
Abstract
Somatosensory pathways convey crucial information about pain, touch, itch and body part movement from peripheral organs to the central nervous system1,2. Despite substantial needs to understand how these pathways assemble and to develop pain therapeutics, clinical translation remains challenging. This is probably related to species-specific features and the lack of in vitro models of the polysynaptic pathway. Here we established a human ascending somatosensory assembloid (hASA), a four-part assembloid generated from human pluripotent stem cells that integrates somatosensory, spinal, thalamic and cortical organoids to model the spinothalamic pathway. Transcriptomic profiling confirmed the presence of key cell types of this circuit. Rabies tracing and calcium imaging showed that sensory neurons connect to dorsal spinal cord neurons, which further connect to thalamic neurons. Following noxious chemical stimulation, calcium imaging of hASA demonstrated a coordinated response. In addition, extracellular recordings and imaging revealed synchronized activity across the assembloid. Notably, loss of the sodium channel NaV1.7, which causes pain insensitivity, disrupted synchrony across hASA. By contrast, a gain-of-function SCN9A variant associated with extreme pain disorder induced hypersynchrony. These experiments demonstrated the ability to functionally assemble the essential components of the human sensory pathway, which could accelerate our understanding of sensory circuits and facilitate therapeutic development.
Similar content being viewed by others
Main
The ascending somatosensory pathways are responsible for conveying environmental and bodily sensory information from the peripheral organs to the central nervous system. Somatosensory neurons, whose cell bodies are located in the dorsal root ganglia (DRG) and trigeminal ganglia (TG), are pseudo-unipolar cells that innervate peripheral organs, including the skin, to detect somatosensory signals and transmit them to the spinal cord and hindbrain. Neurons in the dorsal spinal cord or hindbrain monosynaptically transmit this information to various brain structures, including the thalamus, that further relay these signals to the cerebral cortex1,2. Genetic or environmental disruptions to these pathways can lead or contribute to various neurological disorders, including chronic pain and autism spectrum disorder, and cause both heightened and diminished sensory functions3,4,5,6. Despite the clinical significance, how ascending sensory pathways form during human development and how functional defects arise in pathological conditions during development are still poorly understood. Challenges in bench to bedside translation of pain and sensory research are thought to be related, at least in part, to differences across species. Crucially, the discovery of new treatments and their translation is hindered by the lack of reliable human experimental models that reconstitute the ascending sensory pathway7. In fact, concomitant probing of all components of these circuits through imaging or recording has not yet been achieved in animal models, which limits our understanding of the consequences of various sensory manipulations at the circuit level.
Recent advances in organoid and assembloid technologies hold promise for generating self-organizing stem-cell-based models to study and manipulate human neural circuits ex vivo in the context of development and disease states. We previously developed three-part assembloids to recapitulate the descending cortico-spino-muscle pathway connecting deep layer cortical glutamatergic neurons to cholinergic spinal cord motor neurons to muscle8. The generation of functional circuits in assembloids with more than three components, which would enable the assembly of the ascending sensory pathway from sensory neurons to the cerebral cortex, has not yet been achieved.
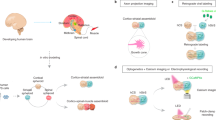
Here we focus on the spinothalamic and trigeminothalamic pathways, which are particularly prominent in humans9 and are crucial for conveying pain and other somatosensory information through the spinal cord and thalamus1. In this circuit, sensory neurons in the DRG or TG connect with neurons in the dorsal spinal cord or hindbrain. These neurons relay information to the thalamus through monosynaptic connections and subsequently to the somatosensory cortex and other cortical areas (Fig. 1a). To reconstitute these pathways from their components, we separately generated somatosensory organoids, dorsal spinal cord organoids, diencephalic (thalamic) organoids and cerebral cortical organoids from pluripotent stem cells and then functionally assembled them to create four-part human ascending somatosensory assembloids (hASAs). We demonstrated that chemical stimulation or glutamate uncaging of sensory neurons elicited responses within hASA. Calcium imaging and electrophysiological recordings of the assembloid revealed synchronized activity across the ascending pathway. Finally, we applied this platform to study circuit-level consequences of loss- and gain-of-function pathogenic variants in the NaV1.7 sodium channel encoded by the SCN9A gene, which have been associated with congenital insensitivity to pain and extreme pain disorder, respectively. We discovered that the loss of NaV1.7 disrupted synchronized activity across the four-part hASA, whereas the gain-of-function mutation resulted in hypersynchrony throughout the sensory pathway in hASA.

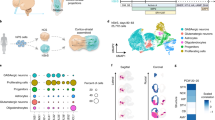
a, Schematic illustrating the key components of the ascending anterolateral sensory pathway. b, Schematic illustrating the generation of hCO, hDiO, hdSpO and hSeO from hiPS cells. c, UMAP visualization of single-cell gene expression of hCO (day 83; n = 8,780 cells from two hiPS cell lines13), hDiO (day 100; n = 12,461 cells from three hiPS cell lines11), hdSpO (days 68–72; n = 20,943 cells from five hiPS cell lines) and hSeO (days 68–72; n = 17,078 cells from four hiPS cell lines). d, UMAP plot coloured by the organoid identity. e, Cell composition in each regionalized neural organoid based on scRNA-seq data. f, Gene expression level for regional identity markers. g, Immunostaining for specific regional markers in hCO, hDiO, hdSpO and hSeO on day 63. h, Schematic illustrating the establishment of an scRNA-seq atlas of human DRG. i, UMAP visualization of scRNA-seq of human DRG (n = 50,813 cells from seven isolated DRG at PCW15). j, Label transfer of the human tissue datasets onto the organoid dataset. UMAP plots were coloured using the prediction score for each brain subregion (top) and organoid identity (bottom). Scale bar, 200 μm (g). CNS, central nervous system; max., maximum; min., minimum; PNS, peripheral nervous system. Credits: b adapted with permission from ref. 11, Elsevier; h adapted from ref. 54, Springer Nature America, Inc.
Generation of key components of the somatosensory pathway
To derive the key parts of the ascending sensory pathway in vitro, we generated regionalized neural organoids from human induced pluripotent stem (hiPS) cells using guided differentiation with small molecules and growth factors (Fig. 1b and Extended Data Fig. 1). We first generated human cortical organoids (hCO) and human diencephalic organoids (hDiO) using previously described and validated methods10,11. To generate human dorsal spinal cord (and hindbrain) organoids (hdSpO), where the soma of spinothalamic (or trigeminothalamic) projection neurons resides, we modified a protocol we have previously reported for deriving the ventral part of cervical spinal cord organoids8 by excluding ventralizing cues (Extended Data Fig. 1). Finally, to derive human somatosensory (also referred to as sensory) organoids (hSeO), which contain somatosensory neurons of the DRG or TG, we developed a protocol that leveraged cues identified in two-dimensional-based methods12 (Extended Data Fig. 1). To verify that we have all key cellular components in the ascending sensory pathway, we combined the single-cell RNA sequencing (scRNA-seq) data of hCO and hDiO that we generated in previous studies11,13 with our newly obtained transcriptional datasets of hdSpO and hSeO (Fig. 1c–e). We found distinct clusters for key cell types in the ascending circuit: cortical glutamatergic neurons (FOXG1+ (ref. 14) and SLC17A7+), thalamic excitatory neurons (TCF7L2+ (ref. 15) and SLC17A6+), dorsal spinal cord and hindbrain projection neurons (HOXB4+ (ref. 16) and PHOX2A+ (ref. 17)) and primary afferent somatosensory neurons (POU4F1+ (ref. 18) and PPP1R1C+ (ref. 19)) (Fig. 1f). We used immunostaining to confirm the protein expression of key region-specific markers in the regionalized organoids (Fig. 1g). To further investigate cell identity in regionalized neural organoids, we performed label transfer analysis20 from a human primary tissue atlas of the whole brain (PCW7–14)21. Given the paucity of publicly available scRNA-seq datasets of developing human DRG22, we procured human primary tissue (at PCW15) and freshly dissected DRG and performed scRNA-seq (Fig. 1h,i and Extended Data Fig. 2a–d). Overall, we found that cells derived from each regionalized organoid resembled the transcriptomic profiles of their corresponding in vivo counterparts (Fig. 1j and Extended Data Fig. 2e).
Characterization of hSeO and hdSpO
To further characterize the cellular identity within hSeO, we inspected the pattern of expression of several canonical markers using scRNA-seq data (Fig. 2a,b and Extended Data Fig. 3a,b). We found that approximately 30–40% of the neurons (STMN2+ (ref. 23)) expressed the peripheral neuron-specific markers POU4F1 and SIX1 (ref. 24) (Fig. 2c), which we independently confirmed in other batches of organoids by reverse transcription–quantitative polymerase chain reaction (RT–qPCR) (Extended Data Fig. 3c). Co-localization of VGLUT2 and NeuN in immunostaining experiments demonstrated the excitatory neuronal identity of hSeO neurons (Extended Data Fig. 3d). We found a substantial proportion of cells expressing neural crest cell markers, such as SOX10 and FOXD3, which is consistent with the neural crest origin of peripheral sensory neurons25 (Fig. 2b,c and Extended Data Fig. 3b,e). We also examined the expression levels of several key sensory receptors26,27,28 (P2RX3, TRPV1, TRPM8, TRPA1, PIEZO1 and PIEZO2) (Fig. 2d and Extended Data Fig. 3f). To further characterize the cellular composition of hSeO, we compared the single-cell transcriptome of hSeO with the scRNA-seq data of the human DRG. Integrated uniform manifold approximation and projection (UMAP) and cell composition proportions revealed similar cellular compositions, except for endothelial cells, macrophages and blood cells, which were absent in hSeO (Extended Data Fig. 3g–k).

a, Generation of hSeO from hiPS cells and scRNA-seq analysis. b, UMAP of scRNA-seq of hSeO on days 68–72 (n = 20,072 cells; the same dataset as in Fig. 1 with optimized QC criteria). c, UMAP of cluster-specific markers. d, Expression of key sensory receptors. e, Calcium imaging for pharmacological studies. f, GCaMP6s expression in hSeO. g, ΔF/Fbase traces showing αβ-MeATP and capsaicin-evoked calcium responses. h, Mean ΔF/Fbase values at baseline and after αβ-MeATP (n = 88 neurons; seven hSeO; three differentiations) and capsaicin (n = 205 cells; ten hSeO; three differentiations) on days 54–82. Wilcoxon matched-pair signed-rank test; ****P < 0.0001. i, Calcium imaging following TNP–ATP and αβ-MeATP exposure of ex vivo mouse DRG and hSeO. j, Calbryte-incubated mouse DRG and GCaMP6s-expressing hSeO. k, Response after αβ-MeATP and TNP–ATP exposure of mouse DRG and hSeO. l,m, Responses after exposure to αβ-MeATP with various TNP–ATP concentrations of mouse DRG (l) (n = 518, 605, 652 and 1,392 cells from three, three, three and four mouse DRG, respectively) and hSeO (m) (n = 110, 99, 50 and 77 cells from three, four, six and four hSeO; two differentiations on days 75–82). Kruskal–Wallis test with Dunn’s test; ****P < 0.0001. n, Generation of hdSpO from hiPS cells and scRNA-seq analysis. o, UMAP of scRNA-seq of hdSpO on days 68–72 (n = 23,718 cells; the same dataset as in Fig. 1 with optimized QC criteria). p, UMAP of cluster-specific markers. q, UMAP showing dorsoventral clusters (circle; dI5 cluster). r, Label transfer of human spinal cord datasets onto hdSpO (UMAP coloured using dI5 prediction score). Data: mean ± s.e.m. Small dots, cells; large dots, organoids or mouse DRG. NS, not significant. Scale bars, 150 μm (f (top)), 20 μm (f (bottom)), 100 μm (j). Pct. exp., percentage expression; avr. exp., averaged expression. Credits: a,n adapted with permission from ref. 11, Elsevier; e,i adapted from ref. 48, Springer Nature America, Inc.
A unique feature of the sensory neurons is their ability to respond to environmental stimuli. To verify whether neurons in hSeO respond to sensory stimuli, we established a live imaging system of intact three-dimensional (3D) organoids expressing a genetically encoded calcium indicator using a chamber equipped to inject chemicals. Consistent with the high abundance of mRNA encoding calcium-permeable P2X purinoceptor 3 (P2RX3) and capsaicin receptor TRPV1 (Fig. 2d), bath application of their agonists, α,β-methyleneATP (αβ-MeATP)26 and capsaicin29, respectively, induced calcium transients (Fig. 2e–h). Interestingly, the dynamics of the responses were similar between hSeO and intact ex vivo primary mouse DRG (Extended Data Fig. 4a–d); αβ-MeATP induced a fast calcium spike, whereas capsaicin induced a slow and prolonged calcium transient, which is consistent with previous pharmacological studies using DRG neurons isolated from rat30,31 or mouse32,33, as well as induced nociceptors in two-dimensional culture34.
There are species-specific differences in the expression and function of some receptors on sensory DRG neurons, which may be responsible for sensory processing differences and challenges in translation from animal models to humans7. For instance, 2′,3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate (TNP–ATP) is a potent antagonist of the rodent P2X3 receptor35; however, primate-specific amino acid substitutions in the P2X3 receptor are thought to modify sensitivity to TNP–ATP in the macaque DRG36. Consistent with previous reports36, we found that TNP–ATP successfully blocked calcium responses in ex vivo mouse DRG in a dose-dependent manner. However, when applied to human sensory neurons in hSeO, TNP–ATP had no significant effect (Fig. 2i–m), which suggests differences in responsiveness across species. These results are consistent with a previous report showing the importance of primate-specific amino acid changes at two residues of the P2X3 receptor (A197 and T202), which influence the TNP–ATP antagonism36.
We further investigated the cellular identity of neurons in hdSpO using scRNA-seq data (Fig. 2n,o and Extended Data Fig. 5a). Similar to the ventral part of the spinal cord organoids8, hdSpO displayed high neuronal diversity (Fig. 2o,p and Extended Data Fig. 5a–c). Relevant to the ascending somatosensory pathway, we found clusters expressing the projection neuronal markers PHOX2A17, LBX1 (ref. 17) and TACR1 (encoding NK1R)37 (Fig. 2p and Extended Data Fig. 5b). We also confirmed the expression of TACR1 and LBX1 by RT–qPCR in hdSpO (Extended Data Fig. 5d). We next annotated the dorsoventral neuronal clusters in hdSpO using combinatorial marker expression, as described previously38,39 (Extended Data Fig. 5e). We found overlap of PHOX2A+ neurons with dI5 cluster, which is consistent with previous reports17,40 (Fig. 2q). We also performed label transfer analysis of scRNA-seq data from the developing human spinal cord dI5 cells38 onto the hdSpO scRNA-seq dataset. We found that the SLC17A6+PHOX2A+TACR1+ neuron cluster demonstrated the highest prediction score values for the developing human dI5 cells, indicating that hdSpO contains a neuronal cluster resembling the transcriptomic identity of human dI5 cells (Fig. 2r). These cells also express another dI5 marker gene, LMX1B40, but not the dI4/6 marker gene PAX2 (ref. 41) (Extended Data Fig. 5b,e). Immunostaining for PAX2 and PHOX2A indicated that the distinct types of spinal neurons in hdSpO were intermingled without a clear spatial organization (Extended Data Fig. 5f). Taken together, these transcriptomic and functional studies indicate that these regionalized organoids contain key cellular components of the developing human ascending sensory pathway.
Projections in hSeO–hdSpO and hdSpO–hDiO
We next examined the specificity of projections between pairs of regionalized organoids. First, we investigated whether the sensory neurons in hSeO could send projections to spinal neurons in hSeO–hdSpO assembloids. We virally labelled hSeO with hSYN1::EYFP and integrated them with hdSpO to form hSeO–hdSpO assembloids (Fig. 3a). We found that the area of EYFP fluorescence on the hdSpO side, corresponding to projections from hSeO neurons, increased progressively over 28 days after fusion (daf) (Fig. 3b,c). These EYFP+ sensory projections co-localized with the axonal marker NF-H (Extended Data Fig. 6a). We next used rabies virus-mediated retrograde tracing combined with Cre recombination in hSeO–hdSpO assembloids (Fig. 3d). On daf 21, we observed tdTomato+GFP+ cells on the hSeO side, indicating successful retrograde tracing of rabies–Cre–GFP resulting in the recombination and expression of DIO-tdTomato (Fig. 3e). These tdTomato+GFP+ cells were not detected in unassembled organoids, which were kept in the same cell culture well and media, confirming projection-dependent retrograde transmission (Extended Data Fig. 6b–d). To investigate the specificity of these projections, we co-immunostained for the sensory neuronal marker BRN3A and found that nearly 80% of tdTomato+ cells were also BRN3A+ (Fig. 3f,g). To further verify functional connectivity between hSeO and hdSpO, we combined optogenetic stimulation with whole-cell electrophysiological recordings. Upon 460-nm light stimulation of channelrhodopsin 2-expressing hSeO, we observed optically evoked excitatory postsynaptic currents (oEPSCs), with a success rate of eight of 30 recorded cells (Fig. 3h,i and Extended Data Fig. 6e,f). Responsive neurons exhibited an average oEPSC amplitude of 52 pA and an onset delay of 32 ms (Extended Data Fig. 6f). These results demonstrate that hSeO sensory neurons can form functional connections with neurons in hdSpO. By contrast, when we generated assembloids by integrating hSeO with hCO, we observed only minimal projections from hSeO that did not increase over time (Extended Data Fig. 6g–i).

a, Generation of hSeO–hdSpO assembloids. b, Gradual increase in sensory–spinal cord projections in hSeO–hdSpO. c, Quantification of EYFP in the hdSpO side of assembloids (n = 10 assembloids; three differentiations; Kruskal–Wallis test with Dunn’s test; *P = 0.0267). d, Retrograde rabies viral tracing in hSeO–hdSpO. e, EGFP+tdTomato+ cells in hSeO side. f, BRN3A+tdTomato+ co-localization in hSeO side. g, Percentage of BRN3A+ cells of tdTomato+ or Hoechst+ cells in hSeO side of assembloids (n = 6 assembloids; three differentiations; Mann–Whitney test; **P = 0.0022). h, Patch-clamp recordings with optogenetic stimulation of hSeO neurons. i, oEPSC traces from three trials in the same neuron. j, Generation of hdSpO–hDiO assembloids. k, Gradual increase in spinothalamic projections in hdSpO–hDiO. l, Quantification of EYFP in the hDiO side of assembloids (n = 10–11 assembloids; three differentiations; Kruskal–Wallis test with Dunn’s test; **P = 0.0050; ***P = 0.0003). m, Retrograde rabies viral tracing in hdSpO–hDiO. n, EGFP+tdTomato+ cells in the hdSpO side of assembloids. o, NK1R+tdTomato+ co-localization in the hdSpO side of assembloids. p, Percentage of NK1R+ cells of tdTomato+ or Hoechst+ cells in the hdSpO side of assembloids (n = 6 assembloids; three differentiations; Mann–Whitney test; **P = 0.0022). q, Calcium imaging of spinothalamic neurons with retrograde viral tracing in hSeO–hdSpO–hDiO. r, GCaMP6f+dsRedExpress+ co-expression in the hdSpO side of assembloids. s, αβ-MeATP-evoked responses and blockade by NBQX and APV (50 μM each). t, Responses to αβ-MeATP with or without NBQX and APV (n = 74 cells; four assembloids; paired t-test; ****P < 0.0001) on daf 70–88. Data: mean ± s.e.m. Small dots, cells; large dots, assembloids. Scale bars, 100 μm (b,e (left),k,n (left)), 50 μm (n (right),r (top)), 30 μm (e (right)), 20 μm (f,o), 10 μm (r (bottom). Credits: a,d,h,j,m,q adapted from ref. 48, Springer Nature America, Inc.
We took a similar approach to investigate spinothalamic projections in hdSpO–hDiO assembloids (Fig. 3j). We observed a significant increase in spinothalamic projections on daf 28 (Fig. 3k,l). Using rabies virus-mediated retrograde tracing (Fig. 3m,n), we found that approximately 40% of tdTomato+ cells co-expressed the spinal cord projection neuronal marker NK1R+ (Fig. 3p). We also observed co-localization of tdTomato and PHOX2A (Extended Data Fig. 6l). In addition, we examined whether hdSpO neurons preferentially project to hDiO compared to hSeO by using three-part hSeO–hdSpO–hDiO assembloids. Quantification of EYFP+ spinal projections within the same assembloid, in both the hDiO and hSeO regions, revealed more projections in hDiO, indicating a preferential for hdSpO targeting the hDiO (Extended Data Fig. 6k–m). These studies, in combination with our previous experiments on thalamocortical projections in hCO–hDiO assembloids11, confirmed that the three pairs of assembloids (hCO–hDiO, hDiO–hdSpO and hdSpO–hSeO) built from the four regionalized neural organoids intrinsically recapitulate some of the basic inter-regional projections.
We next investigated the functionality of these connections in three-part hSeO–hdSpO–hDiO assembloids by combining rabies virus-mediated retrograde tracing, recombinase Flpo-dependent expression of GCaMP6f and live calcium imaging following αβ-MeATP treatment (Fig. 3q). We assembled AAV-EF1α::fDIO-GCaMP6f-infected hdSpO with hSeO to generate hSeO–hdSpO. On daf 43, we infected hDiO with rabies–Flpo–dsRedExpress and AAV-G and fused these onto the hdSpO side of the hSeO–hdSpO assembloid. On daf 70, we detected expression of fDIO-GCaMP6f, which is suggestive of transmission of rabies–Flpo–dsRedExpress from hDiO into hdSpO (Fig. 3r). These GCaMP6f+ neurons demonstrated increased calcium activity after αβ-MeATP exposure, thereby indicating signal transmission from sensory neurons to spinal neurons (Fig. 3s,t). These calcium transients were blocked by the glutamate receptor antagonists 2,3-dioxo-6-nitro-7-sulfamoylbenzo[f]quinoxaline (NBQX) and 2-amino-5-phosphonovaleric acid (APV) (Fig. 3s,t). These results imply that chemical stimulation of sensory neurons in hSeO elicited calcium activity that was transmitted to and induced calcium increase in connected rabies–Flpo-labelled spinothalamic projection neurons in hdSpO. Moreover, hdSpO neurons projected axons into hDiO, as evidenced by rabies tracing.
Functional assembly of four-part hASA
Next, we generated hASA by linearly placing the four key neural regionalized organoids in a tilted cell culture plate (Fig. 4a,b). We measured success assembly rate and evaluated the shapes of assembloids. All samples were successfully assembled, with 87.7% forming linear or curved shapes, which are suitable for functional assays (Extended Data Fig. 7a). We performed viral labelling of each organoid with distinct hSYN1 promoter-driven fluorescent proteins before fusion and confirmed maintenance of distinct domains of these assembloids (Fig. 4c). To verify cellular composition after assembly, we performed scRNA-seq from hASAs and stage- and cell line-matched individual organoids on days 162–229 and found that the composition was comparable between individual organoids and four-part assembloids (Extended Data Fig. 7e,f).

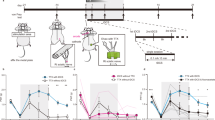
a, Generation of hASA. b,c, Bright-field (b) and fluorescent image (c) of hASA. d,e, Calcium imaging (d) and traces (e) after αβ-MeATP exposure. f, Responses following αβ-MeATP exposure, compared to null value (0), in organoids (hCO, n = 83 cells; hDiO, n = 52 cells; hdSpO, n = 84 cells; hSeO, n = 131 cells from three to four organoids; two differentiations) and assembloids (hCO, n = 91 cells; hDiO, n = 116 cells; hdSpO, n = 98 cells; hSeO, n = 95 cells from six assembloids; one differentiation) on daf 74–78. Wilcoxon signed-rank test; ****P < 0.0001. g,h, Glutamate uncaging after ultraviolet application to hSeO (g) and ΔF/Fbase traces (h). i, Response before and after ultraviolet exposure (hCO, n = 113 cells; hDiO, n = 157 cells; hdSpO, n = 159 cells; hSeO, n = 109 cells; five assembloids; one differentiation; Wilcoxon matched-pair signed-rank test; ****P < 0.0001) on daf 74–99. j, Calcium imaging. k,l, jGCaMP8s expression, spontaneous activity, SCA correlograms (hSeO–hCO pair) and peak percentage correlations in organoids (k) and assembloids (l). Asterisks; synchronous events. m, Correlation values in organoids (O; hCO, n = 96 cells; hDiO, n = 61 cells; hdSpO, n = 63 cells; hSeO, n = 47 cells; seven organoids; three differentiations) and assembloids (A; hCO, n = 208 cells; hDiO, n = 157 cells; hdSpO, n = 156 cells; hSeO, n = 110 cells; ten assembloids; three differentiations) on daf 74–131. Mann–Whitney test; ****P < 0.0001. n,o, Four-part extracellular recordings (n) and traces (o). Asterisks; synchronous events. p, Representative cross-correlograms. q, Co-activation index for hSeO–hdSpO, hSeO–hDiO and hSeO–hCO pairs (n = 223, 180 and 222 channels, respectively; seven to eight assembloids; four differentiations; Wilcoxon matched-pair signed-rank test; ****P < 0.0001) on daf 80–99. Data: mean ± s.e.m. Small dots, cells or channels; large dots, assembloids. Scale bars, 1 mm (b), 500 μm (c), 400 μm (k,l). Veh., vehicle. Credits: a,d,g,j adapted from ref. 48, Springer Nature America, Inc.; n adapted with permission from ref. 11, Elsevier.
We examined functional connectivity within hASA. In one continuous imaging session, we serially treated hASA with either vehicle or αβ-MeATP while recording jGCaMP8s signal at single-cell resolution (Fig. 4d). We calculated the adjusted ΔF/Fbase by subtracting the response to vehicle from the response to αβ-MeATP. We found that αβ-MeATP exposure significantly elevated the calcium levels in hASA neurons in all four regions on daf 74–78 (Fig. 4e,f and Supplementary Video 1). By contrast, in non-assembled organoids, only neurons in hSeO had a significant increase in calcium after the application of αβ-MeATP, whereas neurons in other organoids did not (Fig. 4e,f). To further verify whether specific stimulation of sensory neurons in hSeO elicited responses in other regions of the assembloid, we used 405-nm photostimulation in the presence of 4-methoxy-7-nitroindolinyl (MNI) caged glutamate to trigger rapid and localized release of glutamate in a specific area (Fig. 4g). We performed photostimulation of hSeO within hASA and detected a robust increase in calcium levels, not only in hSeO but also in all other regions, on daf 74–99 (Fig. 4h,i). These experiments suggest transmission of somatosensory signals within the assembloid from sensory neurons through spinal and thalamic neurons to cortical neurons.
We next explored whether the four-part assembly is associated with emergent network properties. We recorded baseline calcium activity at single-cell resolution and compared hASA with four non-assembled organoids placed in close proximity just before imaging (Fig. 4j–l). Notably, we found that neurons in assembloids exhibited synchronous activity across all four regions (Fig. 4l and Supplementary Video 2). We applied scaled correlation analysis (SCA) to minimize the effects of low-frequency noise signals42, which was established in our recent study to quantify synchronous activity across neural circuits43. Representative SCA correlograms from all possible pairs between hSeO and hCO neurons in an assembloid showed that the correlation peaks were distributed around 0 s, whereas representative SCA correlograms from unfused organoids had equal distribution of correlation peaks (Fig. 4k,l). The peak correlation for all combinations of the four regions was significantly higher in assembloids than that in individual non-assembled neural organoids (Fig. 4m).
We further investigated activity patterns through concomitant extracellular recordings with 32 electrodes in each of the four hASA parts (Fig. 4n). We found spontaneous and synchronous activity across the entire assembloid (Fig. 4o). We calculated the co-activation indexes based on cross-correlograms44 (Fig. 4p). When we compared the co-activation index aligned with the activity of hSeO, we found significantly higher values than alignment to random time points (Fig. 4q). Taken together, these results demonstrate that the assembly of intact 3D human regionalized neural organoids resembling the human sensory ganglia, dorsal spinal cord, thalamus and cerebral cortex enables the formation of connections capable of synchronized activity across the pathway and in response to somatosensory like (chemical sensory) stimuli.
SCN9A pathogenic variants alter synchrony in hASA
Finally, we studied whether hASA could be used to identify functional defects in models of neurologic and psychiatric disorders. Pathogenic variants in the gene SCN9A, which encodes the voltage-gated sodium channel NaV1.7, cause pain insensitivity if the variant leads to loss of channel function, or hyperalgesia if the variant causes gain of function6 (Fig. 5a,i). Although defects in channel conductance and excitability of sensory neurons have been proposed as disease mechanisms in cellular and animal models6,45,46,47, the circuit-level consequences have not yet been explored in a human model.

a, SCN9A loss causes clinical absence of pain. b, RT–qPCR of SCN9A (hCO, n = 8 samples; hDiO, n = 8 samples; hdSpO, n = 6 samples; hSeO, n = 10 samples; four to six differentiations; one-way analysis of variance (ANOVA) (F3,28 = 2.691) with Tukey’s test; ***P = 0.0003 for hdSpO versus hSeO; ***P = 0.0010 for hDiO versus hSeO; ****P < 0.0001) on days 45–100. c, Generation of SCN9A KO hSeO. d, Western blot of NaV1.7 (control, n = 7 samples; KO, n = 5 samples; two differentiations; unpaired t-test; **P = 0.0011) on days 48–69. e, Calcium imaging. f,g, jGCaMP8s expression and spontaneous calcium activity in control (f) and SCN9A KO (g) assembloids. h, Correlation values in control (hCO, n = 182 cells; hDiO, n = 199 cells; hdSpO, n = 211 cells; hSeO, n = 218 cells; ten assembloids; three differentiations) and KO (hCO, n = 179 cells; hDiO, n = 202 cells; hdSpO, n = 219 cells; hSeO, n = 208 cells; 12 assembloids; three differentiations) on daf 74–111. Mann–Whitney test; ***P = 0.0005; ****P < 0.0001. i, SCN9A gain-of-function mutation causes excessive pain. j, Generation of SCN9A T1464I hiPS cells. k, Sanger sequencing of hiPS cells. l, Calcium imaging. m,n, jGCaMP8s expression and spontaneous activity in control (m) and SCN9A T1464I (n) assembloids. o, Correlation values in control (hCO, n = 76 cells; hDiO, n = 61 cells; hdSpO, n = 60 cells; hSeO, n = 96 cells; seven assembloids; two differentiations) and T1464I (hCO, n = 97 cells; hDiO, n = 92 cells; hdSpO, n = 81 cells; hSeO, n = 145 cells; eight assembloids; two differentiations) on daf 98. Mann–Whitney test; ***P = 0.0005; ****P < 0.0001. Data: mean ± s.e.m. Small dots, cells; large dots, qPCR, western blot samples or assembloids. Scale bars, 400 μm. Credits: c,e,j,l adapted from ref. 48, Springer Nature America, Inc.
We first verified the expression of SCN9A and found high levels in hSeO (Fig. 5b). We then applied CRISPR-mediated gene editing to induce one or more frameshift variants in the SCN9A gene (Fig. 5c). We confirmed that hSeO derived from pooled SCN9A knockout (KO) iPS cell lines exhibited decreased SCN9A mRNA and NaV1.7 protein levels (Fig. 5d and Extended Data Fig. 8a). The expression of the sensory neuron marker POU4F1 (BRN3A) was not affected by SCN9A KO (Extended Data Fig. 8b–d). Consistent with previous reports of hypo-excitability of sensory neurons in the absence of this key sodium channel45, we found that SCN9A KO hSeO displayed reduced frequency of spontaneous calcium activity (Extended Data Fig. 8e,f). We then generated hASA from isogenic control and SCN9A KO hiPS cells to explore the circuit-level consequences. We recorded spontaneous activity patterns using jGCaMP8s in all regions from control and SCN9A KO hASA (Fig. 5e). We discovered that the synchronous pattern of activity across hASA was reduced in SCN9A KO, although non-synchronized events were found in all regions (Fig. 5f,g and Supplementary Videos 3 and 4). We then performed SCA analysis and found a significant reduction in correlation, indicating that SCN9A KO impaired the emergent synchronization across the assembloid (Fig. 5h and Extended Data Fig. 8g).
In addition, we investigated the circuit-level consequences of the T1464I SCN9A gain-of-function variant, which is associated with paroxysmal extreme pain disorder47 (Fig. 5i). We applied CRISPR–Cas9 gene editing to introduce the heterozygous SCN9A T1464I into a hiPS cell line (Fig. 5j). We confirmed mixed gDNA of control and T1464I SCN9A by Sanger sequencing (Fig. 5k). We then generated hSeO from control and SCN9A T1464I hiPS cells and confirmed that POU4F1 expression level was not altered by genetic engineering (Extended Data Fig. 8h). Calcium activity imaging of control and SCN9A T1464I hSeO demonstrated that the gain-of-function mutation produced hyperexcitability of sensory neurons, which is consistent with previous reports6,47 (Extended Data Fig. 8i,j). To investigate the effects of SCN9A T1464I variant on the emergent synchronized activity of the sensory ascending pathway, we derived hASA from control and SCN9A T1464I hiPS cell lines and imaged spontaneous calcium activity (Fig. 5l). Interestingly, SCA analysis revealed higher peak correlation values across the pairs in hASA, suggesting that SCN9A T1464I resulted in hypersynchrony (Fig. 5m–o and Extended Data Fig. 8k). These results highlight the capability of hASA to capture circuit-level dysfunction associated with the pathogenetic variants of SCN9A and thus its utility to model aberrations in emergent properties within human sensory circuits.
Discussion
Reconstructing the ascending sensory pathway and circuits ex vivo holds significant promise for understanding the mechanisms involved in human sensory system development and thereby unravelling the pathophysiology of sensory-related disorders. We previously demonstrated the potential of assembloids composed of regionalized neural organoids derived from hiPS cells to model circuits in vitro8,48,49. Despite previous efforts to derive neural organoids containing sensory neurons22,50,51, a multiregional assembloid model recapitulating the human somatosensory ascending pathway has not yet been achieved. Here we leveraged this assembloid approach to generate a four-component ascending sensory circuit going from peripheral sensory neurons to cortical neurons. This hASA is an in vitro model encompassing the major components of the human sensory ascending pathway, and includes peripheral sensory neurons, dorsal spinal cord (and hindbrain) projection neurons, thalamic neurons and glutamatergic cortical neurons. This allowed us to visualize transmission of chemically triggered calcium signals through the network following sensory stimulus application and to capture emergent properties of the sensory pathway that cannot be seen in individual organoids, such as synchronous activity across the four parts of the assembloid. We also discovered that SCN9A loss resulted in reduced synchronous activity, whereas gain-of-function SCN9A variant induced increased synchrony across the pathway. Therefore, hASA holds great promise for addressing disease pathophysiology and testing therapeutic approaches. A recent study identified that spontaneous synchronized activity has a crucial role for fine-scaling circuit assembly in neonatal mice, highlighting the importance of synchronized activity during circuit development52. Notably, hASA can also capture human-specific aspects of the sensory system, such as reduced responsiveness to TNP–ATP. Going forward, this multicellular platform can be applied to model a wide range of disorders, including sensory dysfunction in neurodevelopmental disorders or hyperalgesia, and could be helpful for drug screening.
Improvements at several levels would enhance the utility of hASA. First, hiPS cell-derived sensory neurons are still limited in receptor expression and thus in the repertoire of responses to sensory stimulation, which is partially attributable to their developmental stage53. Future studies could improve the specification and maturation of sensory neurons, include other neurons in this pathway and functionally advance this model to study postnatal stages. Future efforts to develop comprehensive transcriptomic atlases of TG and DRG in the developing human will help with further elucidating the molecular identity of sensory neurons. Second, there are several neural pathways involved in sensory perception in addition to the anterolateral spinothalamic tract, which our current hASA models. We envision that these other pathways can be built in parallel with hASA or within the same system. Further studies should be conducted to investigate in detail the specificity of connectivity in these assembloids, including within individual organoids, and the role of activity-dependent processes in establishing and refining these neural networks. These efforts could ultimately be used to dissect the intrinsic rules for establishing connectivity in early human circuits, as well as suggest strategies for accelerating maturation in these in vitro models.
Overall, this new platform holds potential to yield insights into how the human sensory pathways assemble and therefore to develop and screen therapeutics for pain and other sensory system-related disorders.
Methods
Characterization and maintenance of hiPS cells
The hiPS cell lines used in this study were validated using methods described in previous studies. Genome-wide single-nucleotide polymorphisms (SNP) genotyping was performed using the Illumina genome-wide GSAMD-24v2-0 SNP microarray at the Children’s Hospital of Philadelphia. Cultures were tested for and maintained Mycoplasma free. A total of five control hiPS cell lines derived from fibroblasts collected from five healthy participants were used for experiments13 (Supplementary Table 1). Two of the control hiPS cell lines were used to generate SCN9A KO hiPS cell lines, and one control hiPS cell line was used to generate the SCN9A gain-of-function hiPS cell line. Approval for experiments was obtained from the Institutional Review Board panel of Stanford University. Informed consent was obtained from all participants.
Human primary tissue
Human brain tissue samples were obtained under a protocol approved by the Research Compliance Office of Stanford University. PCW15 tissue was delivered on ice, and DRG were dissected immediately after arrival.
Generation of organoids (hCO, hDiO, hdSpO and hSeO) from hiPS cells
For neural differentiation, hiPS cells were cultured on vitronectin-coated plates (A14700; Life Technologies) in Essential 8 Medium (A1517001; Life Technologies). Cells were passaged every 4–5 days with UltraPure 0.5 mM EDTA, pH 8.0 (15575; Thermo Fisher Scientific). For the generation of regionalized neural organoids, hiPS cells were incubated with Accutase (AT104; Innovative Cell Technologies) at 37 °C for 7 min and dissociated into single cells. Optionally, 1–2 days before organoid formation, hiPS cells were exposed to 1% dimethylsulfoxide (472301; Sigma-Aldrich) in Essential 8 Medium. For aggregation into organoids, about 3 × 106 single hiPS cells were seeded per AggreWell 800 plate well in Essential 8 Medium supplemented with the Rho-associated coiled-coil containing kinases (ROCK) inhibitor Y27632 (10 µM; S1049; Selleckchem), centrifuged at 100g for 3 min and then incubated at 37 °C in 5% CO2. On the next day (day 1), organoids consisting of approximately 10,000 cells were collected and transferred into ultra-low attachment plastic dishes (3262; Corning) in Essential 6 Medium (A1516401; Thermo Fisher Scientific) supplemented with patterning molecules, as shown in Extended Data Fig. 1.
The hCO were generated as described previously10,55. For days 1–6, Essential 6 Medium was changed every day and supplemented with dorsomorphin (2.5 µM; P5499; Sigma-Aldrich) and SB-431542 (10 µM; 1614; R&D Systems), and was changed every day. On day 7, the organoids were transferred to neural medium containing Neurobasal-A Medium (10888022; Thermo Fisher Scientific), B-27 Supplement, minus vitamin A (12587010; Thermo Fisher Scientific), GlutaMAX Supplement (1:100; 35050079; Thermo Fisher Scientific), penicillin–streptomycin (1:100; 15070063; Thermo Fisher Scientific), supplemented with FGF2 (20 ng ml−1; 233-FB; R&D Systems) and EGF (20 ng ml−1; 236-EG; R&D Systems) until day 22. From days 23 to 46, the neural medium was supplemented with BDNF (20 ng ml−1; 450-02; PeproTech), NT3 (20 ng ml−1; 450-03; PeproTech), l-ascorbic acid 2-phosphate trisodium salt (200 µM; 323-44822; Wako), N6, 2′-O-dibutyryladenosine 3′,5′-cyclic monophosphate sodium salt (cAMP; 50 μM; D0627; MilliporeSigma) and cis-4, 7, 10, 13, 16, 19-docosahexaenoic acid (10 μM; D2534; MilliporeSigma). From day 47, a neural medium containing Neurobasal-A Medium was used for changes in the medium (every 4 days).
The hDiO were generated as described previously11. For days 1–6, Essential 6 Medium was changed every day and supplemented with dorsomorphin and SB-431542. On day 5, the medium was additionally supplemented with 1 μM CHIR (S1263; Selleckchem). On day 7, organoids were maintained in a neural medium supplemented with CHIR. On day 9 of differentiation, the neural medium was supplemented with 100 nM SAG (days 9–15; 566660-1MG; MilliporeSigma), in addition to the CHIR. Furthermore, on days 12–18 of differentiation, organoids were supplemented with 30 ng ml−1 of BMP7 (120-03P; PeproTech), in addition to the compounds described above. From days 19 to 46, the neural medium was supplemented with BDNF, NT3, l-ascorbic acid 2-phosphate trisodium salt, cAMP and cis-4, 7, 10, 13, 16, 19-docosahexaenoic acid. For days 19–25, DAPT (2.5 μM, 72082; STEMCELL Technologies) was added. From day 47, a neural medium containing Neurobasal-A Medium was used for changes in the medium (every 4 days).
To generate hdSpO, from days 1 to 6, organoids were maintained in Essential 6 Medium supplemented with SB-431542 and dorsomorphin. From days 5 to 6, CHIR (3 μM) was added. On day 7, organoids were transferred to a neural medium supplemented with EGF, retinoic acid (0.1 μM; R2625; Sigma-Aldrich) and CHIR (3 μM; S1263; Selleckchem). On day 22, the medium was supplemented with BDNF, cAMP, l-ascorbic acid 2-phosphate trisodium salt, insulin-like growth factor 1 (IGF-1) (10 ng ml−1; 100-11; PeproTech) until the end of experiments. From days 22 to 28, DAPT was added.
To generate hSeO, from days 1 to 6 in suspension, organoids were maintained in Essential 6 Medium supplemented with SB-431542. From days 1 to 4, BMP4 (5 ng ml; 120-05ET; PeproTech) was added. From days 3 to 5, CHIR (600 nM) was added. On day 6 in suspension, the organoids were transferred to a neural medium supplemented with SB-431542. From day 9, BDNF, GDNF (25 ng ml−1; 450-10; PeproTech) and NGF (25 ng ml−1; 450-01; PeproTech) were supplemented until the end of the experiment. From days 9 to 14, the medium was supplemented with SB-431542. From days 15 to 20, DAPT was included.
Cryosection and immunocytochemistry
Organoids and assembloids were fixed in 4% paraformaldehyde/phosphate-buffered saline (PBS) overnight at 4 °C. They were then washed in PBS and transferred to 30% sucrose/PBS for 2–3 days until the organoids/assembloids sank into the solution. Subsequently, they were rinsed in optimal cutting temperature (OCT) compound (Tissue-Tek OCT Compound 4583; Sakura Finetek) and 30% sucrose/PBS (1:1), embedded and snap-frozen using dry ice. For immunofluorescence staining, 30-µm-thick sections were cut using a Leica CM1860 cryostat. Cryosections were washed with PBS to remove excess OCT compound from the sections and blocked in 10% normal donkey serum (NDS, S30-100ML; MilliporeSigma), 0.3% Triton X-100 (T9284-100ML; MilliporeSigma) and 1% BSA diluted in PBS for 1 h at room temperature. The sections were then incubated overnight at 4 °C with primary antibodies diluted in PBS containing 2% NDS and 0.1% Triton X-100. PBS was used to wash the primary antibodies, and the cryosections were incubated with secondary antibodies in PBS with the PBS containing 2% NDS and 0.1% Triton X-100 for 1 h. The following primary antibodies were used for staining: anti-FOXG1 (rabbit; M227; 1:400 dilution; Takara), anti-TCF7L2 (rabbit; 2569S; 1:200 dilution; Cell Signaling Technology), anti-PHOX2A (rabbit; ab155084; 1:200 dilution; Abcam), anti-BRN3A (mouse; MAB1585; 1:200 dilution; Sigma), anti-mCherry (goat; orb153320; 1:1,000 dilution; Biorbyt), anti-NK1R (rabbit; S8305; 1:200 dilution; Sigma), anti-VGLUT2 (mouse; MAB5504; 1:200 dilution; Millipore), anti-NF-H (mouse; NE1023; 1:200 dilution; Millipore) and anti-PAX2 (goat; AF3364; 1:200 dilution; R&D). The following secondary antibodies were used for staining: Alexa Fluor 647 AffiniPure donkey anti-rabbit IgG (H&L) (711-605-152; 1:1,000 dilution; Jackson ImmunoResearch), Alexa Fluor 568 donkey anti-goat IgG (H&L) (A11057; 1:1,000 dilution; Thermo Fisher Scientific), Alexa Fluor 568 donkey anti-rabbit IgG (H&L) (A10042; 1:200 dilution; Thermo Fisher Scientific), Alexa Fluor 647 donkey anti-mouse IgG (H&L) (A31571; 1:200 dilution; Thermo Fisher Scientific) and Alexa Fluor 568 donkey anti-mouse IgG (H&L) (A10037; 1:1,000 dilution; Thermo Fisher Scientific). Nuclei were visualized with the Hoechst 33258 dye (H3549; 1:10,000 dilution; Life Technologies). Cryosections were mounted for microscopy on glass slides using Aqua-Poly/Mount (18606; Polysciences) or VECTASHIELD (H-1000; Vector Laboratories) and imaged on a confocal microscope. Images were processed and analysed using Fiji (v.2.14.0) and Imaris (Oxford Instruments).
Real-time qPCR
Three to five organoids were collected in the same tube and considered as one sample. The RNA from samples was isolated using the RNeasy Plus Mini kit (74136; QIAGEN). Template cDNA was prepared by reverse transcription using the SuperScript III First-Strand Synthesis SuperMix for qRT-PCR (11752250; Thermo Fisher Scientific). qPCR was performed using the SYBR Green PCR Master Mix (4312704; Thermo Fisher Scientific) on a QuantStudio 6 Flex Real-Time PCR System (4485689; Thermo Fisher Scientific). The primers used in this study are listed in Supplementary Table 2.
Preparation of scRNA-seq library and data analysis
Dissociation of organoids, assembloids and human DRG was performed, as described previously10,49,56,57. For the organoid dissociation, four to five randomly selected organoids were pooled to obtain a single-cell suspension. For assembloids, one to two assembloids were pooled for dissociation. To match the relative cell numbers across different regions, one to two organoids from each of the four regions, totalling four to eight organoids, were pooled for dissociation only when comparing organoids to assembloids. For human primary tissue, one to two DRG were pooled for dissociation. Samples were incubated in 30 U ml−1 of papain enzyme solution (LS003126, Worthington Biochemical) and 0.4% DNase (12,500 U ml−1; LS2007, Worthington Biochemical) at 37 °C for 45 min. After enzymatic dissociation, organoids were washed with a solution containing protease inhibitor and gently triturated to obtain a single-cell suspension. Cells were resuspended in 0.04% BSA/PBS (B6917-25MG, MilliporeSigma) and filtered through a 70-μm Flowmi Cell Strainer (H13680-0070, Bel-Art), and the number of cells were counted. To target 7,000 cells after recovery, approximately 11,600 were loaded per lane on a Chromium Single Cell 3′ chip (Chromium Next GEM Chip G Single Cell Kit; PN-1000127, 10x Genomics), and cDNA libraries were generated with the Chromium Next GEM Single Cell 3′ GEM, Library & Gel Bead Kit v.3.1 (PN-1000128 and PN-1000269, 10x Genomics) according to the manufacturer’s instructions. Each library was sequenced using the Illumina NovaSeq S4 2 × 150 bp (Admera Health) and NovaSeq X 28 + 90 bp (CZ Biohub). Unique molecular identifier (UMI) counting was performed using the ‘count’ function (–include-introns=TRUE) in Cell Ranger (v.7.0.1 and 7.1.0 for organoids; v.7.2.0 for the human DRG and organoid versus assembloid comparison) with GRCh38/Ensembl 98 reference (available at https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2020-A.tar.gz). Further downstream analyses were performed using the R package Seurat (v.4.3.0). Genes on the X or Y chromosome were removed from the count matrix to avoid biases in clustering due to the sex of the hiPS cell lines.
For comparison across four region organoids, hSeO samples (n = 4) and hdSpO samples (n = 5) from this study, along with hDiO samples11 (n = 3; GSE224766) and hCO samples13 (n = 2; GSE145122), were aggregated using the ‘aggr’ function (–normalize=mapped) in Cell Ranger. Cells with more than 7,500 or less than 2,000 detected genes, with less than 2,000 UMI counts or with mitochondrial content higher than 15% were excluded. Genes that were not expressed in at least three cells were not included in the analysis. Gene expression was normalized using a global-scaling normalization method (normalization method; ‘LogNormalize’; scale factor 10,000), and the 2,000 most variable genes were selected (selection method; ‘vst’) and scaled (mean = 0 and variance = 1 for each gene). The top 50 principal components (PCs) were used for clustering (resolution of 1.0) using the ‘FindNeighbors’ and ‘FindClusters’ functions and for visualization with UMAP. Clusters were annotated based on the expression of known marker genes.
For the analysis of hSeO and hdSpO, cells with more than 10,000 or less than 2,500 detected genes, with less than 2,000 UMI counts or cells with mitochondrial content higher than 15% were excluded. Genes that were not expressed in at least three cells in each library were not included. Normalization and variable gene detection were performed, as described above. The hSeO samples (n = 4) and hdSpO samples (n = 5) were each separately integrated using ‘FindIntegrationAnchors’ and ‘IntegrateData’ functions with the default parameter. The top 10 PCs were used for clustering (resolution of 1.0) using the ‘FindNeighbors’ and ‘FindClusters’ functions and for visualization with UMAP. The classification of dorsoventral neuronal cell types of hdSpO was performed based on the combinatorial expression of known markers, as described previously38,39, by using ‘doCellPartition’ function (cell_level_min_step1 = 2, cell_level_min_step2 = 1; available at github.com/juliendelile/MouseSpinalCordAtlas).
For the organoid versus assembloid comparison, cells with more than 10,000 or less than 1,000 detected genes or cells with mitochondrial content higher than 15% were excluded. Genes that were not expressed in at least three cells in each library were not included. After normalization and variable gene detection, individual organoid samples (n = 4) and assembloid samples (n = 4) were integrated and clustered (30 PCs; resolution = 1).
For human DRG data, cells with more than 10,000 or less than 500 detected genes or cells with mitochondrial content higher than 10% were excluded. Genes that were not expressed in at least three cells in each library were not included. After normalization and variable gene detection, human DRG samples (n = 7) were integrated. UMAP visualization and clustering (30 PCs; resolution = 1) were performed. Human DRG samples (n = 7) and hSeO samples (n = 4) were also integrated. After removing putative stressed cells using Gruffi58, an additional round of integration, UMAP visualization and clustering (30 PCs; resolution = 2) were performed.
As a reference for label transfer from the human tissue datasets to four region-specific organoid datasets, the Braun et al.21 reference (available at https://github.com/linnarsson-lab/developing-human-brain) was downsampled, as described in our previous study59, and integrated with human DRG data using each donor as a batch. In the Braun et al. data, subregion labelling of hindbrain, pons and medulla was grouped into one as hindbrain. For label transfer of the dorsoventral subdomain information onto hdSpO, the Rayon et al. developing spinal cord data38 (GSE171892) were quality control (QC) filtered with the same criteria, as described in the original paper, and integrated using each donor as a batch. The prediction scores for each subregion were computed with the ‘FindTransferAnchors’ and ‘TransferData’ functions with 50 PCs. For each cell, the subregion with the highest prediction score was assigned as its predicted subregion. If the highest score was less than 0.5, the predicted subregion was marked as uncertain. Prediction proportions were calculated by dividing the number of cells in each organoid condition with a given subregion prediction by the total number of cells with that subregion prediction.
Generation of assembloids
To generate assembloids, regionalized organoids were differentiated separately and later assembled by positioning them in close proximity. For two-part assembloids, the organoids of interest were fused by placing them in 1.5-ml Eppendorf tubes for 2–3 days inside an incubator. For three- or four-part assembloids, the organoids of interest were integrated by placing them in tilted six-well ultra-low attachment plates (no. 3471; Corning) for 1 week in an incubator. After assembly, the assembloids were maintained in ultra-low attachment plates (24-well or six-well plates) with medium change every 4–7 days. For three- or four-part assembloids, the six-well ultra-low attachment plates were kept tilted in the incubator. The neural medium used for four-part assembloids was supplemented with BDNF (20 ng ml−1; 450-02; PeproTech), GDNF (25 ng ml−1; 450-10; PeproTech), NGF (25 ng ml; 450-01; PeproTech), l-ascorbic acid 2-phosphate trisodium salt (200 µM; 323-44822; Wako), IGF-1 (10 ng ml−1; 100-11; PeproTech) and cAMP (50 μM; D0627; MilliporeSigma). In the course of in vitro development, the timing of neuronal maturation differs along the rostro-caudal axis. This gradient has been taken into consideration especially when generating assembloids composed of organoids resembling spatially separated brain regions, such as hCO and hSpO8. Along the same line, we generally integrated more advanced hCO and hDiO (days 90–120) with earlier-stage hdSpO and hSeO (days 40–70).
Viral labelling and rabies tracing
Viral infection of organoids was performed, as described previously60. In brief, two or three organoids were placed in a 1.5-ml Eppendorf tube containing a 200-μl medium with 0.5 μl of the virus and incubated overnight at 37 °C and 5% CO2. The next day, 800 µl of fresh culture medium was added. The following day, neural organoids were transferred into the fresh culture medium in ultra-low attachment plates. For rabies virus retrograde tracing, organoids representing the ‘presynaptic’ part were labelled with AAV-DIO-tdTomato, and organoids representing the ‘postsynaptic’ part were separately labelled with Rabies-ΔG-Cre-GFP and AAV-EF1α::CVS-G. Two days after viral infection, the organoids were assembled. After 3 weeks of integration, assembloids were fixed with 4% paraformaldehyde and processed for immunocytochemistry. For rabies virus tracing and calcium imaging experiments, hdSpO were labelled with AAV-EF1α::fDIO-GCaMP6f, and hDiO were separately labelled with both Rabies-ΔG-Flpo-dsRedExpress and AAV-EF1α::CVS-G. For GCaMP virus infection of four-part assembloids, one single assembloid was cultured overnight in 200-μl medium containing 0.5 μl of the virus of interest, at 37 °C and 5% CO2. The next day, 4 ml of the fresh culture medium was added.
The viruses used in this study were AAV-DJ-hSYN1::EYFP (GVVC-AAV-16; Stanford University Neuroscience Gene Vector and Virus Core), AAV-DJ-EF1α::CVS-G (produced by Stanford University Neuroscience Gene Vector and Virus Core using Addgene plasmid no. 67528), AAV-DJ-EF1-DIO-tdTomato (GVVC-AAV-169; Stanford University Neuroscience Gene Vector and Virus Core), AAV-DJ-hSYN1::mTurquoise2 (produced by Stanford University Neuroscience Gene Vector and Virus Core using Addgene plasmid no. 99125), AAV-DJ-hSYN1::mCherry (GVVC-AAV-17; Stanford University Neuroscience Gene Vector and Virus Core), Rabies-ΔG-Cre-GFP (Salk), Rabies-ΔG-Flpo-dsRedExpress (Salk using Addgene plasmid no. 32650), AAV1-EF1α::fDIO-GCaMP6f (no. 128315-AAV1; Addgene), Lenti-hSYN1::jGCaMP8s61 (produced by VectorBuilder), AAV1-hSYN1::Cre (no. 105553; Addgene) and AAV-DJ-EF1α::DIO-GCaMP6s (GVVC-AAV-91; Stanford University Neuroscience Gene Vector and Virus Core).
Live calcium imaging from organoids or assembloids
To achieve single-cell-resolution live calcium imaging in intact 3D organoids or assembloids without significant sample movement during chemical injection, we acutely attached samples to ethyleneimine polymer (PEI)-coated coverslips before imaging. Autoclaved 12-mm coverslips were coated with 0.01875% PEI (no. 03880; Sigma) in water for at least 1 h at 37 °C and then washed three times with water before use. An organoid or assembloid was carefully positioned on top of a PEI-coated coverslip, and an L-shaped tubing-type Chamlide CMB chamber (CM-B12-1PB; Live Cell Instrument) was assembled. This chamber was connected to a peristaltic pump (no. 702027; Harvard Apparatus) to establish continuous suction through the outlet hole. To prevent sample drying, a vehicle solution was applied immediately after assembly. Organoids and assembloids with detectable expression of genetically encoded calcium indicators and displaying baseline activity were imaged.
The live imaging chamber was transferred to the confocal microscope stage (Leica STELLARIS 5 or SP8), and the samples were imaged using LAS X software (Leica) under controlled environmental conditions (37 °C and 5% CO2) using a ×5 or ×10 objective. We waited for 5–10 min and then started time-lapse live imaging at approximately 0.436 s per frame, with adjustments for each sample. For imaging spontaneous activity patterns, calcium activity was recorded for 3–5 min. To capture calcium responses after chemical injection, we recorded 60-s baseline activity. Following this, αβ-MeATP (no. 3209; Tocris) or capsaicin (no. 0462; Tocris) dissolved in vehicle was injected over 30 s at a slow speed (approximately 2 ml min−1) to minimize any potential movement of the samples.
For analysis, regions of interest (ROI) were manually drawn, and raw fluorescent intensities were exported using Fiji (v.2.14.0). Using MATLAB (v.R2023a), raw fluorescent intensities were transformed into relative changes in fluorescence: ΔF/Fbase = (F(t) − Fbase)/Fbase, where Fbase is the lower fifth percentile value of the session. The mean amplitudes of ΔF/Fbase from each cell for 60 s were compared before and after chemical injection.
As shown in Fig. 2g,h, a neural medium supplemented with BDNF (20 ng ml−1), GDNF (25 ng ml−1) and NGF (25 ng ml−1) was used as the vehicle solution. After 1 min of baseline imaging, 30 μM of αβ-MeATP or 3 μM of capsaicin was injected into the chamber.
As shown in Fig. 2k,l, a neural medium supplemented with BDNF (20 ng ml−1), GDNF (25 ng ml−1) and NGF (25 ng ml−1) was used as the vehicle solution. The hSeO were incubated with various concentrations of TNP–ATP (10, 100 or 100,000 nM) dissolved in the vehicle solution. After 1 min of baseline imaging, 30 μM of αβ-MeATP was injected into the chamber.
As shown in Fig. 3q,r, assembloids were acutely attached to a PEI-coated 12-well cell culture plate before the imaging. The plate was moved onto the microscope stage, and the imaging was done as the lid of the plate was open. A neural medium supplemented with BDNF, GDNF and NGF was used as the vehicle solution. After 1 min of baseline imaging in 1 ml of the vehicle solution, 500 μl of αβ-MeATP (90 μM) was added during imaging to make 30 μΜ of αβ-MeATP. After 6 min of imaging, the samples were incubated with NBQX (50 μM; no. 0373; Tocris) and APV (50 μM; no. 0106; Tocris) for 30 min in the incubator. Calcium response by αβ-MeATP in the presence of NBQX and APV was performed, as described above.
As shown in Fig. 4e,f, Dulbecco’s phosphate-buffered saline (DPBS) with calcium, magnesium (SH30264.01; Cytiva) or artificial cerebrospinal fluid (aCSF; containing 124 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 1.2 mM MgSO4, 1.5 mM CaCl2, 26 mM NaHCO3 and 10 mM d-(+)-glucose) with the addition of GlutaMAX (35050061; Gibco) was used as the vehicle solution. After 1 min of baseline imaging, 3 μM of αβ-MeATP was injected into the chamber.
As shown in Figs. 4k,l and 5f,g, aCSF was used as the vehicle solution.
The correlation between calcium traces was quantified using SCA42. Traces were first averaged over 1-s intervals and partitioned into 20-s-long segments. Pearson correlation coefficient (r) was computed between corresponding segments and averaged across all segments. This analysis was repeated for multiple time lags within the range of [−20, 20] s for each pair of calcium traces, with 1-s shift step. The peak correlation value across time lags was taken as a measure of synchrony between the calcium signals. This approach yields an accurate correlation estimate between signals on a short timescale, irrespective of common activations over longer periods of time.
Glutamate uncaging in assembloids
Αssembloids were imaged under environmentally controlled conditions (37 °C) using a ×5 objective in a confocal microscope (Leica SP8). For glutamate uncaging experiments, MNI-caged-l-glutamate (1490; Tocris) was used at a final concentration of 2.5 mM in aCSF. The FRAP software module of the Leica SP8 confocal microscope was used to uncage glutamate using ultraviolet light (405 nm). At a frame rate of 2.3 frames per second, one trial consisted of 50 frames for pre-stimulation, three frames of ultraviolet stimulation (in specified ROI) and 100 frames for post-stimulation; three trials were applied for one sample. Calcium responses were calculated by comparing the mean of ΔF/Fbase during 50 frames (21.8 s) before and after ultraviolet illumination, as described above, and averaged values of three trials were used for each sample.
Western blotting
Three to five organoids were collected in the same tube and considered as one sample. Whole-cell protein lysates for samples were prepared using the RIPA buffer system (sc-24948; Santa Cruz Biotechnology). Protein concentration was quantified using the bicinchoninic acid assay (Pierce; catalogue no. 23225; Thermo Fisher Scientific). For electrophoresis, 30 µg of protein per lane was loaded and run on a 4–12% Bis-Tris PAGE gel (Bolt 4–12% Bis-Tris Protein Gel; catalogue no. NW04122BOX; Invitrogen) and transferred onto a polyvinyl difluoride membrane (Immobilon-FL; EMD Millipore). Membranes were blocked with 5% BSA in tris-buffered saline with Tween (TBST) for 1 h at room temperature and incubated with primary antibodies: anti-glyceraldehyde 3-phosphate dehydrogenase (mouse; 1:5,000; GTX627408; GeneTex) for 1 day and anti-NaV1.7 (rabbit; 1:1,000, ASC-008; Alomone Labs) for 6 days. Membranes were washed three times with TBST and then incubated with near-infrared fluorophore-conjugated species-specific secondary antibodies (Goat Anti-Mouse IgG Polyclonal Antibody (IRDye 680RD; 1:10,000; 926-68070; LI-COR Biosciences) or Goat Anti-Rabbit IgG Polyclonal Antibody (IRDye 800CW; 1:10,000; 926-32211; LI-COR Biosciences)) for 1 h at room temperature. Following secondary antibody application, membranes were washed three times with TBST and once with TBS and then imaged using a LI-COR Odyssey CLx imaging system with Image Studio Software (v.5.2; LI-COR). Western blots were analysed using the Image Studio Software or Fiji (v.2.14.0) (see Source data for full blots).
Axon projection imaging and analysis
EYFP+ projections in target regions were imaged under environmentally controlled conditions in intact assembloids using a Leica TCS SP8 or Leica STELLARIS 5 confocal microscope with a motorized stage. Assembloids were transferred to a well in a 24-well glass bottom plate (P24-0-N; Cellvis) in a cell culture medium and incubated in an environmentally controlled chamber for 15–30 min before imaging. Images were taken using a ×10 objective to capture the entire target area at a depth of 100 μm. Projections were quantified using Fiji (v.2.14.0). ROI were manually drawn to cover the target area to be measured in max projection confocal stacks. The percentage of fluorescence-positive pixels over the target region area was calculated in binary images.
Mouse primary tissue dissection and imaging
Mouse tissue samples were obtained using protocols approved by Stanford University’s Administrative Panel on Laboratory Animal Care. Timed pregnant CD1 mice with 11 days of gestation were obtained from Charles River Laboratories and maintained on a 12-h light–dark cycle, an ambient temperature of 20–26 °C and a humidity of 30–70%. We anaesthetized pregnant mice with isoflurane on day 13 of gestation, and E13.5 embryos were used in this study without knowing their sex. DRG were dissected from E13.5 embryos, placed en bloc onto PEI-coated 12-well plates and cultured in DRG culture medium (neural medium supplemented with BDNF, GDNF, NGF and 10% FBS). Calcium imaging was performed the next day after dissection.
As shown in Extended Data Fig. 3a–d, mouse DRG were incubated with Calbryte 520 AM (10 µM; no. 20653; AAT Bioquest). After 45 min, the medium was changed to 1 ml of the DRG culture medium. To image the calcium activity of mouse DRG, a whole-cell culture plate containing mouse DRG was moved to an environment-controlled confocal microscope stage. Imaging was performed when the lid of the plate was opened. After 1 min of baseline line imaging, 500 μl of αβ-MeATP (90 μM) or capsaicin (10 μM) dissolved in the DRG culture medium was added into the well by pipetting without touching the cell culture plate (to prevent XYZ movement). Imaging was performed for 6 min. The final concentrations of the agonists were 30 μM (αβ-MeATP) and 3.3 μM (capsaicin).
As shown in Fig. 2l,m, mouse DRG were incubated with Calbryte 520 AM, as described above, and additionally incubated with various concentrations of TNP–ATP (10, 100 or 100,000 nM; no. 2464; Tocris) dissolved into DRG culture medium for 30 min in the incubator. After 1 min of baseline imaging with TNP–ATP, 500 μl of αβ-MeATP (90 μM) in the DRG culture medium was added into the well. Imaging was performed for 6 min.
Extracellular recordings
Extracellular recordings were performed, as described previously11. Assembloids were embedded into 3% low-melting gel agarose (IB70056; IBI Scientific). Embedded samples were placed on a Brain Slice Chamber-2 (S-BSC2; Scientific Systems Design) and perfused with aCSF (bubbled with 95% O2 and 5% CO2) at 37 °C. The temperature was controlled and retained at 37 °C by connecting to a Proportional Temperature Controller PTC03 (S-PTC03; AutoMate Scientific). Acute 32-channel P-1 probes with two shanks (ASSY-37 P-1; Cambridge NeuroTech) were connected to an Acute probe adaptor; 32-channel Samtec to Omnetics (ADPT A32-Om32; Cambridge NeuroTech). Baseline activities were acquired using the Intan 1024ch recording controller (Intan Technologies) at 30,000 Hz for 5 min using RHX software (v.3.1.0). Raw recording data were processed using Intan Technologies code and analysed using MATLAB (v.R2022a; MathWorks).
To analyse extracellular recording data, band-pass filtering (350–2,000 Hz) was performed followed by common median referencing. A threshold (six times multiplied by s.d.) for each channel was used to count neuronal activities. Activity from hdSpO, hDiO or hCO was counted ± 500 ms of each activity of hSeO channels. Activity counts around the peak area (P) − activity counts in the base area (B)/(P + B) were used as co-activation index44. Parameters (bin size, 5 ms; plot size, ±500 ms) were used to draw the representative cross-correlograms.
Genome editing of hiPS cells to generate SCN9A KO and gain-of-function hiPS cell lines
For generating SCN9A KO cells, three single guide RNAs (sgRNAs) were designed and synthesized by Synthego to induce one or more fragment deletions. The sgRNA sequences are as follows: 5′-AGCUCGUGUAGCCAUAAUCA-3′, 5′-CGUGUGUAGUCAGUGUCCAG-3′ and 5′-UUCUCUUGGUACUCACCUGU-3′. The hiPS cells were dissociated with Accutase, and 0.5 million cells were mixed with 300 pmol sgRNAs and 40 pmol Cas9 protein (SpCas9 2NLS Nuclease (1,000 pmol); Synthego). Nucleofection was performed using the P3 Primary Cell 4D-Nucleofector X Kit S (V4XP-3032; Lonza), a 4D-Nucleofector Core Unit and the X unit (program CA-137; Lonza). Cells were then seeded onto vitronectin-coated six-well plates in Essential 8 Medium supplemented with the ROCK inhibitor Y27632 (10 µM). Essential 8 Medium was used for daily medium change. The genotype of pooled cells was determined by PCR with the primer set Fw, 5′-CGAGAACTACCCATATTATTAGTGATGG-3′; Rv, 5′-CCAAGAACTATCACAAAACGTCTGT-3′, and Sanger sequencing (GENEWIZ) was performed with the reverse primer.
SCN9A gain-of-function cells were generated by Bayspair using two-step scarless genome editing62. Genotyping was performed using PCR with the primer set Fw, 5′-GTGTTTGGAGACCCATGTTTC-3′; Rv, 5′-GAAGTAGTAGTGTCTGAGGGAGATC-3′, and Sanger sequencing (GENEWIZ) was performed with the reverse primer.
Patch-clamp recordings
Patch-clamp recordings were conducted, as described previously11. In brief, hSeO were first infected with Lenti-hSYN1::ChR2-EYFP before assembly and then integrated with hdSpO to create hSeO–hdSpO assembloids. These assembloids were subsequently infected with AAV-DJ-hSYN1::mCherry and placed onto cell culture inserts (0.4-µm pore size; 353090; Corning) positioned in six-well plates to form flattened assembloids. Recordings were typically performed on days 148–164 (daf 99–110). mCherry-positive neurons within the hdSpO were identified using an upright microscope. For optogenetic stimulation, whole-field light-emitting diode illumination (460 nm; 5–20 ms duration; maximal power; CoolLED) was applied through a ×40 objective, and oEPSCs were recorded at a holding potential of −70 mV.
Patch-clamp recordings in whole-cell configuration were performed at room temperature on spinal organoid neurons, which were visualized using an upright microscope (Scientifica) equipped with an INFINITY2 charge-coupled device camera and INFINITY Capture Software (Teledyne Lumenera). The external aCSF recording solution contained 124 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 1 mM MgSO4, 2 mM CaCl2, 26 mM NaHCO3 and 10 mM d-(+)-glucose. During recordings, the assembloids were perfused with the aCSF recording solution (bubbled with 95% O2 and 5% CO2). Thick-walled borosilicate pipettes (resistance of 6–9 MΩ) were filled with an internal solution consisting of 135 mM K-gluconate, 20 mM KCl, 0.1 mM EGTA, 2 mM MgCl2, 2 mM sodium-ATP, 10 mM HEPES and 0.3 mM sodium-GTP (pH adjusted to 7.28 with KOH; approximately 302 mOsm). Data were acquired using a MultiClamp 700B Amplifier (Clampex 10.7; Molecular Devices) and a Digidata 1550B Digitizer (Molecular Devices), low-pass filtered at 2 kHz, digitized at 20 kHz and analysed with Clampfit (v.10.7; Molecular Devices).
Statistics and reproducibility
Data are presented as mean ± s.e.m. unless described otherwise. Raw data were tested for normality of distribution, and statistical analyses were performed by two-tailed unpaired t-test, two-tailed Mann–Whitney test, two-tailed paired t-test, two-tailed Wilcoxon matched-pair signed-rank test, Wilcoxon signed-rank test, one-way ANOVA and Kruskal–Wallis test with multiple comparison tests depending on the dataset. Sample sizes were estimated empirically. GraphPad Prism v.10.0.0 or MATLAB (v.R2023a; MathWorks) was used for statistical analyses. Data shown in representative experiments were repeated with similar results in multiple independent experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Gene expression data have been deposited in the Gene Expression Omnibus under accession number GSE251892. The data in this study are available on request from the corresponding author. Source data are provided with this paper.
Code availability
The codes for SCA were described in a previous study43 and are available at Zenodo (https://doi.org/10.5281/zenodo.13921410)63.
References
Colloca, L. et al. Neuropathic pain. Nat. Rev. Dis. Primers 3, 17002 (2017).
Basbaum, A. I., Bautista, D. M., Scherrer, G. & Julius, D. Cellular and molecular mechanisms of pain. Cell 139, 267–284 (2009).
Han, Q. et al. SHANK3 deficiency impairs heat hyperalgesia and TRPV1 signaling in primary sensory neurons. Neuron 92, 1279–1293 (2016).
Dawes, J. M. et al. Immune or genetic-mediated disruption of CASPR2 causes pain hypersensitivity due to enhanced primary afferent excitability. Neuron 97, 806–822.e10 (2018).
Orefice, L. L. et al. Peripheral mechanosensory neuron dysfunction underlies tactile and behavioral deficits in mouse models of ASDs. Cell 166, 299–313 (2016).
Drenth, J. P. H. & Waxman, S. G. Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J. Clin. Invest. 117, 3603–3609 (2007).
Mogil, J. S. Animal models of pain: progress and challenges. Nat. Rev. Neurosci. 10, 283–294 (2009).
Andersen, J. et al. Generation of functional human 3D cortico-motor assembloids. Cell 183, 1913–1929.e26 (2020).
Kayalioglu, G. in The Spinal Cord (eds Watson, C. et al.) 148–167 (Academic, 2009).
Pasca, A. M. et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 12, 671–678 (2015).
Kim, J. et al. Human assembloids reveal the consequences of CACNA1G gene variants in the thalamocortical pathway. Neuron 112, 4048–4059.e7 (2024).
Zimmer, B. et al. Human iPSC-derived trigeminal neurons lack constitutive TLR3-dependent immunity that protects cortical neurons from HSV-1 infection. Proc. Natl Acad. Sci. USA 115, E8775–E8782 (2018).
Khan, T. A. et al. Neuronal defects in a human cellular model of 22q11.2 deletion syndrome. Nat. Med. 26, 1888–1898 (2020).
Tao, W. & Lai, E. Telencephalon-restricted expression of BF-1, a new member of the HNF-3/fork head gene family, in the developing rat brain. Neuron 8, 957–966 (1992).
Lipiec, M. A. et al. TCF7L2 regulates postmitotic differentiation programs and excitability patterns in the thalamus. Development 147, dev190181 (2020).
Hunt, P. et al. A distinct Hox code for the branchial region of the vertebrate head. Nature 353, 861–864 (1991).
Roome, R. B. et al. Phox2a defines a developmental origin of the anterolateral system in mice and humans. Cell Rep. 33, 108425 (2020).
Gerrero, M. R. et al. Brn-3.0: a POU-___domain protein expressed in the sensory, immune, and endocrine systems that functions on elements distinct from known octamer motifs. Proc. Natl Acad. Sci. USA 90, 10841–10845 (1993).
Cao, J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019).
Braun, E. et al. Comprehensive cell atlas of the first-trimester developing human brain. Science 382, eadf1226 (2023).
Lu, T. et al. Decoding transcriptional identity in developing human sensory neurons and organoid modeling. Cell 187, 7374–7393.e28 (2024).
Grenningloh, G., Soehrman, S., Bondallaz, P., Ruchti, E. & Cadas, H. Role of the microtubule destabilizing proteins SCG10 and stathmin in neuronal growth. J. Neurobiol. 58, 60–69 (2004).
Laclef, C., Souil, E., Demignon, J. & Maire, P. Thymus, kidney and craniofacial abnormalities in Six1 deficient mice. Mech. Dev. 120, 669–679 (2003).
Meltzer, S., Santiago, C., Sharma, N. & Ginty, D. D. The cellular and molecular basis of somatosensory neuron development. Neuron 109, 3736–3757 (2021).
North, R. A. The P2X3 subunit: a molecular target in pain therapeutics. Curr. Opin. Invest. Drugs 4, 833–840 (2003).
Julius, D. TRP channels and pain. Annu. Rev. Cell Dev. Biol. 29, 355–384 (2013).
Murthy, S. E., Dubin, A. E. & Patapoutian, A. Piezos thrive under pressure: mechanically activated ion channels in health and disease. Nat. Rev. Mol. Cell Biol. 18, 771–783 (2017).
Caterina, M. J. et al. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389, 816–824 (1997).
Adamek, P. et al. Dual PI3Kδ/γ inhibitor duvelisib prevents development of neuropathic pain in model of paclitaxel-induced peripheral neuropathy. J. Neurosci. 42, 1864–1881 (2022).
Iwaoka, E. et al. Evodiamine suppresses capsaicin-induced thermal hyperalgesia through activation and subsequent desensitization of the transient receptor potential V1 channels. J. Nat. Med. 70, 1–7 (2016).
Chen, X. et al. Furanocoumarins are a novel class of modulators for the transient receptor potential vanilloid type 1 (TRPV1) channel. J. Biol. Chem. 289, 9600–9610 (2014).
Chen, X. et al. Long-term diabetic microenvironment augments the decay rate of capsaicin-induced currents in mouse dorsal root ganglion neurons. Molecules 24, 775 (2019).
Wainger, B. J. et al. Modeling pain in vitro using nociceptor neurons reprogrammed from fibroblasts. Nat. Neurosci. 18, 17–24 (2015).
Virginio, C., Robertson, G., Surprenant, A. & North, R. A. Trinitrophenyl-substituted nucleotides are potent antagonists selective for P2X1, P2X3, and heteromeric P2X2/3 receptors. Mol. Pharmacol. 53, 969–973 (1998).
Serrano, A. et al. Differential expression and pharmacology of native P2X receptors in rat and primate sensory neurons. J. Neurosci. 32, 11890–11896 (2012).
Todd, A. J. Neuronal circuitry for pain processing in the dorsal horn. Nat. Rev. Neurosci. 11, 823–836 (2010).
Rayon, T., Maizels, R. J., Barrington, C. & Briscoe, J. Single-cell transcriptome profiling of the human developing spinal cord reveals a conserved genetic programme with human-specific features. Development 148, dev199711 (2021).
Delile, J. et al. Single cell transcriptomics reveals spatial and temporal dynamics of gene expression in the developing mouse spinal cord. Development 146, dev173807 (2019).
Ding, Y.-Q. et al. Lmx1b controls the differentiation and migration of the superficial dorsal horn neurons of the spinal cord. Development 131, 3693–3703 (2004).
Gross, M. K., Dottori, M. & Goulding, M. Lbx1 specifies somatosensory association interneurons in the dorsal spinal cord. Neuron 34, 535–549 (2002).
Nikolić, D., Mureşan, R. C., Feng, W. & Singer, W. Scaled correlation analysis: a better way to compute a cross‐correlogram. Eur. J. Neurosci. 35, 742–762 (2012).
Miura, Y. et al. Assembloid model to study loop circuits of the human nervous system. Preprint at bioRxiv https://doi.org/10.1101/2024.10.13.617729 (2024).
Mizuseki, K. & Buzsaki, G. Theta oscillations decrease spike synchrony in the hippocampus and entorhinal cortex. Philos. Trans. R. Soc. London B Biol. Sci. 369, 20120530 (2014).
McDermott, L. A. et al. Defining the functional role of NaV1.7 in human nociception. Neuron 101, 905–919.e8 (2019).
Gingras, J. et al. Global Nav1.7 knockout mice recapitulate the phenotype of human congenital indifference to pain. PLoS ONE 9, e105895 (2014).
Fertleman, C. R. et al. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron 52, 767–774 (2006).
Miura, Y. et al. Generation of human striatal organoids and cortico-striatal assembloids from human pluripotent stem cells. Nat. Biotechnol. 38, 1421–1430 (2020).
Birey, F. et al. Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59 (2017).
Xiao, D. et al. Generation of self-organized sensory ganglion organoids and retinal ganglion cells from fibroblasts. Sci. Adv. 6, eaaz5858 (2020).
Rockel, A. F., Wagner, N., Spenger, P., Ergün, S. & Wörsdörfer, P. Neuro-mesodermal assembloids recapitulate aspects of peripheral nervous system development in vitro. Stem Cell Rep. 18, 1155–1165 (2023).
Matsumoto, N., Barson, D., Liang, L. & Crair, M. C. Hebbian instruction of axonal connectivity by endogenous correlated spontaneous activity. Science 385, eadh7814 (2024).
Sharma, N. et al. The emergence of transcriptional identity in somatosensory neurons. Nature 577, 392–398 (2020).
Andersen, J. et al. Single-cell transcriptomic landscape of the developing human spinal cord. Nat. Neuro. 26, 902–914 (2023).
Yoon, S. J. et al. Reliability of human cortical organoid generation. Nat. Methods 16, 75–78 (2019).
Sloan, S. A. et al. Human astrocyte maturation captured in 3D cerebral cortical spheroids derived from pluripotent stem cells. Neuron 95, 779–790.e6 (2017).
Sloan, S. A., Andersen, J., Pașca, A. M., Birey, F. & Pașca, S. P. Generation and assembly of human brain region-specific three-dimensional cultures. Nat. Protoc. 13, 2062–2085 (2018).
Vértesy, Á. et al. Gruffi: an algorithm for computational removal of stressed cells from brain organoid transcriptomic datasets. EMBO J. 41, e111118 (2022).
Amin, N. D. et al. Generating human neural diversity with a multiplexed morphogen screen in organoids. Cell Stem Cell 31, 1831–1846.e9 (2024).
Miura, Y. et al. Engineering brain assembloids to interrogate human neural circuits. Nat. Protoc. 17, 15–35 (2022).
Zhang, Y. et al. Fast and sensitive GCaMP calcium indicators for imaging neural populations. Nature 615, 884–891 (2023).
Ikeda, K. et al. Efficient scarless genome editing in human pluripotent stem cells. Nat. Methods 15, 1045–1047 (2018).
Kim, J., Pașca, S. & Miura, Y. Assembloid model to study loop circuits of the human nervous system. Zenodo https://doi.org/10.5281/zenodo.13921409 (2024).
Acknowledgements
We thank members of the Pasca laboratory at Stanford University for scientific input and technical support and K. Kaganovsky for support with animal experiments. This work was supported by the Stanford Brain Organogenesis grant from the Wu Tsai Neurosciences Institute (to S.P.P.), NYSCF Robertson Stem Cell Investigator Award (to S.P.P.), Kwan Research Fund (to S.P.P.), Senkut Research Funds (to S.P.P.), Chan Zuckerberg Initiative Ben Barres Investigator Award (to S.P.P.), CZ BioHub Investigator Program (to S.P.P.), National Institutes of Health (R01 MH107800 to S.P.P., R01 NS128028 to S.P.P. and CNCDP-K12 to R.J.L.), Nurturing Next-generation Researchers Program from the National Research Foundation of Korea (to J.-i.K.) and Stanford Medicine Dean’s Fellowship (to K.I.).
Author information
Authors and Affiliations
Contributions
J.-i.K., K.I. and S.P.P. conceived the project and designed the experiments. J.-i.K. carried out the differentiation, characterization of regionalized neural organoids, extracellular recordings and calcium imaging. K.I. performed neural differentiations, single-cell transcriptomics, calcium imaging and SCN9A gene editing. O.J. contributed to the analysis pipelines for synchronized activities. K.W.K. contributed to scRNA-seq analysis. D.W. performed patch-clamp recording experiments. M.V.T. performed neural differentiations, western blotting and characterization of the regionalized neural organoids. Z.H. analysed the calcium imaging results. N.D.A. and R.J.L contributed to the primary tissue experiments. G.S. contributed to experimental design and data analysis. J.-i.K., K.I. and S.P.P. wrote the article with input from all the authors.
Corresponding author
Ethics declarations
Competing interests
Stanford University filed or is filing patent applications covering the generation of regionalized organoids and assembloids.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Protocol for generating regionalized neural organoids.
Protocols to generate hCO, hDiO, hdSpO, and hSeO from hiPS cells.
Extended Data Fig. 2 Single cell transcriptome of human DRG.
a. Table showing the number of single cells per sample. Numbers correspond to total numbers of cells recovered after QC and filtering. b. Heat map showing normalized expression of the top ten unique markers per cell type. c. Violin plots showing total counts (nCount), total number of features (nFeature), percent of mitochondrial (MT) counts, and percent of ribosomal (ribo) counts separated by sample. d. UMAP plots split by sample. e. Proportion of cells with the indicated organoid identity for each brain subdomain label.
Extended Data Fig. 3 Transcriptomic and immunostaining analysis of hSeO.
a. Cell type composition in hSeO colored by cluster (left), and UMAP plots of hSeO colored by the hiPS cell line (right). b. UMAP plots of hSeO samples showing selected cell type markers and receptors. c. RT-qPCR of POU4F1 (hCO, n = 7; hDiO, n = 6; hdSpO, n = 8; hSeO, n = 7 samples, from 3 hiPS cell lines from 2–4 differentiation experiments, one-way ANOVA, F3, 24 = 16.59 with Tukey’s multiple comparisons test, ***P = 0.0001, ****P < 0.0001) and SIX1 (hCO, n = 7; hDiO, n = 6; hdSpO, n = 8; hSeO, n = 7 samples, from 3 hiPS cell lines from 2–4 differentiation experiments, one-way ANOVA, F3, 24 = 15.49 with Tukey’s multiple comparisons test, ****P < 0.0001) expression across four regions at day 45–100. d. Immunostaining of VGLUT2 and NeuN in hSeO (left) and quantification of VGLUT2+/NeuN+ co-localization (right) at days 121–125 (n = 6 organoids from 3 hiPS cell lines from 4 differentiation experiments). e. Dot plot showing selected marker gene expression in each cluster. f. Dot plot showing gene expression of several receptors for sensory stimuli. g. Integrated UMAP visualization of scRNA-seq of human primary DRG (PCW 15) and hSeO (human primary DRG, n = 49,705 cells from 7 DRG samples, one individual; hSeO, day 68–72, n = 18,382 cells from 4 hiPS cell lines, same dataset as in Fig. 1 with optimized QC criteria). h. UMAP plot colored by sample identity. i. Dot plot showing selected marker gene expression in each cluster. j. Cell composition of hSeO and human primary DRG based on scRNA-seq. k. UMAP plots showing selected cell type markers and receptors. Data shown as mean ± s.e.m. Dots indicate qPCR samples or organoids in c, d. Each shape represents a hiPS cell line: Circle, 1205-4; Triangle, 1208-2; Diamond, 0410-1; Square, KOLF2.1 J in c, d.
Extended Data Fig. 4 Calcium imaging of isolated mouse DRG.
a. Schematic of live calcium imaging of isolated mouse DRG following pharmacological treatment. b. Representative image showing GCaMP6s expression in mouse DRG. c. Example ΔF/Fbase traces before and after agonist exposure showing calcium responses evoked by αβ-MeATP (30 μM) and capsaicin (3.3 μM). d. Quantification of mean ΔF/Fbase values at baseline and at 60 s after exposure to αβ-MeATP (left) (n = 384 cells from 3 DRG, Two-tailed Wilcoxon matched-pairs signed rank test, ****P < 0.0001) and capsaicin (right) (n = 762 cells from 10 hSeO, 3 DRG, two-tailed Wilcoxon matched-pairs signed rank test, ****P < 0.0001). Data shown as mean ± s.e.m. Gray dots indicate cells, large colored dots indicate DRG in d. Credit: a adapted from ref. 48, Springer Nature America, Inc.
Extended Data Fig. 5 Transcriptomic and immunostaining analysis of hdSpO.
a. Cell type composition of hdSpO colored by cell clusters (left), and UMAP plots of hdSpO samples colored by the hiPS cell line (right). b. UMAP plots of hdSpO samples showing selected cell type markers. c. Dot plot showing selected marker gene expression in each cluster. d. RT-qPCR of TACR1 (hCO, n = 7; hDiO, n = 6; hdSpO, n = 8; hSeO, n = 7 samples, from 3 hiPS cell lines from 2–4 differentiation experiments, one-way ANOVA, F3, 24 = 17.70 with Tukey’s multiple comparisons test, ***P = 0.0009, ****P < 0.0001) and LBX1 (hCO, n = 7; hDiO, n = 6; hdSpO, n = 8; hSeO, n = 7 samples, from 3 hiPS cell lines from 2–4 differentiation experiments, one-way ANOVA, F3, 24 = 30.50 with Tukey’s multiple comparisons test, ****P < 0.0001) expression across four regions at day 45–100. e. Dot plot showing dorsoventral cluster marker gene expression in each subcluster. f. Representative image showing distribution of PAX2+ and PHOX2A+ neurons in hdSpO (left) and quantification of co-localization of PAX and PHOX2A (n = 5 organoids from 3 hiPS cell lines, 4 differentiation experiments). Data shown as mean ± s.e.m. Dots indicate qPCR samples or organoids in d, f. Each shape represents a hiPS cell line: Circle, 1205-4; Triangle, 1208-2; Diamond, 0410-1; Square, KOLF2.1 J in d, f.
Extended Data Fig. 6 Characterization of projections in assembloids.
a. Immunostaining image showing co-localization of NF-H and EYFP+ projection in the hdSpO side of hSeO–hdSpO assembloids. b. Schematic illustrating rabies tracing experiments in assembloid and unassembled organoids. c. Representative images indicating presence of EGFP+/tdTomato+ cells on the hSeO side of the assembloid and absence in unassembled organoids at daf 21. d. Quantification of tdTomato+ and EGFP+ cells (n = 3 assembloids from 2 hiPS cell lines from 2 differentiation experiments, two-tailed unpaired t test, *P = 0.0155 for tdTomato+ cells and *P = 0.0193 for EGFP+ cells). e. Proportion of responding neurons (8/30 cells). f. Quantification of peak oEPSC amplitude (left) and response latency (right) (n = 8 neurons from 2 hiPS cell lines, 2 differentiation experiments). g. Schematic illustrating the fusion of hSeO and hCO to form hSeO-hCO assembloids. h. Representative images of a hSeO-hCO assembloid showing a limited number of hSeO-derived hSYN1::EYFP projections at 14, 21, and 28 daf. i. Quantification of hSeO-derived EYFP fluorescence in hCO area at 14, 21, and 28 daf in hSeO-hCO assembloids (n = 5–10 assembloids from 3 hiPS cell lines from 3 differentiation experiments, Kruskal-Wallis test with Dunn’s multiple comparisons test, n.s.; not significant). j. Immunostaining image showing co-localization of PHOX2A and tdTomato on the hdSpO side of hSeO–hdSpO assembloids. k. Schematic illustrating the integration of EYFP labeled hdSpO, hSeO and hDiO to form 3–part hSeO–hdSpO–hDiO assembloids. l. Representative image of a hSeO–hdSpO–hDiO assembloid showing preferential spinal projection into hDiO. m. Quantification of hdSpO-derived EYFP fluorescence in hDiO or hSeO at daf29 (n = 9 assembloids from 3 hiPS cell lines from 2 differentiation experiments, two-tailed Wilcoxon matched-pairs signed rank test, **P = 0.0078). Data shown as mean ± s.e.m. Dots indicate assembloids in d, i, m, or neurons in f. Each shape represents a hiPS cell line: Circle, 1205-4; Triangle, 1208-2; Inverted triangle, 8119-1, Square, KOLF2.1 J in d, f, i, m. Credits: b,g,k adapted from ref. 48, Springer Nature America, Inc.
Extended Data Fig. 7 Characterization of hASA.
a Success of assembly (left) and distribution of shapes of assembloids (right). Approximately 87.7% (100/114) hASA were suitable for functional assays. b. Schematic illustrating the scRNA-seq of individual organoids and four-part assembloids. c. UMAP visualization of individual organoids (left) and four-part assembloids at day 162–229 (right) (individual organoids, n = 59,267 cells from 4 samples, 2 differentiation experiments, one hiPS cell line; four-part assembloids, n = 40,700 cells from 4 samples, 2 differentiation experiments, one hiPS cell line). d. Dot plot showing selected marker genes for each cluster of individual organoids (left) and four-part assembloids (right). Credit: b adapted from ref. 48, Springer Nature America, Inc.
Extended Data Fig. 8 Characterization of hSeO with SCN9A pathogenic variants.
a. RT-qPCR analysis of SCN9A expression in control and SCN9A KO hSeO (Control, n = 6; SCN9A KO, n = 4 samples from 2 hiPS cell lines from 2 differentiation experiments, two-tailed unpaired t-test, **P = 0.0082) at day 48–69. b. RT-qPCR analysis of POU4F1 expression in control and SCN9A KO hSeO (Control, n = 6; SCN9A KO, n = 4 samples from 2 hiPS cell lines in 2 differentiation experiments, two-tailed unpaired t-test, n.s.; not significant) at day 48–69. c. Immunocytochemistry images for BRN3A in control and SCN9A KO hSeO. d. Quantification of the percentage of BRN3A+ cells in control and SCN9A KO hSeO (Control, n = 8; SCN9A KO, n = 6 samples from 2 hiPS cell lines from 2 differentiation experiments, two-tailed unpaired t-test, n.s.; not significant). e. Calcium imaging of control and SCN9A KO hSeO f. Quantification of the number of spontaneous calcium responses (Control, n = 28 cells from 5 organoids; SCN9A KO, n = 29 cells from 6 organoids, 2 hiPS cell lines from 2 differentiation experiments, two-tailed Mann-Whitney test, ***P = 0.0006) at day 64-82. g. Representative SCA correlograms between cells from hSeO–hSeO, hSeO–hdSpO, hSeO–hDiO, and hSeO–hCO for a control assembloid (top) and in a SCN9A KO assembloid (bottom). The histograms of peak time-shifts (below each correlograms panel) show peaks around 0 in the control, but not in the SCN9A KO assembloid. h. RT-qPCR analysis of POU4F1 expression in control and SCN9A T1464I hSeO (n = 4 samples from one hiPS cell line from 4 differentiation experiments, two-tailed unpaired t-test, n.s.; not significant) at day 25–31. i. Calcium imaging of control and SCN9A T1464I hSeO j. Quantification of the number of spontaneous calcium activity (CTL, n = 244 cells from 9 organoids; T1464I, n = 149 cells from 8 organoids, one hiPS cell line from 3 differentiation experiments, two-tailed Mann-Whitney test, ****P < 0.0001) at day 103–111. k. Representative SCA correlograms between cells from hSeO–hSeO, hSeO–hdSpO, hSeO–hDiO, and hSeO–hCO for a control assembloid (top) and in a SCN9A T1464I assembloid (bottom). Data shown as mean ± s.e.m. Gray dots indicate cells, large colored dots indicate organoids or qPCR samples in a, b, d, f, h, j. Each shape represents a hiPS cell line: Circle, 1205-4; Triangle, 1208-2; Square, KOLF2.1 J in a, b, d, f, h, j. Credits: e,i adapted from ref. 48, Springer Nature America, Inc.
Supplementary information
Supplementary Information
Supplementary Fig. 1 and Supplementary Tables 1 and 2
Supplementary Video 1
Calcium response to αβ-MeATP exposure in hASA.
Supplementary Video 2
Spontaneous calcium activity in hASA.
Supplementary Video 3
Spontaneous calcium activity in control hASA.
Supplementary Video 4
Spontaneous calcium activity in SCN9A KO hASA.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kim, Ji., Imaizumi, K., Jurjuț, O. et al. Human assembloid model of the ascending neural sensory pathway. Nature 642, 143–153 (2025). https://doi.org/10.1038/s41586-025-08808-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-025-08808-3
This article is cited by
-
Generation and characterization of vestibular inner ear organoids from human pluripotent stem cells
Nature Protocols (2025)
-
Brain tissues, assemble! Inside the push to build better brain models
Nature (2025)
-
Current status and future prospects of toxicity assessment using organoids
Toxicological Research (2025)
