
Abstract
Hematological malignancies are associated with an increased risk of complications during SARS-CoV-2 infections. Primary series or monovalent booster vaccines reduce disease severity, hospitalization, and death among multiple myeloma patients. We characterized virus-neutralizing and spike-binding antibody profiles following monovalent (WA1) or bivalent (WA1/BA.5) SARS-CoV-2 booster vaccination in MM patients. Bivalent vaccination improved the breadth of binding antibodies but not neutralization activity against contemporary variants. Hybrid immunity and immune imprinting impact vaccine-elicited immunity.
Similar content being viewed by others


Introduction
Hematological malignancies are associated with an increased risk of complications during SARS-CoV-2 infections1,2. Although vaccines have led to reduced risk of complications, vaccinated patients with hematologic malignancies still have a higher risk of hospitalization3, as well as developing chronic SARS-CoV-2 infections4,5,6, which have resulted in intrahost viral evolution and the emergence of variants of concern7,8,9. Impaired induction of neutralizing antibodies (nAbs) against SARS-CoV-2 Omicron variants has been reported in multiple myeloma (MM) patients following booster immunization with monovalent vaccines10. Bivalent mRNA vaccines include both the ancestral WA1 and BA.4/5 spike. However, little is known about the breadth of nAbs induced by additional booster immunizations or immunization with monovalent or bivalent vaccines. In this study, we used an in vitro, live virus focus reduction neutralization test (FRNT) to determine the neutralization activity against WA1, BA.1, BA.5, BQ.1.1, and the contemporary XBB.1.5 Omicron subvariant in cohorts of MM patients that received monovalent or bivalent booster immunizations. We also evaluated spike-binding antibody responses against this panel of viruses using a Spike-specific electrochemiluminescence assay11. All the participants provided written informed consent, and the Institutional Review Board of Emory University approved the study.
Results and discussion
The study included two cohorts. The first cohort received anti-SARS-CoV-2 primary immunization and two monovalent booster immunizations, with the last monovalent booster administered 1–6 months after the second dose (n = 101, Supplementary Fig. 1). The second cohort received the anti-SARS-CoV-2 primary immunization, two booster immunizations, and a bivalent booster (containing mRNA for both ancestral and BA.4/5) administered 1–6 months after the third dose (n = 42). The nAb titers against the ancestral strain (WA1) in patients that received the bivalent booster were 2.05-fold higher than neutralization titers in patients that received the monovalent booster (GMT 830 vs 404, Supplementary Fig. 2). nAbs against BA.1 and BA.5 after monovalent or bivalent booster had lower titers compared to WA1, with a reduction in neutralization potency ranging between 7.1-8.2-fold for BA.1 and between 8.9-12.4-fold for BA.5 (Supplementary Fig. 2). Neutralizing antibodies against the newly circulating Omicron variants BQ.1.1 and XBB.1.5 were low or undetectable (ranging between the level of detection and 707 for BQ.1.1 and 512 for XBB.1.5).
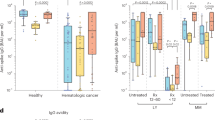
Detection of anti-nucleocapsid (NC) binding antibodies (900 AU/ml as cutoff) was utilized as evidence of recent SARS-CoV-2 exposure12. In the monovalent-boosted group, 45.5% of the monovalent-boosted patients were anti-NC+ (n = 46, out of 101 participants) (Supplementary Fig. 3). Next, we compared neutralization and antibody binding titers between individuals with low/undetectable anti-NC antibodies and those with recent SARS-CoV-2 exposure, defined by NC-binding antibody titers over the cutoff. In the monovalent-boosted group, the neutralization titers against WA1 were 11-fold higher in patients with recent SARS-CoV-2 exposure compared to unexposed patients (Geometric Mean Titer [GMT] 1379 vs 125, P < 0.0001). Neutralization titers against Omicron variants BA.1 (GMT 134 vs. 28, P < 0.0001), BA.5 (GMT 95 vs. 24, P < 0.0001), BQ.1.1 (GMT 38 vs. 20.5, P < 0.0001), and XBB.1.5 (GMT 30 vs. <20, P < 0.0001) were higher in SARS-CoV-2 exposed compared to unexposed patients (Fig. 1A). We then assessed the temporal distribution of sampling among the monovalent-boosted group. In Fig. 1B, antibody titers against WA1 are shown to indicate that most of the samples (69 and 59% in unexposed and exposed patients, respectively) were collected at time points under 50 days after immunization. However, a higher proportion of responders to multiple variants was observed among samples collected from exposed patients compared to unexposed patients (Fig. 1C).

A Comparative response of samples collected from MM patients after monovalent booster immunizations. Black lines, patients without evidence of natural exposure to SARS-CoV-2. Red lines, patients with natural exposure to SARS-CoV-2 by confirmation of positive anti-nucleocapsid binding antibodies. Dark black and dark red lines represent the geometric mean titers (GMT). The focus reduction neutralization test (FRNT50 [the reciprocal dilution of serum that neutralizes 50% of the input virus]) geometric mean titer (GMT) of neutralizing antibodies against the WA1 strain, and each Omicron subvariants is shown at the top of the panel, GMT values in black unexposed patients, GMT values in red patients exposed to SARS-CoV-2 infections. B Time course of FRNT titers against WA1 in patients that received monovalent immunization and were unexposed (gray symbols) or exposed (red symbols) to SARS-CoV-2. C Percentage of responders against individual variants tested, among unexposed (gray bars) or exposed (red bars) to SARS-CoV-2 that received monovalent booster immunization. D Comparative response of samples collected from MM patients after bivalent booster immunizations. Black lines, patients without evidence of natural exposure to SARS-CoV-2. Red lines, patients with natural exposure to SARS-CoV-2 by confirmation of positive anti-nucleocapsid binding antibodies. Dark black and dark red lines represent the GMT of neutralizing antibodies against the virus tested. GMT of neutralizing antibodies is shown at the top of the panel, with GMT values in black unexposed patients and GMT values in red patients exposed to SARS-CoV-2 infections. E Time course of FRNT titers against WA1 in patients that received bivalent immunization and were unexposed (gray symbols) or exposed (red symbols) to SARS-CoV-2. F Percentage of responders against individual variant tested, among unexposed (gray bars) or exposed (red bars) to SARS-CoV-2 that received bivalent booster immunization.
Consistent with high community spread and breakthrough infections with the Omicron variant, we found that 73.8% of the bivalent-boosted patients were anti-NC+ (n = 31, out of 42 participants) (Supplementary Fig. 3). In the bivalent-boosted group, the neutralization titers against WA1 showed modestly elevated titers (1.3 fold) in patients with recent SARS-CoV-2 exposure compared to unexposed patients (GMT 894 vs. 676, P = 0.1722). There was a trend towards increased levels of neutralizing antibodies against Omicron variants BA.1 (GMT 139 vs. 42, P = 0.0438), BA.5 (GMT 87 vs. 32, P = 0.0615), BQ.1.1 (GMT 43 vs. 22, P = 0.0991), and XBB.1.5 (GMT 33.5 vs. <20, P = 0.0936) in SARS-CoV-2-exposed compared to unexposed patients (Fig. 1D). The temporal distribution of sampling among the bivalent-boosted group (Fig. 1E) showed that most of the samples (91 and 87% in unexposed and exposed patients, respectively) were collected at time points over 50 days after immunization. A higher proportion of responders to multiple variants were present among samples collected from exposed patients compared to unexposed patients (Fig. 1F).
We next determined the IgG binding titers using Spike-specific electrochemiluminescence assays11. Nearly all samples across the two cohorts tested positive for SARS-CoV-2 Spike-IgG binding (97%), except seven unexposed SARS-CoV-2 patients receiving a monovalent booster. We tested a panel of pre-Omicron and Omicron Spike variants and found significantly higher binding titers in exposed SARS-CoV-2 patients that received a monovalent booster as compared to unexposed patients (For all the variants tested, P < 0.0001, by Mann-Whitney test; Fig. 2A). In contrast, we did not observe a statistically significant difference in the bivalent vaccinated group (By Mann-Whitney test: WT, P = 0.3079, B.1.1.7, P = 0.3963, B.1.351, P = 0.2567, B.1.617.2, P = 0.2815, BA.1, P = 0.1547; BA.2, P = 0.1309; BA.2.75, P = 0.2333; BA.2.12.1, P = 0.1547; BA.5, P = 0.1309; XBB.1, P = 0.2448; BF.7, P = 0.1633; XBB.1.5, P = 0.2743; BQ.1.1, P = 0.2115; BN.1, P = 0.9303 and BQ.1, P = 0.5663. Fig. 2B). In all the data points tested with sera samples collected from patients that received boosting immunization with monovalent or bivalent vaccines, no differences were observed between binding IgG titers to the Spike protein from early variants (B.1.1.7, B.1.351, and B.1.617.2) and the binding IgG titers to the Spike protein from the ancestral SARS-CoV-2 strain (Supplementary Fig. 4). However, in SARS-CoV-2 unexposed and exposed patients, differences were observed between the IgG antibody response against the Spike protein from the ancestral SARS-CoV-2 strain and the IgG response against most of the Omicron variants (Supplementary Fig. 4).

Antibody responses were measured by electrochemiluminescence using the MesoScale Discovery (MSD) platform. A Comparative response of samples collected from MM patients after monovalent booster immunizations. Black lines, patients without evidence of natural exposure to SARS-CoV-2. Red lines, patients with natural exposure to SARS-CoV-2 by confirmation of positive anti-nucleocapsid binding antibodies (titer >900 AU/ml). Dark black and dark red lines represent the geometric mean titers (GMT). B Comparative response of samples collected from MM patients after bivalent booster immunizations. Black lines, patients without evidence of natural exposure to SARS-CoV-2. Red lines, patients with natural exposure to SARS-CoV-2 by confirmation of positive anti-nucleocapsid binding antibodies (titer >900 AU/ml). Dark black and dark red lines represent the geometric mean titers (GMT). Pre-pandemic plasma samples from healthy individuals were used to set the detection cutoff levels for SARS-CoV-2 Spike-specific IgG antibody titers.
We next analyzed the clinical correlates of neutralizing antibodies against WA1 and the contemporary XBB.1.5 variant as described10,13 (Supplementary Tables 1 and 2). Anti-BCMA therapy correlated with reduced induction of nAb against WA1 in patients who received a monovalent booster (P = 0.007, Supplementary Table 1). nAb against XBB-1 were low in all subsets and not impacted by these variables (Supplementary Table 2). Anti-CD38 therapy correlated with reduced induction of nAb against WA1 in both cohorts of patients that received monovalent and bivalent boosters (P = 0.044 and P = 0.048, Supplementary Table 1).
All the subjects enrolled attended Emory University’s myeloma clinic and were elected without selection bias. Therefore, samples were collected at different time intervals after immunization, with the monovalent group receiving three immunizations and the bivalent group receiving four. This study shows that although bivalent vaccination induced significantly higher nAb titers against the ancestral strain in unexposed individuals, compared to those induced by monovalent boosters, titers against circulating Omicron variants (e.g., XBB.1) were very low or undetectable. Hybrid immunity enabled higher induction of broadly reactive nAbs. However, there was no correlation between Spike-binding antibodies and neutralization against circulating variants, suggesting that vaccination elicited antibodies that are not neutralizing. The inability to elicit antibodies against neutralizing epitopes suggests that a vaccination regimen with repeat boosters or using different vaccine platforms is needed to improve the effect of bivalent booster immunization in MM patients.
We have reported that a single monovalent booster vaccine is associated with increased nAb against the ancestral strain and the B.1.617.2 variant but not the Omicron variants in MM patients10. It has been suggested that immune imprinting provided by prior infection or SARS-CoV-2 vaccination negatively impacts vaccine immunogenicity of booster immunizations14. Consistent with this, we observed preferential boosting of nAb against the ancestral WA1 strain following booster immunization. Booster immunization particularly improved the breadth and longevity of the antibody response in patients with hybrid immunity. These results may be linked to the further expansion of class-switched memory B cells, described previously following monovalent boosters in MM10. As with monovalent vaccines10, consistent with reported data15,16, patients with prior anti-CD38 antibodies also exhibit poor response to bivalent vaccines, which may relate to the adverse impact of these therapies on B/plasma cells and T-follicular-helper cells10,15. The need for multiple vaccines in MM patients may not be SARS-CoV-2-specific, as multiple vaccines were also shown to improve seroprotection following influenza vaccination in a randomized trial in MM17. Overall, our data show that most MM patients lack detectable nAb against Omicron variants circulating early in 2023 at the time of sample collection, despite bivalent booster immunization. In contrast, we have reported that in healthy volunteers, bivalent booster improved neutralization against BQ.1.1 and XBB18, but more recent Omicron variants EG.5.1, HK.3, HV.1, and JN.1 evade antibodies elicited by bivalent booster immunization. Spike-binding IgG antibodies against several Omicron variants were detected, suggesting that while immune imprinting may impact the induction of broadly neutralizing antibodies, reduced nAbs following vaccination in MM are not due to the inability to produce antibodies. High-risk cohorts of MM patients, such as those with prior CD38 or BCMA targeting therapies, may be at particular risk for ongoing reinfections with these viruses and may need consideration of newer approaches, including additional boosters or newer vaccine platforms.
Methods
Patients and patient selection
This study was approved by the Institutional Review Board of Emory University. Per protocol, patients were approached in myeloma clinics of Winship Cancer Institute without any selection bias. Patients who provided informed consent were eligible for a research blood draw. Blood samples were processed to isolate plasma and mononuclear cells.
Nucleocapsid and spike binding assay
SARS-CoV-2 nucleocapsid and spike-specific IgG antibodies were detected using the Meso Scale Discovery (MSD) platform, V-PLEX SARS-CoV-2 Key Variant Spike Panel 1 Kit (catalog number K15651 (IgG)), and Panel 34 (catalog number K15690 (IgG)). Briefly, serum samples were diluted 1:5000 prior to the assay. Kit plates were blocked for 30 min using manufacturer-provided blocking buffer A. Blocking buffer was removed, and plates were washed 3X with wash buffer. Diluted serum samples, controls, and calibrators were added to the plates and incubated for 2 h at room temperature in a plate shaker adjusted at 700 rpm. After a 2-h incubation, plates were washed 3X, loaded with a solution containing MSD Sulfo-Tag anti-human IgG, and incubated for 1 h. After incubation, the plates were washed 3X, and the detection read buffer was added immediately prior to the plate read. Raw data were parsed using Methodical Mind software.
Viruses and cells
VeroE6-TMPRSS2 cells were generated and cultured as previously described19. nCoV/USA_WA1/2020 (WA1), closely resembling the original Wuhan strain, was propagated from an infectious SARS-CoV-2 clone as previously described20. icSARS-CoV-2 was passed once to generate a working stock. The BA.1 isolate has been previously described21. Omicron subvariants were isolated from residual nasal swabs: BA.5 isolate (EPI_ISL_13512579), provided by Dr. Richard Webby (St Jude Children’s Research Hospital), BQ.1.1 isolate (EPI_ISL_15196219) and XBB.1.5 isolate (EPI_ISL_ 16818774) were provided by Dr. Benjamin Pinsky (Stanford University) All variants were plaque purified and propagated once in VeroE6-TMPRSS2 cells to generate working stocks. All viruses used in this study were deep-sequenced and confirmed as previously described22.
Focus reduction neutralization test
FRNT assays were performed as described19,22,23. In duplicate, serum samples were three-fold serially diluted down a plate, then mixed 1:1 with 100–200 PFU of WA, BA.1, BA.5, BQ.1.1 or XBB.1.5 (1:20 starting dilution with the virus). Dilution plates with virus-serum immune complex were incubated at 37 °C for 1 h. At 1 h, the immune complex was loaded onto plates with a VeroE6-TMPRSS2 cell monolayer and incubated at 37 °C for an additional hour. Following incubation, the immune complex was removed from cells and replaced with an 0.85% methylcellulose overlay. Cells were incubated at 37 °C for variant-specific time intervals (16–40 h). At the appropriate time points for each virus, cells were removed from the incubator, washed, and fixed with 2% paraformaldehyde. Cells were permeabilized and incubated with Alexa Fluor-647-conjugated SARS-CoV-2 (AF647-CR3022) for four hours at room temperature or 4 °C overnight. After incubation, cells were washed, placed in Dulbecco’s phosphate-buffered saline, and visualized on an ELISpot reader (CTL Analyzer).
Statistical analysis
Antibody neutralization was quantified by counting the number of foci for each sample using the Viridot program24. The neutralization titers were calculated as 1 - (ratio of the mean number of foci in the presence of sera and foci at the highest dilution of the respective sera sample). Each specimen was tested in duplicate. The FRNT50 titers were interpolated using a 4-parameter nonlinear regression in GraphPad Prism 9.4.1. Samples that do not neutralize at the limit of detection at 50% are plotted at 20 and used for geometric mean and fold-change calculations. The differences between all groups were determined with the Kruskal–Wallis test with Dunn’s correction for multiple comparisons. Binomial logistic regression was used to identify the predictors for neutralizing antibody responses. Among variables statistically significant on univariate analysis, backwards elimination was utilized to identify relevant characteristics for inclusion in the final multivariable models, with an alpha value of 0.20. Unless otherwise noted, all statistical tests were two-sided, and statistical significance was assessed at the 0.05 level.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
Laracy, J. C., Kamboj, M. & Vardhana, S. A. Long and persistent COVID-19 in patients with hematologic malignancies: from bench to bedside. Curr. Opin. Infect. Dis. 35, 271–279 (2022).
Vijenthira, A. et al. Outcomes of patients with hematologic malignancies and COVID-19: a systematic review and meta-analysis of 3377 patients. Blood 136, 2881–2892 (2020).
Becerril-Gaitan, A. et al. Immunogenicity and risk of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) infection after Coronavirus Disease 2019 (COVID-19) vaccination in patients with cancer: a systematic review and meta-analysis. Eur. J. Cancer 160, 243–260 (2022).
Choi, B. et al. Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host. N. Engl. J. Med. 383, 2291–2293 (2020).
Aydillo, T. et al. Shedding of Viable SARS-CoV-2 after Immunosuppressive Therapy for Cancer. N. Engl. J. Med. 383, 2586–2588 (2020).
Lee, C. Y. et al. Prolonged SARS-CoV-2 Infection in Patients with Lymphoid Malignancies. Cancer Discov. 12, 62–73 (2022).
Borges, V. et al. Long-Term Evolution of SARS-CoV-2 in an Immunocompromised Patient with Non-Hodgkin Lymphoma. mSphere 6, e0024421 (2021).
Choudhary, M. C., Crain, C. R., Qiu, X., Hanage, W. & Li, J. Z. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Sequence Characteristics of Coronavirus Disease 2019 (COVID-19) Persistence and Reinfection. Clin. Infect. Dis.: Off. Publ. Infect. Dis. Soc. Am. 74, 237–245 (2022).
Kemp, S. A. et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 592, 277–282 (2021).
Azeem, M. I. et al. Impaired SARS-CoV-2 Variant Neutralization and CD8+ T-cell Responses Following 3 Doses of mRNA Vaccines in Myeloma: Correlation with Breakthrough Infections. Blood Cancer Discov. 4, 106–117 (2023).
Li, F. F. et al. A novel multiplex electrochemiluminescent immunoassay for detection and quantification of anti-SARS-CoV-2 IgG and anti-seasonal endemic human coronavirus IgG. J. Clin. Virol. 146, 105050 (2022).
Chang, A. et al. Antibody binding and neutralization of live SARS-CoV-2 variants including BA.4/5 following booster vaccination of patients with B-cell malignancies. Cancer Res Commun. 2, 1684–1692 (2022).
Nooka, A. K. et al. Determinants of Neutralizing Antibody Response After SARS CoV-2 Vaccination in Patients With Myeloma. J. Clin. Oncol. 40, 3057–3064 (2022).
Reynolds, C. J. et al. Immune boosting by B.1.1.529 (Omicron) depends on previous SARS-CoV-2 exposure. Science 377, eabq1841 (2022).
Aleman, A. et al. Cellular mechanisms associated with sub-optimal immune responses to SARS-CoV-2 bivalent booster vaccination in patients with Multiple Myeloma. EBioMedicine 98, 104886 (2023).
Wagner, A. et al. Breakthrough Infections in SARS-CoV-2-Vaccinated Multiple Myeloma Patients Improve Cross-Protection against Omicron Variants. Vaccines 12, https://doi.org/10.3390/vaccines12050518 (2024).
Branagan, A. R. et al. Tandem high-dose influenza vaccination is associated with more durable serologic immunity in patients with plasma cell dyscrasias. Blood Adv. 5, 1535–1539 (2021).
Davis-Gardner, M. E. et al. Neutralization against BA.2.75.2, BQ.1.1, and XBB from mRNA Bivalent Booster. N. Engl. J. Med. 388, 183–185 (2023).
Edara, V. V. et al. Infection- and vaccine-induced antibody binding and neutralization of the B.1.351 SARS-CoV-2 variant. Cell Host Microbe 29, 516–521 e513 (2021).
Xie, X. et al. An Infectious cDNA Clone of SARS-CoV-2. Cell Host Microbe 27, 841–848 e843 (2020).
Edara, V. V. et al. mRNA-1273 and BNT162b2 mRNA vaccines have reduced neutralizing activity against the SARS-CoV-2 omicron variant. Cell Rep. Med. 3, 100529 (2022).
Edara, V. V. et al. Infection and Vaccine-Induced Neutralizing-Antibody Responses to the SARS-CoV-2 B.1.617 Variants. N. Engl. J. Med. 385, 664–666 (2021).
Vanderheiden, A. et al. Development of a Rapid Focus Reduction Neutralization Test Assay for Measuring SARS-CoV-2 Neutralizing Antibodies. Curr. Protoc. Immunol. 131, e116 (2020).
Katzelnick, L. C. et al. Viridot: An automated virus plaque (immunofocus) counter for the measurement of serological neutralizing responses with application to dengue virus. PLoS Negl. Trop. Dis. 12, e0006862 (2018).
Acknowledgements
This work was supported in part by grants (NIH P51OD011132, 3U19AI057266-17S1, 1U54CA260563, HHSN272201400004C, NIH/NIAID CEIRR under contract 75N93021C00017 to Emory University) from the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), Emory Executive Vice President for Health Affairs Synergy Fund award, the Pediatric Research Alliance Center for Childhood Infections and Vaccines and Children’s Healthcare of Atlanta, COVID-Catalyst-I3 Funds from the Woodruff Health Sciences Center and Emory School of Medicine, Woodruff Health Sciences Center 2020 COVID-19 CURE Award. This work was also supported in part by NCI U54 Seronet award CA260563. MVD is also partly supported by funds from the NIH R35CA197603 and the SCOR award from LLS. KMD is partly supported by funds from NIH CA238471 and AR077926, and LLS. Authors acknowledge the support of Cancer Tissue and Pathology, Data and Technology Applications, and Biostatistics shared resource of the Winship Cancer Institute of Emory University and NIH/NCI under award number P30CA138292. Funders played no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Contributions
A.M., Collected, analyzed, interpreted data, and wrote the original draft. K.M., M.I.A., M.E., and B.W., Performed assays. A.K.N., R.J.M., J.L.K., C.C.H., N.S.J., and S.L., Writing review and editing. J.M.S., Performed statistical analysis. K.M.D, Resources, formal analysis. M.V.D., and M.S.S., Conceptualization, resources, funding acquisition, writing review, and editing.
Corresponding authors
Ethics declarations
Competing interests
A.M. reports grants from NIH during the conduct of the study. J.L.K. reports grants from NIH during the conduct of the study and personal fees from AbbVie, BMS, Janssen, and Incyte outside the submitted work. S.L. reports personal fees from Amgen, grants and personal fees from Takeda, Janssen, and BMS, personal fees from Pfizer, Genentech, AbbVie, and Celgene outside the submitted work; and Board of Directors TG Therapeutics with stock. M.V.D. personal fees from Sanofi, BMS, and Lava Therapeutics outside the submitted work. M.S.S. reports grants and personal fees from Moderna and Ocugen outside the submitted work. No disclosures were reported by the other authors.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Moreno, A., Manning, K., Azeem, M.I. et al. Divergence of variant antibodies following SARS-CoV-2 booster vaccines in myeloma and impact of hybrid immunity. npj Vaccines 9, 201 (2024). https://doi.org/10.1038/s41541-024-00999-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41541-024-00999-6
