
Abstract
Neoantigen (nAg) vaccines can induce anti-tumor specific immunity, and tumor killing promotes further antigen diffusion, which is expected to improve prognosis. However, the mutation of cancer cells under the selective pressure of vaccines and the immunosuppressive tumor microenvironment make the therapeutic effect unsatisfactory. Here, we develop a nanovaccine (nAg-MRDE/Mn) that can deliver nAg and induce in situ cancer vaccination to synergistically promote a personalized immune response, enhance antigen diffusion, and improve the microenvironment by modulating immunosuppressive cells and activating the innate immune response. Experiments show that nAgs are presented by dendritic cells and expressed by T cells, which cooperate with in situ vaccination to stimulate specific immunity. Cells involved in immunosuppression, such as M2 macrophages and regulatory T cells, are down-regulated, while M1 macrophages and natural killer cells are increased. In addition, the hydrogel loaded with chemokines and nAg-MRDE/Mn inhibits postoperative tumor recurrence, and the combination of nAg-MRDE/Mn and αPD-1 improves the therapeutic effect of αPD-1. This study validates the clinical potential of this strategy and provides ideas for improving neoantigen vaccines.
Similar content being viewed by others

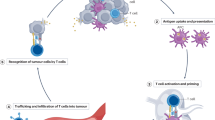
Introduction
Immunotherapy has transformed cancer management, but tumor heterogeneity and immune cold phenotype limit long-term tumor suppression in many patients1,2,3. Personalized neoantigen-based tumor vaccines enrich the endogenous pool of tumor-specific T cells in response to tumor heterogeneity4. However, clinical trials to date have not extensively validated its efficacy, because tumor cells that lack neoantigen expression proliferate under selective pressure, generating a variety of novel epitope mutations5,6. The current strategy is to reduce the likelihood of immune escape by designing cancer vaccines that target multiple neoantigens7,8,9. Of note, both of the positive clinical trials (NCT03897881, NCT04161755) combined immune checkpoint inhibition (ICI) therapy, which further study recognized may be because ICI therapy makes T cells more susceptible to activation by neoantigens and tumor-associated antigens (TAA) released by dying tumor cells, leading to additional tumor-directed immune responses, known as epitope spreading10,11,12. This expansion of the anti-tumor T-cell library often predicts a good prognosis and long progression-free survival13,14,15. Unfortunately, this degree of epitope spreading can still be followed by immune escape after the initial benefit16,17. Therefore, strategies that effectively promote antigen diffusion are needed. In addition, recent studies have speculated that neoantigens combined with immunogenic cell death (ICD) are potential therapeutic strategies for tumor microenvironments (TME) where immunotherapy options are limited due to immune desert phenotype18,19. Inspired by this, we propose that the combination of neoantigens and in situ cancer vaccination can help overcome the immune escape caused by tumor heterogeneity.
The immunosuppressive mechanism in TME not only affects the efficacy of tumor immunotherapy, but also hinders the natural immune response20. Of note, the increase of tumor inflammation and the stimulation of the immune response in the immunosuppressive TME can promote the transformation of cold tumors to hot ones. Increasing the frequency of T cells and neutralizing immunosuppressing cells can further promote the reversal of cold tumors by reprogramming the immunosuppressive TME21. Notably, the activation of the cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathway in cancer cells facilitates the production of type I interferons, which enhances natural killer (NK) cell activity22,23,24. This helps to ensure cross-presentation of antigens for the activation of adaptive immunity and further target heterogeneous tumors with antigenic drift.
Here, we show a nanovaccine (nAg-MRDE/Mn) consisting of STING agonists (Mn2+), dendritic cell-derived exosomes (DEs), and hybrid peptides α-Mel- nAg, which integrated targeting peptides (biomimetic peptide D4F), perforating peptides (melittin, Mel), and nAg. nAgs can be replaced with model neoantigen ovalbumin (OVA). nAg-MRDE/Mn can be loaded into ROS-responsive hydrogel (TSPBA-PVA) containing CXCL10 according to therapeutic requirements (Fig. 1a). The engineered modification confers the ability of nAg-MRDE/Mn to synergistically target tumors through multiple pathways. In vitro and in vivo studies show that the neoantigens are effectively cross-presented, the ICD effector of tumor cells is activated and NK cells are activated through the cGAS-STING pathway, and the TME is improved (Fig. 1b). The combination of the neoantigens with the original cancer vaccine results in a certain increase in the level of anti-tumor specific immunity. Vaccine-loaded hydrogel-based postoperative therapy and vaccine applicability in other heterogeneous tumors such as lung cancer brain metastases and triple-negative breast cancer are explored. Combination therapy with αPD-1 improves response rates to immune checkpoint inhibition25,26,27. The translational potential of the vaccine is explored by simulating clinical conditions. In conclusion, this study has developed a nano-vaccine for treating heterogeneous and immunosuppressive malignancies, offering some thoughts for enhancing the personalized immunotherapy of neoantigen vaccines.

a The steps involved in the preparation of nAg-MRDE/Mn and nAg-MRDE/Mn+CXCL10@Gel. b Schematic diagram of personalized immunotherapy by combining neoantigens with in situ cancer vaccine and converting the tumor from cold to hot. BMDCs, mouse bone marrow-derived DCs; DEs, dendritic cell-derived exosomes; Rh2, ginsenoside Rh2; Mel, melittin; OVA, ovalbumin; PVA, polyvinyl alcohol; BSA, bovine serum albumin; dsDNA, double-stranded DNA; cGAS, cyclic GMP-AMP synthase; cGAMP, cyclic GMP-AMP; STING, stimulator of interferon genes; TBK1, TANK binding kinase 1; IRF3, interferon regulatory factor 3; CTL, cytotoxic T lymphocyte; GLUT1, glucose transporter type 1; SR-BI, scavenger receptor class B type I; ROS, reactive oxygen species.
Results
Synthesis and characterization of nAg-MRDE/Mn and nAg-MRDE/Mn + CXCL10@Gel
Based on the nanovaccine construction strategy, we combined Mn2+ with bovine serum albumin (BSA) to form nanosheets BSA/Mn. DEs were extracted from mouse bone marrow-derived DCs (BMDCs) and then Rh2 was incorporated into the DEs (RDE) by ultrasonic fragmentation. Subsequently, RDE-encapsulated BSA/Mn (RDE/Mn) was used, and the hybridized peptide α-Mel-OVA was conjugated to RDE/Mn by phospholipid bilayer insertion to obtain O-MRDE/Mn (Fig. 2a). In addition to nAg in vaccine design, the engineered DEs could target cancer cells at tumor sites28. Mn2+ can activate NK cells by stimulating the cGAS-STING signaling pathway22. Mel can induce ICD in cancer cells, promoting in situ cancer vaccination29. Ginsenoside Rh2 (Rh2) repolarizes tumor-associated macrophages (TAMs), reduces the percentage of regulatory T cells (Tregs), and reverses the immunosuppressive TME30,31.

a The preparation process of O-MRDE/Mn. b Transmission electron microscope (TEM) images of BSA/Mn, DE, and O-MRDE/Mn. c Flow cytometry detected proteins that are characteristic of DE and O-MRDE/Mn. d Zeta potentials and size distributions. e Hemolysis assay for Mel, α-Mel, α-Mel-OVA, and O-MRDE. f The preparation and ROS responsiveness processes of O-MRDE/Mn+CXCL10@Gel. g Representative morphology and SEM image of hydrogel scaffold loaded with O-MRDE/Mn and CXCL10. h, i Oscillatory strain sweep (h) and cycling strain sweeps (i) of hydrogels. j-l Mass remaining of hydrogels (j) and accumulative release profiles of CXCL10 (k) and Mn (l) from hydrogels under PBS and H2O2 (1 mM) conditions. m Chemotactic migration assay of T cells when treated with different doses of CXCL10. Data in d, e, j–m were mean ± SD (n = 3 independent experiments). Source data are provided as a Source Data file.
The BSA/Mn was formed by Mn2+ binding to aspartic acid or histidine residues in BSA22. As the concentration of Mn2+ increased, the fluorescence emission peaks of BSA gradually diminished, indicating the effective binding between Mn2+ and BSA and showing a two-dimensional nanosheet structure (Fig. 2b, Supplementary Fig. 1). ESI-MS analysis showed that the hybrid peptide synthesis was as expected (Supplementary Fig. 2). DEs and O-MRDE/Mn have a typical cup-shaped vesicle structure with detectable exosome markers CD81, CD63, and CD9 (Fig. 2b, c). The average diameter of O-MRDE/Mn was found to be 139.4 ± 3.1 nm as determined by dynamic light scattering. It maintained good stability at 4 °C (Fig. 2d, Supplementary Fig. 3). The modification efficiency of Rh2 and hybrid peptides on O-MRDE/Mn were 76.95 ± 4.57% and 74.36 ± 1.90%. The encapsulation rate of Mn2+ was 71.45 ± 1.18%, suggesting successful preparation of the nanovaccine. Further examination of the release characteristics showed that O-MRDE/Mn delayed the release of Mn2+ compared to MnCl2 (Supplementary Fig. 4). In addition, hemolytic experiments revealed that O-MRDE/Mn improved the hemolytic reaction produced by the cationic nature of Mel, which preliminarily showed that the nanovaccine has the biosafety (Fig. 2e and Supplementary Fig. 5).
To preserve the drug injected in situ in specific clinical situations, we applied ROS-responsive hydrogels to achieve sustained co-delivery of O-MRDE/Mn and CXCL10 (Fig. 2f and Supplementary Fig. 6). ROS-labile linker N1-(4-boronobenzyl)-N3-(4-boronophenyl)-N1,N1,N3,N3-tetramethylpropane-1,3-diaminium (TSPBA) was used as a cross-linking agent to form a ROS-responsive hydrogel with polyvinyl alcohol (PVA) via phenyl-boron ester bonding reaction. The synthesis of TSPBA by a quaternization reaction was verified by 1H nuclear magnetic resonance (NMR) (Supplementary Fig. 7). The mixed PVA and TSPBA can form a hydrogel within seconds, and scanning electron microscope (SEM) images showed the porous structure of the hydrogel and the trapped O-MRDE/Mn (Fig. 2g). Following this, we examined the rheological characteristics of the hydrogel. Between 0.01 and 100% strain, the value of the storage modulus (G’) of the gel or O-MRDE/Mn+CXCL10@Gel is higher than the value of the loss modulus (G”), proving the existence of a sufficient cross-linked structure to maintain the gelation state (Fig. 2h, i). For cycling strain sweeps, the gel and O-MRDE/Mn+CXCL10@Gel showed recuperation at low strains, highlighting the hydrogel’s fast repair capabilities and flexible cross-linking characteristics (Fig. 2i).
We further examined the ROS-responsive degradation properties of the hydrogels. In the H2O2 environment, the hydrogel volume was reduced faster within 8 days, and O-MRDE/Mn could be delivered as nanoparticles (Fig. 2j and Supplementary Figs. 8, 9). Driven by H2O2, O-MRDE/Mn+CXCL10@Gel released much more CXCL10 (86.09 ± 2.72%) and Mn2+ (83.89 ± 3.25%) in 8 days than in the H2O2-free environment (Fig. 2k, l). DiR-labelled hydrogels were implanted into the brains of healthy mice and tumor resection model mice to examine the responsiveness of the hydrogels in vivo. The results showed that the gel disintegrated faster in the brain compared with the healthy mouse group, suggesting that the hydrogel has a better degradation property in response to ROS in vivo (Supplementary Fig. 10). Meanwhile, we implanted the hydrogel into the brains of Sprague Dawley (SD) rats. The volume of the hydrogel decreased within 10 days and could fully release the encapsulated nanoparticles, further demonstrating the hydrogel’s better in vivo degradability (Supplementary Figs. 11, 12). Furthermore, CXCL10 promoted T cell motility in a time and concentration-controlled mode (Fig. 2m and Supplementary Fig. 13), suggesting that delivering CXCL10 locally via hydrogels for effective T cell recruitment is possible.
O-MRDE/Mn induced immune activation in vitro
To gain insights into the process and mechanism of O-MRDE/Mn in the TME, we performed in vitro examinations of cellular uptake and immune activation. In the design of the nanovaccine, D4F and Rh2 induced endocytosis via scavenger receptor class B type I (SR-BI) and glucose transporter protein 1 (GLUT1), which are highly expressed on tumor cells, and synergistically mediated the uptake of O-MRDE/Mn by the tumor cells with Mel-induced cell membrane penetration32,33,34. The cellular uptake results revealed that the uptake rate of DEs was upregulated after modification with D4F (D4F-DE/Mn), Mel (MDE/Mn), and Rh2 (RDE/Mn). When cells were pretreated with D4F to saturate the SR-BI receptor (defined as MDE/Mn (-)), the uptake rate of MDE appeared down-regulated. Similar results were obtained after blocking GLUT1 using WZB-117 (defined as RDE/Mn(-)). Whereas O-MRDE/Mn showed the highest cellular uptake rate, suggesting that the combination of Mel, D4F, and Rh2 synergistically promotes the internalization of O-MRDE/Mn in GL261 (Fig. 3b and Supplementary Fig. 14). The endocytosis/lysosomal escape ability of Mel was subsequently examined. The red fluorescence of DiD-O-MRDE/Mn overlapped with the green fluorescence of the lysosome at 2 h and diminished at 8 h, indicating a favorable lysosomal escape effect of O-MRDE/Mn (Fig. 3c and Supplementary Fig. 15). It was observed by cytotoxicity assay that O-MRDE/Mn+CXCL10@Gel inhibited the proliferation of GL261 cells. In contrast, there was no significant toxic effect on BMDCs, brain capillary endothelial cells (BCECs), and PC12 cells (Fig. 3d and Supplementary Figs. 16–18). Analysis of the effects of O-MRDE/Mn on BMDCs, BCECs, and PC12 cells may be related to the selective targeted uptake of O-MRDE/Mn.

a Illustrative diagram for the function of each component. b Investigating the mechanism of tumor cellular uptake of O-MRDE/Mn. c Pearson’s correlation coefficient analysis of lysosome escape of D4F-DE/Mn, MDE/Mn, and O-MRDE/Mn. d Cytotoxicity of O-MRDE/Mn+CXCL10@Gel against GL261 cells. e Flow cytometry was used to detect CRT on GL261 cells. f, g Levels of HMGB1 (f) and ATP (g). h, i Expression of CD80, CD86 (h), and CD40 (i) on BMDCs. j Levels of antigen SIINFEKL-presented BMDCs. k Quantitative results of coculture-induced CD8+ T cells activation. l Quantitative results of coculture-induced SIINFEKL-specific CD8+ T cells activation. m Quantitative analysis of the levels of different cytokines using ELISA. n Schematic diagram of innate immune activation of O-MRDE/Mn. o, p Content of cGAMP (o) and IFN-β (p). Data in b–h, j–l, m, o, and (p) were mean ± SD (n = 3 independent experiments). The significance between each of the multiple groups in b, e–h, j–l, o, and (p) was calculated using one-way ANOVA. Source data are provided as a Source Data file.
Subsequently, we examined Mel’s action in triggering ICD. MDE/Mn and O-MRDE/Mn up-regulated the levels of three damage-associated molecular patterns (DAMPs), namely calreticulin (CRT), adenosine triphosphate (ATP), and high mobility group box-1 protein (HMGB1), which would be beneficial for the induction of in situ cancer vaccination (Fig. 3e–g). Meanwhile, O-MRDE/Mn induced high expression of DCs activation maturation-associated CD80, CD86, and CD40 co-stimulatory molecules (Fig. 3h, i and Supplementary Fig. 19). Further analysis revealed increased levels of H-2Kb-SIINFEKL on the surface of CD80+ cells (11.37 ± 0.70%) in the O-MRDE/Mn group, which means the enrichment of the MHC-I/H-2Kb-SIINFEKL complex on the cell membrane of BMDCs (Fig. 3j, Supplementary Figs. 20, 21). These changes were mainly attributed to the cross-presentation of antigens. Mature BMDCs co-cultures stimulated by O-MRDE/Mn-treated tumor cells resulted in a 3.82-fold increase in the expression level of CD8+ T cells, and SIINFEFKL-specific CD8+ T cells were similarly elevated, demonstrating the effective activation of an antigen-specific immune response (Fig. 3k, l and Supplementary Figs. 22, 23). Moreover, examination of cytokine levels in the co-culture model revealed that O-MRDE/Mn up-regulated the levels of tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), interleukin-12 (IL-12), and interferon-γ (IFN-γ), and down-regulated the interleukin-10 (IL-10) levels (Fig. 3m).
Lastly, we sought to determine how Mn2+ regulates innate immunity through tumors. Mn2+ triggers the production of 2'3’-cyclic GMP-AMP (cGAMP) by enhancing the capture of damaged double-stranded DNA (dsDNA) by cGAS and stimulates STING, which induces the generation of type I interferons (IFNs)35,36,37. This process is critical for NK cell activation (Fig. 3n). Compared with the control group, O-MRDE/Mn showed a 5.08-fold increase in the level of cGAMP and a 2.99-fold increase in interferon-β (IFN-β), suggesting that Mn2+ can efficiently initiate the innate immune response (Fig. 3o, p). These explorations revealed that O-MRDE/Mn promotes cross-presentation of model neoantigens, enables in situ cancer vaccination, and facilitates epitope spreading of anti-tumor T cells. In addition, it synergistically exerts anti-tumor effects by inducing a sustained and effective innate immune response by activating the cGAS-STING.
O-MRDE/Mn induces in vivo immune responses to inhibit tumor growth
We further investigated the treatment efficacy of nanovaccines in vivo by constructing a mouse glioblastoma (GBM) model through intracranial injection of GL261-luc cells and GL261-OVA-luc cells, respectively. The course of drug treatment is shown in Fig. 4a, where the pre-treatment group received O-MRDE/Mn 5 days before tumor implantation, and the remaining group was administered 10 days after modeling. Saline (OVA) and O-MRDE/Mn (OVA) represent groups in the GL261-OVA-luc model. Hematoxylin and eosin (H&E) staining illustrated that the saline group exhibited uncontrolled rapid tumor cell growth. In contrast, O-MRDE/Mn exhibited tumor suppression in both models (Fig. 4b). In vivo imaging system (IVIS) results showed the same results, and pretreatment reduced the bioluminescence signal and prolonged the survival of the mice. Although the saline (OVA) group had the strongest tumor signal enhancement, the O-MRDE/Mn (OVA) group still ended up suppressing the proliferation (Fig. 4c–e and Supplementary Fig. 24). Further detection of cytokines revealed that in both models, O-MRDE/Mn induced an increase in the contents of IFN-β, TNF-α, IL-6, IL-12, and a decrease of IL-10 secretion. It can be seen that O-MRDE/Mn can activate the inflammatory response process (Fig. 4f). On the other hand, we also examined the safety risks of nanovaccines. There was no massive body weight loss, and the histological examination of major organs showed no obvious tissue damage. Additionally, there were no substantial changes in liver and kidney function levels and routine blood analysis in all groups, indicating that the nanovaccine has a good in vivo safety profile (Fig. 4g and Supplementary Figs. 25–27).

a Diagram illustrating the experimental setup. b Images of tumor tissues stained with H&E. c Bioluminescence imaging of mice bearing GL261-luc and GL261-OVA-luc tumors. d Quantification of bioluminescent signal intensity. e Survival of mice bearing GL261-luc and GL261-OVA-luc tumors. f Quantitative analysis of cytokine levels using ELISA. g Body weight changes of mice bearing GL261-luc and GL261-OVA-luc tumors. h, i Quantitative analysis of NK cells (CD3-NK1.1+) (h) and mature DCs (CD11c+MHC I+) (i) in tumor. j–l Quantification of CD8+ T cells (CD3+CD8+) (j), IFN-γ+ T cells (CD8+IFN-γ+) (k), and GrB+ T cells (CD8+GrB+) (l) in spleen. m Quantitative analysis of IgM, IgG, and IgA levels using ELISA. Data were mean ± SD (n = 6 mice per group). The significance in (d) between G1 and G2-G7, and between G8 and G9 was calculated using two-way ANOVA. The significance in (e) between G1 and G2–G7, and between G8 and G9 was calculated using Kaplan-Meier survival analysis. The significance between each of the multiple groups in (h–m) was calculated using one-way ANOVA. Source data are provided as a Source Data file.
We subsequently investigated the immune activation levels in vivo to elucidate the mechanism of action of the O-MRDE/Mn. Against the innate immune response, it is interesting to note that the percentage of NK cells in BSA/Mn has been higher than in saline (p < 0.0001), indicating that Mn2+ plays a role. A further increase in NK cell infiltration was observed in tumors injected with MDE/Mn and O-MRDE/Mn, which may be attributed to improved uptake efficiency (Fig. 4h and Supplementary Fig. 28). For the adaptive immune response mediated by cancer vaccination, mature DC levels at tumor sites were upregulated in the O-MRDE/Mn group and O-MRDE/Mn (OVA) group (Fig. 4i and Supplementary Fig. 29). MDE/Mn and O-MRDE/Mn (OVA) increased CD8+ T cell levels while inducing antigen cross-presentation compared to saline. Notably, the O-MRDE/Mn (OVA) group was higher than the O-MRDE/Mn group, probably because of the specific immune response against OVA generated in the GL261-OVA-luc model, and the levels of CD8+ and CD4+ T cells in the tumors were also increased. Of note, there was no significant difference between MRDE/Mn and O-MRDE/Mn, indicating that OVA in the vaccine had no relevant effect in tumor models that do not express OVA (Fig. 4j and Supplementary Figs. 30–32). Further assessment of CD8+ T cell activity revealed that O-MRDE/Mn upregulated the levels of IFN-γ+ and granzyme B+ (GrB+) CD8+ T, suggesting the occurrence of anti-tumor immune response (Fig. 4k, l and Supplementary Figs. 33, 34). Similar results were achieved in the pretreatment group. Finally, specific antibodies in the blood were measured to assess the effectiveness of O-MRDE/Mn. After O-MRDE/Mn treatment, the levels of IgM, IgG and IgA in both model mice were elevated to different degrees, verifying the successful induction of anti-tumor immune responses (Fig. 4m). Therefore, it can be concluded that O-MRDE/Mn inhibits tumor proliferation and prolongs survival by stimulating innate and adaptive immunity, and synergizes with neoantigens to exert specific immune effects, providing effective preventive and therapeutic effects.
O-MRDE/Mn induces antigen-specific immunity and remodels the TME
To further demonstrate that O-MRDE/Mn carrying the model neoantigen OVA could induce personalized anti-tumor-specific immune responses and clarify the need for neoantigens to synergize with in situ cancer vaccination, we established an in situ GBM model based on GL261-OVA-luc. As we anticipated, O-MRDE/Mn inhibited the growth of tumor cells (Supplementary Fig. 35). In addition to DC maturation and CD8+ T cell activation, we also noted that the proportion of OVA peptide (SIINFEKL)-presenting DCs and SIINFEKL-MHC-I tetramer CD8+ T cells was upregulated in MDE/Mn (OVA) and O-MRDE/Mn (OVA) (Fig. 5a, b and Supplementary Figs. 36-39). Meanwhile, compared with the saline, O-MRDE/Mn increased the proportions of IFN-γ+CD8+ T cells, GrB+CD8+ T cells, and CD107a+CD8+ T cells by 5.24, 4.01, and 2.92-fold, respectively (Fig. 5c–e and Supplementary Figs. 40–42). This suggests that the antigen repertoire generated by in situ cancer vaccination can enhance the specific immune response to neoantigens, in which tumor-specific antigens promote endogenous antitumor T cell epitope spreading, enhancing the anti-tumor-specific immune response.

a Levels of antigen SIINFEKL-presented DCs (25d1.16+CD11c+) in tumor. b Levels of SIINFEKL-specific CD8+ T cells from peripheral blood. c, d Levels of IFN-γ+ T cells (CD8+IFN-γ+) (c), and GrB+ T cells (CD8+GrB+) (d) in spleen. e Levels of CD107a+CD8+ T cells in tumor. f Diagrammatic representation of remodeling immunosuppressive TME. g Quantitative analysis of M1/M2 TAMs in tumor. h–l Quantitative analysis of immunofluorescence data of Ki67+ cells (h), CD31+ cells (i), CD44+ cells (j), CD8+ T cells (k) and Tregs (Foxp3+CD4+) (l) in tumors. m Quantitative analysis of CD8+ T cells/Tregs in tumor tissues. n Number of CD3+CD8+ T cells per mg of tumor tissue. o Quantitative analysis of Th1 cells in tumor tissues. p, q Quantitative analysis of immunofluorescence data of Th1 cells (T-bet+CD4+) (p), and Th2 cells (GATA-3+CD4+) (q) in tumor tissues. Data were mean ± SD (n = 6 mice per group). The significance between each of the multiple groups in (a–e), (g–q) was calculated using one-way ANOVA. Source data are provided as a Source Data file.
Subsequently, the prospective application of O-MRDE/Mn in inhibiting TME and enhancing immune cell permeability was validated. Tumor-associated macrophages (TAM) are the richest infiltrating inflammatory cells in TME. Their repolarization is critical for the reversal of immunosuppressive TME38 (Fig. 5f). Flow cytometry analysis of tumor samples showed that O-MRDE/Mn promoted the repolarization of TAM from the M2 phenotype to the M1 phenotype to inhibit tumor progression in both GL261 and GL261-OVA models (Fig. 5g and Supplementary Figs. 43, 44). Tumor tissues were further stained in sections to examine tumor cell proliferation (Ki67), microvessel density (CD31), and metastasis (CD44) to assess the effect of O-MRDE/Mn on TME (Supplementary Fig. 45). Quantitative analysis showed that compared with the saline group, O-MRDE/Mn reduced the levels of Ki67, CD31, and CD44-positive cells in the tumor tissues of mice in the GL261 model by 6.23, 8.41, and 2.53-fold, respectively, and in the GL261-OVA model by 6.97, 10.88, and 10.53-fold, respectively, indicating that O-MRDE/Mn effectively inhibited the proliferation of tumor cells (Fig. 5h-j). Meanwhile, we performed a detailed evaluation of immune cell infiltration. It was shown that one of the main effects of O-MRDE/Mn delaying tumor growth was an increment in CD4+ and CD8+ T cells infiltration and a decrement in Tregs (Fig. 5k–m and Supplementary Fig. 46). Determination of the amount of CD3+CD8+ T cells in the tumor tissue also verified this result (Fig. 5n). Considering the contribution of various subpopulations of helper T (Th) cells in immune response, we further analyzed different Th cells. Th1/Th2 imbalance is an important step in mediating immune escape of tumor cells39. The assay results showed that O-MRDE/Mn could effectively correct the Th1/Th2 imbalance and ensure that the tumor immune microenvironment shifted to Th1 response and maintained a lower level of Th2 response to enhance tumor-specific immune response (Fig. 5o-q and Supplementary Figs. 47, 48). Overall, these findings revealed that O-MRDE/Mn could effectively induce the establishment of personalized anti-tumor immune responses in vivo and reverse immunosuppressive TME by promoting tumor-associated M2-TAM repolarization, improving immune cell infiltration, and reversing Th1/Th2 imbalance.
O-MRDE/Mn + CXCL10@Gel inhibited postoperative recurrence and metastasis
The immunosuppressive microenvironment after tumor resection affects patients’ postoperative consolidation therapy, and we explored the anti-recurrence ability of hydrogel co-delivering O-MRDE/Mn and CXCL10 by establishing a postoperative GBM model. CXCL10 synergistically enhances anti-tumor effects by facilitating migration and adhesion of recruited immune cells into tumor lesions40. The visible tumor was resected 10 days after the model was established, and H&E staining revealed that partial tumor tissue remained around the tumor resection cavity (Fig. 6a). IVIS monitored the process of tumor recurrence. The fluorescence intensity was lowered after O-MRDE/Mn+CXCL10@Gel treatment, and tumor growth was inhibited. Similarly, uncontrolled tumor proliferation was seen in the Saline (OVA) group, whereas tumors were eventually controlled in the O-MRDE/Mn+CXCL10@Gel (OVA) group (Fig. 6b, c and Supplementary Figs. 49, 50). The survival rate of mice was 83.33% within 120 days, the longest survival time, indicating that tumor proliferation was better inhibited (Fig. 6d, e). Meanwhile, Ki67 staining of the resected lesions of the tumors also verified this result (Fig. 6f and Supplementary Fig. 51). The immune activation and infiltration of immune cells were detected 10 days after administration, and compared to the saline (OVA) group, the O-MRDE/Mn+CXCL10@Gel (OVA) group resulted in a 4.28, 3.38, and 6.49-fold increase in the levels of activated NK cells, antigen-presenting cells (CD80+CD86+), and CD8+ T, respectively, showing the activation of anti-tumor immune responses (Fig. 6g–i and Supplementary Figs. 51–53). Further measurement of specific antibodies in the blood revealed that the levels of IgM, IgG, and IgA treated with O-MRDE/Mn+CXCL10@Gel were elevated to varying degrees, corroborating the efficient activation of the immune response (Supplementary Fig. 54). In addition, O-MRDE/Mn+CXCL10@Gel raised the levels of inflammatory cytokines, which in turn effectively induced the inflammatory response process in the TME (Supplementary Fig. 55).

a Diagram of the experimental setup, H&E staining, tumor surgical debulking, and hydrogel cavity implantation. b Representative images of in vivo bioluminescence imaging. c Quantified signal intensity of in vivo bioluminescence imaging. d, e Changes in survival analysis (d) and (e) body weight. f Quantitative analysis of immunofluorescence data of Ki67+ cells in tumor. g, h Levels of NK cells (CD3-NK1.1+) (g) and mature DCs (CD80+CD86+) (h) in tumor. i Levels of CD8+ cells in tumor. j, k Quantitative analysis of M2-TAMs (F4/80+CD206+) (j) and M1-TAMs (F4/80+CD86+) (k) in tumor. l Levels of Tregs (Foxp3+CD4+) in tumor. m, n Quantitative analysis of CD8+ TCM (CD8+CD44+CD62L+) (m), CD4+ TCM (CD4+CD44+CD62L+) (n) in spleen. o Typical images of sections stained with H&E. p Survival analysis of the mice. q Quantitative analysis of immunofluorescence data of Ki67+ cells in tumor. r Quantitative analysis of CD8+ T cells (CD3+CD8+) in spleen and tumor. Data were mean ± SD (n = 6 mice per group). The significance in c between G1 and G2-G8, and between G9 and G10 was calculated using two-way ANOVA. The significance in (d) between G1 and G2-G8, and between G9 and G10 was calculated using Kaplan-Meier survival analysis. The significance between each of the multiple groups in f–n, q, r was calculated using one-way ANOVA. The significance in (p) between G1 and G8, and between G9 and G10 was calculated using Kaplan-Meier survival analysis. Source data are provided as a Source Data file.
Subsequently, we examined the immune mechanism of hydrogel in preventing tumor recurrence in vivo. Compared with the saline group, treatment with O-MRDE/Mn+CXCL10@Gel resulted in a 3.59 and 3.12-fold reduction in M2-TAMs and a 6.19 and 2.53-fold rise in M1-TAMs in GL261-luc and GL261-OVA-luc model mice, which effectively induced the transition from M2-type to M1-type (Fig. 6j, k and Supplementary Fig. 56). Meanwhile, Foxp3+ cell count statistics showed that the hydrogel decreased Tregs proportion and blocked tumor cells’ immunosuppressive effect (Fig. 6l and Supplementary Fig. 51). Further assessment of the protective immune response in mice revealed that O-MRDE/Mn+CXCL10@Gel up-regulated the percentage of CD8+ central memory T cells (CD8+ TCM) and CD4+ TCM in the spleen of mice, providing the basis for the sprotective immune response. Similar results were observed in the O-MRDE/Mn+CXCL10@Gel (OVA) (Fig. 6m, n and Supplementary Figs. 57, 58).
Lastly, we further evaluated whether the hydrogel could prevent tumor re-challenge. After 60 days of the administration, the left striatum was also injected with tumor cells into mice of the hydrogel groups, while age-matched naive mice were injected with tumor cells in the left and right striatum as reference for cancer metastasis and recurrence. H&E staining showed that O-MRDE/Mn+CXCL10@Gel inhibited the growth of GL261-luc and GL261-OVA-luc cells without additional intervention. Whereas mice in the saline group of both models died of tumor load within 32 and 27 days, tentatively indicating the positive efficacy of the hydrogel in preventing tumor recurrence (Fig. 6o, p). Immunofluorescence staining of tumor cells also verified this result. After O-MRDE/Mn+CXCL10@Gel treatment, the percentage of Ki67-positive cell counts was reduced, demonstrating that the tumor cell proliferation was inhibited and ameliorated the problem of tumor recurrence (Fig. 6q and Supplementary Fig. 59). Meanwhile, O-MRDE/Mn+CXCL10@Gel up-regulated CD8+ T cells in the spleen and brain of re-challenge model mice, activating the anti-tumor immune response (Fig. 6r and Supplementary Fig. 60). Collectively, O-MRDE/Mn-centered hydrogel promoted the development of immune memory, thereby preventing tumor recurrence and halting disease progression after surgery.
nAg-MRDE/Mn designs personalized immunotherapy for various tumor models
Extensive molecular and cellular reprogramming during the creation of metastatic niches imparts heterogeneity to metastatic tumors and TME41,42,43. Therefore, we constructed a lung cancer brain metastasis model using LLC and LLC-OVA cells to investigate whether O-MRDE/Mn can promote personalized immune activation by releasing individual epitopes. O-MRDE/Mn was found to reduce the necrotic area of brain tumors and prolong the median survival of mice by H&E staining and survival examination (Fig. 7a and Supplementary Fig. 61). Measurement of immune activation levels in mice revealed that O-MRDE/Mn up-regulated H-2Kb-SIINFEKL levels on DCs and induced antigen-dependent up-regulation of CD8+ T-cell expression, which was 3.59 and 1.75-fold higher than saline group, respectively (Fig. 7b, c and Supplementary Figs. 62-65). Higher levels of infiltration of CD8+ and CD4+ T cells were also observed at the tumor site (Supplementary Figs. 66, 67). Immunofluorescence staining of tumor tissues also verified that O-MRDE/Mn effectively induced infiltration of CD4+ T cells, CD8+ T cells and decreased the percentage of Tregs for favorable anti-tumor immune responses (Fig. 7d-f and Supplementary Fig. 68). After treatment with O-MRDE/Mn, inflammatory cytokines increased, and IgM, IgG, and IgA levels remained high, indicating an anti-tumor immune response (Supplementary Figs. 69, 70).

a H&E staining images of tumor tissue. b Percentage of mature DCs (CD80+CD86+) in tumor. c Levels of CD8+ T cells (CD3+CD8+). d–f Levels of CD4+ T cells (d), CD8+ T cells (e), and Tregs (f) in tumors. g Schematics of the experimental design for the Synergistic immunotherapy effects of M25-MRDE/Mn+CXCL10@Gel plus αPD-1. h Tumor growth profiles of mice. i Tumor inhibition rate of mice during treatment. j Mice survival rate. k, l levels of NK cells (CD3-CD49b+) (k) and CRT (l) after different treatments. m Levels of mature DCs (CD80+CD86+) in tumors. n, o Levels of IFN-γ+ T cells (CD8+IFN-γ+) (n), and GrB+ T cells (CD8+GrB+) (o). p Levels of Tregs (Foxp3+CD4+) in tumors. q Representative images of lung metastases. r Quantitative analysis of lung metastases. Data were mean ± SD (n = 6 mice per group). The significance between each of the multiple groups in (b–f, i, k–p, and r) was calculated using one-way ANOVA. The significance in (h) was calculated using two-way ANOVA. The significance in (j) between G1 and G2-G6 was calculated using Kaplan-Meier survival analysis. Source data are provided as a Source Data file.
Then, we constructed an in situ 4T1 model and used the 4T1-specific neoantigen M25 instead of the model antigen OVA in combination with αPD-1 to further investigate the broad applicability of the combination therapy (Fig. 7g). After hydrogel treatment, M25-MRDE/Mn+CXCL10@Gel showed more potent anti-tumor effects than O-MRDE/Mn+CXCL10@Gel, with prolonged survival time of the mice and an increase in tumor suppression rate from 53.75 ± 8.16% to 67.12 ± 5.72%, which demonstrated the synergistic effect of in situ cancer vaccination with the neoantigen (Fig. 7h–j and Supplementary Fig. 71). NK cell infiltration was enhanced 2.90-fold in tumors in the M25-MRDE/Mn+CXCL10@Gel group compared with the saline group (Fig. 7k and Supplementary Fig. 72). Successful ICD inductionwas illustrated by the increase in CRT expression in tumors, which was further evidenced by the 3.87-fold increase in mature DCs in the M25-MRDE/Mn+CXCL10@Gel group compared with the saline group (Fig. 7l, m and Supplementary Figs. 73, 74). Further subpopulation analysis of CD8+ T cells revealed that M25-MRDE/Mn+CXCL10@Gel+αPD-1 increased the percentage of IFN-γ+CD8+ T cells and GrB+CD8+ T, and decreased Tregs in the tumors compared to the saline group (Fig. 7n-p and Supplementary Figs. 75-77). In addition, we investigated metastatic tumors in the lungs of mice. the M25-MRDE/Mn+CXCL10@Gel combined with αPD-1 treatment group had the smallest metastatic foci and fewer metastatic nodules in the lungs, suggesting that the hydrogel could exert a long-term protective effect through the enhancement of the systemic immune response, and had a significant anti-metastatic effect (Fig. 7q, r). In summary, the above results indicated that the combination of M25-MRDE/Mn+CXCL10@Gel and αPD-1 improved the response of 4T1 tumors to αPD-1, further illustrating that the hydrogel design strategy enhances the broad applicability of the combination therapy.
Potential clinical applicability of nAg-MRDE/Mn
We investigated the translational feasibility of the nanovaccine by establishing a CT-2A xenograft model and a patient-derived xenograft (PDX) tumor model. In particular, the model antigen OVA was replaced by mEpb4 as the neoantigen in the CT-2A model (Fig. 8a). Previous studies have shown that the CT-2A model more closely resembles the immunological characteristics of human GBM than the GL261 model44. We similarly verified that the level of PD-1+ cells in CD8+ T cells was higher in CT-2A model, which was 77.15 ± 3.82%, compared to 56.25 ± 2.15% in the GL261 model (Supplementary Fig. 78). Reassuringly, the mEpb4-MRDE/Mn exhibited a good immunostimulatory effect. The percentage of NK cells in mEpb4-MRDE/Mn was 2.43-fold higher than in the saline group (Fig. 8b and Supplementary Fig. 79). Significant activation of DCs is expected to promote antigen cross-presentation and induce antigen-specific immune activation (Fig. 8c and Supplementary Fig. 80). The infiltration levels of CD8+ and CD4+ T cells were increased at the tumor site (Supplementary Figs. 81, 82). Compared with the saline, mEpb4-MRDE/Mn increased the percentage of IFN-γ+CD8+ T cells, and GrB+CD8+ T cells, by 5.27, and 4.47-folds, respectively (Fig. 8d, e and Supplementary Figs. 83, 84). We further investigated the reversal of TME and the improvement of immune cell infiltration by mEpb4-MRDE/Mn. In tumor samples, mEpb4-MRDE/Mn promoted repolarization of TAMs towards the M1 phenotype and decreased Tregs levels, leading to an ameliorated immunosuppressive TME (Fig. 8f-h and Supplementary Figs. 85-87). Quantification of CD8+ T cells in tumor tissues also confirmed that mEpb4-MRDE/Mn improved immune cell infiltration (Fig. 8i).

a Schematic diagram of the experimental design of the CT-2A model. b, c Quantitative analysis of NK cells (NK1.1+CD3+) (b) and mature DCs (CD80+CD86+) (c) in tumor. d, e Levels of IFN-γ+ T cells (CD8+IFN-γ+) (d) and GrB+ T cells (CD8+GrB+) (e) in the spleen. f, g Quantitative analysis of M2-TAMs (F4/80+CD206+) (f) and M1-TAMs (F4/80+CD86+) (g) in tumor. h Number of CD3+CD8+ T cells per mg of tumor tissue in CT-2A model mice. i Levels of Tregs (Foxp3+CD4+) in tumors. j Schematic diagram of the experimental design of the PDX model. k, l In vivo bioluminescence imaging of PDX model mice (k) and quantification of signal intensity (l). m Survival analysis of the mice. n, o Levels and representative flow cytometry plots of CD4+ T cells (CD3+CD4+) (n) and IFN-γ+ T cells (CD8+IFN-γ+) (o) in PDX model mice after different treatments. Data were mean ± SD (n = 6 mice per group). The significance between each of the multiple groups in (b–i, n, and o) was calculated using one-way ANOVA. The significance in l was calculated using two-way ANOVA. The significance in (m) was calculated using Kaplan-Meier survival analysis. Source data are provided as a Source Data file.
The heterogeneity of human tumors is better reflected in PDX models45 (Fig. 8j). Lymphocyte infiltration was observed in the primary tumor of PDX, which proved that the PDX model in humanized mice had been successfully constructed (Supplementary Fig. 88). The O-MRDE/Mn still exhibits good anti-tumor effects. In brief, IVIS results showed that tumor growth was inhibited in the O-MRDE/Mn group (Fig. 8k, l and Supplementary Fig. 89). Mice in the O-MRDE/Mn survived for a longer period of time, maintaining 100% survival at day 35, while all control mice died (Fig. 8m). In addition, no significant changes in animal weight were observed during the treatment period (Supplementary Fig. 90). Further measurements of immune cell levels indicated a 2.41-fold rise in the percentage of CD4+ T cells and a 2.13-fold increase in the percentage of intratumoral IFN-γ+CD8+ T cells after O-MRDE/Mn treatment, demonstrating the effective activation of specific immune responses (Fig. 8n, o). In summary, we demonstrated that the nAg-MRDE/Mn nanovaccines have good clinical translational potential through CT-2A and PDX modeling applications.
Discussion
In this study, our developed nAg-MRDE/Mn can be absorbed by tumor cells via the synergy of multiple pathways. It can directly kill tumor cells without external stimulation or internal immune activation, and collaborate with tumor neoantigens to activate strong anti-tumor specific immunity. By acting on the cGAS-STING pathway of tumors, it realizes the co-activation of adaptive and innate immunity. Notably, to ensure the effective infiltration of immune cells into tumor tissues, ginsenoside Rh2 and CXCL10 induced by ginsenoside reprogrammed the immunosuppressive TME and further recruited CD8+ T cells. The combined administration of αPD-1 avoids T cell depletion and maintains the forward operation of the immune cycle. These collectively reverse the cold tumor. The ROS-reactive hydrogel TSPBA-PVA was introduced to co-deliver nAg-MRDE/Mn and CXCL10 to meet the clinical needs of preventing recurrence and metastasis after surgery, enhancing the accumulation of immune cells, eliminating the safety risk of chemokines, and achieving long-term retention and continuous release of drugs. The clinical translational potential of nAg-MRDE/Mn was confirmed by further pharmacodynamic evaluation. In conclusion, nAg-MRDE/Mn as a neoantigen delivery platform can play a role in tumor heterogeneity and cold tumors, which provides some inspiration for personalized immunotherapy for heterogeneous malignant tumors.
Methods
Ethical regulations
All research conformed to all pertinent ethical stipulations. The luciferin-expressing human GBM stem-like cells (GSCs) GBM#01 were derived from a GBM surgical specimen in the Department of Neurosurgery, Qilu Hospital, whose relevant studies were approved by the Research Ethics Committee of Shandong University and the Ethics Committee of Qilu Hospital (Shandong, China). The experiments complied with relevant guidelines and regulations. Consent forms were signed by the patients after they had been informed (permit nos. SDULCLL2021-1-17). All in vivo experiments with Animals in this study were carried out in strict accordance with the ARRIVE guidelines 2.0 and the Guidelines for the Care and Use of Laboratory Animals Ethics Committee of Nanjing University of Chinese Medicine (NO.202310A053 and NO.202411A063). The maximal tumor burden was strictly limited to 1.2 cm in diameter for mice, with a total tumor weight not exceeding 10% of the mice body weight. No animals exceeded these limits during the study.
Materials
Anhui Guoping Pharmaceutical Co. Ltd. synthesized the D4F peptide (Ac-FAEKFKEAVKDYFAKFWD, a biomimetic peptide of apolipoprotein A-I), the α-Mel peptide (DWFKAFYDKVAEKFKEAF-GSG-GIGAVLKVLTTGLPALISWIKRKRQQ-NH2), the α-Mel-OVA peptide (DWFKAFYDKVAEKFKEAF-GSG-GIGAVLKVLTTGLPALISWIKRKRQQ-GSG-SIINFEKL-NH2), the α-Mel-M25 peptide (DWFKAFYDKVAEKFKEAF-GSG-GIGAVLKVLTTGLPALISWIKRKRQQ-GSG-KDYTAAGFSSFQKLRLDLTSMQIITTD-NH2), and the α-Mel-mEpb4 (DWFKAFYDKVAEKFKEAF-GSG-GIGAVLKVLTTGLPALISWIKRKRQQ-GSG-ELEQFESTIGFKLPNLRAAKRLWK-NH2) with a purity of 90%. Sigma-Aldrich provided manganese dichloride (MnCl2·4H2O) and NaOH. Yuanye Bio-Technology Co. Ltd (Shanghai) sold BSA and ginsenoside Rh2. Gibco Co., Ltd. (USA) provided PBS (pH 7.4), FBS, high-glucose DMEM, and RPMI-1640. Beyotime Biotechnology (Shanghai, China) provided FITC, DMSO, L-glutamine, DiD, Hoechst33342, anti-fluorescence quencher (including DAPI), and BCA Protein Assay Kit. ELISA Kits were acquired from Elabscience Biotechnology Co, Ltd. Anti-CD11c-FITC (E-AB-F0991C), Anti-mouse CD86-APC (E-AB-F0994E), Anti-CD80-PE/Cyanine7 (E-AB-F0992H), Anti-CD40-APC (E-AB-F1028E), Anti-CD3-PE (E-AB-F1013D), Anti-CD4-PE/Cyanine7 (E-AB-F1097H), Anti-CD8a-FITC (E-AB-F1104C), Anti-F4/80-APC (E-AB-F0995E), Anti-CD86-PE/Cyanine7 (E-AB-F0994H), Anti-CD206-FITC (E-AB-F1135C), Anti-IFN-γ-APC (E-AB-F1101E), Anti-CD3-APC (E-AB-F1013E), Anti-CD44-PE (E-AB-F1100D), Anti-CD62L-PE/Cyanine7 (E-AB-F1011H), Anti-Foxp3-PE (E-AB-F1238D), Anti-CD16/32 (E-AB-F0997A), and Anti-CD107a-PE/Cyanine7 (E-AB-F1254H) for mice, and Anti-CD45-FITC (E-AB-F1137C), Anti-CD3-PE (E-AB-F1001D), Anti-CD8a-PE/Cyanine7 (E-AB-F1110H), Anti-CD4-APC (E-AB-F1109E), and Anti-IFN-γ-APC (E-AB-F1196E) for humans were purchased from Elabscience. Anti-IL-4-APC (504105), Anti-CD63-APC (143905), Anti-CD81-PE (104905), and Anti-CD9-APC (124811) for mice were purchased from BioLegend.
Cell lines and primary cells
GL261-luc, GL261-OVA-luc, and LLC-OVA cell lines were acquired from Zhenjiang Vigen Biotech Company Limited. GL261, CT-2A, LLC, 4T1, BCEC, and PC12 cell lines were acquired from Shanghai Cell Bank. Cells were characterized before use by short tandem repeat DNA fingerprinting (STR)-PCR DNA profiling and were determined to be free of mycoplasma contamination. GL261, GL261-OVA-luc, GL261-luc, BCECs, PC12, and LLC cells were grown in DMEM medium (KeyGEN) supplemented with 10% FBS (Vazyme). 4T1 cells were grown in RPMI-1640 medium (KeyGEN) supplemented with 10% FBS (Vazyme). GL261-luc cells were chosen by applying 10 μg/mL puromycin (Gibco). BMDCs were cultured in RPMI-1640 containing 20% FBS, GM-CSF (MedChem Express, 10 ng mL-1), and IL-4 (MedChem Express, 10 ng mL-1). The luciferase-expressing human GBM stem-like cells (GSCs) GBM#01 were cultured in serum-free Neurobasal™ medium (Gibco, 21103049) supplemented with B-27™ Neuro Mix (Thermo Fisher, 17504044), epidermal growth factor (Thermo Fisher, PHG0314), and basic fibroblast growth factor (PeproTech, 100-18B). Tumorspheres were split using Accutase™ (Thermo Fisher, A1110501) to expand GSCs. All cells were cultured in a humidified atmosphere containing 5% CO2 at 37 °C.
Animal models
Female mice of C57BL/6 strain (18–22 g, 6–8 weeks) and Balb/c strain (18–22 g, 6–8 weeks), and Male SD rats (180–220 g, 6–8 weeks) were sourced from Qinglongshan Animal Breeding Field (Nanjing, China). Female NOG mice (18–22 g, 6–8 weeks) were obtained from Weitong Lihua Experimental Animal Center (China). The cerebral cortex was excised at the right cerebral hemisphere (−1.0 mm, 3.0 mm) of the rats (strain SD), and the drug-loaded hydrogel was injected intracavely. For brain tumor modeling, the cell suspension was injected into the right cerebral hemisphere of mice (strain C57BL/6) at a depth of 2 mm (−0.6 mm, 1.8 mm). For breast cancer modeling, the cell suspension was inoculated into the mammary fat pad of mice (strain Balb/c). Animals with their wound sterilized and seamed were carefully housed in a specific pathogen-free (SPF) environment. The temperature of the animal room lies between 20 and 26 °C, the warm humidity is within 40 to 70%, light and darkness alternate for 12 h, and all mice can freely obtain food and water.
Preparation of nAg-MRDE/Mn
Serum-free medium for exosome isolation was obtained by centrifugation, and DEs were isolated from medium using ultracentrifugation techniques. BSA (10 mg mL−1) and MnCl2 (15 mg mL−1) were mixed and stirred, followed by adding aqueous solutions of NaOH (40 mg mL−1) to a pH of 11. After incubation at 37 °C for 2 h, free Mn2+ was removed by 30 kDa cutoff centrifugal filter device (Amicon). BSA was mixed with different concentrations of Mn2+ and the fluorescence spectra were scanned in the range of 290-450 nm using an excitation wavelength of 280 nm, and Gen5 (v2.0) was used for data collection.
Hybrid peptides were monitored by LC-ESI-MS/MS analysis, and Xcalibur (v4.0) and Masshunter Workstation Data Acquisition software (vB.08.00) were used for data collection. The Rh2 (1.5 mg) dissolved in DMSO was added to the DEs (400 μg mL−1). It was then incubated at 37 °C for 1 h, RDE was prepared. Following electroporation, BSA/Mn solution (1 mL) was added dropwise and incubated for an additional h, RDE/Mn was prepared. Subsequently, the α-Mel-nAg PBS solution (1 mg mL−1, 0.5 mL) was added dropwise and stirred magnetically for 1 h. Afterwards, the resultant blend was centrifuged employing an 30 kDa ultrafiltration tube to eliminate impurities. Then, PBS was added to achieve a final volume of 1 mL, nAg-MRDE/Mn was prepared.
Characterization of O-MRDE/Mn
The OVA-Mel-D4F was labeled with FITC and O-MRDE/Mn was prepared. Ultrafiltration was used to separate the unbound proteins and other impurities, the fluorescence intensity was measured using a microplate reader (PerkinElmer) and the loading efficiency of the hybridized peptides was calculated. The loading rate of Rh2 was determined by HPLC (Thermo) after O-MRDE/Mn was treated with methanol. The chromatographic conditions were as follows: mobile phase: acetonitrile: water = 60:40, column: BDS HYPERSIL C18 (250 mm × 4.6 mm, 5 μm), detection wavelength: 203 nm, column temperature: 30 °C. The encapsulation rate of Mn was determined by ICP-MS (PerkinElmer).
The typical morphology and structure of BSA/Mn, DEs, and O-MRDE/Mn were observed using the Hitachi HT7800 transmission electron microscope (Hitachi, Japan). The expression levels of CD81, CD63, and CD9 in DEs and O-MRDE/Mn were analyzed by flow cytometry. The Malvern Zetasizer Nano-ZS90 device (Malvern Zetasizer) was utilized to measure the particle size and zeta potential, and Malvern zetasizer (v7.13) was used for data collection. In order to examine the placement stability of O-MRDE/Mn, the aforementioned preparations were shielded from light and kept at a temperature of 4 °C. Particle size, zeta potential and PDI were conducted daily.
O-MRDE/Mn and MnCl2 were placed in a dialysis bag and agitated in phosphate buffer (pH 7.4) at 37 °C. Samples were taken and replenished with equal amounts of freshly released media at different time points, detected using an ICP-MS instrument, and the cumulative release rate calculated. Syngistix (v3.3) was used for data collection.
For the hemolysis assays, various concentrations of Mel, α-Mel, α-Mel-OVA, and O-MRDE/Mn were prepared. Plasma was eliminated from mice blood through centrifugation, followed by the adjustment of blood cell concentration to 2%. Subsequently, the blood was incubated with the drug for a duration of 3 h at a temperature of 37 °C. The absorbance of the supernatant after centrifugation at 540 nm was measured.
Preparation of O-MRDE/Mn + CXCL10@Gel
A clear solution was obtained by mixing PVA (72 kDa; 98% hydrolyzed; 5 g) and deionized water (100 ml), and stirring them at 90 °C. O-MRDE/Mn and CXCL10 were introduced into the PVA water-based solution. Hydrogel was formed by mixing TSPBA (5 wt% in H2O, 2 ml) and PVA (5 wt% in H2O, 2 ml) together.
Characterization of O-MRDE/Mn + CXCL10@Gel
1H NMR (Varian Unity Inova 500, Palo Alto) was used to examine TSPBA, and MestReNova Mnova (v15.0) was used for data collection. Teneo SEM (Thermo Fisher) was used to acquire SEM images. Rheology experiments were performed using the Anton Paar MCR 302 rheometer (Anton Paar), and Rheocompass (v1.22) was used for data collection. Take 6 data points of each order of magnitude for analysis during amplitude sweeping. The strain of the shear recovery test was selected as 1% and 500% and maintained for 15 s and 10 s, respectively. The cycle was repeated twice, and one data point was taken every second.
ROS responsiveness of O-MRDE/Mn + CXCL10@Gel
To detect ROS-responsive degradation of hydrogels in vitro, two gels of identical size were subjected to PBS and PBS + 1 mM H2O2. The visual appearance of the gels was recorded daily for 9 days, and the weight loss was measured to calculate the degradation rate. The release of CXCL10 and Mn from O-MRDE/Mn+CXCL10@Gel was studied using a dialysis bag at room temperature in PBS and PBS + 1 mM H2O2. 200Μl of hydrogel was taken for the experiment, 0.5 mL of release PBS was taken daily for eight days, and fresh PBS was added. The particle size of O-MRDE/Mn before and after hydrogel degradation was measured with NS300 (NanoSight), and NTA software (v3.4.4) was used for data collection. DIR dye (MCE) was used to label the DEs, and DIR-O-MRDE/Mn+CXCL10@Gel was prepared. It was implanted into the brains of healthy mice and post-tumor-surgery mice, respectively, and the fluorescence signal intensity of the hydrogel in the mouse brain was observed by using a small-animal in vivo imaging system, and quantitative analyses were carried out to examine the responsive degradation properties of the hydrogel. To detect ROS-responsive degradation of hydrogels in vivo, part of the cortex on the right side of SD rats was removed under the microscope to mimic the surgical wound. O-MRDE/Mn+CXCL10@Gel (120 μL) was injected. Every two days, three rats were euthanized. The hydrogel was weighed, and the degradation rate was calculated, then gently homogenized. ELISA (Elabscience) was used to determine the release of CXCL10, while ICP-MS (PerkinElmer) was employed to determine the presence of Mn.
Recruitment of CD8+ T cells in vitro
T cells were placed in the upper compartment of a Transwell, while the lower compartment received different concentrations of the drug. Following a 24 h incubation, the cells from the lower chamber were collected, and the chemotaxis index was calculated. After incubating of O-MRDE + CXCL10 (5 ng mL-1) for 6, 12, 24, and 36 h, the cells from the lower chamber were collected, and the chemotaxis index was calculated.
Cell uptake studies
Different preparations were prepared using FITC-labeled BSA/Mn (BSA/MnFITC). The administration groups were categorized as DE/MnFITC, D4F-DE/MnFITC, MDE/MnFITC, MDE(-)/MnFITC, Rh2-DE/MnFITC, Rh2-DE(-)/MnFITC, and O-MRDE/MnFITC. Where MDE(-)/MnFITC and Rh2-DE(-)/MnFITC denote the pre-treatment of GL261 cells with D4F and WZB 117 (MedChemExpress), respectively. After 2 h of treatment with the different preparations, the nuclei were visualized by Hoechst staining, and the images were captured using a fluorescence microimaging system (Keyence), and BZ-X800 Wide Image Viewer (v1.0) was used for data collection. In addition, DMSO was introduced to lyse the cells and the intracellular fluorescence intensity was measured using a microplate reader. (Fitc: λEx/λEm = 488/525 nm)
Confocal imaging analyses on the endo-lysosomal escape
Dye D4F-DE/Mn, MDE/Mn, and O-MRDE/Mn with DiD. The GL261 cells were cultured on cell climbing slices in 12-well plates. The GL261 cells underwent treatment with D4F-DE/Mn, MDE/Mn, and O-MRDE/Mn for 0.5, 2, and 8 h respectively. Staining with 75 nM LysoTracker Green (Meilun Biotechnology) and DAPI (Beyotime) was performed. The Leica TCS-SP8 (Leica Microsystems) was utilized for the examination, and Leica Application Suite (v3.6) was used for data collection.
Evaluation of in vitro safety
Cell viability was evaluated with the CCK8 assay Kit. In short, GL261, BCEC, BMDCs, and PC12 cells were placed in 96-well plates. The medium was then substituted with either serum-free medium, serum-free medium containing gel, or O-MRDE/Mn+CXCL10@Gel. The absorbance was recorded at 450 nm.
Detection of ICD biomarkers
GL261 cells were treated with different formulations, namely BSA/Mn, DE/Mn, MDE/Mn, and O-MRDE/Mn. The cells were labeled with anti-mouse CRT antibody (Bioss) at 4 °C for 30 min and Alexa Fluor 568 Donkey Anti-Rabbit IgG secondary antibody (Proteintech) at 4 °C for 30 min. The HMGB1 and ATP Chemiluminescence Assay Kit (Elabscience) were used to analyze the content of HMGB1 and ATP released by GL261 cells after different treatments.
Mechanisms of specific immune activation
To assess antigen presentation, O-MRDE/Mn and physical mixtures (pure physical mixing of each component) were co-incubated with BMDCs separately, and the cells were fixed, closed and labeled by addition of APC anti-mouse H-2Kb bound to SIINFEKL Antibody. Nuclei were visualized by Hoechst staining and images were captured using a fluorescence microimaging system (Keyence).
GL261 cells, in 24-well plates, were treated with various formulations, namely BSA/Mn, DE/Mn, MDE/Mn, and O-MRDE/Mn. Afterwards, cancer cells and supernatant were gathered and co-cultured with BMDCs in 24-well dishes. Cells were subjected to flow cytometer analysis after being incubated with anti-CD11c, anti-CD40, anti-CD80, anti-CD86, and H-2Kb-restricted SIINFEKL. After a 36 h incubation, BMDCs can be gathered and introduced into the 24-well plate of T cells (1: 10). The cells were stained with anti-CD3, anti-CD4, and anti-CD8, and H-2Kb OVA Tetramer-SIINFEKL. The supernatant obtained was utilized for ELISA experiments to measure TNF-α, IL-6, IL-12, IFN-γ, and IL-10 (Elabscience).
Activation of the innate immune system mechanisms
GL261 cells, in 24-well plates, were treated with various formulations, and The medium was gathered to evaluate IFN-β using ELISA Kit (Elabscience). Simultaneously, cells were harvested to measure cGAMP levels using the cGAMP ELISA Kit (Elabscience).
Antitumor study in orthotopic GBM model
3 × 105 GL261-luc cells and GL261-OVA-luc cells were injected into the right striatum of C57BL/6 mice. The pre-treatment was given O-MRDE/Mn on day 5, while the other group was dosed on day 10. Both groups were orthotopically injected at the surgical cavity of mice (0.4 μL g-1). Randomly selected brains from each group were removed after administration for H&E staining, and Slide Viewer (v2.6) was used for data collection. The weight of mice was measured, and GBM growth was monitored by IVIS imaging system (PerkinElmer), and Living Image (v4.4) used for data collection. Mouse survival was tracked to obtain Kaplan-Meier survival curves. The biochemical assays were conducted using the serum collected on day 11 post-administration. Major organs were harvested for H&E staining. Serum levels of cytokines and antibodies were determined according to ELISA Kit instructions (Elabscience). 8 days after administration, samples were treated with anti-CD16/32, followed by staining with anti-CD3, anti-NK1.1, anti-CD11c, anti-MHC-I, anti-CD4, anti-CD8, anti-IFN-γ, and anti-GrB. Kaluza (v2.2.1) was used for data collection.
Activation of antigen-specific immunity and reprogramming of the TME in orthotopic GBM models
To examine antigen-specific responses, 3 × 105 GL261-OVA cells were administered into the right striatum of C57BL/6 mice. Each group was dosed on day 10. Mice were euthanized 8 days after administration. Flow samples were treated with anti-CD16/32, followed by staining with anti-CD11c, anti-CD40, anti-CD80, anti-CD86, H-2Kb-restricted SIINFEKL, anti-CD3, anti-CD4, anti-CD8, H-2Kb OVA Tetramer-SIINFEKL, anti-IFN-γ, anti-GrB, and anti-CD107a.
After orthotopic GBM models were euthanized, tumor tissues were collected and treated with anti-CD16/32, followed by staining with anti-F4/80, anti-CD86, and anti-CD206, anti-CD3, anti-CD8, anti-CD4, anti-IFN-γ, anti-IL-4, and anti-Foxp3. The brain was stained with Ki67, CD31, CD44, CD3, CD8, CD4, GATA-3, T-bet, and Foxp3 antibodies, respectively, and DAPI.
Antitumor study in postoperative GBM model and GBM rechallenge model
Ten days after inoculation, mice carrying in situ GL261-luc or GL261-OVA-luc cells were operated on and administered. According to the change in bioluminescence intensity, whether the tumor is effectively removed can be judged. Subsequently, the mice were treated with saline, blank gel, BSA/Mn@Gel, DE/Mn@Gel, MDE/Mn@Gel, O-MRDE/Mn@Gel, and O-MRDE/Mn+CXCL10@Gel. The weight of mice was measured, and GBM growth was monitored by the IVIS imaging system. Mouse survival was tracked to obtain Kaplan-Meier survival curves. Mice were sacrificed 10 days after administration. Serum levels of cytokines and antibodies were determined according to the ELISA Kit (Elabscience) instructions. Immunofluorescence staining of Ki67, CD8, and Foxp3 was performed on sections of the brain. Flow samples were treated with anti-CD16/32, followed by staining with anti-CD3, anti-NK1.1, anti-CD11c, anti-CD80, anti-CD86, anti-F4/80, anti-CD206, anti-CD3, anti-CD8, anti-CD4, anti-CD44, and anti-CD62L. After 60 days of administration, the surviving mice of the O-MRDE/Mn+CXCL10@Gel were injected with 3 × 105 GL261 or GL261-OVA cells at the left striatum. Blank mice of the same week age were injected with GL261 (3 × 105) at the right and left striatum. Mouse survival was tracked to obtain Kaplan-Meier survival curves. Ten days after establishing the GBM rechallenge model, sections were labeled with H&E and Ki67. Flow cytometry was used for CD8+ T cells in the spleen and tumor.
Antitumor study in lung cancer brain metastasis model
1 × 106 LLC cells were administered into the right striatum of C57BL/6 mice. Mouse survival was tracked to obtain Kaplan-Meier survival curves. After 8 days of administration, blood was collected by removing the eyeball after mice were anesthetized with isoflurane. Serum levels of cytokines and antibodies were determined according to the ELISA Kit instructions (Elabscience). Treated with anti-CD16/32, flow samples of tumors were followed by staining with anti-CD11c, anti-CD80, anti-CD86, H-2Kb-restricted SIINFEKL, anti-CD3, anti-CD4, and anti-CD8. Flow samples of spleens were followed by staining with anti-CD3 and anti-CD8. Flow samples of peripheral blood were followed by staining with anti-CD3, anti-CD8, H-2Kb OVA Tetramer-SIINFEKL. Brain tissues were removed 8 days after administration for H&E and immunofluorescence staining of CD4, CD8, and Tregs.
Antitumor study of M25-MRDE/Mn + CXCL10@Gel in combination with αPD-1 in the orthotopic 4T1 models
5 × 105 4T1 cells were administered to the right mammary fat pad of BALB/c mice. αPD-1 injection: 20 mg kg-1 intraperitoneally. Tumor size and weight were measured. Mouse survival was tracked to obtain Kaplan-Meier survival curves. Tumor samples were obtained 20 days post inoculation with 4T1 cells. The tumor cells were labeled with anti-mouse CRT antibody (Proteintech) at 4 °C for 30 min and Alexa Fluor 568 Donkey Anti-Rabbit IgG secondary antibody (Proteintech) at 4 °C for 30 min. Flow samples were first processed with anti-CD16/32, and then labeled with anti-CD3, anti-CD49b, anti-CD80, anti-CD86, anti-CD8, anti-IFNγ, anti-GrB, anti-CD4, anti-Foxp3. Then, the dyed cells were quantified using a flow cytometer (Beckman). The lungs were immobilized with Bouin solution (G-CLONE) for 4 h, then white metastatic nodules were counted, and finally H&E staining was conducted.
Anti-tumor studies in CT-2A and PDX model
To further evaluate the translational feasibility, 3 × 105 CT-2A cells were administered into the right striatum of C57BL/6 mice. Flow samples were treated with anti-CD16/32, followed by stained with anti-CD3, anti-NK1.1, anti-CD11c, anti-CD80, anti-CD86, anti-CD8, anti-PD-1, anti-IFN-γ, anti-GrB, anti-F4/80, anti-CD86, and anti-CD206, anti-CD4, and anti-Foxp3.
To further evaluate the antitumor efficacy against PDX model mice, we established an orthotopic humanized mouse model by intracranial injection of luciferase-expressing human GSCs GBM#01 into NOG mice. In brief, 0.2 mL (1 × 106 peripheral blood mononuclear cells (PBMC)) of cell suspension was administered via the tail vein in each mouse. Human Peripheral Blood CD14+ Cells were obtained from IPHASE Biotechnology (Suzhou). To obtain PBMV-derived DEs, PBMC were cultured in medium containing 100 ng mL-1 human GM-CSF (PeproTech) and 20 ng mL-1 human IL-4 (InvivoGen) for 48 h, and DCS were harvested by culturing in medium containing 10 ng mL-1 TNF-α (InvivoGen) for 3 days and further cultured to obtain DEs. 3 weeks later, GBM#01 cells were intracranially implanted. On day 4, mice injected with or without PBMC were euthanized. Successful construction of the PDX model was checked by immunofluorescence staining (CD45, Servicebio, GB300644-H) and flow cytometric analyses. The progression of the tumor was observed using IVIS imaging, and the survival was recorded. To evaluate the degree of activation in the immune system, spleens and tumors were isolated and processed. CD4+ T cells in spleen were staining with anti-CD45, anti-CD3, anti-CD4, and IFN-γ+ T cells in tumor were staining with anti-CD45, anti-CD3, anti-CD8, anti-IFN-γ. The Animal Ethics Committee of Nanjing University of Chinese Medicine granted approval for the experiments (permit NO.202411A063).
Statistics and reproducibility
We used a variety of tumor cell lines and mouse breeds for experimental design. FlowJo (v10.9.0) was used to analyze the obtained flow cytometry data. Graphpad Prism (v9.0.0) was used for statistical analysis throughout this study. One-way analysis of variance was used for comparison among multiple groups. P < 0.05 was considered statistically significant. The values and scatter in the bar graphs represent the mean ± SD and the number of individual replicates, respectively. Experiments performed in biological replicates are provided in the corresponding legends. No statistical method was used to predetermine sample size. No data were excluded from the analyses. The experiments were not randomized. The Investigators were not blinded to allocation during experiments and outcome assessment.
Figures and artwork
Graphic elements in Fig. 1a, b, Fig. 2a, f, Fig. 3a, n, Fig. 5f, and Fig. 7g were created using ChemDraw Professional (v16.0), 3ds Max (v2022) and Adobe Illustrator (v2021).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
NMR data of recombinant proteins are summarized in Supplementary Fig. 2. Source data for main and Supplementary Figures are provided with this paper and are also available in Figshare (https://doi.org/10.6084/m9.figshare.28789718.v3). The remaining data are available within the Article, Supplementary Information or Source data file. Source data are provided with this paper.
References
Kraehenbuehl, L., Weng, C. H., Eghbali, S., Wolchok, J. D. & Merghoub, T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat. Rev. Clin. Oncol. 19, 37–50 (2022).
Yang, K., Halima, A. & Chan, T. A. Antigen presentation in cancer - mechanisms and clinical implications for immunotherapy. Nat. Rev. Clin. Oncol. 20, 604–623 (2023).
Antonarelli, G. et al. Therapeutic cancer vaccines revamping: technology advancements and pitfalls. Ann. Oncol. 32, 1537–1551 (2021).
Blass, E. & Ott, P. A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 18, 215–229 (2021).
Katsikis, P. D., Ishii, K. J. & Schliehe, C. Challenges in developing personalized neoantigen cancer vaccines. Nat. Rev. Immunol. 24, 213–227 (2024).
Dagogo-Jack, I. & Shaw, A. T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 15, 81–94 (2018).
Gainor, J. F. et al. T-cell responses to individualized neoantigen therapy mRNA-4157 (V940) alone or in combination with pembrolizumab in the phase 1 KEYNOTE-603 study. Cancer Discov. 14, 2209–2223 (2024).
Hsiue E. H. et al. Targeting a neoantigen derived from a common TP53 mutation. Science 371, eabc8697 (2021).
Verdegaal, E. M. et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature 536, 91–95 (2016).
Khattak, A. et al. Abstract CT001: a personalized cancer vaccine, mRNA-4157, combined with pembrolizumab versus pembrolizumab in patients with resected high-risk melanoma: efficacy and safety results from the randomized, open-label phase 2 mRNA-4157-P201/Keynote-942 trial. Cancer Res 83, CT001 (2023).
Rojas, L. A. et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 618, 144–150 (2023).
Hu, Z. et al. Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat. Med 27, 515–525 (2021).
Corbiere, V. et al. Antigen spreading contributes to MAGE vaccination-induced regression of melanoma metastases. Cancer Res 71, 1253–1262 (2011).
Liu, J. et al. Cancer vaccines as promising immuno-therapeutics: platforms and current progress. J. Hematol. Oncol. 15, 28 (2022).
Ott, P. A. et al. A phase Ib trial of personalized neoantigen therapy plus anti-PD-1 in patients with advanced melanoma, non-small cell lung cancer, or bladder cancer. Cell 183, 347–362.e324 (2020).
Lang, F., Schrors, B., Lower, M., Tureci, O. & Sahin, U. Identification of neoantigens for individualized therapeutic cancer vaccines. Nat. Rev. Drug Discov. 21, 261–282 (2022).
Dunn, G. P., Bruce, A. T., Ikeda, H., Old, L. J. & Schreiber, R. D. Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. 3, 991–998 (2002).
Lybaert, L. et al. Challenges in neoantigen-directed therapeutics. Cancer Cell 41, 15–40 (2023).
Hornburg, M. et al. Single-cell dissection of cellular components and interactions shaping the tumor immune phenotypes in ovarian cancer. Cancer Cell 39, 928–944.e926 (2021).
Galon, J. & Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 18, 197–218 (2019).
Ochoa de Olza, M., Navarro Rodrigo, B., Zimmermann, S. & Coukos, G. Turning up the heat on non-immunoreactive tumours: opportunities for clinical development. Lancet Oncol. 21, e419–e430 (2020).
Wang, X. et al. A protein-based cGAS-STING nanoagonist enhances T cell-mediated anti-tumor immune responses. Nat. Commun. 13, 5685 (2022).
Zhao, X. et al. Herpesvirus-Mimicking DNAzyme-Loaded Nanoparticles as a Mitochondrial DNA Stress Inducer to Activate Innate Immunity for Tumor Therapy. Adv. Mater. 34, e2204585 (2022).
Poznanski, S. M. et al. Metabolic flexibility determines human NK cell functional fate in the tumor microenvironment. Cell Metab. 33, 1205–1220.e1205 (2021).
Weiss, S. A. et al. Epigenetic tuning of PD-1 expression improves exhausted T cell function and viral control. Nat. Immunol. 25, 1871–1883 (2024).
Cillo, A. R. et al. Blockade of LAG-3 and PD-1 leads to co-expression of cytotoxic and exhaustion gene modules in CD8(+) T cells to promote antitumor immunity. Cell 187, 4373–4388.e4315 (2024).
Chow, A., Perica, K., Klebanoff, C. A. & Wolchok, J. D. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat. Rev. Clin. Oncol. 19, 775–790 (2022).
Zitvogel, L. et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat. Med 4, 594–600 (1998).
Duan, X. et al. Melittin-incorporated nanomedicines for enhanced cancer immunotherapy. J. Control Release 375, 285–299 (2024).
Huang, M. Y. et al. Ginsenoside Rh2 augmented anti-PD-L1 immunotherapy by reinvigorating CD8(+) T cells via increasing intratumoral CXCL10. Pharm. Res 198, 106988 (2023).
Hong, C. et al. One stone four birds: a novel liposomal delivery system multi-functionalized with ginsenoside Rh2 for tumor targeting therapy. Nanomicro Lett. 12, 129 (2020).
Li, Q. Q. et al. Selective inhibition of cancer cells by enzyme-induced gain of function of phosphorylated melittin analogues. Chem. Sci. 8, 7675–7681 (2017).
Xia, J. et al. Versatile ginsenoside Rg3 liposomes inhibit tumor metastasis by capturing circulating tumor cells and destroying metastatic niches. Sci. Adv. 8, eabj1262 (2022).
Ning, D. et al. Lipid-centric design of plasma membrane-mimicking nanocarriers for targeted chemotherapeutic delivery. Adv. Mater. 36, e2306808 (2024).
Sun, X. Q. et al. Amplifying STING activation by cyclic dinucleotide-manganese particles for local and systemic cancer metalloimmunotherapy. Nat. Nanotechnol. 16, 1260–1270 (2021).
Lv, M. Z. et al. Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res. 30, 966–979 (2020).
Dvorkin, S., Cambier, S., Volkman, H. E. & Stetson, D. B. New frontiers in the cGAS-STING intracellular DNA-sensing pathway. Immunity 57, 718–730 (2024).
Cassetta, L. & Pollard, J. W. A timeline of tumour-associated macrophage biology. Nat. Rev. Cancer 23, 238–257 (2023).
Guo, W. et al. Tumor draining lymph nodes connected to cold triple-negative breast cancers are characterized by Th2-associated microenvironment. Nat. Commun. 15, 8592 (2024).
Limagne, E. et al. MEK inhibition overcomes chemoimmunotherapy resistance by inducing CXCL10 in cancer cells. Cancer Cell 40, 136–152.e112 (2022).
Wang, R. P. et al. Single-cell dissection of intratumoral heterogeneity and lineage diversity in metastatic gastric adenocarcinoma. Nat. Med. 27, 141–151 (2021).
Bejarano, L. et al. Interrogation of endothelial and mural cells in brain metastasis reveals key immune-regulatory mechanisms. Cancer Cell 42, 378–395.e310 (2024).
Zhang, Q. et al. The spatial transcriptomic landscape of non-small cell lung cancer brain metastasis. Nat. Commun. 13, 5983 (2022).
Liu, C. J. et al. Treatment of an aggressive orthotopic murine glioblastoma model with combination checkpoint blockade and a multivalent neoantigen vaccine. Neuro Oncol. 22, 1276–1288 (2020).
Zanella, E. R., Grassi, E. & Trusolino, L. Towards precision oncology with patient-derived xenografts. Nat. Rev. Clin. Oncol. 19, 719–732 (2022).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 82274104 received by R.W., 82074024 received by L.D., 82374042 received by L.D.), National Ten Thousand Talents Program for Young Top-notch Talents (received by R.W.), the Natural Science Foundation of Jiangsu Province (No. BK20240144 received by R.W.), the Innovation Projects of State Key Laboratory on Technologies for Chinese Medicine Pharmaceutical Process Control and Intelligent Manufacture (Grant No. NZYSKL240103 received by R.W.), Nanjing University of Chinese Medicine’s Project (No.RC202407 received by R.W.).
Author information
Authors and Affiliations
Contributions
R.W. conceived the research and designed the experiments. K.F., X.Z., and J.L. performed the experiments. M.H. provided human GSCs GBM#01 cell line and technical support in the construction of the PDX tumor model. J.W., F.C., and Z.Y. analyzed the data. L.D. and K.F. wrote the manuscript. R.W. supervised the project. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jutaek Nam and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Feng, K., Zhang, X., Li, J. et al. Neoantigens combined with in situ cancer vaccination induce personalized immunity and reshape the tumor microenvironment. Nat Commun 16, 5074 (2025). https://doi.org/10.1038/s41467-025-60448-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60448-3
