
Abstract
Direct seawater electrolysis at ampere-level current densities, powered by coastal/offshore renewables, is an attractive avenue for sustainable hydrogen production but is undermined by chloride-induced anode degradation. Here we demonstrate the use of hexafluorophosphate (PF₆⁻) as an electrolyte additive to overcome this limitation, achieving prolonged operation for over 5,000 hours at 1 A cm−2 and 2300 hours at 2 A cm−2 using NiFe layered double hydroxide (LDH) as anode. Together with the experimental findings, PF₆⁻ can intercalate into LDH interlayers and adsorb onto the electrode surface under an applied electric field, blocking Cl⁻ and stabilizing Fe to prevent segregation. The constant-potential molecular dynamics simulations further reveal the accumulation of high surface concentrations of PF6− on the electrode surface that can effectively exclude Cl−, mitigating corrosion. Our work showcases synchronous interlayer and surface engineering by single non-oxygen anion species to enable Cl− rejection and marks a crucial step forward in seawater electrolysis.
Similar content being viewed by others


Introduction
Water electrolysis is pivotal to achieving global net-zero targets by 2050, yet the increasing hydrogen demand strains already scarce freshwater supplies1,2,3,4,5,6,7. As Earth’s most abundant water source, seawater offers a promising alternative8,9,10,11. Alkaline seawater electrolysis bypasses energy-intensive desalination and extensive pretreatment, improving process efficiency12,13,14. When coupled with coastal or offshore renewable energy, it further reduces operational costs and facilitates scalable green hydrogen production15,16. Nonetheless, the high Cl⁻ concentration (~0.5 M) promotes electrode corrosion through metal chloride–hydroxide pathways (Supplementary Note 1), accelerating anode degradation17,18,19,20. Moreover, while the oxygen evolution reaction (OER) is thermodynamically preferred in alkaline media, it is a four-electron process with sluggish kinetics. In contrast, the competing chlorine evolution reaction (ClER) proceeds via a two-electron pathway with lower kinetic barriers, generating toxic chlorine gas21,22. Although seawater alkalinization shifts the OER potential below that of ClER by 480 mV23,24,25,26, achieving stable ampere-level current densities (j) remains a challenge due to increased resistance and persistent corrosion.
Despite incorporating precious metals, current anodes remain constrained by resource limitations, hindering the integration of catalytic activity, selectivity, and Cl⁻ tolerance27,28,29,30,31. Recently, researches on transition-metal sulfides32, phosphides33, nitrides34, or Ni(Co)Fe layered double hydroxides (LDHs)35,36,37,38,39,40, have shown that in situ formation of oxyanion-rich layers (e.g., SO₄²⁻, PO₄³⁻, NO3⁻) or fabricating chloride-repellent coatings37,38 can repel Cl⁻, enabling efficient and long-lasting seawater oxidation. LDHs are particularly attractive due to their layered structures, compositional tunability, and strong OER activity in alkaline media41,42,43,44, but their resistance to Cl⁻-induced degradation remains inadequate36,37. Strategies include creating physical shielding layers35,45, adding oxyanion species (SO₄²⁻, PO₄³⁻) that can adsorb onto the anode surface46,47, or forming surface chloride-immobilizing layers (AgCl, IrCl)48,49 have been proposed to exclude Cl− via Coulombic repulsion or the common ion effect. The layered and positively charged nature of LDHs also supports anion intercalation, and numerous studies confirm that intercalated oxyanions can block chloride penetration50,51,52,53. Extending this concept, some studies have combined surface and interlayer engineering with dual species54,55. The Zou group54 intercalated CO₃²⁻ into CoFe LDH and attached graphene quantum dots on the surface, achieving 2800 h of long-term durability at ~1.25 A cm−2 in simulated seawater. Our group55 further introduced OH⁻-enriching Cr₂O₃ onto NiFe LDH surfaces and Cl⁻-repelling CrO₄²⁻ into the interlayers, enabling 1000 h at 1 A cm−2 followed by 1500 h at 2 A cm−2. Despite these advancements indicating considerable progress, developing simple and effective strategies to sustain prolonged electrolysis (>5000 h) under high j for anode materials is still a major challenge.
In this work, a NiFe LDH anode, operated with hexafluorophosphate (PF₆⁻) as an electrolyte additive, delivers stable operation for over 5000 h at 1 A cm−2 and 2300 h at 2 A cm−2. Ex situ/in situ evidences show that PF₆⁻, driven by the electric field, can facilely intercalate into the LDH layers and adsorb onto the surface, facilitating a single-ion-driven surface-interlayer synergy that excludes Cl⁻. This additive also improves the durability of CoFe LDH, enabling continuous operation at 2 A cm−2 for 1200 h, thereby rendering CoFe LDH/NiFe LDH as top contenders for industrial-level seawater electrolysis. Constant-potential molecular dynamics simulations further verify that surface-accumulated PF₆⁻ efficiently excludes Cl⁻. Beyond its durability, the NiFe LDH anode coupled with a Pt/C cathode also delivers notable performance in an alkaline seawater electrolyzer.
Results
Enhanced alkaline seawater oxidation (ASO) performance with PF₆⁻ additive
The NiFe LDH nanosheet array, consisting of two-dimensional (2D) layers, was fabricated on nickel foam (NiFe LDH/NF) through a one-step hydrothermal approach56. Extensive characterizations via X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), high-resolution TEM (HRTEM), and high-angle annular dark field scanning TEM (HAADF-STEM) verify the successful synthesis of NiFe LDH/NF with a Ni:Fe atomic ratio of ~3:1 (Supplementary Figs. 1–5, Supplementary Table 1).
A standard three-electrode configuration was employed to evaluate the OER performance of NiFe LDH/NF in alkaline seawater, either containing 20 mM PF₆⁻ or without any additive. As shown in the polarization curves (Fig. 1a, Supplementary Fig. 6), the presence of PF₆⁻ reduces the overpotentials required to obtain the j of 200, 500, and 1000 mA cm−2 (209, 247, 282 mV) compared to the corresponding values of 282, 315, and 343 mV in PF₆⁻-free seawater. NiFe LDH/NF in PF₆⁻-containing seawater also outperforms bare NF, benchmark RuO₂ and IrO₂ catalysts, ranking among the top-tier catalysts reported to date (Supplementary Table 2). Turnover frequency (TOF) analysis (inset of Fig. 1a, Supplementary Fig. 7) further shows enhanced intrinsic activity in the presence of PF₆⁻. Electrochemical double-layer capacitance (Cdl) measurements (Supplementary Fig. 8) indicate a higher electrochemically active surface area (ECSA) with PF₆⁻, in line with its enhanced OER activity. Even after normalizing j to ECSA (Supplementary Fig. 9), NiFe LDH/NF in PF₆⁻-containing seawater maintains higher activity than in the PF₆⁻-free counterpart. Tafel slope analysis based on polarization curves obtained at a slow scan rate of 1 mV s−1 (Supplementary Fig. 10, Fig. 1b) confirms that PF₆⁻ addition improves the reaction kinetics for NiFe LDH/NF, outperforming RuO₂ and IrO₂. Electrochemical impedance spectroscopy over the potential range of 1.405–1.485 V (Supplementary Fig. 11) further reveals that NiFe LDH/NF in PF₆⁻-containing seawater exhibits lower charge-transfer resistance than in PF₆⁻-free seawater, supporting accelerated interfacial electron transfer. To assess corrosion resistance, Tafel scanning was performed (Fig. 1c), showing that the corrosion potential of NiFe LDH/NF shifts positively in PF₆⁻-containing seawater, indicative of improved corrosion stability. Moreover, Faradaic efficiency for oxygen evolution at 1 A cm−2 reaches nearly 100% with NiFe LDH/NF in PF₆⁻-containing seawater (Supplementary Fig. 12), confirming selective four-electron O₂ evolution. At an industrial-level j of 1.0 A cm−2, NiFe LDH/NF in PF₆⁻-containing seawater operates stably for over 5000 h (Fig. 1d), a 41.6-fold improvement in stark contrast to its rapid failure within 120 hours in PF₆⁻-free seawater (inset of Fig. 1d). Ultraviolet–visible spectroscopy (Supplementary Fig. 13) verifies minimal active chlorine generation with PF₆⁻ present, in stark contrast to high chlorine levels in the PF₆⁻-free seawater electrolyte. Even under a higher j of 2.0 A cm−2, NiFe LDH/NF demonstrates stable operation in PF₆⁻-containing seawater for over 2300 h (Fig. 1e). In light of these results, using NiFe LDH/NF anode with PF₆⁻ as an electrolyte additive facilitates a notable advance for seawater electrolysis at high j, beyond that of currently explored anionic species (Supplementary Table 3), and showing promise for industrial applications.

a Polarization curves at a scan rate of 10 mV s⁻1 and b corresponding Tafel slopes of NiFe LDH/NF in PF6−-free and PF6−-containing seawater, alongside RuO2/NF, IrO2/NF and NF in PF6−-free seawater with 100% iR correction. c Corrosion behavior curves for NiFe LDH/NF in PF6−-free and PF6−-containing seawater. d Chronopotentiometry curves at a j of 1 A cm−2 for NiFe LDH/NF in PF6−-free and PF6−-containing seawater without iR correction. The inset magnifies a section of the chronopotentiometry curve for NiFe LDH/NF in PF₆⁻-free seawater. e Chronopotentiometry curve at a j of 2 A cm−2 for NiFe LDH/NF in PF6−-containing seawater without iR correction. Source data are provided as a Source Data file.
Investigation of activity enhancement and corrosion resistance
Operando Raman spectroscopy was performed using a custom-designed electrochemical cell (Supplementary Fig. 14a) to monitor structural changes for NiFe LDH from open-circuit potential (OCP) to 1.75 V vs. the reversible hydrogen electrode (RHE). In PF₆⁻-free seawater, Raman spectra collected at potentials below 1.35 V show peaks at 454 and 527 cm−1, assigned to the A1g stretching modes of Ni–OH and Ni–O, respectively (Fig. 2a). Upon increasing the applied potential, two additional peaks emerge at 475 and 556 cm−1, corresponding to the Eg bending and A1g stretching vibrations of Ni–O for NiOOH44,57,58,59. Concurrently, the disappearance of the Ni–OH lattice vibration (~300 cm−1) and attenuation of signals associated with CO₃²⁻/NO₃⁻ species (~727 cm−1) further support the occurrence of dehydration and oxidation processes during OER60,61,62. In PF₆⁻-containing seawater, apart from the progressive blue shifts of Ni–O bands (454/527 to 475/556 cm−1), an additional Eg vibration of PF₆⁻ is observed at increasing potentials, potentially due to its intercalation into LDH layers. As the applied potential continues to rise, the intensification of the A1g vibration indicates extensive PF₆⁻ accumulation on the electrode surface driven by a stronger applied electric field (Fig. 2b, Supplementary Fig. 14b). To verify electric-field-driven intercalation, NiFe LDH/NF electrode was held at 1.15 V for 10 min in PF₆⁻-containing seawater. Structural analyses, including XRD, SEM, TEM, HRTEM, and HAADF-STEM (Supplementary Figs. 15–19), collectively confirm the successful incorporation of PF₆⁻ into the NiFe LDH interlayers. Given its large ionic radius, PF₆⁻ displaces native interlayer anions, expanding the interlayer spacing, exposing more active sites, thus promoting OER activity. Raman spectroscopy after intercalation (Supplementary Fig. 20) further confirms this process, as evidenced by the disappearance of characteristic CO₃²⁻/NO₃⁻ peaks and the emergence of a pronounced Eg vibration mode of PF₆⁻. Additionally, we observed a slight blue shift for NiFe LDH Raman peaks following PF₆⁻ intercalation (Supplementary Fig. 21), contrary to the usual red shifts induced by cation-driven Ni–O bond elongation58,63,64. Operando Raman spectroscopy of PF₆⁻-intercalated NiFe LDH in PF₆⁻-free seawater (Fig. 2c) shows delayed γ-NiOOH formation, consistent with the observations in PF₆⁻-containing seawater. With increasing potential, partial deintercalation of PF₆⁻ occurs, accompanied by interlayer contraction due to shortening of Ni–O bonds. A subsequent chronopotentiometry test at 2 A cm−2 shows that PF₆⁻-intercalated NiFe LDH/NF maintains stable operation for about 520 h in PF₆⁻-free seawater (Supplementary Fig. 22), substantially less than the 2300 h achieved in PF₆⁻-containing seawater. These observations underscore the crucial role of surface-accumulated PF₆⁻ in enhancing resistance to chloride-induced corrosion under prolonged high j conditions.

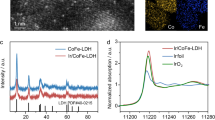
Operando Raman spectra collected from OCP to 1.75 V vs. RHE for NiFe LDH/NF in a PF₆⁻-free and b PF₆⁻-containing seawater. c Operando Raman spectra under the same potential range for PF₆⁻-intercalated NiFe LDH/NF in PF₆⁻-free seawater. d Normalized Ni K-edge XANES spectra for PF₆⁻-intercalated NiFe LDH/NF, NiFe LDH, NiO, and Ni foil. e Normalized Fe K-edge XANES spectra for PF₆⁻-intercalated NiFe LDH, NiFe LDH, Fe₂O₃, and Fe foil. f First-derivative Fe K-edge spectra of PF₆⁻-intercalated NiFe LDH, NiFe LDH, Fe₂O₃, and Fe foil. g TOF-SIMS mapping of PF₆⁻, Cl⁻, OH⁻, Ni, Fe, and K fragments on PF₆⁻-intercalated NiFe LDH/NF following a 120-h test. h Schematic of the proposed PF₆⁻ intercalation and surface adsorption mechanisms for Cl⁻ rejection. Source data are provided as a Source Data file.
X-ray photoelectron spectroscopy analyses also confirm the successful incorporation of PF₆⁻, quantified at 5.93 wt.% by inductively coupled plasma mass spectrometry (ICP-MS; Supplementary Table 4), with distinct P 2p and F 1s peaks (Supplementary Fig. 23a, b), alongside shifts toward lower and higher binding energies for Ni 2p and Fe 2p, respectively (Supplementary Fig. 23c, d). X-ray absorption near-edge structure (XANES) analyses further reinforce these findings, revealing a reduced oxidation state for Ni and elevated oxidation state for Fe after PF₆⁻ intercalation (Fig. 2d, e). First-derivative Fe K-edge spectra of the PF₆⁻-intercalated NiFe LDH reveal enhanced Fe–M (M = Fe or Ni) coordination (Fig. 2f), supporting the idea that Fe experiences stronger bonding and electron withdrawal. Conversely, Ni–O coordination weakens (Supplementary Fig. 24), and Ni becomes more reduced, suggesting that PF₆⁻ does not directly remove electrons from Ni but instead facilitates electron transfer from Fe to Ni, which potentially account for the delayed γ-NiOOH formation observed for PF₆⁻-intercalated NiFe LDH and NiFe LDH in PF₆⁻-containing seawater. Furthermore, the slight contraction of the Ni–O bond upon PF₆⁻ intercalation accounts for the blue shift observed in Raman spectra (Supplementary Fig. 21).
We quantified Fe segregation after 120-hour chronoamperometry test at 1 A cm−2. The Fe concentration leached from NiFe LDH/NF in PF₆⁻-containing seawater is notably lower (0.291 µg mL−1) compared to PF₆⁻-free seawater (1.5 µg mL−1). Correspondingly, the Fe retained in PF₆⁻-containing conditions is markedly higher (263.5 µg cm−2 vs. 14.5 µg cm−2; Supplementary Fig. 25). Time-of-flight secondary ion mass spectrometry (TOF-SIMS) further confirms the retention of Fe, abundant PF₆⁻, and minimal Cl⁻, verifying the effective Cl⁻ exclusion by PF₆⁻ (Fig. 2g). Morphological characterizations reveal that NiFe LDH/NF in PF₆⁻-containing seawater preserves its NF skeleton and 2D nanosheet arrays, while the PF₆⁻-free electrode suffers severe structural failure after 120 hours (Supplementary Figs. 26, 27), affirming the efficacy of PF₆⁻ in enhancing mechanical durability and mitigating chloride-induced corrosion. Furthermore, EDS mapping and XPS analyses (Supplementary Figs. 28, 29) conducted after the long-term test confirm the presence of P, F, Ni, Fe, and O throughout the nanosheets, alongside extensive NiOOH formation at the surface, also supporting that PF₆⁻ incorporation stabilizes Fe and suppresses its leaching. Raman spectroscopy further verifies the surface phase transformation and the retention of PF₆⁻ within the interlayers (Supplementary Fig. 30).
Through combined ex situ and in situ analyses, we verify that PF₆⁻ can intercalate into the NiFe LDH interlayers and accumulate on the electrode surface under applied electric field (Fig. 2h). This dual regulation of interlayer species and surface adsorption by a single non-oxygen anion contributes to Fe stabilization by suppressing segregation, and efficiently excludes Cl⁻, enabling durable ampere-level seawater oxidation.
Catalytic durability with PF₆⁻ additive: insights from experiment and molecular dynamics
Our strategy is also applicable to other LDHs for ASO catalysis, as demonstrated using CoFe LDH as a model. Operando Raman spectra collected from OCP to 1.70 V also validate the intercalation and surface adsorption behavior of PF₆⁻ (Supplementary Fig. 31), with additional evidence from XPS P 2p and F 1 s spectra supporting PF₆⁻ incorporation (Supplementary Fig. 32). Electrochemical tests show that CoFe LDH/NF in PF₆⁻-containing seawater exhibits lower overpotentials than in PF₆⁻-free seawater (Supplementary Fig. 33), achieving 1 A cm−2 at only 373 mV, outperforming many reported CoFe-based ASO anodes (Supplementary Table 5). Stability tests at 2.0 A cm−2 show over 1200 h of stable operation in the presence of PF₆⁻, compared to failure after 65 h without it (Fig. 3a), representing an 18.5-fold improvement. The long-term durability achieved under high current density also enders CoFe LDH/NF as the most robust CoFe-based ASO anode currently known (Supplementary Table 6). Notably, the PF₆⁻ additive enables synchronous interlayer and surface engineering of benchmark CoFe LDH/NiFe LDH catalysts under an applied electric field, offering a simple and more effective Cl⁻ exclusion strategy compared to physical shielding, surface chloride immobilization, oxyanion-based electrostatic repulsion, or dual-species interlayer/surface modifications (Supplementary Table 7, Fig. 3b).

a Chronopotentiometry curves at a j of 2 A cm−2 for CoFe LDH/NF in PF6−-free and PF6−-containing seawater without iR correction. b Comparison of various state-of-the-art electrodes and Cl− repelling strategies for ASO. c–e Theoretical calculations for the interactions between PF6− and Cl− at the electrolyte/electrode interface at a positive potential of 1.5 V. f The density distribution of Cl− at the electrolyte/electrode interface. Source data are provided as a Source Data file.
To further elucidate the mechanism by which PF₆⁻ enhances corrosion resistance, we employed advanced constant-potential molecular dynamics (CPMD) simulations to investigate the interactions between PF₆⁻ and Cl⁻ at the electrolyte-electrode interface under a positive potential of U = 1.5 V65. This approach provides an accurate simulation of atomic-scale dynamics at the electrocatalyst/electrolyte interface under operational conditions65. The calculation results indicate that the introduction of a single PF₆⁻ into the electrolyte has no marked effect on Cl⁻ at the electrode surface (Fig. 3c, d), because the density distribution of Cl⁻ on the electrode surface does not change considerably. Further increasing the number of PF6− to three generates a higher concentration of PF6− on the electrode surface, and we observe that the density of Cl⁻ moves substantially away from the electrode surface by about 2 Å (Fig. 3e, f). These results support the operando Raman spectroscopy and electrochemical durability results, where the accumulation of PF6− at the catalyst/electrolyte interface under high j correlates with enhanced long-term stability.
Practical electrolysis applications
To evaluate the practical application, we constructed a membrane electrode assembly (MEA) electrolyzer with an anion exchange membrane (AEM, PiperION-A60) (Fig. 4a). In this configuration, anions and water migrate through the AEM, facilitating electron transfer at the NiFe LDH/NF anode and the Pt/C/NF cathode. Concurrently, H₂, O₂, and discharged seawater, are expelled from the electrolyzer chamber. For comparison, a RuO₂/NF||Pt/C/NF configuration was also tested. The NiFe LDH/NF||Pt/C/NF electrolyzer employes a dual-feed approach with 6 M KOH+ seawater served as the catholyte and the anolyte consisted of 6 M KOH+ seawater with the addition of PF₆⁻. In contrast, the RuO₂/NF||Pt/C/NF electrolyzer uses 6 M KOH+ seawater as both the anolyte and catholyte. All experiments were meticulously conducted at a high temperature of 60 °C to simulate industrial operational conditions. The performance metrics of the MEA with NiFe LDH/NF||Pt/C/NF exhibits notable electrocatalytic activity, outperforming that of the RuO₂/NF||Pt/C/NF counterpart (Fig. 4b). Specifically, the NiFe LDH/NF||Pt/C/NF electrolyzer requires only 2.02 V to achieve a j of 1.0 A cm−2. Furthermore, the energy consumption at the j of 0.2 and 0.5 A cm−2 is calculated to be 3.60 and 4.04 kWh m⁻³ H₂, respectively, outperforming conventional electrolyzer that demand 4.47 kWh m⁻³ at 0.2 A cm−2. Stability tests were carried out to evaluate the long-term robustness of the electrocatalytic system. The NiFe LDH/NF‖Pt/C/NF electrolyzer operating in 6 M KOH + seawater with PF₆⁻ additives maintain stable performance for over 1000 h at a j of 1.0 A cm−2 (Fig. 4c). In contrast, the same MEA electrolyzer in PF₆⁻-free seawater experiences rapid degradation after only 31 hours (Supplementary Fig. 34), further underscoring the critical role of PF₆⁻ in sustaining electrode stability. A preliminary economic analysis (Supplementary Note 2) indicates that, under a typical electricity rate of US$0.02 kWh−1, the hydrogen production cost is approximately US$1.07 kg−1 H₂ at 1.0 A cm−2 and 2.02 V. Additionally, Supplementary Table 8 and Fig. 4d illustrates that the incorporation of PF₆⁻ additives into the NiFe LDH/NF anode not only provides a competitive cell voltage at high j but also ensures prolonged electrolysis stability. Specifically, the MEA electrolyzer with NiFe LDH/NF anode and PF₆⁻ additives exhibit outstanding activity and durability, paving an important step toward practical deployment of seawater electrolysis for hydrogen production.

a Schematic of the flow electrolytic cell with asymmetric seawater feed. b Polarization curves of NiFe LDH/NF‖Pt/C/NF and RuO₂/NF‖Pt/C/NF without iR correction, with energy consumption values indicated for NiFe LDH/NF‖Pt/C/NF. c Chronopotentiometry curve at a j of 1.0 A cm−2 for NiFe LDH/NF‖Pt/C/NF. Inset shows a photograph of two electrode electrolyzer. d Comparison of cell voltages and long-term electrolysis tolerance for NiFe LDH/NF with PF₆⁻ additives against recently reported ASO catalysts. Source data are provided as a Source Data file.
Discussion
Our study identifies PF₆⁻ as a crucial electrolyte additive that markedly enhances the durability of NiFe LDH anode, achieving over 5000 h of stable operation at 1 A cm−2 and 2300 h at 2 A cm−2. Comprehensive ex situ/in situ studies and CPMD simulations reveal the mechanisms behind this improvement. Firstly, under a low applied electric field, PF₆⁻ intercalates into the LDH layers, expanding interlayer spacing and exposing more active sites, thereby boosting ASO activity. Concurrently, PF₆⁻ weakly coordinates with Fe sites, preventing segregation and preserving electrode integrity. Secondly, PF₆⁻ also adsorbs and accumulates on the electrode surfaces under a high applied electric field, effectively blocking Cl⁻. In a two-electrode MEA electrolyzer, this configuration requires ~19.46% less electrical energy than conventional electrolyzer and maintains electrolysis durability for over 1000 h. This study pioneers the efficacy of single-anion engineering in enabling interlayer/surface modifications, providing LDH structures with structural integrity and Cl⁻-repelling capabilities, thereby paving the way for sustainable and large-scale hydrogen production.
Methods
Materials
All chemicals used in this study are listed below with their respective purity and sources. Nickel nitrate hexahydrate (Ni(NO₃)₂·6H₂O, AR), cobalt nitrate hexahydrate (Co(NO₃)₂·6H₂O, AR), potassium hydroxide (KOH, 96 wt.%), ammonium fluoride (NH₄F, AR), urea (CO(NH₂)₂, AR), potassium hexafluorophosphate (KPF₆, AR), ruthenium oxide (RuO₂, AR), iridium oxide (IrO₂, AR), platinum on carbon (Pt/C, 20 wt.%), Nafion (5 wt.%), sodium hypochlorite (NaClO, AR), and iron nitrate nonahydrate (Fe(NO₃)₃·9H₂O, AR) were obtained from Aladdin Industrial Co. Hydrochloric acid (HCl, 98 wt.%) and anhydrous ethanol were sourced from Beijing Chemical Corp. Nickel foam (NF, 0.2 mm thick) was purchased from Qingyuan Metal Materials Co., Ltd. in Xingtai. Seawater samples were collected from Huangdao District, Qingdao City. All aqueous solutions were prepared using deionized water with a resistivity of 18.3 MΩ·cm.
Synthesis of NiFe LDH/NF
NiFe LDH was grown on NF (2 cm × 3 cm) via a hydrothermal process. An aqueous solution containing 0.7 mmol Fe(NO₃)₃·9H₂O, 2.1 mmol Ni(NO₃)₂·6H₂O, 4 mmol NH₄F, and 10 mmol urea was transferred to a Teflon-lined autoclave along with NF and heated at 120 °C for 6 h. After cooling to room temperature, the NiFe LDH/NF was rinsed with deionized water, dried at 60 °C. The loading mass of NiFe LDH is ~1.93 mg cm−2.
Synthesis of PF₆⁻-intercalated NiFe LDH/NF
NiFe LDH/NF electrode was held at 1.15 V for 10 min in PF₆⁻-containing seawater to obtain PF₆⁻-intercalated NiFe LDH/NF, with Hg/HgO and graphite serving as reference and counter electrodes, respectively. The sample was then removed, rinsed with deionized water, dried at 60 °C. The resulting material has a loading mass of ~2.02 mg cm−2.
Synthesis of CoFe LDH/NF
CoFe LDH was prepared on NF (2 cm × 3 cm) by dissolving 3 mmol Co(NO₃)₂·6H₂O, 1 mmol Fe(NO₃)₃·9H₂O, and 3 mmol urea in 30 mL deionized water. This mixture and the nickel foam substrate were placed in a Teflon-lined autoclave and heated at 120 °C for 6 h. After cooling, the CoFe LDH/NF was thoroughly rinsed, dried at 60 °C. The loading mass of CoFe LDH is ~1.59 mg cm−2.
Preparation of IrO2/NF and RuO2/NF
The IrO₂/NF and RuO₂/NF electrodes were prepared following the procedures reported in our previous work without modification21. Commercial RuO₂ and IrO₂ powders were loaded onto NF with the same mass loading as that of NiFe LDH/NF.
Characterizations
The crystal phases of the synthesized materials were identified using X-ray diffraction (XRD) with a Bruker D8 Advance instrument. Surface and structural morphologies were examined via scanning electron microscopy (SEM) on a Gemini SEM 300 (ZEISS) and transmission electron microscopy (TEM) alongside high-resolution TEM (HRTEM) using a JEM-F200 (JEOL). Elemental and chemical state analyses were carried out using X-ray photoelectron spectroscopy (XPS) on an ESCALABMK II spectrometer equipped with an Mg source. X-ray absorption fine structure (XAFS) data were collected at room temperature at the Shanghai Synchrotron Radiation Facility (beamline BL11B). The metal content of the catalysts and the extent of Fe leaching in alkaline seawater were measured using inductively coupled plasma mass spectrometry (ICP-MS, Agilent 5110). Optical absorbance was assessed with a SHIMADZU UV-1800 spectrophotometer, and detailed surface composition was analyzed using time-of-flight secondary ion mass spectrometry (TOF-SIMS) on a PHI TRIFT V nano TOF system.
Electrochemical tests
ASO evaluations were performed at room temperature using a CHI 660E electrochemical workstation, and electrode stability was tested with a LANHE battery tester (Wuhan, China) under steady current conditions. A typical three-electrode arrangement was implemented, using NiFe LDH/NF, CoFe LDH/NF, RuO₂/NF, IrO₂/NF, or NF as the working electrodes. The reference electrode was Hg/HgO, and a graphite rod served as the counter electrode. The electrolytes used were either 1 M KOH + seawater or seawater containing 20 mM PF₆⁻, with a total volume of 50 mL. To reduce magnesium and calcium ion concentrations, natural seawater was treated with sodium carbonate (Na₂CO₃, AR) prior to use21. To prepare 1 M KOH + 20 mM PF₆⁻ seawater electrolytes, 56.11 g of KOH and 3.69 g of KPF₆ were dissolved in 1 L of treated seawater, stirred, and sonicated. The electrolytes were used within 1 h of preparation, kept at room temperature. The pH was measured to be 13.97 ± 0.11. Electrode potentials were recalibrated against the RHE using the equation: ERHE = EHg/HgO + 0.098 + 0.059 × pH. The Hg/HgO electrode was calibrated in H₂ using a Pt wire as the working electrode. The iR-compensated potential (Ecorr) was determined using the relation Ecorr = E − iR, where E is the measured potential, i is the applied current, and R is the solution resistance determined at OCP, measured to be 1.544 ± 0.01 Ω. Electrochemical impedance spectroscopy measurements were performed over a frequency range of 10 kHz to 0.01 Hz with a perturbation amplitude of 5 mV. Prior to long-term durability evaluation, the electrode was subjected to chronopotentiometric activation to facilitate PF₆⁻ intercalation. Throughout the test, alkaline seawater was continuously replenished to counteract electrolyte loss.
TOF calculation
TOF was determined using the formula TOF = Aj/(4Fm), where A represents the geometric area of the electrode, j is the current density, 4 corresponds to the number of electrons per mole of O₂ produced, and F is the Faradaic constant (96,485 C mol−1). The parameter m denotes the active site concentration in moles. To calculate m, the slope of the oxidation peak current versus scan rate was analyzed using the equation Slope = n²F²m/4RT, with n = 1 assumed for a single-electron process.
ECSA and ECSA-normalized j
The specific activity was determined by normalizing the j to ECSA. The ECSA was calculated based on the Cdl, measured through CV curves from 1.09 to 1.19 V vs. RHE without iR correction, where no Faradaic current occurs. Cdl was plotted as Δj/2 at 1.14 V vs. RHE against scan rates to obtain the slope. ECSA was then derived using the formula ECSA = ACdl/Cs, where Cs is the specific capacitance (0.04 mF·cm−2)55.
Operando Raman tests
Raman spectra were recorded using a LabRAM HR Evolution confocal Raman spectrometer with a 532 nm laser and a 50× objective lens. Operando experiments were conducted using a custom-designed Raman cell, where NiFe LDH, PF₆⁻-intercalated NiFe LDH or CoFe LDH served as the working electrode. A platinum wire was utilized as the counter electrode, and Hg/HgO acted as the reference electrode. The electrolyte solution comprised 1 M KOH + seawater or PF6−-containing seawater. Voltage-time tests were executed on a CHI 660E electrochemical workstation at OCP–1.75 V vs. RHE (step: 0.05 V).
Constant potential molecular dynamics calculations
Constant potential molecular dynamics (CPMD) is an effective methodology for examining the dynamics of electrochemical interfaces in explicit solvent environments under fixed potential conditions. This approach is well-suited for systems of moderate size, allowing comprehensive investigation of interfacial properties and electrochemical interactions65. The simulation procedure began by arranging 40 explicit water molecules to create a density of 1 g cm⁻3. Following this, three Cl⁻ were placed randomly in the aqueous phase to ensure uniform distribution. Finally, we achieved the variation of different concentrations by introducing different numbers of PF6⁻ on the surface of the catalyst. In our CPMD simulations, Langevin dynamics with a friction coefficient of 0.2 was utilized to regulate the system temperature at 300 K66. A simulation timestep of 1 femtosecond (fs) was adopted, with hydrogen atoms assigned a mass of 2 atomic mass units (u). The simulation trajectories were conducted with eigenstate settings of 1.0e−4, density settings of 1.0e−5, and energy settings of 1e−6, ensuring accurate representation of the electrochemical processes.
Fabrication of MEA
The MEA was constructed using NiFe LDH/NF as the anode and Pt/C/NF with a geometric area of 1 cm² as the cathode. An anion exchange membrane (60 μm thickness, 1.2 cm × 1.2 cm in size) was pre-treated in 1 M KOH for over 12 h to convert it into the hydroxide form, followed by thorough rinsing with deionized water. During operation, alkaline seawater electrolyte was continuously circulated through the cell at a constant flow rate of 50 mL min−1, and full-cell performance was evaluated at 60 °C using a GW Instek PSW 80-13.5.
Data availability
The source data generated in this study are provided in the Source Data file. Source data are provided with this paper.
Change history
19 June 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41467-025-61100-w
References
Turner, A. et al. Sustainable hydrogen production. Science 305, 972–974 (2004).
Ram, R. et al. Water-hydroxide trapping in cobalt tungstate for proton exchange membrane water electrolysis. Science 384, 1373–1380 (2024).
Pandya, R. et al. Concurrent oxygen evolution reaction pathways revealed by high-speed compressive Raman imaging. Nat. Commun. 15, 8362 (2024).
Goldthau, A. et al. The G20 must govern the shift to low-carbon energy. Nature 546, 203–205 (2017).
Lagadec, M. F. & Grimaud, A. Water electrolysers with closed and open electrochemical systems. Nat. Mater. 19, 1140–1150 (2020).
Lee, S. et al. Tracking high-valent surface iron species in the oxygen evolution reaction on cobalt iron (oxy)hydroxides. Energy Environ. Sci. 15, 206–214 (2022).
Li, L. et al. Ruthenium containing molecular electrocatalyst on glassy carbon for electrochemical water splitting. Dalton Trans. 51, 7957–7965 (2022).
Service, R. F. Seawater splitting could help green hydrogen grow. Science 379, 1075 (2023).
Xie, H. et al. A membrane-based seawater electrolyser for hydrogen generation. Nature 612, 673–678 (2022).
Tong, W. et al. Electrolysis of low-grade and saline surface water. Nat. Energy 5, 367–377 (2020).
Liu, T. et al. In-situ direct seawater electrolysis using floating platform in ocean with uncontrollable wave motion. Nat. Commun. 15, 5305 (2024).
Jin, H. et al. Emerging materials and technologies for electrocatalytic seawater splitting. Sci. Adv. 9, eadi7755 (2023).
Xu, X. et al. Corrosion-resistant cobalt phosphide electrocatalysts for salinity tolerance hydrogen evolution. Nat. Commun. 14, 7708 (2023).
Hu, H. et al. Efficient and durable seawater electrolysis with a V2O3-protected catalyst. Sci. Adv. 10, eadn7012 (2024).
Hu, H. et al. Engineered nickel–iron nitride electrocatalyst for industrial-scale seawater hydrogen production. Adv. Mater. 37, 2415421 (2025).
Farràs, P. et al. Water electrolysis: direct from the sea or not to be? Joule 5, 1921–1923 (2021).
Guo, J. et al. Direct seawater electrolysis by adjusting the local reaction environment of a catalyst. Nat. Energy 8, 264–272 (2023).
Malek, A. et al. Dynamically restructuring NixCryO electrocatalyst for stable oxygen evolution reaction in real seawater. Angew. Chem. Int. Ed. 62, e202309854 (2023).
Qi, L. et al. Controlled growth of metal atom arrays on graphdiyne for seawater oxidation. J. Am. Chem. Soc. 146, 5669–5677 (2024).
Li, P. et al. Common-ion effect triggered highly sustained seawater electrolysis with additional NaCl production. Research 2020, 2872141 (2020).
He, X. et al. Structural evolution and self-reconstruction of nickel hexacyanoferrate Prussian blue analogues for long-lasting ampere-current seawater oxidation. Nano Today 58, 102454 (2024).
Li, Z. et al. Carbon oxyanion self-transformation on NiFe oxalates enables long-term ampere-level current density seawater oxidation. Angew. Chem. Int. Ed. 63, e202316522 (2024).
Zhang, S. et al. Concerning the stability of seawater electrolysis: a corrosion mechanism study of halide on Ni-based anode. Nat. Commun. 14, 4822 (2024).
Liu, W. et al. Ferricyanide armed anodes enable stable water oxidation in saturated saline water at 2 A/cm2. Angew. Chem. Int. Ed. 62, e202309882 (2023).
Liang, J. et al. Electrocatalytic seawater splitting: nice designs, advanced strategies, challenges and perspectives. Mater. Today 69, 193–235 (2023).
Wang, X. et al. Electrosynthesis of transition metal coordinated polymers for active and stable oxygen evolution. Angew. Chem. Int. Ed. 63, e202409628 (2024).
Liu, D. et al. Efficient and ultrastable seawater electrolysis at industrial current density with strong metal-support interaction and dual Cl−-repelling layers. Adv. Mater. 36, 2408982 (2024).
You, H. et al. Monolayer NiIr-layered double hydroxide as a long-lived efficient oxygen evolution catalyst for seawater splitting. J. Am. Chem. Soc. 144, 9254–9263 (2022).
Kang, X. et al. A corrosion-resistant RuMoNi catalyst for efficient and long-lasting seawater oxidation and anion exchange membrane electrolyzer. Nat. Commun. 14, 3607 (2023).
Hu, H. et al. Metal nitrides for seawater electrolysis. Chem. Soc. Rev. 53, 163–203 (2024).
Yu, H. et al. Advancing direct seawater electrocatalysis for green and affordable hydrogen. One Earth 6, 267–277 (2023).
Kuang, Y. et al. Solar-driven, highly sustained splitting of seawater into hydrogen and oxygen fuels. Proc. Natl Acad. Sci. Usa. 116, 6624–6629 (2019).
Wang, S. et al. Synthesis of 3D heterostructure Co-doped Fe2P electrocatalyst for overall seawater electrolysis. Appl. Catal. B Environ. 297, 120386 (2021).
Yu, L. et al. Non-noble metal-nitride based electrocatalysts for high-performance alkaline seawater electrolysis. Nat. Commun. 10, 5106 (2019).
Wang, Z. et al. MnOx film-coated NiFe-LDH nanosheets on Ni foam as selective oxygen evolution electrocatalysts for alkaline seawater oxidation. Inorg. Chem. 61, 15256–15265 (2022).
Liu, H. et al. High-performance alkaline seawater electrolysis with anomalous chloride promoted oxygen evolution reaction. Angew. Chem. Int. Ed. 62, e202311674 (2023).
Liang, J. et al. Expanded negative electrostatic network-assisted seawater oxidation and high-salinity seawater reutilization. ACS Nano 19, 1530–1546 (2025).
Yang, C. et al. Poly(3-thiophenemalonic acid) modified NiFe layered double hydroxide electrocatalyst for stable seawater oxidation at an ampere-scale current density. ACS Mater. Lett. 6, 5248–5255 (2024).
Chen, H. et al. Stable seawater electrolysis over 10000 h via chemical fixation of sulfate on NiFeBa-LDH. Adv. Mater. 36, 2411302 (2024).
Liu, W. et al. Self-protecting CoFeAl-layered double hydroxides enable stable and efficient brine oxidation at 2 A cm−2. Nat. Commun. 15, 4712 (2024).
Hunter, B. M. et al. Effect of interlayer anions on [NiFe]-LDH nanosheet water oxidation activity. Energy Environ. Sci. 9, 1734–1743 (2016).
Zhou, D. et al. Layered double hydroxide-based electrocatalysts for the oxygen evolution reaction: Identification and tailoring of active sites, and superaerophobic nanoarray electrode assembly. Chem. Soc. Rev. 50, 8790–8817 (2021).
Li, Z. et al. Seed-assisted formation of NiFe anode catalysts for anion exchange membrane water electrolysis at industrial-scale current density. Nat. Catal. 7, 944–952 (2024).
Bai, L., Lee, S. & Hu, X. Spectroscopic and electrokinetic evidence for a bifunctional mechanism of the oxygen evolution reaction. Angew. Chem. Int. Ed. 60, 3095–3103 (2021).
Zhou, L. et al. A restricted dynamic surface self-reconstruction toward high-performance of direct seawater oxidation. Nat. Commun. 15, 2481 (2024).
Ma, T. et al. The critical role of additive sulfate for stable alkaline seawater oxidation on nickel-based electrodes. Angew. Chem. Int. Ed. 60, 22740–22744 (2021).
Yu, M. et al. Anionic formulation of electrolyte additive towards stable electrocatalytic oxygen evolution in seawater splitting. J. Energy Chem. 72, 361–369 (2022).
Xu, W. et al. Ag nanoparticle-induced surface chloride immobilization strategy enables stable seawater electrolysis. Adv. Mater. 36, 2306062 (2024).
Duan, X. et al. Dynamic chloride ion adsorption on single iridium atom boosts seawater oxidation catalysis. Nat. Commun. 15, 1973 (2024).
Sun, X. et al. Corrosion-resistant NiFe anode towards kilowatt-scale alkaline seawater electrolysis. Nat. Commun. 15, 10351 (2024).
Yu, L. et al. High-performance seawater oxidation by a homogeneous multimetallic layered double hydroxide electrocatalyst. Proc. Natl Acad. Sci. USA 119, e2202382119 (2022).
Qi, H. et al. Boosting direct seawater electrolysis through intercalation engineering of layered double hydroxides. Ind. Eng. Chem. Res. 62, 19674–19682 (2023).
Zhang, B. et al. High corrosion resistance of NiFe-layered double hydroxide catalyst for stable seawater electrolysis promoted by phosphate intercalation. Small 18, 2203852 (2022).
Fan, R. et al. Ultrastable electrocatalytic seawater splitting at ampere-level current density. Nat. Sustain. 7, 158–167 (2024).
Cai, Z. et al. Stabilizing NiFe sites by high-dispersity of nanosized and anionic Cr species toward durable seawater oxidation. Nat. Commun. 15, 6624 (2024).
Song, J. et al. Citrate ions-modified NiFe layered double hydroxide for durable alkaline seawater oxidation. J. Colloid Interf. Sci. 679, 1–8 (2025).
Diaz-Morales, O., Ferrus-Suspedra, D. & Koper, M. T. The importance of nickel oxyhydroxide deprotonation on its activity towards electrochemical water oxidation. Chem. Sci. 7, 2639–2645 (2016).
Lee, S., Bai, L. & Hu, X. Deciphering iron-dependent activity in oxygen evolution catalyzed by nickel–iron layered double hydroxide. Angew. Chem. Int. Ed. 59, 8072–8077 (2020).
Kim, S. et al. Plasma-engineered 2D Ni nanoplates as advanced oxygen evolution reaction electrocatalysts for direct seawater electrolysis. ACS Appl. Energy Mater. 8, 1101–1111 (2025).
Yan, L. et al. Visible-light-driven electrocatalytic oxygen evolution reaction: NiFe2O4/NiFe–layered double hydroxide Z-scheme heteronanosheet as a model. Energy Technol. 8, 2000607 (2020).
Luo, Yang et al. NiFe-layered double hydroxide synchronously activated by heterojunctions and vacancies for the oxygen evolution reaction. ACS Appl. Mater. Interfaces 12, 42850–42858 (2020).
Bujdosó, T. et al. Structural characterization of arsenate ion exchanged MgAl-layered double hydroxide. Appl. Clay. Sci. 44, 75–82 (2009).
Garcia, A. C. et al. Enhancement of oxygen evolution activity of nickel oxyhydroxide by electrolyte alkali cations. Angew. Chem. Int. Ed. 58, 12999–13003 (2019).
Jia, H. et al. Unveiling the electrolyte cations dependent kinetics on CoOOH-catalyzed oxygen evolution reaction. Angew. Chem. Int. Ed. 62, e202313886 (2023).
Melander, M. M. et al. Constant inner potential DFT for modelling electrochemical systems under constant potential and bias. npj Comput. Mater. 10, 5 (2024).
Korpelin, V. et al. Addressing dynamics at catalytic heterogeneous interfaces with DFT-MD: Anomalous temperature distributions from commonly used thermostats. J. Phys. Chem. Lett. 213, 2644–2652 (2022).
Acknowledgements
X.S. thanks the funding support from the Free Exploration Project of Frontier Technology for Laoshan Laboratory (No. 16-02). F.L. acknowledges the funding support from the Science and Technology Program of Tibet (No. XZ202201ZY0002G). T.W. acknowledges the funding support from the Natural Science Foundation of China (No. 52202214). The numerical calculations in this paper have been done on Computing Center in Xi’an. The authors thank BL11B beamline of the Shanghai Synchrotron Radiation Facility (SSRF) for providing the XAFS beamtime.
Author information
Authors and Affiliations
Contributions
X.S. designed this research. X.H. and X.S. wrote the manuscript. X.H. and Y.Y. conducted material synthesis, characterizations, and performance tests. X.H., Y.Y. and L.Z. conceived and completed all the schematic drawings. H.W. performed the operando Raman tests. Y.L. performed the SEM tests. H.T., W.J., Y.R. and J.N. participated in discussions. T.W. conducted theoretical calculations. F.L., B.T. and X.S. supervised the research. All authors contributed and reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Bolong Huang and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
He, X., Yao, Y., Zhang, L. et al. Hexafluorophosphate additive enables durable seawater oxidation at ampere-level current density. Nat Commun 16, 4998 (2025). https://doi.org/10.1038/s41467-025-60413-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60413-0
