
Abstract
There is an urgent need to identify predictive biomarkers of Parkinson’s disease (PD) with cognitive impairment (PDCI) in order to individualize patient management, ensure timely intervention, and improve prognosis. The aim of this study was to screen for these biomarkers by comparing the plasma proteome and metabolome of PD patients with or without cognitive impairment. Proteomics and metabolomics analyses were performed on a discover cohort. A machine learning model was used to identify candidate protein and metabolite biomarkers of PDCI, which were validated in an independent cohort. The predictive ability of these biomarkers for PDCI was evaluated by plotting receiver operating characteristic curves and calculating the area under the curve (AUC). Moreover, we assessed the predictive ability of these proteins in combination with neuroimaging. In the discover cohort (n = 100), we identified 25 protein features with best results in the machine learning model, including top-ranked PSAP and H3C15. The two-proteins were used for model construction, achieving an Area under the curve (AUC) of 0.951 in the train set and AUC of 0.981 in the test set. Similarly, the model gives a rank list of endogenous metabolite features, Glycocholic Acid and 6-Methylnicotinamide were two top features. Combining these two markers further got the AUC of 0.969 in train set and 0.867 in the test set. To validate the performance of the protein biomarkers, we performed targeted analysis of selected proteins (H3C15 and PSAP) and proteins likely associated with PDCI (NCAM2 and LAMB2) using parallel reaction monitoring in validation cohort (n = 116). The AUC of the classifier built with H3C15 and PSAP is 0.813. Moreover, when combining H3C15, PSAP, NCAM2, and LAMB2, the model achieved AUC of 0.983 in the train set, AUC of 0.981 in the test set, and AUC of 0.839 in the validation set. Furthermore, we verified that these protein markers we discovered can improve the predictive effect of neuroimaging on PDCI: the classifier built with neuroimaging features had AUC of 0.833, which improved to 0.905 when combined with H3C15. Taken together, our integrated proteomics and metabolomics analysis successfully identified potential biomarkers for PDCI. Additionally, H3C15 showed promise in enhancing the predictive performance of neuroimaging for cognitive impairment.
Similar content being viewed by others

Introduction
Parkinson’s disease (PD) is often accompanied by cognitive impairment (PDCI), which worsens disability and elevates the risk of dementia1. Identifying PD patients who are at a higher risk of cognitive impairment is crucial for personalized management, timely interventions, and even preventive measures. However, reliable predictors of PDCI are currently lacking. Firstly, potential predictors of PDCI include motor and non-motor symptoms, but they lack sensitivity and specificity2. Secondly, temporal-parietal atrophy on T1Magnetic resonance imaging (MRI)3, and hypo-metabolism in the medial frontal and parietal regions observed in FDG-PET are associated with impaired executive and memory function in PD4. However, these findings are not consistently replicated in other studies5,6. Moreover, some studies have reported decreased concentrations of Aβ1-42, increased levels of total tau, and higher or similar levels of phosphorylated tau (p-tau) in PD patients with dementia (PDD) compared to those without dementia and healthy controls4,7. However, recent research by Chiu et al. indicated that plasma Aβ1−42 and p-tau levels did not increase in PDCI patients as observed in patients with Alzheimer’s disease (AD)8. Plasma neurofilament light chain (NfL) has been proposed as a potential biomarker for PDCI and predicting the clinical progression from PDCI to PDD9. However, elevated NfL levels have also been observed in patients with AD and other cognitive disorders10. Hence, none of the proposed markers appear to exhibit the necessary sensitivity and specificity required for clinical application. Although a meta-analysis demonstrated the efficacy of cholinesterase inhibitors, rivastigmine, and donepezil, in treating dementia in PD and AD patients11, these two neurodegenerative diseases associated with cognitive impairment possess distinct pathological mechanisms. Therefore, there is an urgent need to develop novel drugs for the treatment of PD, as well as identify disease markers that can predict cognitive impairment and dementia in PD patients.
Proteomics and metabolomics are robust techniques for screening disease markers in plasma and cerebrospinal fluid (CSF). The detection of markers in blood or CSF is convenient in clinical settings, providing more objective results compared to neuroimaging. Proteomics studies have successfully identified potential biomarkers for Lewy body-associated dementia12, multiple sclerosis13, and plasma proteins capable of distinguishing between different endophenotypes of AD14. Furthermore, some research groups have identified proteomic and metabolomic differences between PD patients and healthy controls15,16, although the association between cognitive impairment and alterations in the proteome and metabolome of PD patients remains unclear.
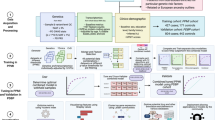
The objective of this study was to investigate changes in the proteome and metabolome associated with cognitive impairment in PD. To achieve this, we compared the plasma proteomics and metabolomics profiles of PD patients without cognitive impairment, those with mild impairment, and those with dementia to identify potential biomarkers for cognitive impairment. The obtained results were then subjected to analysis using a machine-learning model and subsequently validated in an independent cohort. Additionally, we explored the predictive capability of the identified protein biomarkers, both individually and in combination with neuroimaging features, for detecting cognitive impairment. An overview of the framework employed for proteomics and metabolomics analyses, model development, model evaluation, and model validation is illustrated in Fig. 1.

Proteomics and metabolomics analyses were performed on a discover cohort. A machine learning model was used to identify candidate protein and metabolite biomarkers of Parkinson’s disease with cognitive impairment (PDCI), which were validated in an independent cohort. The predictive ability of the proteins in combination with neuroimaging was also analyzed. The figure were performed with Adobe Illustrator 2019. C, control healthy subjects; DC degree centrality, fMRI functional Magnetic resonance imaging, GMV Gray matter volume, PD Parkinson’s disease, PDD Parkinson’s disease patients with dementia, PDMCI Parkinson’s disease patients with mild cognitive impairment, PDNC Parkinson’s disease patients with normal cognition.
Results
Characteristics of the PD cohort for proteomics
A total of 216 blood samples from both the discovery cohort (40 females and 60 males) and the validation cohort (52 females and 64 males) were analyzed. There were no significant differences observed among the groups in terms of age, although the PDD group consisted of slightly older patients compared to the healthy controls. Table 1 summarizes the demographic and clinical information for both cohorts.
PD subjects exhibited an altered plasma proteome compared to healthy controls
Although PD primarily affects the central nervous system, it can potentially impact peripheral tissues and alter the proteomes of urine, CSF, and other biological fluids15,17. We utilized a 4D label-free proteomics assay to identify differentially expressed proteins (DEPs) in the plasma of healthy controls and PD patients. Following depletion of highly abundant proteins such as albumin, we detected 1892 unique proteins in the plasma samples, with 1709 being quantifiable. The heatmap shown in Fig. 2a illustrates that the proteomic profiles of the three PD groups differed from those of the control group, PD with mild cognitive impairment (PDMCI) and PD dementia (PDD) exhibiting more distinct patterns. Notably, the proteomes of the PDMCI and PDD groups were distinctly separate from that of the PD with normal cognitive (PDNC) group (Fig. 2a). Through pairwise comparisons, we identified multiple DEPs associated with neurodegenerative diseases, including S100A9, HEBP1 and RPL22 (Fig. 2b). Compared to the control group, the pro-inflammatory protein S100A9, known to modulate alpha-synuclein aggregation18, was upregulated in the PDNC group. Heme-binding protein 1(HEBP1), a potential sensitizer for neurotoxicity in the early stage of AD19, was upregulated in PDMCI subjects. Additionally, the PDD group exhibited significant upregulation of RPL22, with higher transcript levels detected in the frontal cortex of PD patients20. The lysosome-associated protein prosaposin (PSAP) showed downregulation in all PD subjects. Gene variants of PSAP have been associated with the risk of PD, consistent with its role in regulating alpha-synuclein degradation and progranulin transport21.

a, b Volcano plot and heatmap showing proteins that are downregulated (blue) or upregulated (red) in PD patients relative to controls, proteins above the gray dotted line are statistically significant (P < 0.05). c Venn diagram showing differentially expressed proteins in the pairwise comparisons, and their overlap. d Enrichment of differentially expressed proteins in Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. Dot sizes reflect the number of proteins in each pathway, and color reflects the P value for enrichment.
Overall, we found that 282 proteins exhibited significantly altered expression levels in PD patients compared to healthy controls, among which 42 proteins were consistently altered across all PD subgroups, suggesting their involvement in disease progression. Additionally, 50 DEPs were common to both the PDMCI and PDD groups but not the PDNC group, indicating their potential association with cognitive impairment (Fig. 2c). KEGG pathway analysis of the dysregulated proteins in all PD groups revealed a significant enrichment of pathways related to neurodevelopment and the maintenance of normal neurological function. Any disruption in these pathways could potentially initiate the pathological process of PD. Notably, the inositol phosphate metabolism pathway was particularly enriched in the PDD and PDMCI groups compared to the control group (Fig. 2d). Metabolites of inositol phosphate play a role in the biosynthesis of phosphoinositides, which mediate neuroinflammation and regulate the progression of dementia22.
Correlation of protein expression pattern with the severity of PD-related cognitive impairment
To further investigate the relationship between the plasma proteome of PD subjects and the severity of cognitive impairment, we employed the Mfuzz clustering method. Mfuzz is a technique used to identify potential time series patterns in expression profiles and cluster genes with similar patterns23. Our Mfuzz analysis classified eight distinct gene expression patterns, among which clusters 1, 3, and 5 showed associations with the severity of cognitive impairment (Fig. 3a), encompassing 95, 86, and 97 unique proteins, respectively. Proteins within clusters 1 and 5 exhibited a positive correlation with cognitive impairment, as evidenced by their increased expression levels in PDMCI and PDD subjects compared to both the control and PDNC subjects. Conversely, cluster 3 displayed a negative correlation with cognitive impairment, as the expression levels of its proteins decreased with cognitive decline (Fig. 3a). Moreover, we observed group-specific clusters. For instance, proteins in cluster 2 were highly expressed in the plasma of PDNC subjects but showed low levels in PDMCI and PDD subjects. Similarly, proteins in clusters 4 and 6 were specific to the PDD and PDMCI groups, respectively (Supplementary Fig. 1a).

a Fuzzy c-means clustering identified 3 representative protein clusters among C, PDNC, PDMCI, and PDD samples. The profiles are color-coded according to their distance from the respective cluster center (red is close to the center, blue is further away from the center). b Protein-protein interaction networks within clusters 1, 3, and 5. Node size reflects its degree, while node color reflects its membership. c Enrichment of proteins within each cluster in Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways.
Abnormal protein-protein interactions (PPIs) can disrupt normal cellular processes and contribute to aggregation-related disorders such as AD, PD and other neurodegenerative diseases24. Therefore, we conducted a PPIs network analysis to gain a better understanding of the characteristics of proteins within each cluster (Fig. 3b, Supplementary Fig. 1b). In cluster 1, the main network consisted of immunoglobulin family proteins, including IGHG1, IGLL5, and IGKC. In contrast, cluster 3 showed a smaller network with fewer interactions. Cluster 5 contained three sub-networks: one comprised of keratin proteins (e.g., KRT13, KRT14, KRT19), another comprising ribosomal proteins (e.g., RPL26, RPL11, RPS19), and a third consisting of proteins associated with AD (such as TLN122 and ITGB323), although with fewer interactions (Fig. 3b). KEGG pathway analysis revealed distinct patterns across clusters. Cluster 1 exhibited enrichment in infection- and immunity-related pathways, while cluster 3 did not show any significantly enriched pathways. In contrast, cluster 5 displayed significant enrichment in dopaminergic synapses, estrogen signaling, and Rap1 signaling pathways, all of which are associated with PD25. Necroptosis pathways, known to play a significant role in neurodegenerative disease, were also enriched in cluster 1 (Fig. 3c). GOCC analysis indicated protein localization in the distal axons of neuronal cells for cluster 1 (Supplementary Fig. 1c). GOBP and GOMF analysis for cluster 3 suggested involvement in the regulation of receptor activity and stimulus response regulation (Supplementary Fig. 1c, e). GOBP analysis for cluster 5 revealed proteins associated with the aging process (Supplementary Fig. 1c), while GO cellular component analysis highlighted their presence in chromatin, anchoring junctions, and intermediate filaments (Supplementary Fig. 1d). Cluster 2, specific to PD, showed significant enrichment in pyruvate metabolism, which aligns with the role of pyruvate metabolism and metabolites in PD progression. For the PDD-specific cluster 4, proteins were primarily enriched in oxidative phosphorylation and ribosome-related processes. However, no distinct pathways were enriched in the PDMCI-specific cluster 6 (Fig. 3c).
Identification of protein modules associated with clinical manifestations of PD
To investigate protein co-expression patterns and identify disease protein modules associated with cognitive impairment in PD, we performed weighted gene co-expression network analysis (WGCNA). After adjusting for common confounders such as age, sex, and education background, we identified seven modules of strongly co-expressed protein groups, color-coded as described previously26 (Fig. 4a). These protein modules were significantly enriched in PD-related pathways, including neuroactive ligand-receptor interaction, PPAR signaling, inositol phosphate metabolism, estrogen signaling, and Rap1 signaling pathways (Fig. 4b). Notably, the latter three pathways were also identified in our previous analyses (Figs. 2d, 3c).

a Dendrogram showing seven co-expression modules, each labeled with a color. b Barplot showing the five Kyoto Encyclopedia of Genes and Genomes pathways most significantly enriched in each protein module. The x-axis indicates Fisher’s exact p value after –log10 transformation. c Heatmap of correlations between protein expression in each module (far left) and clinical features of PD (bottom). Red indicates a positive correlation; blue, a negative one. HAMA Hamilton Anxiety Scale, HAMD Hamilton Depression Scale, H-Y Hoehn–Yahr scale, LEDD levodopa-equivalent daily dose, MoCA Montreal Cognitive Assessment, RBD-Q Rapid Eye Movement Sleep Behavior Disorder Screening Questionnaire, UPDRSIII Part III of the Unified Parkinson’s Disease Rating Scale. d Scatter plot showing Pearson’s correlation between expression of proteins in the “blue” module and scores on the MoCA. The proteins above the gray dotted line are statistically significant (P < 0.05). e Scatter plot showing Pearson’s correlation between expression of metabolite and scores on the MoCA. The proteins above the gray dotted line are statistically significant (P < 0.05).
The brown, yellow, blue, green, and red modules showed significant correlations with the PD status (Fig. 4c). The brown and yellow modules were positively associated with Hamilton Depression Scale (HAMD) and Hamilton Anxiety Scale (HAMA) scores, while the green module exhibited a negative correlation. The blue module displayed the strongest association with cognitive impairment, showing a negative correlation with Montreal cognitive assessment (MoCA) scores (Fig. 4c), indicating that proteins within the blue module have the potential to serve as predictive markers for cognitive impairment in PD. Therefore, we examined the relationship between MoCA scores and individual proteins within the blue module across the three PD groups. As depicted in Fig. 4d, MYOM3, H3C15, and PSAP exhibited the strongest correlation with MoCA scores.
Integrated proteomics and metabolomics analysis of the PD groups
Metabolic abnormalities, including mitochondrial dysfunction and oxidative damage, contribute to the pathological progression of PD. In line with this, we identified pathways involved in cholesterol metabolism, inositol phosphate metabolism, and carbohydrate metabolism within PD-related protein modules (Fig. 4b). Therefore, we also investigated alterations in the metabolome associated with cognitive impairment following the onset of PD. Interestingly, we found a significant negative correlation between Glycocholic acid, a downstream metabolite of cholesterol, and MoCA scores (Fig. 4e). Additionally, 6-Methylnicotinamide, which is negatively correlated with MoCA scores (Fig. 4e), has been previously linked to impaired neurological function during the acute stage of intracerebral hemorrhage27. Cluster analysis of DEPs revealed similar protein expression patterns between the PDMCI and PDD groups, distinguishing them from the PDNC group (Fig. 5a, left). We identified 77 DEPs, with 43 proteins upregulated (Cluster 1) and 34 downregulated (Cluster 2). Upregulated proteins were enriched in glycan-related metabolism, while downregulated proteins were enriched in neuroactive receptor interaction (Fig. 5b, Up).

a Heatmap showing differential expression of proteins (left) and metabolites (right) in plasma. Hierarchical clustering divided the proteins and metabolites into clusters 1 and 2. b Enrichment of differentially expressed proteins (above) and metabolites (below) from clusters 1–2 in Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. The x-axis indicates Fisher’s exact p value after -log10 transformation. c KEGG pathways simultaneously enriched in differentially expressed proteins (blue) and metabolites (gold). d Network of Spearman’s correlations among differentially expressed proteins and metabolites. Only correlations whose coefficients had an absolute value > 0.4 are shown.
The PDD group exhibited a distinct metabolome compared to the PDNC and PDMCI groups. The number of metabolites in cluster 1 was higher in the PDD subjects than in the PDNC and PDMCI subjects (Fig. 5a, right). Furthermore, most metabolites in cluster 2 were lower in the PDD and PDMCI groups but higher in the PDNC group. KEGG analysis demonstrated that differentially expressed metabolites were enriched in neurological pathways such as glutamatergic synapse and long-term depression (Fig. 5b, down). The integration of differentially expressed proteins and metabolites revealed 32 pathways, including those related to neurodegenerative diseases (PD, GABAergic synapse, Dopaminergic synapse: KIF5A, GNAI2, L-Glutamic Acid, L-Glutamine, L-Tyrosine), as well as metabolic pathways (Fig. 5c). Notably, amino acids, unsaturated fatty acids, and inositol metabolites that are known to be altered in PD patients or animal models were among the identified metabolites16. L-Glutamic Acid, L-Glutamine, and L-Tyrosine are important neurotransmitters or neurotransmitter precursors. Previous studies have demonstrated the potential of Glutamic Acid and Glutamine to enhance cognitive function through their anti-inflammatory properties27,28, while increased tyrosine dosages lead to enhanced dopamine synthesis in the brain, thereby improving cognitive performance29. These results indicate a significant association between the changes observed in the proteomic and metabolomic profiles in the plasma of PD patients. Co-expression network analysis highlighted a negative correlation between H3C15, a member of the H3 histone family, and metabolites involved in amino acid metabolism. Furthermore, the metabolites Dioxindole, 2,4-Dichlorophenol, and 4-tert-butylbenzoic-acid exhibited strong co-expression with a subset of proteins associated with PD pathways, including KIF5A and GNAI2 (Fig. 5d).
Machine learning-based identification of biomarkers for cognitive impairment
The U map plot revealed similarities in molecular patterns between PDNC and PDMCI_PDD samples (Supplementary Fig. 2), underscoring the importance of feature selection using machine learning techniques. Hence, we employed a machine learning model to identify robust biomarkers based on proteomic and metabolic profiles for discriminating between PD patients with and without cognitive impairment. A random forest approach was utilized for feature selection based on mean decrease accuracy. Both proteomics (Fig. 6a) and metabolomics (Fig. 6b) datasets were inputted for model training, respectively. The diagnostic performances of protein and metabolite markers were assessed using ROC analysis.

Identification of (a) protein and (b) metabolite biomarkers. The mean decrease in accuracy is shown at the left, receiver operating characteristic curves for combined biomarkers in the center, and the classification error matrix on the right. Areas under the operating curves are shown within the curve plots. b, c Abundance of the protein and metabolite biomarkers in plasma. ***P < 0.001; **P < 0.01; *P < 0.05; P-values from student’s t-test (protein) and Wilcoxon rank-sum test (metabolite) are shown. Lines indicate median and interquartile range. n = 24 (PDNC), 50 (PDMCI_PDD).
We identified 25 protein markers, with PSAP and H3C15 ranking among the top markers (Fig. 6a). The two-proteins were used for model construction, achieving an Area under the curve (AUC) of 0.951 in the train set and AUC of 0.981 in the test set. As for metabolic markers, Glycocholic acid and 6-methylnicotinamide emerged as the top markers. Combining these two markers further got the AUC of 0.969 in train set and 0.867 in the test set (Fig. 6b). To validate the potential of these features as high-confidence biomarkers, we applied a classification error matrix assay on the discovery cohort subjects. Using the protein classifier, we correctly distinguished 14 out of 15 PDMCI and PDD subjects, as well as 6 out of 7 PDNC subjects in the test set (Fig. 6a). Similarly, the metabolite markers accurately differentiated 14 out of 15 PDMCI and PDD subjects, along with 6 out of 7 PD subjects (Fig. 6b). The abundance of the protein and metabolite biomarkers is depicted in Fig. 6c.
Validation of protein biomarkers in an independent cohort
To confirm the effectiveness of H3C15 and PSAP as biomarkers, we conducted targeted analysis using parallel reaction monitoring (PRM)30 on an independent cohort consisting of 62 PDCI and 35 PDNC subjects. Additionally, we measured the expression levels of NCAM2, LAMB2, and GCLC, which have previously been associated with cognitive dysfunction in relevant studies31,32,33, within the validation cohort. Consistent with the findings from the discovery cohort, we observed higher expression levels of H3C15 and lower levels of PSAP, NCAM2, and LAMB2 in the PDMCI and PDD groups compared to the PDNC group (Fig. 7a, b; Supplementary Fig 3a, b). Conversely, GCLC exhibited opposite trends (Supplementary Fig 3c, d). The two-proteins classifier constructed with H3C15 and PSAP achieved an AUC of 0.813 and accurately identified 61 out of 62 PD subjects with cognitive impairment (Fig. 7c). Moreover, when combining H3C15, PSAP, NCAM2, and LAMB2, the model achieved AUC of 0.983 in the train set, AUC of 0.981 in the test set, and AUC of 0.839 in the validation set (Fig. 7d).

Abundance of the protein biomarkers (a) H3C15 and PSAP and (b) NCAM2 and LAMB2. *P < 0.05; P-values from student’s t-test are shown. Lines indicate median and interquartile range. n = 35 (PDNC), 62 (PDMCI_PDD). Receiver operating characteristic curves for the combination of (c) H3C15 and PSAP or (d) all four protein biomarkers on the left, and the classification error matrices on the right. Areas under the operating curves are shown within the curve plots.
Combination of protein biomarkers with neuroimaging to predict cognitive dysfunction in PD
We also investigated the ability of the protein biomarkers, in combination with neuroimaging, to distinguish between PD patients with and without cognitive impairment. In the validation cohort, H3C15 exhibited a significant negative correlation with MoCA scores (P < 0.01, r = –0.52, Fig. 8a), while PSAP, NCAM2, and LAMB2 did not show any significant correlation with MoCA. Neuroimaging analysis revealed significantly decreased gray matter volume (GMV) in the right temporal and right insula regions in PDCI patients compared to PDNC patients (GRF correction, voxel level p < 0.005, and cluster level p < 0.05, Supplementary Table 1, Fig. 8b up, c). Additionally, PDCI individuals showed a significant decrease in degree centrality (DC) in the left fusiform and left cerebellum regions, as well as remarkably increased DC in the left middle frontal gyrus when compared to PDNC subjects (GRF correction, voxel level p < 0.005, and cluster level p < 0.05, Supplementary Table 1, Fig. 8b down, c). Finally, we constructed a logistic regression classifier using neuroimaging features (GMV + DC), resulting in an AUC of 0.833. When combined with H3C15, the AUC improved to 0.905 (Fig. 8d). It indicates that the protein biomarkers identified in this study can enhance the predictive power of neuroimaging for cognitive impairment in PD patients.

a Search for potential correlations of protein biomarker levels with scores on the Montreal Cognitive Assessment (MoCA). Red line is statistically significant (P < 0.05). b, c Brain regions showing significant differences in gray matter volume (GMV) and degree centrality (DC). ***P < 0.001; **P < 0.01; *P < 0.05; P-values from student’s t-test are shown. Lines indicate median and interquartile range. n = 28 (PDNC), 51 (PDMCI_PDD). d Receiver operating characteristic curves to evaluate the ability of H3C15 alone, neuroimaging alone, or the two together to predict cognitive decline. Areas under the operating curves are shown within the plot.
Discussion
The objective of this study was to identify biomarkers of cognitive impairment in PD patients by integrating proteomics and metabolomics data. We discovered that several proteins and associated metabolic pathways are altered in PD patients with mild cognitive impairment or dementia. These findings have potential implications for screening high-risk PD patients, optimizing management strategies, and developing targeted treatments for cognitive decline. Additionally, the protein biomarkers identified in this study improved the predictive power of neuroimaging data for cognitive impairment in PD patients. Our results shed light on the metabolic and signaling pathways involved in driving cognitive impairment and dementia in PD.
Two prominent protein biomarkers associated with cognitive dysfunction in PD were H3C15 and PSAP. Each marker demonstrated the ability to distinguish between PD patients with and without cognitive impairment, and their combined use enhanced discriminatory capabilities. H3C15 belongs to the histone family of basic nuclear proteins, which play a role in maintaining chromosomal structure in eukaryotic cells. In a Drosophila model, α-synuclein was found to directly bind to histones, inhibiting acetyltransferase activity and promoting neurotoxicity34. Moreover, studies have observed increased acetylation of histone H3 at specific sites (H3K14 and H3K18) in the primary motor cortex of PD patients, leading to overall hyperacetylation of histone H335. PSAP encodes saposin, a multifunctional protein involved in neuroprotection, neurotrophism, and regulation of lysosomal function. Dysregulation of PSAP expression can disrupt lysosomal function, contributing to the aggregation of alpha-synuclein and phosphorylated Tau36. Furthermore, recent research has suggested that loss of PSAP leads to increased reactive oxygen species (ROS) production and lipid peroxidation in neurons, ultimately resulting in neuronal ferroptosis, which underlies both PD and cognitive impairment37.
The candidate metabolite biomarkers of cognitive impairment identified through machine learning were glycocholic acid and 6-methylnicotinamide. Glycocholic acid is a conjugated bile acid derived from glycine and cholic acid and has been associated with microbiome dysbiosis, inflammation, and α-synuclein misfolding in PD38. Notably, our study appears to be the first to establish a link between 6-methylnicotinamide and cognitive impairment in PD. It is known to inhibit oxidative phosphorylation in mitochondria39, and is found to be elevated during the acute stage of cerebral hemorrhage27. Therefore, further investigations are warranted to elucidate the role of this metabolite in cognitive dysfunction during PD progression.
In addition to H3C15 and PSAP, we quantified the abundance of NCAM2, LAMB2, and GCLC, which have previously been implicated in cognitive dysfunction in PD patients31,32,33, across different PD subgroups. Consistently, in both the discovery and validation cohorts, the expression level of H3C15 was higher in the PDCI group compared to the PDNC group, while PSAP exhibited lower expression levels. Furthermore, the classifier built with H3C15 and PSAP achieved an AUC of 0.765 in the validation cohort, confirming their ability to distinguish between PD patients with and without cognitive impairment. NCAM2 and LAMB2 were downregulated in both cohorts, while GCLC showed an opposite trend. Importantly, the classifier incorporating H3C15, PSAP, NCAM2, and LAMB2 demonstrated a high AUC of 0.956 in the validation cohort.
NCAM2 belongs to the immunoglobulin superfamily of cell adhesion molecules and plays a regulatory role in critical neuron-specific processes such as neuronal differentiation, synaptogenesis, and memory formation40. Growing evidence supports the notion that cognitive impairment in PD is predominantly associated with the loss of neurons and synapses41. It has been reported that NCAM2 mediates synaptic adhesion, and proteolysis of NCAM2 by Aβ can result in synaptic disassembly31. On the other hand, LAMB2, a subunit of laminin, a major component of the basal lamina, is involved in the elimination of Aβ and other soluble brain metabolites through the basement membranes within the walls of cerebral arteries, which represent the brain’s lymphatic drainage pathways32. Hence, the loss of NCAM2 and LAMB2 may be closely associated with cognitive decline. Overall, the diagnostic panel comprising these four proteins holds promise for predicting cognitive impairment in PD.
Neuroimaging holds the potential to directly indicate both structural and functional changes in the brain, making it a valuable indicator of cognitive impairment42. In our study, the results from structural and functional imaging suggested that PD patients with cognitive decline may exhibit dysfunctional visual and executive networks compared to those without cognitive decline (Supplementary Fig. 4). Previous studies have consistently demonstrated that cognitive decline in PD primarily manifests as executive dysfunction and visuospatial disturbance43. Additionally, among the four proteins (H3C15, PSAP, NCAM2, and LAMB2), H3C15 exhibited a significant negative correlation with MoCA scores (Fig. 8a) and improved the predictive ability of neuroimaging for PDCI. These findings highlight the promising potential of combining imaging and blood markers to predict PDCI.
However, there were certain limitations to consider in our study. Firstly, the study design was single-center and cross-sectional, necessitating verification on a multi-center cohort with longitudinal samples to confirm the results. Secondly, there was a lack of common and reliable methods for validating the selected metabolites, which prevented confirmation of the metabolic findings in the validation cohort. Furthermore, it is important to note that we excluded patients with a family history of AD during the recruitment of PD patients to minimize the potential impact of AD, but our study did not involve AD screening for AD markers such as cerebrospinal fluid Aβ and tau levels44. Therefore, we cannot completely rule out the possibility of AD and PD overlapping in an exceptionally small number of patients. Finally, the potential impact of medications on the proteomic and metabolomic profiles was not considered in our study and should be addressed in future investigations.
To the best of our knowledge, this was the most comprehensive analysis of the plasma proteome and metabolome associated with cognitive dysfunction in PD patients. We have demonstrated the predictive value of plasma protein and metabolism biomarkers for cognitive impairment and highlighted how these protein markers can enhance the performance of neuroimaging. Further exploration of the relationship between altered plasma protein profiles and changes in brain structure and function is warranted to deepen our understanding of the pathogenesis of PD. In summary, our findings laid the groundwork for the development of a high-performance, non-invasive blood-based test for screening and monitoring PD patients with cognitive decline and provide multiple protein targets for future therapeutic development.
Methods
Study design and patients
Two cohorts, namely the “discovery cohort” and the “validation cohort,” consisted of healthy control subjects (C), PD patients with normal cognition (PDNC), PD patients with mild cognitive impairment (PDMCI), and PDD. These individuals were recruited from the First Affiliated Hospital of Kunming Medical University. The discovery cohort (n = 100) was enrolled between January 2022 and June 2022, while the validation cohort (n = 116) was recruited between July 2022 and December 2022.
The study was approved by the ethics committee of the First Affiliated Hospital of Kunming Medical University (2022-L-46). All participants provided informed consent and that the study conformed to the principles set out in the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report.
Healthy control participants had normal cognitive function as determined by the Montreal cognitive assessment (MoCA) test. They had no history of psychological or neurological disorders, nor did they have any current chronic conditions. PD/PDNC, PDMCI, and PDD patients were diagnosed according to the criteria set forth by the Movement Disorder Society45,46,47. Exclusion criteria for patients included the following: (1) presence of atypical or secondary symptoms associated with PD, such as multiple progressive supranuclear palsy, multiple system atrophy, cortico-basal ganglionic degeneration, vascular parkinsonism, or neuroleptic agent-related parkinsonism; (2) history of additional neurological or psychological disorders; (3) cognitive dysfunction caused by other factors and patients with a family history of AD; (4) contraindications to magnetic resonance imaging; (5) left-handedness; or (6) history of intracranial surgery for PD. Motor symptom severity in patients was assessed using the Hoehn–Yahr scale and Part III of the Unified PD Rating Scale (UPDRS-III). The levodopa-equivalent daily dose was calculated as recommended. Non-motor symptoms were evaluated using the Rapid Eye Movement Sleep Behavior Disorder Screening Questionnaire (RBDSQ), Hamilton Depression Scale (HAMD), and Hamilton Anxiety Scale (HAMA).
Regarding the cognitive status assessment, each patient’s cognitive evaluation was conducted independently by two associate chief physicians or above. This rigorous approach was undertaken to ensure reliable diagnoses and minimize potential selection bias in the study. Furthermore, all scale assessments were carefully reviewed by two attending physicians who underwent standardized training to ensure accurate measurements.
Plasma collection
For each study subject, a standard 12-h clinical biochemistry fasting protocol was followed. The fasting blood sample was collected into 10-mL K2EDTA Vacutainer tubes (BD 366643). Then K2EDTA tubes were then inverted serval times to mix the anticoagulant additive with blood followed by the centrifugation at 2500 × g for 15 min at room temperature to obtain plasma. The supernatant plasma was carefully transferred into new 15 mL tubes, mixed by inverting three times and then aliquoted into 1.5 mL tubes. Plasma samples were snap-frozen in liquid nitrogen and stored at −80 °C for further analysis.
Protein preparation and trypsin digestion
600 μg of plasma from each sample were first treated to remove the highest abundance plasma proteins using Pierce™ Top 14 Abundant Protein Depletion Spin Columns Kit (Thermo Fisher) according to the manufacturer’s instructions. The samples were then inactivated at room temperature for 2 h. Subsequently, the samples reduced with 5 mM dithiothreitol for 30 min at 56 °C and alkylated with 11 mM iodoacetamide for 15 min at room temperature in darkness. The samples were digested by trypsin (Promega, V5117) at an enzyme-to-protein mass ratio of 1:50 overnight at 37 °C, and the peptides were then extracted and dried (SpeedVac, Eppendorf).
Plasma proteomics LC-MS/MS analysis and database search
The proteomics LC-MS/MS assay was performed by a commercial company with quantitative proteomic service (PTM Bio., Zhejiang, CN). The tryptic peptides were dissolved in solvent A, directly loaded onto a Home-made reversed-phase analytical column (100 μm i.d. × 25 cm) packed with 1.9 μm/120 Å ReproSil-PurC18 resins (Dr. Maisch GmbH, Ammerbuch, Germany) The mobile phase consisted of solvent A (0.1% formic acid, 2% acetonitrile/in water) and solvent B (0.1% formic acid, 90% acetonitrile/in water). Peptides were separated with the following gradient: 0–68 min, 9%–25%B; 68–82 min, 25–35%B; 82–86 min, 35%–80%B; 86–90 min, 80%B, and all at a constant flow rate of 300 nl/minon a EASY-nLC 1200 UPLC system (Thermo Scientific). The separated peptides were analyzed in Orbitrap Exploris 480 (Thermo Scientific) with a nano-electrospray ion source. The electrospray voltage applied was 2000 V. FAIMS compensate voltage (CV) was set as –45 V, –70 V. Precursors and fragments were analyzed at the Orbitrap detector. The full MS scan resolution was set to 60,000 for a scan range of 400–1500 m/z. The MS/MS scan was fixed first mass as 100 m/z at a resolution of 15,000 with the TurboTMT was set as off. Up to 20 most abundant precursors were then selected for further MS/MS analyses with 30 s dynamic exclusion. The HCD fragmentation was performed at a normalized collision energy (NCE) of 28%. Automatic gain control (AGC) target was set at 100%, with an intensity threshold of 2E4 ions/s and a maximum injection time of 100 ms. MS data were acquired by Xcalibur and processed by Proteome Discoverer (v2.4.1.15). Tandem mass spectra were searched against the human Uniprot database (2021-07-21; 78,120 entries) concatenated with reverse decoy database. Trypsin was specified as cleavage enzyme allowing up to 2 missing cleavages. Min peptide length was set as 6 and max number of modification per peptide was set as 3. Mass error was set to 10 ppm for precursor ions and 0.02 Da for fragment ions. Carbamidomethyl on Cys was specified as fixed modification. Oxidation on Met, acetylation on protein N-terminal, met-loss on Met and met-loss+acetyl on Met were specified as variable modification. False discovery rate (FDR) of protein, peptide and PSM was adjusted to <1%.
Plasma metabolomics extraction and LC-MS/MS analysis
All steps for this protocol are to be done on ice. Plasma samples (50 μl) were thawed for 30 min on ice, then 300 μl of prechilled extraction solvent (20% acetonitrile in MeOH) was added to each sample. The samples were vortex for 3 min and centrifuged at 4 °C, 12000 rpm/min for 10 min. A volume of 200 μL supernatant was transferred into a new 1.5 ml tube and placed at –20 °C for 30 min. The samples were then centrifuged again at 4 °C, 12,000 rpm/min for 3 min. Finally, 180 μl supernatant was collected for LC-MS/MS analysis.
Metabolomic LC-MS/MS was performed on a QTRAP tandem mass spectrometer with a ExionLC AD UPLC system (AB Sciex Instruments) by commercial company (Metware Ltd, Wuhan, CN). Sample was load on the reverse-phase HSS T3 UPLC column (Waters, 1.8 µm, 2.1 mm*100 ml) with solvent A (0.1% formic acid in water) and separated with solvent B (0.1% formic acid in ACN). The gradient of B was as follows: 0–11 min, 5%–90% B; 11–12 min, 90% B; 12–12.1 min, 95% B; 12.1–14 min, 95% B. Ions were generated using the following parameters: ion source voltage, 5500 V; ion source (ESI) temperature, 500 °C. MS data was acquired and processed by SCIEX Analyst v1.6.3 software. Metabolite mass spectra were searched against a customized metabolites database named metware database (MWDB).
KEGG and go ontology analysis
Differential proteomics analysis between the C, PDNC, PDMIC, and PDD groups was performed on all quantifiable proteins. The student’s t-test was used to assess the statistical significance of protein expression difference between paired samples. Proteins with a P value < 0.05 and fold change >1.5 or <0.67 were considered significantly upregulated or downregulated proteins. The Kyoto Encyclopedia of Genes and Genomes (KEGG) database was applied to annotate enriched protein pathways using a two-tailed Fisher’s exact test to evaluate the enrichment of the differentially expressed protein against all identified proteins. Pathways with a corrected P value < 0.05 were considered significant. Gene Ontology (GO)48 annotation proteome was derived from the UniProt-GOA database49 (http://www.ebi.ac.uk/GOA/). Protein IDs were converted to UniProt IDs before mapping to GO IDs. For proteins not annotated by UniProt-GOA database, the InterProScan50 soft were used to annotated proteins’ GO functional based on protein sequence alignment method. For the prediction of subcellular localization, WoLF PSORT51, an updated version of PSORT/PSORT II for the prediction of eukaryotic sequences, were applied.
Protein expression pattern and protein interaction network analysis
The protein abundance profile used in KEGG analysis was also employed for gene expression pattern analysis. Mfuzz v.2.46.0R package23 was used to detect different sub-clustering pattern of protein expression among C, PDNC, PDMCI, PDD groups. R v.3.6.2 was used to implement Fisher’s exact test. String52 version 11 was used for protein–protein interaction network analysis.
Weighted gene co-expression network analysis
In brief, we first constructed a gene-gene similarity network by their correlation coefficient which was calculated by Person correlation. The similarity network was then divided into modules in which the gene expression level was similar. Next, the correlation between phenotypic traits and different modules was calculated to identify the drive gene within the module. This analysis was performed using the R package26.
Machine learning
Data processing and machine learning was performed in R (version3.6.2). Proteins with missing values in more than 50% of the samples were excluded. Subsequently, the missing values were imputed using the KNN algorithm and protein intensities were z-score normalized using scale function in R. Samples from PDNC, PDCI (PDMCI + PDD) were used to set up a two-stratified classifier. The processed data were divided into train set (70%) and test set (30%) using the createDataPartition function. Features were selected using a random forest model via the mean decrease accuracy in the train set. The hyper-parameters of the random forest model were optimized by the train function in the caret package. To evaluate the performance of the classifier model, tenfold cross-validation was applied to the train set, yielding the sensitivity (true positive rate) of and specificity (true negative rate). Additionally, the classification error matrix assay was used to assess the performance of the classifier. The prediction results from the test and validation sets were used to assess the classifier’s generalization ability. In addition, protein features finally used for model construction are complete with no missing value.
Performing parallel reaction monitoring (PRM) mass spectrometry
After trypsin digestion, the tryptic peptides were dissolved in 0.1% formic acid (solvent A), directly loaded onto a home-made reversed-phase analytical column. The gradient was comprised of an increase from 6% to 23% solvent B (0.1% formic acid in 98% acetonitrile) over 38 min, 23% to 35% in 14 min and increase to 80% in 4 min then holding at 80% for 4 min. The solvent flow rate was 700 nL/min on an EASY-nLC 1000 UPLC system. The peptides were subjected to NSI source followed by tandem mass spectrometry (MS/MS) in Q ExactiveTM Plus (Thermo Scientific). The electrospray voltage was set to 2.0 kV and the M/Z scan range was 350 to 1000 in a full scan mode. The intact peptides were detected in the orbitrap at a resolution of 35k. Targeted peptides were then selected for MS/MS using NCE setting as 27% and the fragments were detected in the Orbitrap at a resolution of 17,500. A data-independent procedure that alternated between one MS scan followed by 20 MS/MS scans. Automatic gain control was set at 3E6 for full MS and 1E5 for MS/MS. The isolation window for MS/MS was set at 2.0 m/z. The resulting MS data were processed in Skyline (v.3.6) software. In the Skyline, peptide settings were set to Trypsin [KR/P], max missed cleavage set as 2, peptide length was set as 8–25, fixed modification was set as Carbamidomethyl on Cys and oxidation on Met. Transition settings: precursor charges were set as 2, 3, ion charges were set as 1, 2, ion types were set as b, y, p. The product ions were set as from ion 3 to last ion, the ion match tolerance was set as 0.02 Da.
Neuroimaging
MRI and resting-state functional MRI were performed on 28 PD patients without cognitive impairment and 51 PD patients with cognitive impairment in the validation cohort. Gray matter volume (GMV) was analyzed using voxel-based morphometry, and functional connectivity was assessed based on degree centrality (DC)53.
All MRI was conducted with a 3-T whole-body scanner (Discovery 750w, GE Healthcare, USA) using standard transmitting and receiving head coils. Before scanning, all participants put on earplugs and were asked to lie down, relax, not think about anything in particular, not fall asleep and keep their head still.
Three-dimensional, T1-weighted, magnetization-prepared rapid gradient echo sagittal images were acquired using the follow parameters: number of slices, 156; slice thickness, 1 mm; gap, none; voxel size, 1 × 1 × 1 mm; repetition time, 1900 ms; echo time, 2.0 ms; inversion time, 450 ms; flip angle, 12°; field of view, 256 mm × 256 mm; matrix dimensions, 256 × 256. Resting-state functional MRI was performed as echo planar imaging with the follow parameters: number of slices, 36; slice thickness, 3 mm; gap, none; voxel size, 3.5 × 3.5 × 4 mm; number of volumes, 240; repetition time, 2000 ms; echo time, 30 ms; flip angle, 90°; field of view, 224 mm; matrix dimensions, 64 × 64; Scans were removed if they contained clinically significant abnormalities, substantial head movement or were otherwise of poor quality.
Magnetic resonance imaging data processing
The retained scans were preprocessed using SPM12 (http://www.fil.ion.ucl.ac.uk/spm/) in MATLAB 2013b (MathWorks, Natick, MA, USA)54. Unified segmentation was applied to the structural T1-weighted images, and the resulting probability maps for gray and white matter were spatially normalized to the Montreal Neurological Institute template using a high-level, non-linear warping algorithm involving diffeomorphic anatomical registration based on exponentiated Lie algebra55. Modulated volumes were smoothed using a Gaussian kernel featuring a full width at half-maximum of 6 mm.
Resting-state functional MRI images were analyzed using Data Processing and Analysis of Brain Imaging 4.5 (DPABI, http://www.rfmri.org/dpabi), which was based on SPM1254. To remove potential artifacts due to scanner calibration and subjects’ adaptation to the scanning environment, we immediately removed the first 10 scans, then corrected the remaining images for slice timing with the middle slice as a reference. All images were realigned to correct for head motion, and images were removed if motion exceeded 2 mm or rotation along any direction exceeded 2°. The T1-weighted anatomical image was coregistered to the mean functional image using rigid-body transformation, then structural images were segmented into gray matter, white matter, and cerebrospinal fluid using diffeomorphic anatomical registration based on exponentiated Lie algebra55, which was also used to normalize data to the Montreal Neurological Institute template. The normalized data were resampled to voxels measuring 3 × 3 × 3 mm and spatially smoothed using an isotropic Gaussian kernel with a full width at half-maximum of 6 mm. Low-frequency drifts and physiological high-frequency noise were removed using linear detrending and temporal bandpass filtering (0.01–0.08 Hz). Nuisance linear regression was performed on the white matter, cerebrospinal fluid, and 24 head motion parameters56.
Degree centrality calculation
Based on preprocessed data, voxel-wise DC value calculations were performed using the DPABI software. We extracted the BOLD time series of each voxel and computed Pearson’s correlation coefficients (r) between any pair of brain voxels within the whole-brain gray matter mask. Then, Pearson’s correlation data were normalized with Fisher’s r-to-z transformation to obtain the Z-score DC value map, and the whole-brain functional network was mapped with the threshold r > 0.25 in accordance with previous studies57.
Statistical analysis
Data of general demographic information and clinical assessment were analyzed using SPSS version 25.0 (IBM, Armonk, NY, USA). Intergroup differences were assessed as mentioned in the respective tables or figure legends. The Kolmogorov–Smirnov test can be used to assess whether the continuous variable follows a normal distribution. For normally distributed data, the mean and standard deviation was reported, P-values were derived from multivariate analysis of variance. For skewed data, the median and 25–75% percentiles were reported, and P-values were derived from Kruskal–Wallis test. Frequencies were expressed as percentages, and P-values were derived from Chi-square. Pearson’s correlation analysis was performed to identify proteins or metabolites associated with cognitive impairment in the discovery cohort. The ability of potential biomarkers to predict cognitive impairment in the validation cohort was determined by receiver operating characteristic (ROC) curve analysis, and calculated as the area under the curve (AUC). P < 0.05 was considered statistically significant.
Maps of smoothed GMV and DC were compared between PDNC and PDCI (PCMCI + PDD) using SPM12, and differences in GMV and DC were assessed for significance using a two-samples t test. The threshold level was set at p < 0.05. Gaussian random field (GRF) was used for multiple comparison correction(voxel level p < 0.005, and cluster level p < 0.05). Age, gender, years of education have been used as covariates in the above analysis.
Data availability
The data supporting the conclusions of this article are included within the article and its additional files. Additional data are available from the corresponding author upon request.
References
Foltynie, T., Brayne, C. E., Robbins, T. W. & Barker, R. A. The cognitive ability of an incident cohort of Parkinson’s patients in the UK. The CamPaIGN study. Brain 127, 550–560 (2004).
Chung, S. J. et al. Factor analysis-derived cognitive profile predicting early dementia conversion in PD. Neurology 95, e1650–e1659 (2020).
Hogue, O., Fernandez, H. H. & Floden, D. P. Predicting early cognitive decline in newly-diagnosed Parkinson’s patients: a practical model. Parkinsonism Relat. Disord. 56, 70–75 (2018).
Montine, T. J. et al. CSF Aβ(42) and tau in Parkinson’s disease with cognitive impairment. Mov. Disord. Off. J. Mov. Disord. Soc. 25, 2682–2685 (2010).
Bai, X. et al. Microstructural but not macrostructural cortical degeneration occurs in Parkinson’s disease with mild cognitive impairment. NPJ Parkinsons Dis. 8, 151 (2022).
Meyer, P. T., Frings, L., Rücker, G. & Hellwig, S. 18F-FDG PET in Parkinsonism: differential diagnosis and evaluation of cognitive impairment. J. Nuclear Med. 58, 1888–1898 (2017).
Lin, J. et al. Evolution and predictive role of plasma Alzheimer’s disease-related pathological biomarkers in Parkinson’s disease. J. Gerontol. 78, 2203–2213 (2023).
Chiu, M. J. et al. Synergistic association between plasma abeta(1-42) and p-tau in Alzheimer’s disease but not in Parkinson’s disease or Frontotemporal dementia. ACS Chem. Neurosci. 12, 1376–1383 (2021).
Aamodt, W. W. et al. Neurofilament light chain as a biomarker for cognitive decline in Parkinson disease. Mov. Disord. 36, 2945–2950 (2021).
Moscoso, A. et al. Longitudinal associations of blood phosphorylated Tau181 and neurofilament light chain with neurodegeneration in Alzheimer disease. JAMA Neurol. 78, 396–406 (2021).
Wang, H. F. et al. Efficacy and safety of cholinesterase inhibitors and memantine in cognitive impairment in Parkinson’s disease, Parkinson’s disease dementia, and dementia with Lewy bodies: systematic review with meta-analysis and trial sequential analysis. J. Neurol. Neurosurg. Psychiatry 86, 135–143 (2015).
van Steenoven, I. et al. Identification of novel cerebrospinal fluid biomarker candidates for dementia with Lewy bodies: a proteomic approach. Mol. Neurodegener. 15, 36 (2020).
Borràs, E. et al. Protein-based classifier to predict conversion from clinically isolated syndrome to multiple sclerosis. Mol. Cell. Proteom. 15, 318–328 (2016).
Jiang, Y. et al. Large-scale plasma proteomic profiling identifies a high-performance biomarker panel for Alzheimer’s disease screening and staging. Alzheimers Dement. 18, 88–102 (2022).
Karayel, O. et al. Proteome profiling of cerebrospinal fluid reveals biomarker candidates for Parkinson’s disease. Cell Rep. Med. 3, 100661 (2022). 100661.
Shao, Y. & Le, W. Recent advances and perspectives of metabolomics-based investigations in Parkinson’s disease. Mol. Neurodegener. 14, 3 (2019).
Virreira Winter, S. et al. Urinary proteome profiling for stratifying patients with familial Parkinson’s disease. EMBO Mol. Med. 13, 13257 (2021). e13257.
Horvath, I. et al. Co-aggregation of pro-inflammatory S100A9 with alpha-synuclein in Parkinson’s disease: ex vivo and in vitro studies. J. Neuroinflammation 15, 172 (2018).
Yagensky, O. et al. Increased expression of heme-binding protein 1 early in Alzheimer’s disease is linked to neurotoxicity. eLife, 8, e47498 (2019).
Garcia-Esparcia, P. et al. Altered machinery of protein synthesis is region- and stage-dependent and is associated with alpha-synuclein oligomers in Parkinson’s disease. Acta Neuropathol. Commun. 3, 76 (2015).
Lin, Z. H. et al. PSAP intronic variants around saposin D ___domain and Parkinson’s disease. Brain 144, e3–e3 (2021).
Ernest James Phillips, T. & Maguire, E. Phosphoinositides: roles in the development of microglial-mediated neuroinflammation and neurodegeneration. Front. Cell Neurosci. 15, 652593 (2021).
Kumar, L. & M, E. F. Mfuzz: a software package for soft clustering of microarray data. Bioinformation 2, 5–7 (2007).
Ryan, D. P. & Matthews, J. M. Protein-protein interactions in human disease. Curr. Opin. Struct. Biol. 15, 441–446 (2005).
Villa, A., Vegeto, E., Poletti, A. & Maggi, A. Estrogens, neuroinflammation, and neurodegeneration. Endocr. Rev. 37, 372–402 (2016).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Hu, E. et al. Temporal metabolomic alteration in rat brains of experimental intracerebral hemorrhage. Brain Res. Bull. 170, 234–245 (2021).
Rahn, K. A., Slusher, B. S. & Kaplin, A. I. Glutamate in CNS neurodegeneration and cognition and its regulation by GCPII inhibition. Curr. Med. Chem. 19, 1335–1345 (2012).
Hensel, C. et al. Influence of nutritional tyrosine on cognition and functional connectivity in healthy old humans. NeuroImage 193, 139–145 (2019).
Rauniyar, N. Parallel reaction monitoring: a targeted experiment performed using high resolution and high mass accuracy mass spectrometry. Int. J. Mol. Sci. 16, 28566–28581 (2015).
Leshchyns’ka, I. et al. Abeta-dependent reduction of NCAM2-mediated synaptic adhesion contributes to synapse loss in Alzheimer’s disease. Nat. Commun. 6, 8836 (2015).
Matejas, V. et al. Mutations in the human laminin beta2 (LAMB2) gene and the associated phenotypic spectrum. Hum. Mutat. 31, 992–1002 (2010).
Wang, T. et al. Neuroprotective effects of Danshensu on rotenone-induced Parkinson’s disease models in vitro and in vivo. BMC Complement. Med. Ther. 20, 20 (2020).
Kontopoulos, E., Parvin, J. D. & Feany, M. B. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 15, 3012–3023 (2006).
Gebremedhin, K. G. & Rademacher, D. J. Histone H3 acetylation in the postmortem Parkinson’s disease primary motor cortex. Neurosci. Lett. 627, 121–125 (2016).
Mendsaikhan, A., Tooyama, I., Serrano, G. E., Beach, T. G. & Walker, D. G. Loss of lysosomal proteins progranulin and prosaposin associated with increased neurofibrillary tangle development in Alzheimer disease. J. Neuropathol. Exp. Neurol. 80, 741–753 (2021).
Hambright, W. S., Fonseca, R. S., Chen, L., Na, R. & Ran, Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 12, 8–17 (2017).
Li, P. et al. Gut microbiota dysbiosis is associated with elevated bile acids in Parkinson’s disease. Metabolites 11, 29 (2021).
Willets, J. M., Lunec, J., Williams, A. C. & Griffiths, H. R. Neurotoxicity of nicotinamide derivatives: their role in the aetiology of Parkinson’s disease. Biochem. Soc. Trans. 21, 299S (1993).
Parcerisas, A., Ortega-Gasco, A., Pujadas, L. & Soriano, E. The hidden side of NCAM family: NCAM2, a key cytoskeleton organization molecule regulating multiple neural functions. Int. J. Mol. Sci. 22, 10021 (2021).
Emre, M. Dementia associated with Parkinson’s disease. Lancet Neurol. 2, 229–237 (2003).
Zhu, Y. et al. Cortical atrophy is associated with cognitive impairment in Parkinson’s disease: a combined analysis of cortical thickness and functional connectivity. Brain Imaging Behav. 16, 2586–2600 (2022).
Goldman, J. G. & Sieg, E. Cognitive impairment and dementia in Parkinson disease. Clin. Geriatr. Med. 36, 365–377 (2020).
Zhang, H. et al. Interaction between Aβ and Tau in the pathogenesis of Alzheimer’s disease. Int. J. Biol. Sci. 17, 2181–2192 (2021).
Postuma, R. B. et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 30, 1591–1601 (2015).
Litvan, I. et al. Diagnostic criteria for mild cognitive impairment in Parkinson’s disease: movement disorder society task force guidelines. Mov. Disord Off. J. Mov. Disord. Soc. 27, 349–356 (2012).
Emre, M. et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov. Disord. Off. J Mov. Disord. Soc. 22, 1689–1707 (2017).
Gene Ontology Consortium. Gene ontology consortium: going forward. Nucleic Acids Res. 43, D1049–D1056 (2015).
Dimmer, E. C. et al. The UniProt-GO annotation database in 2011. Nucleic Acids Res. 40, D565–D570 (2012).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Horton, P. et al. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 35, W585–W587 (2007).
Szklarczyk, D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–d613 (2019).
Zuo, X. N. et al. Network centrality in the human functional connectome. Cereb. Cortex 22, 1862–1875 (2012).
Yan, C. G., Wang, X. D., Zuo, X. N. & Zang, Y. F. DPABI: data processing & analysis for (resting-state) brain imaging. Neuroinformatics 14, 339–351 (2016).
Ashburner, J. A fast diffeomorphic image registration algorithm. NeuroImage 38, 95–113 (2007).
Gallea, C. et al. Antisaccade, a predictive marker for freezing of gait in Parkinson’s disease and gait/gaze network connectivity. Brain 144, 504–514 (2021).
Buckner, R. L. et al. Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 29, 1860–1873 (2009).
Acknowledgements
This study was funded by the Applied Basic Research Foundation of Yunnan Province [Grant Numbers 202301AS070045, 202101AY070001-115]; National Natural Science Foundation of China [Grant number 81960242]; The Major Science and Technology Special Project of Yunnan Province [Grant number 202102AA100061].
Author information
Authors and Affiliations
Contributions
Conceptualization: Y.B.Y., Z.Y.Y., X.Z., P.A.L. and Y.X.L. Supervision: X.Z., P.A.L. and Y.X.L. Data curation: Y.B.Y., Z.Y.Y., L.K.L., W.F., L.B., Z.Q., T.Y.C., and L.Z.C. Writing—original draft: Y.B.Y. and Z.Y.Y. Review and editing: Y.L., B.R.Q., L.C.Y., R.H., X.Z., P.A.L. and Y.X.L. Investigation: Y.B.Y., Z.Y.Y., L.K.L., W.F., L.B., Z.Q., T.Y.C., L.Z.C., Y.L., B.R.Q., L.C.Y., and R.H. All co-authors read and approved the document.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, B., Zhu, Y., Li, K. et al. Machine learning model base on metabolomics and proteomics to predict cognitive impairment in Parkinson’s disease. npj Parkinsons Dis. 10, 187 (2024). https://doi.org/10.1038/s41531-024-00795-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-024-00795-y
