capture. Most conventional redox-active CO2 sorbents are O2 sensitive in their reduced states, causing undesired parasitic reactions21,29,31. In stark contrast, 1 exhibits high robustness towards O2, suggested by CV under simulated flue gas, which contains 10% CO2 and 3% O2 (Supplementary Fig. 19). In a DMSO solution with 100 mM LiTFSI, pure O2 exhibits an onset reduction potential of −1.2 V (vs. Fc+/0), giving rise to LiO2/Li2O2 products. In the presence of CO2, the reduction potential of O2 shifts anodically to −1.1 V (vs. Fc+/0), probably due to the formation of peroxocarbonate or percarbonate species. This chemical property challenges the design of redox-tunable CO2 sorbents for EMCC under practical operating conditions, as the O2 reduction reaction may occur alongside or even prior to sorbent reduction/activation. Redox-active CO2 sorbents can also act as redox mediators to further promote O2 reduction47. So far, sorbents with the most positive redox potentials are 4,4’-azopyridine (−1.1 V vs. Fc+/0 in DMSO) and isoindigos (−1.2 to −0.9 V vs. Fc+/0 in DMF)23,24. To our delight, CV reveals that 1 possesses an onset reduction potential of −0.75 V (vs. Fc+/0) in the presence of MEA in DMSO and has no redox mediation effect on O2 reduction even with CO2 present. Furthermore, 1 is thermodynamically robust against O2. 1H NMR shows 1 maintains its chemical integrity after being purged and stored in a pure O2 atmosphere in DMSO after 7 days (Supplementary Fig. 20a).
As an additional note on sorbent stability, the RAMAR chemistry minimizes parasitic reactions by decoupling electron transfer and CO2 complexation. That is, amines are exclusively responsible for CO2 capture/release, while RAs serve as proton acceptors/donors modulated by electrochemical potentials. This is fundamentally different from conventional redox-active CO2 sorbents11,12,16, where electron transfer and CO2 complexation occur at the same reaction site, causing sorbent reversibility and O2 stability issues.
To facilitate efficient, flow-based EMCC technologies, RAs need to be highly soluble in the electrolyte solvent to ensure practical CO2 separation capacity (Supplementary Fig. 20b). The solubility of different RAs is summarized in Supplementary Table 2. It is noteworthy that 1 possesses a high solubility of 2.68 M in pure DMSO, and the coexistence of supporting salts and amines has little impact on its solubility. For example, the solubility of 1 is maintained at 2.71, 2.43, and 1.40 M in DMSO solutions comprising 0.5 M LiTFSI, 0.5 M LiTFSI with 0.5 M MEA, and 0.5 M LiTFSI with 5 M MEA, respectively. The solubility of RAs can be further improved by molecular engineering; replacing phenyl groups at N-substitutes with sec-butyl groups gives rise to 3, which is a liquid at room temperature and fully miscible with the electrolyte.
In addition to RAs, optimizing the physicochemical properties of amine absorbents and their CO2 adducts is equally important for RAMAR practicality. We expanded the chemical space of amine absorbents beyond MEA by measuring the CV of 1 in the presence of different amines, including diethanolamine (DEA), morpholine (Mph), tetrahydroisoquinoline (THIQ), piperazine (Pz), and 2,2′-(ethylenedioxy)bis(ethylamine) (DODA) (Fig. 4). To our delight, these amines exhibit CV curves similar to that of MEA, suggesting the generalizability of the RAMAR chemistry. The reduction/oxidation peaks shift slightly for each amine due to their different basicity, which are summarized in Supplementary Table 3.

a Monoethanolamine (MEA). b Diethanolamine (DEA). c Morpholine (Mph). d Tetrahydroisoquinoline (THIQ). e Piperazine (Pz). f 2,2′-(ethylenedioxy)bis(ethylamine) (DODA). CV of 1 (5 mM) was measured in DMSO containing amine (10 mM), NBu4PF6 (100 mM) as the support salt under N2, 10% CO2, and pure CO2, respectively. 1 was scanned anodically at a rate of 50 mV s−1. Source data for Fig. 4 are provided as a Source Data file.
Amines with sp3-N are usually considered susceptible to oxidation. Therefore, the electrochemical stability windows of amine solutions were determined by linear sweep voltammetry under both N2 and CO2 (Supplementary Fig. 21). Primary amines possess greater oxidative stability than secondary amines in N2 atmosphere, and CO2 stabilizes amines from oxidative decomposition due to adduct formation, as indicated by the positively shifted oxidation potentials. As one of the most readily available amines, MEA displays a stability window from −2.72 to 0.9 V (vs. Fc+/0) under CO2, which is sufficient to cover the redox range of 1. Furthermore, we observed that monoamines exhibit decent solubility in their CO2 adduct forms. Using MEA and THIQ as examples, they are liquid and fully miscible with pure DMSO or DMSO with 0.5 M LiTFSI. When saturated with CO2, the amine solutions (amine:DMSO = 1:1 v/v) exhibit no obvious precipitation (Supplementary Fig. 20c). On the contrary, we found that the CO2 adducts of diamines, like Pz and DODA, possess poor solubility in DMSO (Supplementary Fig. 20d). White precipitates quickly formed when purging 10% CO2 into a solution of Pz or DODA (0.5 M) in DMSO. This is probably caused by the formation of zwitterionic species or hydrogen bonded polymers of anionic carbamates and cationic ammoniums48. Therefore, primary monoamines are considered the best candidates for RAMAR.
Finally, we tested the kinematic viscosity of the RA-amine mixtures under practical CO2 absorption conditions (Supplementary Table 4). For the case of 0.5 M LiTFSI, 0.5 M 1, 0.1 M 1-ox, 0.2 M HTFSI, and 0.5 M MEA in DMSO, the 10% CO2 saturated mixture displays as a free-flow liquid with a kinematic viscosity of 6.76 mm2 s−1 at 25 °C, which is ~3 times that of pure DMSO and ~5 times that of aqueous MEA solution (0.5 M, saturated with 10% CO2). We also determined the viscosity of highly concentrated amine solutions (5 M MEA) saturated with 10% CO2. A DMSO solution containing 5 M MEA and 0.5 M LiTFSI and an aqueous 5 M MEA solution exhibit comparable kinematic viscosities of 4.28 and 2.44 mm2 s−1 at 25 °C, respectively. The decent viscosity of our RAMAR formulation ensures reasonable pumping costs and favourable mass transfer kinetics for CO2 capture.
Determining the CO2 absorption capacity in RAMAR
Although the reaction between CO2 and amines in aqueous environments has been extensively studied49, the amine-CO2 adducts are less understood in nonaqueous media. A previous report revealed that amines, such as 2-ethoxyethylamine, can form both carbamic acid and carbamate adducts with CO2 in DMSO50. The former involves a one-amine-one-CO2 formation process, while the latter requires two amine equivalents and one captured CO2 to generate the carbamate-ammonium dyads (Fig. 5a). Therefore, it is of great importance to understand the amine adduct composition to determine the CO2 absorption capacity in RAMAR.

a Regeneration of carbamic acid adduct via 1H+−1CO2 pathway and carbamate adduct via 2H+−1CO2 pathway. b 1H NMR study of the amine-CO2 reaction pathways in DMSO as a factor of amine concentration, supporting salt, and CO2 partial pressure. Black, MEA (50 mM) under N2; red, MEA (50 mM), HTFSI (50 mM) under N2; blue, MEA (50 mM) saturated with 100% CO2; green, MEA (50 mM) in DMSO with LiTFSI (500 mM) saturated with 100% CO2; purple, MEA (500 mM) in DMSO with LiTFSI (500 mM) saturated with 100% CO2; yellow, MEA (500 mM) in DMSO with LiTFSI (500 mM) saturated with 10% CO2. c Illustration of the titration experiment setup. The solution was purged by a stream of 20% CO2 (balanced with N2) at a flow rate of 10 sccm. The CO2 level at the exit of the septum-capped vial was monitored by an IR-based CO2 sensor. For the CO2 absorption experiment, various amounts of MEA were added to the vial containing either neat DMSO or DMSO with 0.5 M LiTFSI. For the CO2 desorption experiment, a solution of MEA (500 mM) and LiTFSI (500 mM) in DMSO was saturated with 20% CO2 and then added with various amounts of TfOH. For both experiments, the CO2 level was allowed to return to 20% before the next addition. d CO2 absorption capacity at various MEA concentrations. Grey, pure DMSO solvent; red, DMSO with 500 mM LiTFSI as the supporting salt. e CO2 desorption capacity with various concentrations of TfOH added. Source data for Fig. 5 are provided as a Source Data file.
We investigated the interaction between MEA and CO2 in DMSO via a combination of NMR spectroscopy and titration experiments. 1H NMR reveals that the carbamic acid (a’, 6.57, b’, 3.36, and c’, 2.98 ppm) pathway dominates in dilute MEA solution (50 mM) without supporting salts under 100% CO2 due to the absence of ammonium species (b, 3.54 and c, 2.82 ppm) (Fig. 5b). To mimic RAMAR working conditions, LiTFSI (500 mM) was added as the supporting salt to examine its effect on CO2 absorption pathways. As evidenced by the appearance of ammonium species in 1H NMR, Li+ acts as a Lewis acid to stabilize the carbamate adduct, which lowers the CO2 absorption capacity40,50. Similarly, increasing the MEA concentration (from 50 mM to 500 mM) also induces the carbamate pathway via deprotonating the carbamic acid by free amines. Lastly, reducing the CO2 partial pressure (from 100% to 10%) promotes the formation of carbamate-ammonium mixture due to decreased CO2 availability in the solution. Under simulated RAMAR working conditions with a 10% CO2 feed, the DMSO solution (with 500 mM MEA and 500 mM LiTFSI) exhibits a nearly 1:1 mole ratio of the carbamic acid and carbamate adduct by comparing the integration of the carbamic acid/carbamate peak (c’, 2.98 ppm) and ammonium peak (c, 2.82 pm), equivalent to 0.75 CO2 absorbed per amino group.
Titration experiments (Fig. 5c) were conducted to quantitatively assess the CO2 absorption capacity of MEA and the subsequent CO2 release facilitated by triflic acid (TfOH). Under a constant influx of 20% CO2 (balanced with N2) at a flow rate of 10 standard cubic centimetres per minute (sccm), various amounts of MEA were injected into pure DMSO or DMSO containing 0.5 M LiTFSI (pre-saturated with 20% CO2). The CO2 concentration at the outlet was monitored using an IR-based CO2 sensor to determine the net amount of CO2 absorbed. Similarly, for CO2 desorption, various amounts of TfOH were injected into a DMSO solution containing 0.5 M MEA and 0.5 M LiTFSI (pre-saturated with 20% CO2), followed by monitoring of the outlet CO2 level. As depicted in Supplementary Fig. 22, immediate changes in CO2 readings were observed upon the addition of MEA or TfOH, indicating CO2 capture and release, respectively. The total amounts of CO2 captured/released per equivalent of MEA/TfOH are plotted against the final concentrations of MEA/TfOH in Fig. 5d, e.
Consistent with NMR, titration experiments suggest a lower CO2 absorption capacity in DMSO containing LiTFSI compared to pure DMSO, alongside a notable reduction in CO2 absorption capacity with increasing MEA concentration. Figure 5e reveals that the concentration of superacid has a negligible impact on CO2 release capacity. Specifically, a DMSO solution with 0.5 M MEA and 0.5 M LiTFSI can release 0.6 equivalent of CO2 for every equivalent of TfOH added. According to Fig. 3b, 1-ox’s conjugate acid exhibits a comparable pKa to TfOH. Therefore, we posit that our RAMAR formulation (0.5 M MEA and 0.5 M LiTFSI in DMSO) has a theoretical capacity to separate 0.6 CO2 per monoamine. That is, each electron input to RAs will generate one proton, which can liberate 0.6 CO2. This theoretical value will be used subsequently to assess the actual performance of our RAMAR prototype.
Evaluating RAMAR performance in symmetric flow cells
A prominent challenge facing nonaqueous EMCC devices is the crossover of active species through the ion exchange membrane, stemming from the lack of membranes compatible with organic electrolytes16,51,52, which compromises the cycling stability23,24. Previous studies from others and us21,23,24,28 utilize counter electrolytes such as ferrocene, leading to low electrochemical (Coulombic) efficiency (typically <90%) and incomplete regeneration of redox-active sorbents, largely due to counter electrolyte crossover. The use of counter electrolytes also gives rise to large overpotentials (energy input) between CO2 capture and release steps. Symmetric flow cells, which utilize the same material in different redox states as both catholyte and anolyte, provide a plausible solution for mitigating crossover. Nevertheless, such a concept has yet to be demonstrated in nonaqueous EMCC due to the chemical susceptibility of redox-active CO2 carriers in their reduced state. Consequently, prior to assembling symmetric cells, conventional redox-active CO2 carriers must undergo electrochemical reduction against counter electrolytes in flow cells, a process that is time-consuming, complicates reactor design, introduces undesired impurities from counter electrolyte crossover, and increases the overall cost.
In contrast, our RA molecules are stable and isolatable in both reduced and oxidized forms. The synthetic feasibility of the oxidized counterparts of RAs streamlines the assembly of symmetric flow cells. Furthermore, we compared the crossover rate of 1 and ferrocene in LiTFSI DMSO solution across Nafion 212, a commonly used membrane in EMCC flow cells (Supplementary Fig. 23 and Supplementary Table 5)23. Interestingly, the crossover rate of 1 is 20 times slower than ferrocene, a characteristic that is conducive to achieving high CO2 separation efficiency in symmetric cells.
As a proof of concept, we built a bench-scale symmetric flow cell to evaluate the performance of RAMAR via cyclic CO2 capture-release operations (Supplementary Fig. 24). In the working electrode tank (WE), the electrolyte (a mixture of RA in the reduced and oxidized forms) was first reduced and activated for CO2 capture. In the counter electrode tank (CE), the electrolyte was first oxidized and deactivated for CO2 release. The polarity of the applied potential was switched periodically.
We started performance evaluation using RA 1 and MEA (Fig. 6, 125 mM 1, 125 mM 1-ox, 500 mM MEA). In a typical cycle, under an influx of 10% CO2 at a flow rate of 2 sccm, WE and CE were first reduced and oxidized at 10 mA (2 mA cm−2) for 90 min, respectively. The CO2 readings at the gas outlet of WE and CE confirmed CO2 capture and release. Subsequently, the CO2 readings of WE and CE were allowed to return to the baseline by halting the current for 90 min. WE and CE were then oxidized and reduced at 10 mA for 90 minutes, respectively, followed by another 90 min of rest.

a CO2 readings at the outlet of the WE tank (blue) and CE tank (red), respectively, over 21 repeating cycles for 126-h operation. The CO2 sensor at the CE tank lost connection to the computer at ~40 h, resulting in a data loss of ~2 h during the 7th cycle of CO2 capture. b, c CO2 readings for selected capture-release cycles with the corresponding cumulative amount of CO2 captured/released in WE (b) and CE (c). The yellow regions highlight the capture and release steps. For each separation cycle, both sorbent tanks were reduced/oxidized at 10 mA (2 mA cm−2) for 90 min followed by a 90 min rest. d, e Performance metrics of WE (d) and CE (e). WE and CE were composed of 10 ml DMSO solution containing 125 mM 1, 125 mM 1-ox, 250 mM HTFSI, 500 mM MEA, and 500 mM LiTFSI. Both tanks were stirred and continuously purged with 10% CO2 (balanced with N2) at a flow rate of 2 sccm. Source data for Fig. 6 are provided as a Source Data file.
Our initial test exhibited consistent CO2 capture-release behaviours over repeated cycling (Fig. 6a–c). 1 demonstrated fully reversible operation with an electrochemical efficiency of 100% (Fig. 6d, e). Furthermore, the system exhibited CO2 capacity utilization efficiency close to unity for both WE and CE, which is defined as the actual amount of CO2 separated relative to the theoretical value (See detailed calculations in Supplementary Note 2). The high reversibility of the CO2 capture-release process is evidenced by the ~100% release/capture efficiency. Overall, the system reveals an average work of separation of 62.3 ± 3.8 kJ mol−1 CO2 over 21 cycles (Supplementary Fig. 25a), nearly half of the numbers previously reported for nonaqueous EMCC systems. A summary of benchmark EMCC performance is shown in Supplementary Table 6.
We also evaluated the EMCC performance of RA 3 (Supplementary Fig. 26). Over 41 cycles (205 h), the symmetric cell exhibited an average work of separation of 90.6 ± 3.8 kJ mol−1 CO2, an average CO2 capacity utilization of ~85%, a release/capture efficiency of ~100%, and an electrochemical efficiency of ~100%.
The promising results from our initial tests motivated us to push the limit of this system further. Leveraging the low cost and synthetic feasibility of 1 and 1-ox, we upscaled the system by raising the sorbent concentration (0.5 M 1, 0.05 M 1-ox, 0.5 M MEA). Using the same symmetric flow cell configuration, we tested the EMCC performance at various current densities (1–10 mA cm−2, Supplementary Fig. 27). By elevating the current density, the system exhibited an increasing rate of CO2 capture and release, as evidenced by the sharper and deeper changes in CO2 readings (Supplementary Fig. 27a). The three metrics of performance, including CO2 capacity utilization efficiency, release/capture efficiency, and electrochemical efficiency, are summarized in Supplementary Fig. 27b. The release/capture and electrochemical efficiencies remained constant with increasing current, and the CO2 capacity utilization displayed a moderate decrease to ~75% as the current was raised to 10 mA cm−2. The work of separation gradually increased from 66 ± 0.8 to 177.6 ± 6.4 kJ mol−1 CO2 from 1 to 10 mA cm−2 (Supplementary Fig. 27c, d).
Subsequently, we investigated the effect of water on the performance of 1 by adding 10% (v/v) of water to the DMSO electrolyte. As shown in Supplementary Fig. 28, the RAMAR flow cell maintained stable cycling over 16 cycles (112 h) with performance comparable to the water-free conditions. The system exhibited an average CO2 capacity utilization efficiency of 98.8 ± 0.9%, an electrochemical efficiency of 100%, and an average work of separation of 57.1 ± 1.0 kJ mol−1 CO2. The slight decrease in energetics compared to the dry conditions (Fig. 6) may be attributed to the reduced impedance of the electrolyte in the presence of water.
Further, we examined the RAMAR performance of 1 under two aerobic conditions (10% CO2 with 2% O2 and 21% O2) for 73 cycles (365 h) and 83 cycles (415 h), respectively (Supplementary Fig. 29 and Fig. 7). Remarkably, 1 maintained its chemical integrity without side products in both WE and CE, confirmed by the crude 1H NMR after long-term cycling (Supplementary Figs. 30 and 31). At a current of 12 mA (2.4 mA cm−2), the former condition (2% O2) exhibited an average CO2 capacity utilization of ~90%, a release/capture efficiency of ~100%, and a work of separation of 84.5 ± 9.6 kJ mol−1 CO2. The latter condition (21% O2) revealed an average CO2 capacity utilization of ~83%, a release/capture efficiency of ~100%, and a work of separation of 84.6 ± 30 kJ mol−1 CO2. These values align with the results obtained in our current step experiment (2 mA cm−2) under O2-free conditions, indicating that O2 has a minimal impact on RAMAR chemistry (Supplementary Fig. 27c).

a CO2 readings at the outlet of the WE tank (blue) and CE tank (red), respectively, over 83 repeating cycles for 415-h operation. b, c CO2 readings for selected capture-release cycles with the corresponding cumulative amount of CO2 captured/released in WE (b) and CE (c). For each separation cycle, both sorbent tanks were reduced/oxidized at 12 mA (2.4 mA cm−2) for 60 min followed by a 90 min rest. d, e Performance metrics of WE (d) and CE (e). WE and CE were composed of 10 ml DMSO solution with 0.5 M 1, 0.05 M 1-ox, 0.1 M HTFSI, 0.5 M MEA, and 0.5 M LiTFSI. Both tanks were stirred and continuously purged with 10% CO2 + 21% O2 (balanced with N2) at a flow rate of 2.5 sccm. Source data for Fig. 7 are provided as a Source Data file.
Lastly, we conducted a deep CO2 removal test on 1 using 25 cm2 flow fields, which are five times larger than those used in prior experiments (Fig. 8). Despite the absence of gas contactors facilitating CO2 absorption, our prototype cell achieved a single pass removal of > 85 % under a 10% CO2 feed, with an average work of separation of 150.6 ± 17.6 kJ mol−1 CO2 over 20 cycles (200 h). 1 maintained high purity after cycling, confirmed by crude 1H NMR (Supplementary Fig. 32). Both WE and CE exhibited faster CO2 release than capture (evidenced by the sharper gas desorption profile). This phenomenon is attributed to insufficient mass transport in our prototype absorber, which leads to a CO2 concentration gradient and variations in the amine-CO2 addition/elimination pathways during capture/release (Fig. 5a). Specifically, the 2H+−1CO2 pathway dominates under extremely low CO2 concentration during capture, reducing the absorption capacity and kinetics. Conversely, the 1H+−1CO2 pathway prevails under high CO2 concentration during release, facilitating a more effective desorption process. In practical operation, this challenge can be addressed by enhancing CO2 mass transport via device and process optimization.

a CO2 readings at the outlet of the WE tank (blue) and CE tank (red), respectively, over 20 repeating cycles for 200-h operation. b, c CO2 readings for selected capture-release cycles with the corresponding cumulative amount of CO2 captured/released in WE (b) and CE (c). For each separation cycle, both sorbent tanks were reduced/oxidized at 60 mA (2.4 mA cm−2) for 60 min followed by a 240 min rest. d, e Performance metrics of WE (d) and CE (e). WE and CE were composed of 15 ml DMSO solution with 0.5 M 1, 0.125 M 1-ox, 0.25 M HTFSI, 0.5 M MEA, and 0.5 M LiTFSI. Both tanks were stirred and continuously purged with 10% CO2 (balanced with N2) at a flow rate of 1.2 sccm. Flow fields of 25 cm2 were used in this experiment. Source data for Fig. 8 are provided as a Source Data file.
Discussion
In conclusion, by harnessing the synergy between RAs and classic amines, our RAMAR mechanism presents an effective strategy to address the practical limitations of many current EMCC absorbents, including limited stability under aerobic conditions, susceptibility to parasitic reactions during electrochemical cycling, high synthetic cost, and incompatibility with ion-exchange membranes. The Wurster-type RAs exhibit a wide electrochemical pKa swing with a range of > 20 units in organic media, allowing them to effectively and reversibly regenerate amines and potentially other basic absorbents for carbon capture. Through a combination of experimental and DFT studies, we provide fundamental insights into the PCET pathways of RAs, the molecular design principles for finetuning RA properties, and the underlying mechanisms of CO2–amine interactions in nonaqueous systems. The synthetic versatility of RAs, coupled with their exceptional (electro)chemical stability, enable the development of a symmetric flow-based carbon capture prototype with superior stability (>80 cycles and 400 h) under aerobic conditions (21% O2). Overall, this work facilitates the process engineering of sustainable and reliable electrochemical amine carbon capture systems for complex gas environments at the molecular level.
Methods
Materials
Unless otherwise stated, all the chemicals were purchased from Sigma Aldrich in ACS reagent grade and used without further purification. N1,N4-Diphenylbenzene−1,4-diamine (1) (CAS No.: 74-31-7, purity: 98%) and N1,N4-di(naphthalen-2-yl)benzene−1,4-diamine (2) (CAS No.: 93-46-9, purity: 98%) were purchased from Ambeed. N1,N4-Di-sec-butylbenzene−1,4-diamine (3) (CAS No.: 101-96-2, purity: >98%) was purchased from TCI. DMSO (anhydrous, ≥99.9%) was purchased from Sigma Aldrich for cyclic voltammetry experiments.
Synthesis of N 1 ,N 4-diphenylcyclohexa-2,5-diene−1,4-diimine (1-ox)
To a stirred solution of FeCl3 • 6H2O (16.2 g, 60 mmol) in water (500 ml) was added a solution of 1 (5.2 g, 20 mmol) in acetone (100 ml) dropwisely. The mixture was stirred for 5 h at room temperature and the precipitate was filtered, washed with copious amounts of water and cooled diethyl ether. The orange powder was recrystallized in toluene and dried in vacuo at 60 °C to give a brown coloured crystal (3.48 g, 67%). 1H NMR (400 MHz, DMSO) δ 7.41 (dt, J = 13.3, 7.8 Hz, 4H), 7.24–7.15 (m, 2H), 7.13 (d, J = 2.1 Hz, 1H), 7.04 (dd, J = 10.2, 2.2 Hz, 1H), 6.87 (ddd, J = 12.5, 10.1, 5.4 Hz, 5H), 6.78–6.75 (m, 1H). 13C NMR (101 MHz, DMSO) δ 157.83, 157.78, 149.69, 137.64, 136.32, 129.07, 129.04, 125.27, 125.05, 125.00, 124.31, 120.35, 120.21. The NMR agrees well with previous literatures52,53.
Synthesis of N 1 ,N 4-di-sec-butylcyclohexa-2,5-diene−1,4-diimine (3-ox)
To a stirred solution of 3 (1.8 ml, 7.68 mmol) in acetone (100 ml) was added silver oxide (2.66 g, 5.76 mmol) in portions. The mixture was stirred at room temperature for 1 h and filtered through a Celite cake. The filtrate was dried in vacuo to give a dark red oil, which solidified in ambient condition after a few days to afford a red crystalline solid (1.44 g, 85%). 1H NMR (400 MHz, DMSO) δ 7.27–6.99 (m, 2H), 6.84–6.58 (m, 2H), 3.85 (dd, J = 12.5, 6.2 Hz, 2H), 1.64–1.40 (m, 4H), 1.09 (d, J = 6.1 Hz, 6H), 0.77 (t, J = 7.4 Hz, 6H). 13C NMR (101 MHz, DMSO) δ 156.96, 156.73, 137.59, 135.24, 121.99, 120.69, 56.65, 56.57, 30.78, 30.75, 21.97, 21.84, 10.77.
Synthesis of diethyl 2,5-bis(phenylamino)terephthalate (4)
To a mixture of diethyl 2,5-dioxocyclohexane−1,4-dicarboxylate (512.5 mg, 2 mmol) and p-toluenesulfonic acid (100 mg, 0.58 mmol) in toluene (20 ml) was added aniline (0.548 ml, 6 mmol) under argon. The mixture was refluxed for 1 day using a Dean-Stark trap before being allowed to cool to room temperature. The organic phase was washed with water for three times, dried over Na2SO4, and evaporated to dryness to give an orange powder. The crude product was recrystallized in dichloromethane/hexane to afford an orange crystal. The orange product was dissolved in tetrahydrofuran (50 ml). The solution was added with trifluoroacetic acid (0.05 ml) and heated at 50 °C for 8 h in the air. The tetrahydrofuran solvent was removed in vacuo and the crude product was dissolved in ethyl acetate. The organic layer was washed with sat. NaHCO3 (aq) for three times and brine and dried over Na2SO4. The solvent was evaporated to dryness to give a red powder (678 mg, 84%). 1H NMR (400 MHz, DMSO) δ 8.56 (s, 2H), 7.81 (s, 2H), 7.30 (t, J = 7.7 Hz, 4H), 7.10 (d, J = 7.6 Hz, 4H), 6.95 (t, J = 7.2 Hz, 2H), 4.24 (dd, J = 14.0, 7.0 Hz, 4H), 1.21 (t, J = 7.1 Hz, 6H).
Electrochemical measurements
Electrochemical measurements were performed with a BioLogic VSP potentiostat from BioLogic Science Instruments. Cyclic voltammetry (CV) was measured using a standard three-electrode cell with a glassy carbon electrode (3 mm diameter) as the working electrode, a platinum wire as the counter electrode, and a silver wire as the quasi-reference electrode, with ferrocene as the internal reference. In a standard CV test, redox-active molecules (5 mM) were dissolved in anhydrous DMSO with 100 mM NBu4PF6 as the supporting electrolyte. CV were typically recorded at a scanning rate of 50 mV s–1. The electrochemical data were gathered and analysed by EC-lab V11.50.
Bulk electrochemical oxidation and reduction of 1
Constant current bulk electrolysis of 1 was carried out using a setup consisting of a three-neck round-bottom flask (RBF), with the three necks for the working, counter, and reference electrodes, respectively. Carbon felt (CT GF030; Fuel Cell Store) was used as the working electrode and a silver wire was used as the reference electrode. The counter electrode was separated from the solution 1 with a fritted electrode chamber (MR−1196; Bioanalytical Systems). LiClO4 (0.5 M) in DMSO-d6 was used as the electrolyte. A piece of carbon felt (1 cm2) coated with lithium-titanate-oxide (LTO) (64 mg) was used as the counter electrode, which was immersed in the electrolyte (1 ml). In a standard condition, 1 (26 mg, 0.1 mmol) was stirred and oxidized in the presence of monoethanolamine (12.1 μl, 0.2 mmol) at a constant current of 1.5 mA in DMSO-d6 (4 ml, 0.5 M LiClO4) under continuous 10% CO2 bubbling for 3.6 h. After the oxidation was complete, 0.8 ml of the electrolyte in the working electrode chamber was withdrawn for crude NMR analysis without further purification. Subsequently, the solution left in the RBF was stirred and reduced at a constant current of −1.5 mA until the voltage was below −1.6 V under continuous 10% CO2 bubbling. Similarly, another 0.8 ml of the electrolyte in the working electrode chamber was withdrawn for crude NMR analysis without further purification.
Treatment of Nafion membrane23
The Nafion membranes (Nafion 212, 2.5 × 3 cm2, 50 μm in thickness) were heated in boiling 3% H2O2 (aq) for 1 h. The membranes were then heated in 0.25 M sulfuric acid (aq) for 1 h and cleaned in deionized water for 30 min (twice) at 100 °C. The membranes were heated in 0.25 M NaOH (aq) for 1 h and cleaned in deionized water for 30 min (twice) at 100 °C. The membranes were dried under vacuum at 80 °C for 2 d and stored in DMSO.
EMCC symmetric flow cell
In a standard setup, two scintillation vials (20 ml) with septum caps, serving as the sorbent tank of WE and CE, were continuously purged with CO2 feed gas (balanced with N2) at a controlled flow rate using Alicat mass flow controllers. Simulated flue gas conditions were mimicked using a 10% CO2 and 2% O2 mix, balanced by argon. Ambient oxygen conditions were mimicked using a 10% CO2 and 21% O2 mix, balanced by N2. CO2 levels exiting the WE and CE tanks were continuously monitored using an infrared-based CO2 sensor (SprintIR-W 100%), with data recorded via Labview 2021. The flow cell incorporated two graphite plates with 5 or 25 cm2 interdigitated flow fields, pressing against three pieces of carbon paper electrodes (Sigracet 28 AA) on each side of the graphite plates. WE and CE electrolytes were circulated at a flow rate of 10- or 40-ml min−1 (for 5 or 25 cm2 flow fields, respectively) through a commercial flow cell (Scribner) by a two-channel peristaltic pump (Masterflex). A treated Nafion 212 membrane, flanked by Celgard 3501, was placed between the carbon electrodes. The cell was sealed by Kalrez fluoropolymer elastomer gaskets (0.51 mm thick). CO2 capture/release was conducted in constant current mode. The cycling protocols of each experiment are provided in the corresponding figure captions. The temperature of the flow cell was kept at 25 °C using a pair of cartridge heaters connecting to a Digi-Sense TC6000 temperature controller.
Data availability
The data generated or analyzed during this study are included in the manuscript and its Supplementary Information. The source data underlying Figs. 2–8 and Supplementary Figs. 10, 15, 16, 18, 19, 21–23, and 25–29 are provided as a Source Data file. The atomic coordinates of the optimized computational models have been deposited in the Zenodo Database [https://doi.org/10.5281/zenodo.15132348]. Source data are provided with this paper.
References
Rochelle, G. T. Amine scrubbing for CO2 capture. Science 325, 1652–1654 (2009).
Boot-Handford, M. E. et al. Carbon capture and storage update. Energy Environ. Sci. 7, 130–189 (2014).
Khan, U. et al. Assessing absorption-based CO2 capture: research progress and techno-economic assessment overview. Carbon Capture Sci. Technol. 8, 100125 (2023).
Cantu, D. C. et al. Molecular-level overhaul of γ-aminopropyl aminosilicone/triethylene glycol post-combustion CO2-capture solvents. ChemSusChem 13, 3429–3438 (2020).
Kortunov, P. V., Siskin, M., Paccagnini, M. & Thomann, H. CO2 reaction mechanisms with hindered alkanolamines: control and promotion of reaction pathways. Energy Fuels 30, 1223–1236 (2016).
Heldebrant, D. J. et al. Water-lean solvents for post-combustion CO2 capture: fundamentals, uncertainties, opportunities, and outlook. Chem. Rev. 117, 9594–9624 (2017).
Leclaire, J. et al. Tetrameric self-assembling of water-lean solvents enables carbamate anhydride-based CO2 capture chemistry. Nat. Chem. 16, 1160–1168 (2024).
Seo, S. et al. Chemically tunable ionic liquids with aprotic heterocyclic anion (AHA) for CO2 capture. J. Phys. Chem. B 118, 5740–5751 (2014).
Zhu, P. et al. Continuous carbon capture in an electrochemical solid-electrolyte reactor. Nature 618, 959–966 (2023).
Li, H. et al. Capturing carbon dioxide from air with charged-sorbents. Nature 630, 654–659 (2024).
Sharifian, R., Wagterveld, R. M., Digdaya, I. A., Xiang, C. & Vermaas, D. A. Electrochemical carbon dioxide capture to close the carbon cycle. Energy Environ. Sci. 14, 781–814 (2021).
Barlow, J. M. et al. Molecular design of redox carriers for electrochemical CO2 capture and concentration. Chem. Soc. Rev. 51, 8415–8433 (2022).
Liu, Y., Lucas, É., Sullivan, I., Li, X. & Xiang, C. Challenges and opportunities in continuous flow processes for electrochemically mediated carbon capture. iScience 25, 105153 (2022).
Rahimi, M., Khurram, A., Hatton, T. A. & Gallant, B. Electrochemical carbon capture processes for mitigation of CO2 emissions. Chem. Soc. Rev. 51, 8676–8695 (2022).
Diederichsen, K. M. et al. Electrochemical methods for carbon dioxide separations. Nat. Rev. Methods Primers 2, 68 (2022).
Li, X., Mathur, A., Liu, A. & Liu, Y. Electrifying carbon capture by developing nanomaterials at the interface of molecular and process engineering. Acc. Chem. Res. 56, 2763–2775 (2023).
Zito, A. M. et al. Electrochemical carbon dioxide capture and concentration. Chem. Rev. 123, 8069–8098 (2023).
Renfrew, S. E., Starr, D. E. & Strasser, P. Electrochemical approaches toward CO2 capture and concentration. ACS Catal. 10, 13058–13074 (2020).
Massen-Hane, M., Diederichsen, K. M. & Hatton, T. A. Engineering redox-active electrochemically mediated carbon dioxide capture systems. Nat. Chem. Engin. 1, 35–44 (2024).
Liu, Y., Ye, H.-Z., Diederichsen, K. M., Van Voorhis, T. & Hatton, T. A. Electrochemically mediated carbon dioxide separation with quinone chemistry in salt-concentrated aqueous media. Nat. Commun. 11, 2278 (2020).
Diederichsen, K. M., Liu, Y., Ozbek, N., Seo, H. & Hatton, T. A. Toward solvent-free continuous-flow electrochemically mediated carbon capture with high-concentration liquid quinone chemistry. Joule 6, 221–239 (2021).
Barlow, J. M. & Yang, J. Y. Oxygen-stable electrochemical CO2 capture and concentration with quinones using alcohol additives. J. Am. Chem. Soc. 144, 14161–14169 (2022).
Li, X., Zhao, X., Liu, Y., Hatton, T. A. & Liu, Y. Redox-tunable Lewis bases for electrochemical carbon dioxide capture. Nat. Energy 7, 1065–1075 (2022).
Li, X. et al. Redox-tunable isoindigos for electrochemically mediated carbon capture. Nat. Commun. 15, 1175 (2024).
Jin, S., Wu, M., Jing, Y., Gordon, R. G. & Aziz, M. J. Low energy carbon capture via electrochemically induced pH swing with electrochemical rebalancing. Nat. Commun. 13, 2140 (2022).
Seo, H., Rahimi, M. & Hatton, T. A. Electrochemical carbon dioxide capture and release with a redox-active amine. J. Am. Chem. Soc. 144, 2164–2170 (2022).
Kim, S. et al. Asymmetric chloride-mediated electrochemical process for CO2 removal from oceanwater. Energy Environ. Sci. 16, 2030–2044 (2023).
Pang, S. et al. A phenazine-based high-capacity and high-stability electrochemical CO2 capture cell with coupled electricity storage. Nat. Energy 8, 1126–1136 (2023).
Seo, H. & Hatton, T. A. Electrochemical direct air capture of CO2 using neutral red as reversible redox-active material. Nat. Commun. 14, 313 (2023).
Bui, A. T., Hartley, N. A., Thom, A. J. W. & Forse, A. C. Trade-off between redox potential and the strength of electrochemical CO2 capture in quinones. J. Phys. Chem. C 126, 14163–14172 (2022).
Scovazzo, P., Poshusta, J., DuBois, D., Koval, C. & Noble, R. Electrochemical separation and concentration of <1% carbon dioxide from nitrogen. J. Electrochem. Soc. 150, D91 (2003).
Wilm, L. F. B. et al. Photoswitchable nitrogen superbases: using light for reversible carbon dioxide capture. Angew. Chem. Int. Ed. 61, e202112344 (2022).
Liao, Y. Design and applications of metastable-state photoacids. Acc. Chem. Res. 50, 1956–1964 (2017).
Wimberger, L. et al. Large, tunable, and reversible pH changes by merocyanine photoacids. J. Am. Chem. Soc. 143, 20758–20768 (2021).
Stern, M. C., Simeon, F., Herzog, H. & Hatton, T. A. Post-combustion carbon dioxide capture using electrochemically mediated amine regeneration. Energy Environ. Sci. 6, 2505–2517 (2013).
Wang, M., Herzog, H. J. & Hatton, T. A. CO2 capture using electrochemically mediated amine regeneration. Ind. Eng. Chem. Res. 59, 7087–7096 (2020).
Rahimi, M. et al. An electrochemically mediated amine regeneration process with a mixed absorbent for postcombustion CO2 capture. Environ. Sci. Technol. 54, 8999–9007 (2020).
Mao, Y. et al. Stability improvement of the advanced electrochemical CO2 capture process with high-capacity polyamine solvents. Appl. Energy 369, 123597 (2024).
Wu, X. et al. Electrochemically-mediated amine regeneration of CO2 capture: from electrochemical mechanism to bench-scale visualization study. Appl. Energy 302, 117554 (2021).
Kuo, F.-Y., Jerng, S. E. & Gallant, B. M. Dual salt cation-swing process for electrochemical CO2 separation. ACS Cent. Sci. 9, 1750–1757 (2023).
Jin, S., Wu, M., Gordon, R. G., Aziz, M. J. & Kwabi, D. G. pH swing cycle for CO2 capture electrochemically driven through proton-coupled electron transfer. Energy Environ. Sci. 13, 3706–3722 (2020).
Watkins, J. D. et al. Redox-mediated separation of carbon dioxide from flue gas. Energy Fuels 29, 7508–7515 (2015).
Moreno, E., Nolasco, L. A., Caggiano, L. & Jackson, R. F. W. Synthesis of orthogonally protected biaryl amino acid derivatives. Org. Biomol. Chem. 4, 3639–3647 (2006).
McCarthy, B. D. & Dempsey, J. L. Decoding proton-coupled electron transfer with potential–pKa diagrams. Inorg. Chem. 56, 1225–1231 (2017).
Oldacre, A. N. & Young, E. R. Electrochemical proton-coupled electron transfer of an anthracene-based azo dye. RSC Adv. 10, 14804–14811 (2020).
Hansch, C., Leo, A. & Taft, R. W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 91, 165–195 (1991).
Han, X.-B. & Ye, S. Structural design of oxygen reduction redox mediators (ORRMs) based on anthraquinone (AQ) for the Li–O2 battery. ACS Catal. 10, 9790–9803 (2020).
Sim, J. et al. Isolation and crystal structure determination of piperazine dicarbamate obtained from a direct reaction between piperazine and carbon dioxide in methanol. B. Korean Chem. Soc. 37, 1854–1857 (2016).
Said, R. B., Kolle, J. M., Essalah, K., Tangour, B. & Sayari, A. A unified approach to CO2–amine reaction mechanisms. ACS Omega 5, 26125–26133 (2020).
Kortunov, P. V., Siskin, M., Baugh, L. S. & Calabro, D. C. In situ nuclear magnetic resonance mechanistic studies of carbon dioxide reactions with liquid amines in non-aqueous systems: evidence for the formation of carbamic acids and zwitterionic species. Energy Fuels 29, 5940–5966 (2015).
Lehmann, M. L. et al. Membrane design for non-aqueous redox flow batteries: current status and path forward. Chem. 8, 1611–1636 (2022).
Sandberg, M. & Hjertberg, T. E/Z isomerism in polyaniline, a model study. Synth. Met. 29, 257–264 (1989).
Han, C.-C., Balakumar, R., Thirumalai, D. & Chung, M.-T. The different electronic natures displayed by the alkylthio groups in simple and higher conjugated aniline systems. Org. Biomol. Chem. 4, 35116–3516 (2006).
Acknowledgements
The authors acknowledge financial support from the Johns Hopkins University, the David and Lucile Packard Foundation, and the Arnold and Mabel Beckman Foundation. A.L. and Y.L. acknowledge financial support from the National Science Foundation (award number 2237096). X.L. acknowledges the financial support from the City University of Hong Kong (grant number 9382002). C.B.M. and W.A.G. acknowledge support from the Liquid Sunlight Alliance, which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Fuels from Sunlight Hub under Award No. DE-SC0021266. This research used resources of the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 using NERSC award BES-ERCAP0024109.
Author information
Authors and Affiliations
Contributions
X.L and Y.L. conceived the project and designed the experiments. X.L. conducted the experiments and analyzed the data. C.B.M. performed the DFT calculations under the supervision of W.A.G. A.L. measured the solubility of RAs. J.M. tested the CV with different amines. J.Z. measured the crossover rates. Y.L. supervised the project. X.L. and Y.L. co-wrote the manuscript. All authors discussed the results and revised or commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, X., Musgrave, C.B., Liu, A. et al. Electrifying amine carbon capture with robust redox-tunable acids. Nat Commun 16, 4339 (2025). https://doi.org/10.1038/s41467-025-59732-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59732-z
