
Abstract
Aptamers, nucleic acid ligands targeting specific molecules, have emerged as drug candidates, sensors, imaging tools and nanotechnology building blocks. The predominant method for their discovery, systematic evolution of ligands by exponential enrichment, while successful, is laborious, time-consuming and often results in candidates enriched for unintended criteria. Here we present UltraSelex, a noniterative method that combines biochemical partitioning, high-throughput sequencing and computational signal-to-background rank modeling for discovering RNA aptamers in about 1 day. UltraSelex identified high-affinity RNA aptamers capable of binding a fluorogenic silicon rhodamine dye and two protein targets, the SARS-CoV-2 RNA-dependent RNA polymerase and HIV reverse transcriptase, enabling live-cell RNA imaging and efficient enzyme inhibition, respectively. From the ranked sequences, minimal aptamer motifs could be easily inferred. UltraSelex provides a rapid route to reveal new drug candidates and diagnostic tools.

This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
27,99 € / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
269,00 € per year
only 22,42 € per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Data availability
NGS raw data that support the findings of this study are available for academic use in the online repository (https://www.ultrarnalab.com). A mirror data repository can be accessed on the National Center for Biotechnology Information BioProject (PRJNA1216547). UltraSelex library construction and the aptamer sequences selected for downstream applications are compiled in the Supplementary Information. Source data are provided with this paper.
Code availability
The web server for UltraSelex deep sequencing data analysis is freely available and will be routinely updated on the site (https://www.ultrarnalab.com). A mirror code repository can be accessed on Zenodo (https://zenodo.org/records/14164763)45. Other data analysis scripts used in this study will be made available to registered users via this website.
References
Hogan, M. J. & Pardi, N. mRNA vaccines in the COVID-19 pandemic and beyond. Annu. Rev. Med. 73, 17–39 (2022).
Polack, F. P. et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N. Engl. J. Med. 383, 2603–2615 (2020).
Cao, Y. et al. Imprinted SARS-CoV-2 humoral immunity induces convergent Omicron RBD evolution. Nature 614, 521–529 (2023).
Kurhade, C. et al. Low neutralization of SARS-CoV-2 Omicron BA.2.75.2, BQ.1.1 and XBB.1 by parental mRNA vaccine or a BA.5 bivalent booster. Nat. Med. 29, 344–347 (2023).
Paige, J. S., Wu, K. Y. & Jaffrey, S. R. RNA mimics of green fluorescent protein. Science 333, 642–646 (2011).
Zhou, J. & Rossi, J. Aptamers as targeted therapeutics: current potential and challenges. Nat. Rev. Drug Discov. 16, 181–202 (2017).
Sunbul, M. et al. Super-resolution RNA imaging using a rhodamine-binding aptamer with fast exchange kinetics. Nat. Biotechnol. 39, 686–690 (2021).
Valero, J. et al. A serum-stable RNA aptamer specific for SARS-CoV-2 neutralizes viral entry. Proc. Natl Acad. Sci. USA 118, e2112942118 (2021).
Xu, Y. & Zhu, T. F. Mirror-image T7 transcription of chirally inverted ribosomal and functional RNAs. Science 378, 405–412 (2022).
Tong, Y. et al. Programming inactive RNA-binding small molecules into bioactive degraders. Nature 618, 169–179 (2023).
Mullard, A. FDA approves second RNA aptamer. Nat. Rev. Drug Discov. 22, 774 (2023).
Litke, J. L. & Jaffrey, S. R. Highly efficient expression of circular RNA aptamers in cells using autocatalytic transcripts. Nat. Biotechnol. 37, 667–675 (2019).
Takahashi, M. et al. High throughput sequencing analysis of RNA libraries reveals the influences of initial library and PCR methods on SELEX efficiency. Sci. Rep. 6, 33697 (2016).
Gholamalipour, Y., Karunanayake Mudiyanselage, A. & Martin, C. T. 3′ end additions by T7 RNA polymerase are RNA self-templated, distributive and diverse in character-RNA-seq analyses. Nucleic Acids Res. 46, 9253–9263 (2018).
Verwilt, J., Mestdagh, P. & Vandesompele, J. Artifacts and biases of the reverse transcription reaction in RNA sequencing. RNA 29, 889–897 (2023).
Davis, J. H. & Szostak, J. W. Isolation of high-affinity GTP aptamers from partially structured RNA libraries. Proc. Natl Acad. Sci. USA 99, 11616–11621 (2002).
Famulok, M. Molecular recognition of amino acids by RNA-aptamers: an L-citrulline binding RNA motif and its evolution into an L-arginine binder. J. Am. Chem. Soc. 116, 1698–1706 (2002).
Wirth, R., Gao, P., Nienhaus, G. U., Sunbul, M. & Jäschke, A. SiRA: a silicon rhodamine-binding aptamer for live-cell super-resolution RNA imaging. J. Am. Chem. Soc. 141, 7562–7571 (2019).
Meiser, L. C. et al. DNA synthesis for true random number generation. Nat. Commun. 11, 5869 (2020).
Mayer, G. et al. Fluorescence-activated cell sorting for aptamer SELEX with cell mixtures. Nat. Protoc. 5, 1993–2004 (2010).
Campagnola, G., Govindarajan, V., Pelletier, A., Canard, B. & Peersen, O. B. The SARS-CoV nsp12 polymerase active site is tuned for large-genome replication. J. Virol. 96, e0067122 (2022).
Gao, Y. et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 368, 779–782 (2020).
Hillen, H. S. et al. Structure of replicating SARS-CoV-2 polymerase. Nature 584, 154–156 (2020).
Sun, L. Z., Jiang, Y., Zhou, Y. & Chen, S. J. RLDOCK: a new method for predicting RNA–ligand interactions. J. Chem. Theory Comput. 16, 7173–7183 (2020).
Zhang, Y. et al. Structural mechanisms for binding and activation of a contact-quenched fluorophore by RhoBAST. Nat. Commun. 15, 4206 (2024).
Will, S., Reiche, K., Hofacker, I. L., Stadler, P. F. & Backofen, R. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput. Biol. 3, e65 (2007).
Bernhart, S. H., Hofacker, I. L., Will, S., Gruber, A. R. & Stadler, P. F. RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinformatics 9, 474 (2008).
Van Zundert, G. C. P. et al. The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 428, 720–725 (2016).
Lee, C. W., Li, L. & Giedroc, D. P. The solution structure of coronaviral stem–loop 2 (SL2) reveals a canonical CUYG tetraloop fold. FEBS Lett. 585, 1049–1053 (2011).
Liu, P. et al. A U-turn motif-containing stem–loop in the coronavirus 5′ untranslated region plays a functional role in replication. RNA 13, 763–780 (2007).
Alam, K. K. et al. Poly-target selection identifies broad-spectrum RNA aptamers. Mol. Ther. Nucleic Acids 13, 605–619 (2018).
Tuerk, C., MacDougal, S. & Gold, L. RNA pseudoknots that inhibit human immunodeficiency virus type 1 reverse transcriptase. Proc. Natl Acad. Sci. USA 89, 6988–6992 (1992).
Bühler, B. et al. Avidity-based bright and photostable light-up aptamers for single-molecule mRNA imaging. Nat. Chem. Biol. 19, 478–487 (2023).
Jarmoskaite, I., AlSadhan, I., Vaidyanathan, P. P. & Herschlag, D. How to measure and evaluate binding affinities. eLife 9, e57264 (2020).
Szeto, K. et al. RAPID-SELEX for RNA aptamers. PLoS ONE 8, e82667 (2013).
Wang, H. et al. Decoding the RNA interactome by UltraGen. Preprint at Research Square https://doi.org/10.21203/rs.3.rs-4461517/v1 (2024).
Chang, D. et al. A high-dimensional microfluidic approach for selection of aptamers with programmable binding affinities. Nat. Chem. 15, 773–780 (2023).
Hofacker, I. L. & Stadler, P. F. Memory efficient folding algorithms for circular RNA secondary structures. Bioinformatics 22, 1172–1176 (2006).
Zhang, J., Wang, L., Jäschke, A. & Sunbul, M. A color-shifting near-infrared fluorescent aptamer-fluorophore module for live-cell RNA imaging. Angew. Chem. Int. Ed. Engl. 60, 21441–21448 (2021).
Sunbul, M. & Jäschke, A. SRB-2: a promiscuous rainbow aptamer for live-cell RNA imaging. Nucleic Acids Res. 46, e110 (2018).
Watkins, A. M., Rangan, R. & Das, R. FARFAR2: improved de novo rosetta prediction of complex global RNA folds. Structure 28, 963–976 (2020).
Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 (2003).
O’Boyle, N. M. et al. Open Babel: an open chemical toolbox. J. Cheminform. 3, 33 (2011).
Mathews, D. H. et al. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl Acad. Sci. USA 101, 7287–7292 (2004).
Zhang, Y. UltraSelex_SGREELI_code_package_v1. Zenodo https://doi.org/10.5281/zenodo.14164763 (2024).
Esling, P., Lejzerowicz, F. & Pawlowski, J. Accurate multiplexing and filtering for high-throughput amplicon-sequencing. Nucleic Acids Res. 43, 2513–2524 (2015).
Chen, X. et al. Visualizing RNA dynamics in live cells with bright and stable fluorescent RNAs. Nat. Biotechnol. 37, 1287–1293 (2019).
Acknowledgements
We thank Jäschke Lab members for discussions, R. Wirth (Heidelberg University) for the cDNA resource of published work and SiR dye, M. Sunbul (Heidelberg University) and R. Völk (Heidelberg University) for RNA resources, M. Knop (Heidelberg University) for access to TECAN, W. Nickel (Heidelberg University) for access to BLI-Octet, M. Brunner (Heidelberg University) for access to qPCR, V. Benes (European Molecular Biology Laboratory) for access to HTS, G. Mayer (University of Bonn) for T7 Y639F RNA polymerase plasmid, Y. Wu (Heidelberg University) for aesthetic figures, SDS@hd for data storage and bwHPC (bwForCluster MLS&WISO and bwUniCluster) for cluster computation resources.
Author information
Authors and Affiliations
Contributions
Y.Z. designed and synthesized the UltraSelex and other HTS libraries and developed and carried out rank modeling analysis. Y.Z., Y.J. and S.F. synthesized and purified RNA. Y.Z., Y.J., S.F. and K.W. measured ligand affinities. D.K., Y.Z. and Y.J. conducted protein purification and inhibition activity assays. W.H., S.D., H.W., Y.Z., Q.Y. and Z.T. contributed to the web server construction and code review. W.L. and Y.Z. simulated RNA–ligand interaction. S.F. and J.Z. measured the fluorescence turn-on. S.F. imaged RNA in live cells. P.D. synthesized the biotinylated mock-bead compound. D.I. multiplexed and supported sequencing. Y.Z. and A.J. conceptualized the study, wrote the original draft of the manuscript and provided supervision. All authors did the reviewing and editing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
Y.Z. has filed a patent application (PCT/CN2023/099222) relating to this work. A.A.H. is a founder of Dewpoint Therapeutics and a member of the board as well as a shareholder in Caraway Therapeutics. The other authors declare no competing interests.
Peer review
Peer review information
Nature Chemical Biology thanks Jia Song, Julian Tanner and William Thiel for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Design features of the UltraSelex RNA libraries.
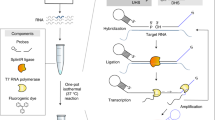
a, Conventional SELEX scheme. The input dsDNA pool (round 0) is in vitro transcribed into RNA to yield the initial RNA pool, which is incubated with the immobilized target. After removal of unbound RNA by washing, the bound fraction is eluted, reverse-transcribed into cDNA and PCR-amplified. The obtained enriched dsDNA pool is used as input for the next iteration, and this cycle is repeated until binders dominate. Binders are then identified, based on their abundance, by sequencing. b, Design of the partially structured RNA pool used in all UltraSelex experiments reported here, and stepwise processing to yield the barcoded sequencing library. The RNA pool contained two constant primer-binding regions (purple and blue)17, two randomized stretches of 26 nt each (N26) and a constant 12 nt internal hairpin (H12) thought to increase the abundance of high-affinity aptamers in the starting pool16. An aliquot of the same RNA pool was used previously for the isolation of SiR-binding aptamers by conventional SELEX18. The offset PCR system was designed to reduce bias in the sequencing of templates with variable and constant regions, thereby achieving balanced per-base nucleotide compositions46. Equimolar amounts of seven forward primers (Frw) and seven reverse primers (Rev) containing 0–6 inserted offset nucleotides (golden) were mixed and used for PCR amplification, generating length-heterogenous sequencing libraries. After offset PCR with the primer mixture, the Illumina primers, indices and barcodes were attached in another PCR reaction (barcode PCR). Before sequencing, a custom balancer oligonucleotide was added. For sequences, see Supplementary Table 2. c, Detailed architecture of the complete sequencing template.
Extended Data Fig. 2 Features and application of the SGREELI algorithm.
a, Number of HTS reads per RNA species in the UltraSelex input library. Three independent replicates. b, Decreasing sequence diversity with increasing group number e1–11 in the SiR-B UltraSelex library. The x axis indicates the log2 of the number of HTS reads per species, while the y axis denotes the number of RNA species with the specified abundance. The partitioning eluate (e) number is individually denoted. c, Percentage of RNA species with ≥2 HTS reads in the different partitioning groups (e1–11) of UltraSelex library SiR-B. The dashed gray line represents 1% as a reference. d, Formula and simulation of γ functions, assigning γi values to different partitioning groups gi for i ≥ 2. c = 0.5, eluate_nums, number of partitioning eluates (here 11). Colors in the line plot represent different exponents θ from 0.1 to 10. e, Illustration of the SGREELI calculation protocol, comprising seven steps. After sequencing and quality control, fold-change (FC) values were calculated for each eluate (e2–11 for FC2–FC11) by normalizing read counts to their abundance in eluate e1. An RNA species that ranks at exactly the top 1% by FC value in each eluate is defined as the reference RNA (FCref), with this step performed for each eluate group (ref2–ref11). The fold-change values of these reference RNAs are log2-transformed (log2 FCref). Each eluate group is then assigned a specific γ value, here based on a linear function with exponent θ = 1, reflecting the expected elution patterns of strong binders in later fractions. The log2 FCref value is then divided by γ to derive a normalization factor for each eluate group. All RNA species in a given eluate group have their FC values log2 transformed and adjusted by this normalization factor, yielding intra- and inter-group adjusted fold-change (aFC) values. Finally, the area under the curve (AUC) is computed by integration of the aFC values for each species, using the corresponding γ values as the x-axis parameters.
Extended Data Fig. 3 Partitioning effects during the selection process.
a, Spike-in SiR binder RNA and non-binding RNA were enriched differently under various washing conditions. Sample preparation followed Fig. 2b. Washing condition: ‘1× ASB’ (20 mM HEPES (pH 7.4), 125 mM KCl and 5 mM MgCl2), ‘HEPES only’ (20 mM HEPES (pH 7.4)); and ‘HEPES + EDTA’ (20 mM HEPES (pH 7.4) and 5 mM EDTA18). Partitioning included 14 wash eluates (e1–14) and a final release eluate (e15). RT–qPCR signals were normalized to initial abundance prior to selection. Continuous bands denote standard deviation, with the mean at the center (n = 3). b, Box plot showing the abundance of previously SELEX-identified aptamers in UltraSelex libraries. The x axis indicates partitioning groups, while the y axis denotes species abundance in reads per million qualified reads (RPM). Each colored dot or triangle represents a previously identified aptamer: red dots indicate RNA species with higher abundance than SiR1, while gray triangles represent those lower or equal to SiR1. The percentage of gray dots in each eluate is shown above. SiR-A (n = 39), SiR-B (n = 37) and SiR-C (n = 38). The box plots are defined as in Fig. 2f. c, Abundance of three known SiR-binding aptamers in UltraSelex sequence data from SiR-A and SiR-C, as in Fig. 2d. d, Plot of the aFC values of the same three aptamers, as in Fig. 2c. e, Rank distribution of turn-on ('On') and non-turn-on ('Non') SELEX RNA aptamers in UltraSelex libraries. The y axis represents aptamer rank in each library. Turn-on and non-turn-on groups are shown in purple and gray, respectively. SiR-A (n = 39), SiR-B (n = 37) and SiR-C (n = 38). Box plot definition follows b. f, Rank analysis of SiR-A and SiR-C UltraSelex and SELEX libraries, following Fig. 2f. SiR-A: e1–11 and AUC (n = 39), On (n = 19), Non (n = 20). SiR-C: e1–11 and AUC (n = 38), On (n = 19), Non (n = 19). SELEX: r4–14 and AUC (n = 39), On (n = 19), Non (n = 20). The box plots are defined as in Fig. 2f.
Extended Data Fig. 4 Analysis of SiR UltraSelex aptamers.
a, Fluorescence turn-on of a silicon rhodamine dye by the 25 top-ranked RNA aptamers from UltraSelex SiR-A (left) and SiR-C (right). Error bars represent the mean ± SD from technical triplicates. All other details are as in Fig. 3d. b, The measured turn-on fluorescence and KD values of six UltraSelex SiR-B RNA aptamers with binding affinities in the nanomolar range. c, Quantification of the SiR fluorescence intensity of 50 aptamer-transfected HEK-239T cells with plasmids encoding self-circularizing (Tornado) overexpressed RNA constructs of SiR-B7 and SiR-B15 aptamers and SELEX-derived SiR-binding aptamer18 in comparison with a SiR-unrelated insert47. The confocal imaging procedure was similar to that in Fig. 3f, except the cells were incubated with 1 µM SiR-(PEG)2-NH2 for 45 min. The cytosolic fluorescence intensity of aptamer-transfected cells (red dots) was compared with that of control cells (gray dots). Control cells were transfected with the same plasmid type, carrying the SiR-unrelated RNA insert. Fluorescence values were compared between the groups by p values: ****p(SiR-B7/control) = 2.42 × 10−12 and ****p(SiR-B15/control) = 0.0001 and ****p(SELEX/control) = 1.73 × 10−17 using a two-sided Mann–Whitney U test. The box plots are defined as in Fig. 2f. d, Confocal images of live HEK-239T cells transfected with plasmids encoding the aptamers in c. The punctuate fluorescence is likely due to lysosomal accumulation of the dye over time. The image channels were identical to those in Fig. 3f. Scale bars = 10 µm. e, Sankey plot displaying the enrichment route of the top-ranked SiR-A and SiR-C UltraSelex aptamers. All details are as in Fig. 3g.
Extended Data Fig. 5 Enrichment of RNA aptamers targeting nsp12 in UltraSelex libraries.
a, BLI KD measurement for the reaction mixture between RNA starting pool and nsp12 proteins. b, Percentage of non-stochastic RNA species (≥2 HTS reads) in the different partitioning groups (e1–11) of UltraSelex libraries Nsp-A, Nsp-B and Nsp-C. PCR cycles were normalized. The dashed gray line represents 1% as a reference. c, Plot of the aFC values of three representative nsp12 aptamers, calculated using a top percentile value of 1.0% and a linear γ function. All details are as in Fig. 2e, but using nsp12 UltraSelex RNA aptamers. d, Sankey plot displaying the enrichment route of the top-ranked nsp12 UltraSelex aptamers. In the upper part, column AUC represents the top 0.001% of the sequences ranked by SGREELI (golden bucket), while columns e1–11 rank sequences by read abundance (golden: top 0.001%; orange: sequence observed, but ranked below 0.001%, dark green: sequences not observed in that group. The gray lines indicate the flow of the sequences between the nodes. In the magnified bottom part, the yellow bucket stands for the top 25 sequences, the gold for the top 25–top 0.001% and the orange and dark green buckets are as in the upper part.
Extended Data Fig. 6 RNA aptamers in UltraSelex and SELEX libraries.
a, Presence of the 47 UltraSelex top aptamers (representing the top 25 from SiR-A, SiR-B and SiR-C each) in the final round of the conventional SELEX library18. Only 5/47 sequences were found (pie chart) with ranks (by read abundance) between 3 and 484 (box plot). The box pots are defined as in Fig. 2f. b, BLI KD measurement for the reaction mixture between the RNA from SELEX round 11 pool and nsp12 proteins. The concentration of RNA pool was 1000 nM. c, The rank order distribution of the 76 high-affinity RNA aptamers in three UltraSelex libraries (all 76 species found) vs three SELEX libraries against nsp12 (9 species were absent in SELEX-A, 11 in SELEX-B and 11 species in SELEX-C, ranked by their abundance after 11 rounds of selection). The box pots are defined as in Fig. 2f.
Extended Data Fig. 7 Optimized SiR UltraSelex ranking, taking into account mock-bead binding.
a, Chemical structure of the SiR original (blue and black part) and mock (black part only) compound for RNA aptamer selection. b, High correlation of the normalized AUC values among the UltraSelex experimental replicates against the SiR mock target. The AUC values from the top 2000 species of each replicate were analyzed. The raw AUC values were normalized by the min–max method using (AUC − min_AUC)/(max_AUC − min_AUC). Each species is denoted by a blue dot, and the reference line x = y = z is represented by the dashed gray line. c, Fraction of identical sequences between the triplicate UltraSelex datasets from b with increasing top rank of aptamers. d, Comparison of the rank order distribution of turn-on ('On') and non-turn-on ('Non') SELEX RNA aptamers in the UltraSelex libraries before (w/o mock) and after (mock) computational filtering for mock-bead binding. The RNA species were ranked by the ratio of the normalized AUC in SiR-B to SiR mock target. The y axis represents the rank order of the RNA aptamer in the specified UltraSelex library. The group of turn-on and non-turn-on RNA aptamers are shown with a purple box and a gray box, respectively. SiR-A (n = 39), SiR-B (n = 37) and SiR-C (n = 38). The box pots are defined as in Fig. 2f. e, Venn diagram showing the intersection of the 25 top-ranked RNA aptamers derived from the three mock-bead-optimized UltraSelex experiments SiR-A, SiR-B and SiR-C.
Extended Data Fig. 8 Characterization of SiR RNA aptamers by focused UltraSelex.
a, Consensus secondary structure of the SiR-A-like species in the top 7500 UltraSelex SiR-B library, containing the tetranucleotide UGAA (purple) in the loop and UAUC (green) in the bulge. In total, 1293 species harbor both tetranucleotides. b, Revised consensus secondary structure of the predominant species with a three base-pair stem embedding the UAUC tetraloop. c, Distribution of lengths and base pairing types of the stem embedding the UAUC loop of the RNA species in a and b. ‘|’ indicates a base pair (A–U, G–C and G–U), while ‘.’ symbolizes unpaired bases. The predominant stem type with three base pairs and two/four base pairs are shown in light and dark green, respectively, while the minor variants are depicted in blue. d, Detailed sequence information for the subgroup with a 3 bp-stem in c. Wobble pairs (G–U) are denoted by red color. ‘#’ symbolizes the UAUC loop. e, Correlation between the length of the stem embedding the ‘UAUC’ and the measured turn-on fluorescence and KD values. Seven UltraSelex SiR-B RNA aptamers containing the ‘UAUC’ motif are depicted with their ranking order. Symbols indicate different stem types. KD values above 2000 nM are shown as ‘2000 nM’. f, Abundance distribution of the UltraSelex SiR-B top 7500 RNA species in the mutagenized focused UltraSelex library. The left box plot panel illustrates (in triplicates) the distribution of these 7500 progenitor species from the original synthesized DNA library (input DNA) to mutagenesis PCR products, Taq PCR products to in vitro transcribed RNA. The right box plot panel shows the abundance of these progenitor species in the focused UltraSelex partitioning eluates (e1–11). The box pots are defined as in Fig. 2f.
Extended Data Fig. 9 Characterization of Nsp RNA aptamer binding features.
a, The CUUGA motif is the most abundant pentamer in the random region of 45 high-affinity Nsp aptamer (KD < 10 nM). b, Consensus secondary structure of four main binding subtypes for nsp12 aptamers using LocARNA26 and RNAalifold27. Degenerate bases: R(A/G), Y(C/U), M(A/C), K(G/U), S(G/C), W(A/U), H(A/T/C), B(G/T/C), V(G/A/C) and D(G/A/U). Base-pair types are color-coded; lowercase letter indicates a gap at the alignment position with a low frequency. Subtype 1: Nsp-B5, B6, B19, B21, B43, B63, B137 and B138; subtype 2: Nsp-B9, B59, B71, B76, B100 and B113; subtype 3: Nsp-B2, B8, B28, B39, B58 and B69; subtype 4: Nsp-B16, B30, B32, B68 and B79. c, Predicted secondary structures of picomolar binder Nsp-B2 (KD = 827 pM, RdRp inhibitor) and Nsp-B58 (KD = 892 pM, RdRp non-inhibitor) using RNAfold38 with default parameters. Both belong to subtype 3 in b. The right panel highlights a structural difference in Nsp-B58 caused by a single-nucleotide deletion at position 79 in Nsp-B2. d, Illustration of classical Nsp-B113 aptamer (subtype 2 in b) truncation. The head region (113-54H) is shaded in red, while the tail region (113-51T) is enclosed by a red circle frame. The overlap area contains both features. e, Nsp-B113 active truncation variant Nsp-B113-56H+T (56 nt), concatenating the head and tail region, retains nanomolar binding affinity (KD = 3 nM). Removing parts of primer A (‘AGCUCAGCCUUCACUGC’, Nsp-B113-56H + T) or B (‘UCGGAUCCAC’, Nsp-B113-50H+T, KD = 4 nM in Fig. 5f) maintains nanomolar affinity. Unlike SiR aptamers, where primer A generally pairs with primer B (Extended Data Fig. 8a,b), in nsp12 aptamers primer A and primer B typically pair with parts of the randomized region (d; Supplementary Fig. 6a). f, BLI KD measurements for Nsp-113 truncation as described in Fig. 5c.
Extended Data Fig. 10 Discovery of 2′-F-modified RNA aptamers that inhibit HIV-1 reverse transcriptase by UltraSelex.
a, Inhibition of template extension by HIV-1 reverse transcriptase (RT, HXB2; Addgene, 153311) using UltraSelex-derived 2′-F-modified RNA aptamers. An 18-nt fluorescent 5′-Cy3-labeled DNA primer (10 nM) and 31-nt DNA template (100 nM)31 were incubated with the indicated 2′-F CTP/UTP-modified RNA aptamer (10 nM) and RT (10 nM) for 15 min. Controls included reaction without RT (-Enzyme), RNA aptamer (PC), or dNTP (-dNTP), while the reported unmodified strong broad spectrum of RT inhibitor unmodified RNA 70N89 (ref. 31) served as reference. Selected 2′-F-modified RNA aptamers containing GCCU and CGGG motifs32 were designated as the ‘cg’ group. b, Quantification of the inhibition effect from a. The gray dashed line represents the inhibition level of 70N89 aptamer. RNA aptamers with more than 10% inhibition (high) are marked with an asterisk (*), while those with similar inhibition to 70N89 were classified as ‘intermediate’ group. Error bars represent the mean ± s.d. from technical triplicates. c, Consensus secondary structure of RNA aptamers with ‘high’ inhibition in b using LocARNA26 alignment with default parameters. Degenerated bases are annotated as in Extended Data Fig. 9b. d, Alignment annotated with the LocARNA26 consensus structure of the ‘high’ (top) and ‘intermediate’ group (bottom) from b. The percentage of inhibition higher than 70N89 is shown in the last column. e, Predicted secondary structure of the RNA aptamer RT-F6, which exhibited the highest inhibition capability, using Mfold42 with default parameters. f, Box plot distribution of normalized AUC values of RT RNA aptamers from b. The AUC value with the weight of heptamer was min–max normalized into range between 0 and 1. The box plot is defined as in Fig. 2f.
Supplementary information
Supplementary Information
Supplementary Figs. 1–6, Supplementary Tables 1–5, Supplementary Methods and Supplementary Notes 1 and 2.
Supplementary Data 1
Statistical supporting data for Supplementary Figs. 1–6.
Supplementary Data 2
Gel image supporting data for Supplementary Fig. 3e,f.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 3
Microscopy images for Fig. 3f.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 4
Gel images for Fig. 4g,i.
Source Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 4
Microscopy images for Extended Data Fig. 4d.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 9
Statistical source data.
Source Data Extended Data Fig. 10
Statistical source data.
Source Data Extended Data Fig. 10
Gels images for Extended Data Fig. 10a.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, Y., Jiang, Y., Kuster, D. et al. Single-step discovery of high-affinity RNA ligands by UltraSelex. Nat Chem Biol 21, 1118–1126 (2025). https://doi.org/10.1038/s41589-025-01868-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41589-025-01868-6
This article is cited by
-
Sorting high-affinity aptamers in a single selection round
Nature Chemical Biology (2025)
