
Abstract
Lysosomes, central hydrolytic organelles, are regulated by ion flow, including calcium and protons, via transporters and channels to maintain an acidified lumen for hydrolytic activity. TRPML1, a lysosomal ion channel, effluxes cations upon activation, promoting rapid conjugation of ATG8 proteins to the lysosomal membrane in a process known as conjugation of ATG8 to single membranes (CASM). However, our understanding of how TRPML1 activation reorganizes the lysosomal proteome is poorly understood. Here, we identify DMXL1 as a key regulator of lysosomal homeostasis through quantitative proteomics of lysosomes during TRPML1 activation by the agonist MLSA5. DMXL1 is recruited to lysosomes and Salmonella-containing vacuoles, both in a CASM-dependent manner. As the mammalian ortholog of yeast Rav1, DMXL1 assembles with Rav2 ortholog ROGDI and WDR7, and associates with V0 and V1 subunits of the lysosomal V-ATPase. TRPML1 activation drives V1 subunit recruitment to lysosomes in a DMXL1- and DMXL2-dependent manner. DMXL1- and DMXL2-deficient cells display reduced V1-ATPase recruitment, increased lysosomal pH and diminished hydrolytic capacity. Using AlphaFold modeling supported by cross-linking proteomics, we identify interaction interfaces within the DMXL1–ROGDI–WDR7 complex, as well as an ATP6V1A binding interface in DMXL1, whose mutation affects interaction and function. Our findings suggest CASM-dependent DMXL1 recruitment, coupled with V-ATPase assembly, is critical for maintaining lumenal pH and lysosomal function in response to TRPML1 activation.
Similar content being viewed by others


Main
Lysosomes are crucial membrane-bound organelles that support proteostasis in eukaryotic cells through degradation of intracellular cargo, including damaged organelles and protein aggregates1. Optimal lysosomal hydrolytic activity depends on maintenance of an acidic lumenal pH between 4.5 and 5 (ref. 2). Acidification is driven, in part, by functionally assembled V-ATPases, which use ATP hydrolysis to translocate protons into the lumen2. The assembly of a membrane-embedded V0 complex and a cytosolic V1 complex results in a functional V-ATPase2. Alongside the V-ATPase, lysosomal transporters and channels have been identified to contribute to the stabilization of the lysosomal proton gradient through dynamic influx and efflux of metabolites and ions across the lysosomal membrane3,4.
Transient receptor potential channel mucolipin (TRPML) proteins are endolysosomal cation channels that efflux mono- and divalent cations into the cytosol upon activation5,6,7. TRPML channel activity is involved in various cellular processes, from endolysosomal trafficking to transcriptional activation3,8,9,10,11. Of the three human TRPML genes (MCOLN1–MCOLN3), MCOLN1 (or TRPML1) is the most broadly expressed and best studied, and mutations in this gene cause mucolipidosis type IV, a lysosomal storage disease associated with muscular dystrophy and neurodegeneration12. TRPML1 forms a functional channel as a tetramer13,14,15 and is predominantly localized on lysosomes where phosphatidylinositol 3,5-bisphosphate (PI(3,5)P2), an endogenous activator, is enriched16,17,18,19.
Previous studies have shown that activation of TRPML1 by agonists, such as MLSA5, induces conjugation of ATG8 proteins (in the LC3 and GABARAP family) to the cytosolic face of the lysosomal membranes—a process termed CASM. CASM employs the ATG8-conjugation system from canonical autophagy but circumvents the upstream ULK1–FIP200–ATG13 and VPS34 kinase complexes20,21. ATG8ylation of lysosomes and other vesicles is implicated in TFEB activation, STING signaling and bacterial vacuole formation8,22,23,24,25,26,27,28. Evidence indicates CASM is triggered, upon proton gradient dissipation, through the interaction between the ATP6V1H subunit and the WD40 ___domain of ATG16L1, a core component of the ATG8 conjugation machinery29,30,31,32,33,34,35,36.
Despite these advances, how specific signals, such as cation efflux, drive CASM or organelle homeostasis remains unclear20. Here, we combined lysosome immunoprecipitation (Lyso-IP)37 with quantitative proteomics to identify proteins recruited to lysosomes upon TRPML1 activation. Among them was DMXL1 (DmX-like protein 1), an ~340-kDa protein with three WD40 domains and a helical region, which are conserved with its paralog DMXL2. In yeast, its ortholog Rav1 is part of the regulator of the H+-ATPase of vacuolar and endosomal membranes (RAVE) complex, which regulates assembly of the V-ATPase through unclear mechanisms38,39,40,41. In mammals, the DMXL1 coprecipitates with V-ATPase components42,43,44,45, and its depletion by RNA interference (RNAi) impairs lysosomal re-acidification following bafilomycin A1 (BafA1) washout42. However, its role in V-ATPase assembly remains poorly defined.
Under basal conditions, we find that DMXL1 localizes to the cytosol and Golgi. However, upon TRPML1 activation, DMXL1 rapidly translocates to lysosomes in a CASM-dependent manner. Interaction proteomics revealed DMXL1’s assembly with ROGDI (Rav2 in yeast) and WDR7, consistent with previous studies44, and cross-linking proteomics supports structural models of the DMXL1–ROGDI–WDR7 complex predicted by AlphaFold 3 (AF3)46. Additionally, we observed interaction between DMXL1 and V-ATPase subunits. AF3 predicts DMXL1 binding to ATP6V1A through its Rav1-C ___domain, and substitutions at this interface disrupt interaction and lysosomal hydrolase processing. DMXL1 and DMXL2 (DMXL1/2) loss impairs recruitment of V1 subunits to lysosomes upon TRPML1 activation, leading to defects in lumenal pH, lysosome hydrolase processing and cargo degradation. Collectively, these data position DMXL1 as a key component of V-ATPase trafficking and assembly machinery and reveal that its recruitment of V1 subunits is essential for maintaining acidic lysosomal function in response to TRPML1 activation.
Results
TRPML1 activation recruits DMXL1 to lysosomes
To capture changes to the lysosomal proteome upon TRPML1 activation, we used U2OS cells as our cellular model because TRPML1 is the predominantly expressed TRPML channel47. Indeed, TRPML1-deficient cells failed to induce LC3B lipidation, a marker for CASM, and to express the TFEB-target gene GPNMB22,48 upon treatment with MLSA5 for 24 h (Extended Data Fig. 1a,b). This suggests that the agonist MLSA5 is on-target for TRPML1. We generated U2OS cells stably expressing the Lyso-IP affinity tag TMEM192-mRFP-3×HA and performed a time-course experiment in which cells were either untreated or treated with MLSA5 for 0.5, 1, 2 or 4 h, and Lyso-IP samples were analyzed by Tandem Mass Tag (TMT)-based proteomics (Fig. 1a,b, Extended Data Fig. 1c and Supplementary Table 1). As expected, ATG8 proteins (GABARAP, GABARAPL1, GABARAPL2 and MAP1LC3B2, subsequently referred to as LC3B), as well as ATG5, a component of the ATG8ylation machinery, were among the most highly enriched proteins on lysosomes at all four time points (Fig. 1b and Extended Data Fig. 1c), consistent with MLSA5-dependent activation of CASM. DMXL1 was also enriched on lysosomes, with consistent levels throughout the time course as indicated by the ion intensity of the TMT reporter (Extended Data Fig. 1d). DMXL1, and its paralog DMXL2, are thought to represent mammalian orthologs of Rav1 in budding yeast40,49,50. Similar enrichment of the ATG8 conjugation machinery and DMXL1 were observed in A101D melanoma cells treated with MLSA5 (Extended Data Fig. 1e,f and Supplementary Table 2).

a, Scheme depicting the experimental design for identification of proteins recruited to lysosomes in response to TRPML1 activation. n = 3 for each condition. b, Volcano plot (log2(FC) versus –log10(q)) for Lyso-IP from U2OS cells that were treated with MLSA5 for 0.5 h or were untreated. ATG8 proteins are indicated by red circles. DMXL1, DMXL2, ROGDI and WDR7 are indicated by blue circles. Autophagy proteins are indicated by yellow circles. c, Schematic outlining experimental conditions, and the number of replicates, to determine ATG16L1-dependent lysosomal proteome changes during TRPML1 activation by MLSA5 (2 h). d, Volcano plot (log2(FC) versus –log10(q)) for Lyso-IP from WT or ATG16L1-KO U2OS cells treated with MLSA5 for 2 h compared with untreated cells. ATG8 proteins are indicated by red circles, and DMXL1 and WDR7 are indicated by blue circles. e, Immunoblotting validation of MLSA5-dependent recruitment of DMXL1 and LC3B-II to lysosomes isolated by Lyso-IP. MLSA5 treatment was 2 h. Whole-cell extracts are included as controls. The blot is representative of three independent experiments. Also see Extended Data Figures 1 and 2.
CASM-dependent DMXL1 recruitment to lysosomes
We next examined the potential role of CASM in DMXL1 recruitment to lysosomes in response to TRPML1 activation20,22. A previous study8 reported that in cells lacking ATG7, which encodes an essential component of the ATG8ylation machinery, treatment with the TRPML1 agonist MLSA1 failed to lead to a robust increase in the cytosolic Ca2+ concentration, as measured by Fura-2 calcium imaging. Such a dependence of Ca2+ efflux from TRPML1 on ATG7 (and presumably ATG8ylation) would confound any analysis of a role for CASM in processes downstream of TRPML1 activation. Therefore, to directly examine whether the absence of the ATG8ylation machinery affects TRPML1 activation, we generated ATG16L1-knockout (ATG16L1-KO) U2OS cells (Extended Data Fig. 2a,b) and used organelle electrophysiology to measure current activation by the TRPML1 agonist MLSA5 on endolysosomes isolated from both wild-type (WT) and ATG16L1-KO cells. Our recordings revealed no significant difference in MLSA5-activated currents between both types of endolysosomes (Extended Data Fig. 2c,d and Supplementary Table 3), consistent with previous findings using a different TRPML1 agonist, C8 (ref. 21). Thus, mutations in the ATG8ylation machinery can be used to examine downstream effects of cation efflux mediated by TRPML1.
We investigated whether ATG16L1 is required for DMXL1 enrichment on lysosomes in response to TRPML1 activation as an initial test of CASM involvement. We performed proteomic analysis on Lyso-IP samples from WT and ATG16L1-KO cells treated with MLSA5 (2 h) (Fig. 1c, Extended Data Fig. 2e,f and Supplementary Table 1). Whereas ATG8 and DMXL1 were enriched on lysosomes in WT cells, these proteins were not enriched on lysosomes from ATG16L1-KO cells (Fig. 1d,e and Extended Data Fig. 2e,f). Consistent with the loss of CASM in ATG16L1-KO cells, immunoblotting revealed that LC3B-II was not generated upon TRPML1 activation, and proteomics indicated that ATG8 proteins were not enriched (Fig. 1d,e and Extended Data Fig. 2b). Thus, ATG8 and DMXL1 enrichment on lysosomes in response to TRPML1 activation displayed the expected dependencies in a correlation plot of ATG16L1 dependence versus lysosomal enrichment (Extended Data Fig. 2e).
We conducted confocal microscopy to examine the spatial parameters associated with DMXL1 recruitment to lysosomes following TRPML1 activation. Quantitative proteomics in several cell lines indicated that the DMXL1 protein copy number is low (8,600 molecules per cell in HeLa cells)51,52. Therefore, to examine the dynamics of DMXL1 recruitment, we stably expressed DMXL1–mNeonGreen (DMXL1–mNG) in either WT, ATG16L1-KO or MCOLN1-KO cells (Fig. 2a–d and Extended Data Fig. 3a). Under basal conditions, DMXL1–mNG exhibited a largely diffuse distribution in the cytoplasm, with noticeable puncta overlapping with a subset of mScarlet-LAMP1, a marker for endolysosomes (Fig. 2a). However, in WT cells, the addition of MLSA5 led to rapid accumulation of DMXL1–mNG on mScarlet–LAMP1-positive structures, as revealed by live-cell imaging, with the mean fluorescence intensity of mNG on these structures peaking within 10 min (Fig. 2a,e and Supplementary Video 1). This effect reflected the stimulation of cation efflux by MLSA5 because cells lacking TRPML1 failed to recruit DMXL1–mNG to lysosomes (Extended Data Fig. 3a,b).

a,b, Live-cell imaging of WT (a) or ATG16L1-KO (b) U2OS cells expressing DMXL1–mNG and mScarlet–LAMP1 to mark lysosomes upon treatment with MLSA5. Images taken at 2-min intervals for 10 min are shown. c, As in a, but after pretreatment with BafA1 to block CASM. d, Live-cell imaging of WT U2OS cells stably expressing DMXL1–mNG and mScarlet–LAMP1 to mark lysosomes, upon treatment with the mTOR inhibitor, Torin1. Images taken at 2-min intervals for 10 min are shown. e, Fold change in DMXL1–mNG mean fluorescence intensity over 20 min. Quantification was performed on cells from three biological replicates: WT, n = 21; ATG16L1 KO, n = 17; pretreatment BafA1, n = 12; Torin1, n = 16; pretreatment BFA, n = 11. Data are presented as mean values ± s.d. Two-way repeated measures ANOVA with the Geisser–Greenhouse correction followed by Dunnett’s multiple comparison test was used to assess significance. The indicated statistical significance is for the 20-min timepoint. ****P ≤ 0.0001; n.s., not significant. Scale bar, 10 µm; inset scale bar, 3 µm. f, Immunoblots from ATG16L1-KO U2OS cells reconstituted with either WT ATG16L1 or ATG16L1-K490A, with or without MLSA5 (2 h), were probed with the indicated antibodies. α-GAPDH was used as a loading control. The blot is representative of three independent experiments. g, The indicated cell lines, with or without MLSA5 (4 h), were fixed before confocal microscopy to detect colocalization of DMXL1–mNG and LAMP1. Scale bar, 10 µm; inset scale bar, 3 µm. h, Quantification of colocalization between α-LAMP1 for lysosomes and DMXL1–mNG using Mander’s colocalization coefficient for data in g. Quantification was performed on the number of cells indicated on the plot, from three independent experiments. Boxes show the 25th to 75th percentiles, whiskers extend from minimum to maximum values and the line represents the median. Statistical significance for each cell line was assessed by unpaired two-sided Student’s t-test. ****P ≤ 0.0001. Also see Extended Data Figures 3 and 4 and Supplementary Video 1.
Several additional lines of evidence indicate that DMXL1 recruitment to lysosomes requires CASM. First, we found that cells lacking ATG16L1 were defective in DMXL1–mNG recruitment to lysosomes in live cells (Fig. 2b,e and Supplementary Video 1). Second, pretreatment of WT cells with BafA1, which is known to block CASM20, resulted in complete loss of DMXL1–mNG recruitment to lysosomes (Fig. 2c,e). Third, to directly examine the specificity of CASM in DMXL1 recruitment, we reconstituted ATG16L1-KO cells with WT ATG16L1 or the CASM-defective ATG16L1K490A mutant (Fig. 2f). Consistent with a role for CASM, expression of WT ATG16L1, but not ATG16L1K490A, significantly increased DMXL1–mNG colocalization with lysosomes upon TRPML1 activation (Fig. 2g,h). As expected, we found that WT ATG16L1, but not ATG16L1K490A, rescued MLSA5-dependent LC3B lipidation (Fig. 2f). Furthermore, we found that the mTOR inhibitor Torin1, which induces canonical autophagy, does not induce rapid DMXL1–mNG translocation to lysosomes, consistent with the idea that DMXL1 recruitment is not a general response to alterations of lysosomal signaling (Fig. 2d,e). This result contrasts with the conclusions of a previous study53.
To further validate these results, we assessed the subcellular localization of endogenous DMXL1; however, commercially available antibodies to endogenous DMXL1 were unreliable for immunofluorescence microscopy. Therefore, we gene-edited U2OS cells to add a hemagglutinin (HA)-dTAG to the amino terminus of DMXL1 (Extended Data Fig. 4a–c). Immunostaining with an antibody to HA showed cytosolic staining, and some staining overlapped with LAMP1-positive puncta. The overlap was further enhanced upon TRPML1 activation (Extended Data Fig. 4d,e). We also observed colocalization of HA-dTAG–DMXL1 with TGN46- and GM130-postitive structures, indicating Golgi localization under basal and MLSA5-treated conditions (Extended Data Fig. 4d). Consistent with this result, U2OS cells expressing DMXL1–mNG also revealed a Golgi-associated pool of DMXL1–mNG when cells were permeabilized (Extended Data Fig. 4f), possibly through the loss of the cytosolic pool enhancing the signal from the Golgi-associated pool. This further raises the question of which pool(s) of DMXL1 might be mobilized for lysosomal recruitment during TRPML1 activation. To address the Golgi as a possible source, we disrupted Golgi trafficking by pretreatment with brefeldin A (BFA) and found that DMXL1–mNG retained the ability to translocate to lysosomes, albeit with a reduced mean fluorescence intensity (Fig. 2e and Extended Data Fig. 4g). We also found that pretreatment with nocodazole, which inhibits microtubule-based vesicular trafficking, did not prevent DMXL1–mNG recruitment to lysosomes in response to TRPML1 activation (Extended Data Fig. 4h). Immunoblotting of cytosol and membrane fractions from U2OS cells and DMXL1–mNG-expressing cells revealed the presence of both cytosolic and membrane-bound pools for both endogenous and ectopically expressed DMXL1, with levels increased in the membrane fraction in response to TRPML1 activation (Extended Data Fig. 4i). Taken together, these data indicate that TRPML1 activation results in a rapid recruitment of DMXL1 to lysosomes, primarily from a cytoplasmic pool, although a contribution of a Golgi pool cannot be fully ruled out.
Mobilization of DMXL1 to Salmonella-containing vacuoles
CASM can occur on diverse types of subcellular compartments, including a modified phagosome known as Salmonella-containing vacuoles (SCVs), which are formed upon entry of Salmonella into mammalian cells20,22,33. We therefore tested whether DMXL1–mNG is recruited to SCVs. U2OS cells stably expressing GFP–LC3B or DMXL1–mNG were infected with mCherry-expressing Salmonella and then analyzed by confocal microscopy (Fig. 3a–f). In WT cells, approximately 60% of SCVs colocalized with GFP–LC3B or DMXL1–mNG signals (Fig. 3a,c,d,f). We also assessed whether infection with Salmonella lacking the sopF gene (ΔsopF) enhanced DMXL1–mNG colocalization with SCVs, as has been previously reported for GFP–LC3B36. The sopF gene encodes a type III secretion system effector protein with ADP-ribosyltransferase activity targeting the ATP6V0C subunit. This disrupts ATG16L1-mediated ATG8ylation on SCVs36. As expected, the percentage of GFP–LC3B-positive SCVs increased in the context of ΔsopF Samonella, and a similar effect was observed for DMXL1–mNG, although it did not reach statistical significance (Fig. 3b,c,e,f). Consistent with a role for CASM, the recruitment of both GFP–LC3B and DMXL1–mNG to SCVs was largely blocked in cells lacking ATG16L1, but deletion of MCOLN1 did not alter this recruitment (Fig. 3a–f). In the absence of ATG16L1, the few SCVs that exhibited DMXL1 localization (Fig. 3f) could arise from ATG8 lipidation mediated by the recently identified TECPR1–ATG5–ATG12 complex54. Together, these results suggest that DMXL1 can be recruited to distinct endocytic compartments involving ATG8ylation.

a,b, U2OS cells of the indicated genotypes expressing GFP–LC3B were infected with mCherry-expressing WT (a) or ΔsopF (b) Salmonella (1 h) and then subjected to confocal microscopy to detect mCherry, GFP or immunofluorescence for LAMP1. Line traces (right side) indicate overlapping signals (AU, arbitrary units) for the indicated proteins or Salmonella. Scale bars, 10 µm. c, Percentage of SCVs positive for GFP–LC3B from n = 3 biological replicates. The total number of SCVs quantified: 157 in WT cells, 160 in ATG16L1-KO cells, 109 in MCOLN1-KO cells (a); 126 in WT cells, 102 in ATG16L1-KO cells, 103 in MCOLN1-KO cells (b). Data are presented as mean values ± s.d. For statistical comparison within each Salmonella strain, ordinary one-way ANOVA followed by Dunnett’s multiple-comparison test was used. For statistical comparison between WT and ΔsopF Salmonella, within each U2OS genotype, unpaired two-sided Student’s t-test was used. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001; n.s., not significant. d,e, U2OS cells of the indicated genotypes expressing DMXL1–mNG were infected with mCherry-expressing WT (d) or ΔsopF (e) Salmonella (1 h) and then subjected to confocal microscopy to detect mCherry, mNG, or immunofluorescence for LAMP1. Line traces (right side) indicate overlapping signals (AU, arbitrary units) for the indicated proteins or Salmonella. Scale bars, 10 µm. f, Percentage of SCVs positive for DMXL1–mNG from n = 3 biological replicates. Total number of SCVs quantified: 100 in WT cells, 91 in ATG16L1-KO cells, 98 in MCOLN1-KO cells (d); 106 in WT cells, 155 in ATG16L1-KO cells, 87 in MCOLN1-KO cells (e). Data are presented as mean values ± s.d. Statistical analysis was performed as described in c.
DMXL1 interaction proteomics
Previous studies have found that DMXL1 coprecipitates with ROGDI and WDR7, whereas WDR7 or ROGDI immune complexes contain DMXL1 and/or DMXL2 (refs. 42,44,55), leading to the conclusion that DMXL1–ROGDI–WDR7 is analogous to the Rav1–Rav2 V-ATPase chaperone in budding yeast40,42,44. Whereas DMXL1 and/or DMXL2 can be detected in immune complexes from a small subset of V1-ATPase subunits42,44,55, the DMXL1’s repertoire of known interactions is likely incomplete. Therefore, we initially used U2OS cells stably expressing DMXL1–mNG for interaction proteomics through tandem mass tag mass spectrometry (TMT-MS), with mNG as an affinity handle for the pull-down (Fig. 4a and Supplementary Table 4). In addition to ROGDI and WDR7, as expected, this analysis revealed the presence of all eight subunits of the soluble V1-ATPase complex, as well as A1–A3, C, D1 and ATP6AP1 subunits of the membrane-embedded V0-ATPase complex, which together form the V-ATPase assembly (Fig. 4b). Previous studies have demonstrated that endogenously tagged DMXL1 complexes contain WDR7 and ROGDI, but V-ATPase subunits were not substantially enriched44. Similarly, affinity purification of endogenously tagged HA-dTAG–DMXL1, followed by proteomic analysis, revealed an enrichment of WDR7 and, to a lesser extent, ROGDI (Fig. 4c and Supplementary Table 14). As in earlier studies44, the detected V-ATPase subunits displayed a rightward skew in the volcano plot, but did not reach statistical significance as enriched proteins (Fig. 4c and Supplementary Table 4). Notably, the copy number of components in the DMXL1/2 complex in U2OS or HeLa cells is >15-fold lower than the average V1-ATPase subunit copy number51,52, which could complicate the detection of V1-ATPase subunits in association with DMXL1 at endogenous levels through immunoprecipitation. We suggest that ectopic expression of DMXL1 facilitates detection of associated V-ATPase subunits, and moreover that the presence of all subunits suggests that DMXL1 can associate with a fully intact V1-ATPase complex in the context of detergent extracts (Fig. 4a,b).

a, U2OS cells with or without stable expression of DMXL1–mNG were subjected to immunoprecipitation using a nanobody against mNG, followed by TMT-MS analysis (3 biological replicates per condition). Volcano plot (log2(FC) versus –log10(q)) for DMXL1–mNG complexes relative to cells not expressing DMXL1–mNG is shown, with V-ATPase subunits shown in red and DMXL1–ROGDI–WDR7 proteins in blue. b, Summary of V-ATPase and other subunits identified in association with DMXL1–mNG. c, Isolation of HA–dTAG–DMXL1 at endogenous levels. Cells gene-edited to express an N-terminal HA–dTAG-tagged DMXL1, along with untagged cells, were subjected to α-HA immunoprecipitation, followed by TMT-MS analysis (3 biological replicates per condition). DMXL1 and WDR7 were enriched, and V-ATPase was not significantly enriched. d, Domain organization of Rav1 and Rav2 proteins from budding yeast and DMXL1/2, ROGDI and WDR7 in humans. The ___location of the Rav1-C ___domain identified in PFAM is shown. e, AF3 models for budding yeast Rav1–Rav2 (left), DMXL1–ROGDI (middle) and DMXL1–ROGDI–WDR7 (right). The locations of Lys–Lys cross-links identified in DMXL1–ROGDI–WDR7 complexes immunoprecipitated from U2OS cells stably expressing DMXL1–mNG (Supplementary Table 4) are indicated by red lines, for cross-links within structured elements, and cyan lines, for cross-links on linear segments. The ___location of cross-links between DMXL1–ROGDI and ROGDI–WDR7 are indicated by red and black arrows, respectively. ipTM scores for pairwise combinations are indicated. Also see Extended Data Figure 5 and Supplementary Video 2. f, Summary of inter- and intra-protein cross-links for DMXL1–mNG–ROGDI–WDR7 complexes isolated from U2OS cells. The legend indicates the categories of cross-links identified. Positions of cross-links (XL) and distances within AF3 predictions are listed in Supplementary Table 4. g, Summary of inter- and intra-protein DSSO cross-links involving Dmxl2, Rogdi or Wdr7 identified in mouse synaptic vesicles from a prior study59, mapped on the AF3-predicted structure. The legend in f also applies to g. Positions of cross-links and distances within AF3 predictions are provided in Supplementary Table 4.
Molecular architecture of the DMXL1/2-ROGDI-WDR7 complex
Yeast Rav1 and human DMXL1/2 are characterized by two N-terminal WD40 domains followed by a helical ___domain, with sequences in the second WD40 and helical domains conserved between human and yeast proteins (referred to as the Rav1-C ___domain in the Protein Families (PFAM) database) (Fig. 4d). Additionally, DMXL1/2 contain a carboxy-terminal WD40 ___domain that is not found in Rav1. Previous deletion-mapping studies have identified interacting regions between Rav1 and Rav2, including an interaction of Rav2 with the first WD40 ___domain of Rav1 (Fig. 4d)41. We generated predictive models of DMXL1–ROGDI-WDR7 and Rav1–Rav2 using AF3 (ref. 46). Consistent with previous biochemical results41, Rav2 was predicted to interact with the first WD40 propeller (interface-predicted template modeling score, ipTM = 0.77) (Fig. 4e, left) and a very similar structure was predicted for DMXL1–ROGDI (ipTM = 0.74) (Fig. 4e, middle). The N-terminal seven-blade WD40 ___domain interacts with ROGDI and a partial four-blade WD40 ___domain packs against the N-terminal and extreme C-terminal WD40 ___domain of DMXL1 (Fig. 4e, middle). A recently published Rav1–Rav2 structure56, released during the revision of this study, revealed a nearly identical interaction interface to the AF3 Rav1–Rav2 prediction. However, the N-terminal 20 residues of the Rav2 model that were not seen in the cryo-EM structure appear to interact extensively with the WD40 propeller (Extended Data Fig. 5a). The PFAM-defined Rav1-C homology region contains the C-terminal half of the second WD40 ___domain (WD40_2), which is shared by both Rav1 and DMXL1/2, as well as a portion of the helical ___domain. AF3 predicts that the structures of the core of the helical ___domain are similar in Rav1 and DMXL1/2, although DMXL1 has a large insert (residues 1351–1496) that is absent in Rav1 (Extended Data Fig. 5b). In the context of a predicted DMXL1/2–ROGDI–WDR7 complex, the helical ___domain is positioned between WD40_2 and WD40_3 from DMXL1/2, and the relative orientations of WD40_1 and WD40_3 are stabilized by interactions with WDR7 and ROGDI (Extended Data Fig. 5b,c).
As an approach to support these predictions, we performed cross-link mass spectrometry (XL-MS) on the complex obtained from DMXL1–mNG immunoprecipitation (Supplementary Table 4). Using the disuccinimidyl sulfoxide (DSSO) cross-linker, this approach can identify structural regions at which two lysine residues have Cα carbon atoms within approximately 35 Å of each other57. Using stringent cross-link search parameters (Methods)58, we identified several interprotein and intraprotein cross-links that support core aspects of the AF3 prediction (Fig. 4e,f, Supplementary Table 4 and Supplementary Video 2). First, interprotein cross-links between ROGDIK38 and DMXL1K215 and between ROGDIK137 and WDR7K321 fit distance constraints in the model (Fig. 4e,f and Supplementary Table 4). Second, multiple intraprotein cross-links are consistent with structural predictions at the interface of the second WD40 ___domain in DMXL1 and a subcomponent of the central helical ___domain (Fig. 4e,f and Supplementary Table 4). Multiple intraprotein cross-links are consistent with the predicted Rav1-C ___domain, although several cross-links outside the distance constraint are present in unstructured regions (Fig. 4e,f and Supplementary Table 4). To extend cross-link analysis to DMXL2, we mined previous cross-linking data from mouse synaptic vesicles59, in which Dmxl2 is known to be expressed, for XLs between Dmxl2, Rogdi and Wdr7. This analysis revealed analogous interprotein cross-links between Dmxl2–Rogdi and Rogdi–Wdr7, similar to those observed in the human DMXL1 complex, as well as additional intraprotein cross-links in Dmxl2 that support components of the predicted WD40 and Rav1-C domains (Fig. 4g, Extended Data Fig. 5c and Supplementary Table 4). Together, these data support AF3’s overall structural predictions, although several regions within human DMXL1 and mouse Dmxl2 are predicted to be unstructured.
DMXL1/2 support lysosomal acidification upon TRPML1 activation
Recruitment of DMXL1 to lysosomes in response to TRPML1 activation, coupled with the ability of DMXL1 to associate with the V-ATPase, led us to examine the control of lysosomal pH. Using CRISPR–Cas9, we generated U2OS cells lacking DMXL1 as well as cells that lacked DMXL1 and were heterozygous knockout for DMXL2 (Extended Data Fig. 6a–c). We could not generate cells lacking all copies of DMXL1 and DMXL2, suggesting that DMXL proteins might be essential for viability of U2OS cells. In DMXL1−/− and DMXL1−/−DMXL2+/− cells, LC3B lipidation induced by TRPML1 activation remained intact, suggesting that CASM is upstream or independent of DMXL1/2 function. We also observed that TFEB activation in response to TRPML1 activation was maintained in DMXL1/2-deficient cells (Extended Data Fig. 6d–f). Given these results, we investigated lysosomal pH using a lysosome-targeted pH-sensitive dye. Lysosomal pH in DMXL1−/− and DMXL1−/−DMXL2+/− cells was not significantly altered compared with that in WT cells under basal conditions (Fig. 5a,b). In WT cells, there was a small increase in lysosomal pH from ~5.0 to ~5.2 upon TRPML1 activation (Fig. 5a,b). By contrast, lysosomal pH strikingly increased to ~5.7 in DMXL1−/− cells and further to ~6.0 in DMXL1−/−DMXL2+/− cells (Fig. 5a,b). By comparison, WT cells treated with BafA1 to block V-ATPase function displayed a lysosomal pH of ~7.5, indicating that the dynamic range of the pH sensor was sufficient to capture changes in lysosomal pH in response to MLSA5 (Fig. 5b and Extended Data Fig. 6g). These data support a model wherein recruitment of the DMXL1 complex, together with the V1-ATPase, suppresses an increase in lysosomal pH that results from acute TRPML1 activation.

a, Confocal images showing the fluorescence intensity of the lysosomal pH sensor, pseudo-colored to scale, in the indicated U2OS cells after treatment with MLSA5 (2 h). Scale bar, 20 µm. b, Lysosomal pH measurements for the indicated U2OS cells, with or without MLSA5, or BafA1 treatment (2 h). Quantification was performed on the following total number of fields of view from three independent experiments: control: WT n = 23, DMXL1−/− n = 19, DMXL1−/−DMXL2+/− n = 23; MLSA5: WT n = 21, DMXL1−/− n = 18, DMXL1−/−DMXL2+/− n = 20; BafA1: WT n = 16. Each field of view contained between 50 and 70 cells. Data are presented as mean values ± s.d. A one-way ANOVA followed by Dunnett’s multiple-comparisons test was used for each treatment group. An unpaired two-sided Student’s t-test was used for comparison between WT control and WT treated with MLSA5. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. c, Schematic of TMT-MS experimental design for analysis of V-ATPase recruitment to lysosomes in response to MLSA5 treatment. d, Volcano plot (log2(FC) versus –log10(q)) for Lyso-IP of U2OS cells of the indicated genotypes treated with MLSA5 for 4 h or left untreated. V1-ATPase subunits are indicated by red points, and V0-ATPase subunits are indicated by blue points. MLSA5 treatment leads to enrichment of V1 but not V0-ATPase subunits on lysosomes in WT but not in DMXL1−/− or DMXL1−/−DMXL2+/− cells. e, Immunoblot analysis of whole-cell lysates or Lyso-IP samples from the indicated U2OS cells, with or without MLSA5 treatment (2 h). The data are representative of two independent experiments. Also see Extended Data Figure 6.
DMXL1/2 support V1-ATPase recruitment upon TRPML1 activation
To investigate whether DMXL1 mediates recruitment of the V1-ATPase to promote V-ATPase assembly, we expressed the Lyso-IP handle in WT, DMXL1−/− and DMXL1−/−DMXL2+/− cells and performed TMT-MS analysis on Lyso-IP samples prepared from untreated and MLSA5-treated (4 h) conditions (Fig. 5c, and Supplementary Table 5). Volcano plots comparing WT MLSA5-treated cells and untreated cells revealed enrichment of all subunits of the V1-ATPase, but not V0-ATPase subunits (Fig. 5d). By contrast, in both DMXL1−/− and DMXL1−/−DMXL2+/− cells, V1-ATPase subunits were not appreciably enriched after MLSA5 treatment (Fig. 5d). These results were further validated by immunoblotting of Lyso-IP samples with anti-ATP6V1B2 and anti-ATP6V1D antibodies (Fig. 5e). Together, these data are consistent with a model in which DMXL1/2 promote recruitment of the V1-ATPase to lysosomes in response to TRPML1 activation.
DMXL1/2 support lysosomal hydrolase activity
The increase in lysosomal pH in DMXL1/2-deficient cells led us to examine whether lysosomal hydrolytic activity was altered. Hydrolytic processing of Legumain (LGMN) from a precursor form to the mature form was reduced in both DMXL1−/− and DMXL−/−DMXL2+/− cells when compared with WT cells upon TRPML1 activation (Fig. 6a,b). Similarly, DMXL1/2-deficient cells displayed reduced levels of the mature form of Cathepsin B under TRPML1 activation (Fig. 6a,b). We next directly examined lysosomal hydrolytic activity by measuring the fluorescence intensity of DQ-BSA Red. In intact DQ-BSA, the fluorophores are quenched, but upon endocytosis and trafficking to the lysosomal lumen, the hydrolysis of DQ-BSA alleviates this quenching, resulting in an increase in fluorescence intensity. Under basal conditions, the DQ-BSA Red signals in WT, DMXL1−/− and DMXL1−/−DMXL2+/− cells exhibited comparable mean fluorescence intensities (Fig. 6c,d and Extended Data Fig. 7b). By contrast, cells treated with MLSA5 (4 h) displayed progressively less hydrolytic activity in DMXL1−/− and DMXL1−/−DMXL2+/− cells than in WT cells (Fig. 6c,d and Extended Data Fig. 7b). As expected, BafA1 suppressed hydrolytic activity by >90% (Fig. 6d and Extended Data Fig. 7a,b). Moreover, expression of DMXL1-mNG in DMXL1−/−DMXL2+/− cells rescued the defect in hydrolytic activity observed under MLSA5 treatment (Extended Data Fig. 7c,d). Taken together, these data are consistent with a model in which, upon TRPML1 activation, DMXL1/2 facilitate the recruitment of V1-ATPase to lysosomes to support lysosomal acidification and hydrolytic activity.

a, U2OS cells of the indicated genotypes were untreated or treated with MLSA5 for 4 h, and cell lysates were analyzed by immunoblotting with the indicated antibodies to detect unprocessed and mature hydrolytic enzymes. Blots were probed with α-Hsp90 as loading control. Representative of three independent experiments. b, Quantification of three independent experiments, as shown in a. Data are presented as mean values ± s.d. An ordinary one-way ANOVA followed by Dunnett’s multiple-comparisons test was used for statistical comparison. c, Confocal images showing fluorescence intensity of DQ-BSA Red in the indicated U2OS cells after treatment with MLSA5 (4 h). Scale bar, 20 µm. d, DQ-BSA Red fluorescence intensity in experiments equivalent to that shown in c. Quantification was performed on the following total number of cells from three biological replicates: control: WT n = 28, DMXL1−/− n = 28, DMXL1−/−DMXL2+/− n = 28; MLSA5: WT n = 28, DMXL1−/− n = 28, DMXL1−/−DMXL2+/− n = 30. Quantification for BafA1 treatment represents n = 14 cells from one biological replicate. Arb. u., arbitrary units. Treatment with BafA1 was used to measure the maximum inhibition of lysosomal activity as measured with DQ-BSA Red. Data are presented as mean values ± s.d. An ordinary one-way ANOVA followed by Dunnett’s multiple comparison test was used to compare genotypes within the control group and Brown Forsyth and Welch’s ANOVA followed by Dunnett’s T3 test was used to compare genotypes within MLSA5 treatment group. Unpaired two-sided Student’s t-test was used to compare between WT control and WT MLSA5-treated conditions. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001; n.s., not significant. Also see Extended Data Figure 7.
Structural basis for DMXL1–V-ATPase complex formation
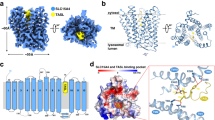
To investigate the interactions between DMXL1 and the V-ATPase, we performed systematic pairwise AF3 predictions of DMXL1 with each V1 subunits as well as the major cytosolic-facing V0 subunit ATP6V0A, with a particular focus on interactions that occur within the conserved Rav1-C helical ___domain. The highest-scoring pairs were DMXL1–ATP6V1A and DMXL1–ATP6V1C (ipTM = 0.76 and 0.73, respectively); similar predictions were found with the equivalent pairs in the context of the Rav1-C helical ___domain from DMXL1 (ipTM = 0.75 and 0.67, respectively) (Extended Data Fig. 8a). The V1-ATPase A and C subunits are predicted to bind to distinct surfaces in the DMXL1 Rav1-C helical ___domain (Extended Data Fig. 8b). Notably, AF3 predicts similar interfaces between the Rav1-C ___domain of Rav1 and the analogous Vma1 and Vma5 subunits of the budding yeast proteins (ipTM = 0.64 and 0.42, respectively), including conserved candidate interface residues found in DMXL1-ATP6V1A (Fig. 7a and Extended Data Fig. 8c). To examine the relevance of this predicted interface for the interaction of DMXL1 with the V-ATPase, we performed IP-immunoblot analysis of DMXL1WT–mSG2–HA (mSG2 refers to monomeric StayGold)60 and DMXL17A–mSG2–HA, a mutant lacking seven residues predicted to be at the interface (K1679A, L1682A, F1686A, L1689A, F1701A, D1709A, K1717A) (Fig. 7a), expressed at equivalent levels in DMXL1−/−DMXL2+/− cells. Whereas ATP6V1A was enriched in the DMXL1WT immune complex above background levels, this was not seen for the DMXL17A interface mutant, consistent with a loss of interaction (Fig. 7b). Unlike DMXL1WT, the DMXL17A interface mutant was not recruited to membranes in a crude lysosome preparation, a method previously used to examine recruitment of V1-ATPase to lysosomes53 (Fig. 7c). To examine the functional consequences of the interface mutant, we examined LGMN processing. DMXL1WT rescued the LGMN processing defect in DMXL1−/−DMXL2+/− cells, but the DMXL17A mutant failed to do so, indicative of a compromised ability to acidify lysosomes in response to TRPML1 activation (Fig. 7d).

a, Left, AF3 prediction of Rav1 helical ___domain (blue) and Vma1 (lime) highlighting conserved or similar interface residues. Right, AF3 prediction of the DMXL1 helical ___domain (green) together with ATP6V1A (yellow) highlighting interface residues, including those that are conserved or similar in Rav1–Vma1. The conserved residue A1685 in DMXL1 that is mutated in DMXL2 (A1712V) in DEE81 is shown in light blue. b, U2OS DMXL1−/−DMXL2+/− cells were reconstituted with DMXL1WT–mSG2–HA or DMXL17A–mSG2–HA, and α-HA immune complexes were analyzed by immunoblotting with the indicated antibodies. Relative abundance was quantified from three independent experiments. Data are presented as mean values ± s.d. A paired two-sided Student’s t-test was used for statistical comparison; **P ≤ 0.01. c, Crude lysosomal fractions53 using cells from b with or without MLSA5 (2 h) were immunoblotted with the indicated antibodies. The results are representative of three independent experiments. d, Immunoblot of LGMN processing in U2OS DMXL1−/−DMXL2+/− cells expressing DMXL1WT–mSG2–HA or DMXL17A–mSG2–HA, with or without MLSA5 (4 h). Representative of three independent experiments. e, AF3 prediction of SidK (residues 1–278, lavender) and ATP6V1A (yellow) interaction overlayed with the analogous DMXL1 helical ___domain AF3 prediction (green). SidK and DMXL1 interact with an overlapping region of ATP6V1A, albeit with distinct interfaces. f, Volcano plot (log2(FC) versus –log10(q)) for DMXL1–mNG IP from U2OS cells expressing mCherry or SidK–mCherry. Three biological replicates per condition. V-ATPase subunits (red); DMXL1/2, ROGDI and WDR7 (blue). g, Immunoblot of DMXL1–mNG complexes or cell extracts from cells expressing either mCherry or SidK–mCherry from f. h, Model for DMXL1–ROGDI–WDR7–V-ATPase based on the AF3 prediction of the DMXL1 helical ___domain association with ATP6V1A aligned on bovine V-ATPase (PDB: 6XBW). Left, one set of peripheral ATP6V1E1/ATP6V1G and ATP6V1C subunits removed to mimic the Rav1–Rav2–partial V-ATPase cryo-EM structure56. Red box, ___location of the DMXL1 helical ___domain–ATP6V1A interaction interface, as shown in a (rotated horizontally by ~180°). Right, same as left but including ATP6V1E1, ATP6V1G and ATP6V1C (cyan) subunits. i, Overlay of Rav1–Vma1 AF3 model (blue/lime subunits, respectively) with the yeast Rav1–Rav2–Skp1–partial V1-ATPase complex structure (PDB: 9COP)56. The proposed56 localization relative to the membrane is shown.
Previous studies indicate that the Legionella pneumophila effector protein SidK binds the same interface on ATP6V1A that is predicted to interact with DMXL1 (refs. 61,62), and the AF3 model for the pairwise combinations (Fig. 7e) indicated that SidK might interfere with the interaction between DMXL1 and the V-ATPase. Therefore, as an independent approach, we examined the effects of SidK overexpression on DMXL1–V-ATPase interaction. We performed TMT-MS analysis on DMXL1–mNG immunoprecipitates from cells overexpressing either mCherry or SidK–mCherry (Methods). The association of the detected V-ATPase subunits was decreased in the context of SidK–mCherry overexpression, with the ATP6V1A and ATP6V1B2 subunits displaying the greatest degree of reduction (Fig. 7f and Supplementary Table 6). The reduction in ATP6V1A association was validated by immunoblotting (Fig. 7g). Consistent with SidK’s established role in maintaining a neutral pH in the phagosomal lumen during the early phase of infection63, SidK–mCherry overexpression completely blocked LGMN processing (Fig. 7g). Together, these data are consistent with a functional interaction between DMXL1 and the ATP6V1A subunit.
We next assessed the overall orientation of the DMXL1–ROGDI–WDR7 complex in association with the V-ATPase. Alignment of the AF3-predicted DMXL1–ROGDI–WDR7–ATP6V1A complex structure with one of the three ATP6V1A subunits in the context of a full V-ATPase complex positions WD40_3 from DMXL1 as well as the C-terminal region of WDR7 near the outer leaflet of the membrane (Fig. 7h). During manuscript revision, a cryogenic electron microscopy (cryo-EM) structure of Rav1–Rav2–Skp1 bound to a partial assembly of the V1-ATPase from budding yeast lacking 5 of 16 subunits was reported56. In this structure, the Rav1-C ___domain of Rav1 directly interacts with the Vma1 subunit (corresponding to ATP6V1A) through the same surface identified by AF3. This was confirmed by overlaying the AF3 prediction for the Rav1-C ___domain of Rav1 with Vma1 onto the Rav1–Rav2–Skp1-V–ATPase cryo-EM structure (Fig. 7i)56. The overall orientation places Rav2 in a position expected to be proximal to the membrane during facilitated delivery of V1 to V0 (Fig. 7i)56. In this model, DMXL1 is predicted to clash with the peripheral stalk helical domains of ATP6V1E and G subunits (Fig. 7h, right panel). In this regard, the Rav1–Rav2 complex with a partial V-ATPase from yeast also lacks the peripheral stalk adjacent to the Vma1 subunit that engages Rav1 (ref. 56), suggesting the potential for alternative conformations of DMXL1 or V1-ATPase subunits in the context of the preloaded complex (see ‘Discussion’).
Mechanism of CASM-driven recruitment of DMXL1 to lysosomes
ATG8 proteins interact with effector proteins through short LC3-interacting region (LIR) motifs of the form [W/F/Y]xx[L/I/V], sometimes preceded by acidic residues. We employed two parallel approaches to search for candidate LIR motifs in DMXL1 that could promote recruitment to lysosomes in response to TRPML1 activation. First, we screened 53 candidate LIR sequences in DMXL1 identified by the iLIR algorithm64 as 36-residue peptides for AF3-based structural predictions with GABARAP, an approach that has previously been shown to facilitate identification of functional LIRs65 (Supplementary Table 4). The majority of LIR sequences either had sub-threshold ipTM scores (<0.4) or were localized in highly folded regions of DMXL1, rendering them inaccessible for interaction with GABARAP (Fig. 8a). However, one candidate LIR—residues 2695–2698 (FLVI)—was found in an unstructured region of DMXL1, near the C-terminal WD40 ___domain. The LIR-containing peptide interacted with GABARAP, as evidenced by AF3 analysis (ipTM = 0.55) (Supplementary Table 4). This LIR was also preceded by an acidic ED dipeptide. As a follow-up, we performed an unbiased AF3 prediction using GABARAP and DMXL1. Remarkably, all five models positioned GABARAP in association with the FLVI sequence described above, with the highest-scoring model achieving an ipTM of 0.69 (Fig. 8a). The position of the hydrophobic LIR residues in the LIR-docking site of GABARAP was virtually identical to the crystal structure of the LIR motif from TEX264 (residues 273–276, FEEL)66. Notably, this LIR motif is predicted to be near the lipidated and membrane-associated GABARAP protein during CASM, as modeled with DMXL1′s Rav1-C ___domain bound to ATP6V1A (Fig. 7h).

a, Predicted interaction of candidate DMXL1 LIR (residues 2695–2698, FLVI) and GABARAP. Locations of other candidate LIR motifs are shown in magenta. Locations of the Phe and Val residues in the DMXL1 LIR overlap with the LIR of TEX264 bound to GABARAP (PDB: 7VEC)66. b,c, Analysis of lysosomal localization of DMXL1–mSG2–HA WT, ΔLIR and ΔWD40_3 proteins in response to TRPML1 activation. c shows confocal images of cells subjected to immunofluorescence with or without MLSA5 treatment (2 h). Scale bar, 10 µm; inset scale bar, 3 µm. b shows quantification of colocalization between DMXL1 and LAMP1. Quantification was based on the number of cells indicated on the plot, from three independent experiments. The box represents the 25th to 75th percentiles, whiskers extend from the minimum to maximum values and the line represents the median. Statistical significance was assessed using unpaired two-sided Student’s t-test. ****P ≤ 0.0001; n.s., not significant. d, Crude lysosome fractions from the indicated cell lines, with or without MLSA5 (2 h), were probed with the indicated antibodies. The result is representative of three independent experiments. e, Immunoblot analysis of LGMN processing in U2OS DMXL1−/−DMXL2+/− cells reconstituted with WT, ΔLIR or ΔWD40_3 DMXL1–mSG2–HA, with or without MLSA5 (4 h). The result is representative of three independent experiments. f, Working model. TRPML1 in the closed form is unable to allow passage of cations. Binding of the agonist MLSA5 enables cations release, which promotes recruitment of the ATG8ylation machinery to pre-existing V-ATPase on lysosomes. The ATG8ylation machinery (composed of ATG16L1 (ATG16), the E1 enzyme ATG7 and the ATG12-ATG5 conjugate, referred to as 12-5/7) promotes ATG8 conjugation to the lysosomal limiting membrane (CASM), which is a prerequisite for the recruitment of DMXL1–V1-ATPase complexes to the lysosome where it facilitates assembly of V1-ATPase on pre-existing V0-ATPase, forming functional proton pumps to maintain lysosomal pH. Currently, whether DMXL1 directly binds ATG8 proteins in response to CASM is unclear, as indicated by ‘??.’ After V-ATPase assembly, the DMXL1 complex is presumably recycled to the cellular pool. The signaling mechanisms underlying lysosomal recruitment as well as release of the DMXL1 complex from a fully assembled V-ATPase are unclear.
To test whether this sequence in DMXL1 is important for recruitment to lysosomes, we stably expressed DMXL1ΔLIR–mSG2–HA, in which the LIR residues are replaced with alanine, or DMXL1ΔWD40_3–mSG2–HA, in which the C-terminal WD40 ___domain, including the preceding candidate LIR, is deleted, in WT U2OS cells (Fig. 8b,c). Confocal microscopy revealed that DMXL1WT was recruited to lysosomes following TRPML1 activation, whereas the ΔLIR and ΔWD40_3 mutants did not display significant lysosomal recruitment (Fig. 8b,c). As an alternative approach to examining recruitment to lysosomes, we analyzed crude lysosome fractions from DMXL1−/−DMXL2+/− cells stably expressing WT, ΔLIR or ΔWD40_3 DMXL1–mSG2–HA, with or without MLSA5 treatment. WT DMXL1, but neither of the mutants, was enriched in the crude lysosome preparation, consistent with a requirement of these sequences for recruitment upon TRPML1 activation (Fig. 8d). Furthermore, WT, but not the ΔLIR or ΔWD40_3 mutants, rescued LGMN processing in DMXL1−/−DMXL2+/− cells, supporting a functional role for the candidate LIR. (Fig. 8e). Taken together, these results suggest that the C-terminal region of DMXL1, including the candidate LIR sequence, functions in recruitment to lysosomes in response to TRPML1 activation.
Discussion
TRPML1 is a lysosomal cation channel critical for various aspects of endolysosomal function. Substitutions in TRPML1 cause mucolipidosis type IV, a recessive lysosomal storage disorder characterized by delayed development and vision impairment12,67,68. Altered TRPML1 function has been linked to disruptions in key endolysosomal processes, including trafficking and hydrolytic activity3,4,10,17. Thus, understanding how TRPML1 activation supports endolysosomal homeostasis is essential for advancing the development of small molecules that leverage TRPML1 function20,22,69.
Using the Lyso-IP lysosome enrichment method with quantitative proteomics, we found DMXL1 recruitment to lysosomes following TRPML1 activation. Our working model is outlined in Figure 8f. In the closed state, TRPML1 restricts cation flux. Binding of MLSA5 opens the channel, activating efflux of Ca2+ and other ions3,4,6,7,17. Earlier studies have shown that TRPML1 activation triggers the recruitment of ATG8ylation machinery, including ATG16L1, to V-ATPase assemblies, promoting ATG8 conjugation to the lysosomal membrane (CASM)20,22. Our results confirmed the enrichment of ATG8 protein on lysosomes following TRPML1 activation (Fig. 1). DMXL1 recruitment is dependent on CASM, as indicated by loss of recruitment in the context of ATG16L1K490A, or BafA1 treatment, both of which inhibit CASM (Figs. 1 and 2). DMXL1 recruitment is presumably coupled to assembly of V1 subunits with pre-existing V0 subunits present in the lysosomal membrane, thereby sustaining lysosomal acidification and the activity of lumenal hydrolytic enzymes (Fig. 8f). This is supported by Lyso-IP proteomics and measurements of lysosomal pH, degradation and hydrolase processing in WT and DMXL1/2-deficient cells (Figs. 5 and 6). Our findings support the emerging understanding that CASM is responsive to endolysosomal pH changes31,33,34,36; furthermore, they suggest that the role of CASM is not only to recognize proton-gradient dissipation, but also to mitigate this dissipation by promoting endolysosomal acidification through DMXL1–V1-ATPase recruitment (Fig. 8f). DMXL1 recruitment occurs in parallel with TFEB activation, as DMXL1/2-deficient cells still exhibit TFEB nuclear translocation and GPNMB upregulation (Extended Data Fig. 6d–f)22,48. We conclude that TRPML1 activation drives lysosomal ATG8ylation, triggering independent homeostatic programs, including TFEB activation and V1-ATPase recruitment, through DMXL1/2 (Fig. 8f). DMXL1 is also present in the Golgi (Extended Data Fig. 4), and further studies are required to elucidate any specific functions of this DMXL1 population.
AF3 predictions coupled with functional studies support a working model for the mechanism of DMXL1 action (Fig. 8f). DMXL1 is predicted to bind ATP6V1A and ATP6V1C through distinct surfaces, and substitutions in DMXL1′s Rav1-C ___domain reduce ATP6V1A interaction (Fig. 7a,b). This interface resembles the Rav1–Vma1 interaction in a budding yeast V-ATPase structure reported while this manuscript was being revised56. A similar ATP6V1A interface also binds SidK, and SidK overexpression disrupts DMXL1–ATP6V1A binding and LGMN processing (Fig. 7e–g). When bound to ATP6V1A and associated with an assembled V-ATPase, the C terminus of DMXL1 and its interaction partners, ROGDI and WDR7, would be near the membrane (Figs. 7h and 8f), as is the yeast Rav2 protein56 (Fig. 7i). However, this model displays a clash between the Rav1-C ___domain of DMXL1 and the ATP6V1E and ATP6V1G peripheral stalk (Fig. 7h). Interestingly, the Rav1–Rav2 structure resembles rotational state 3 but lacks one such stalk56, suggesting that DMXL1 could recognize specific V1-ATPase sub-assemblies. Furthermore, in yeast, Vma5 (ATP6V1C ortholog) joins the V-ATPase after Rav1–Rav2 binding39,56. Our results demonstrate possible interaction between DMXL1 and ATP6V1C (Fig. 4c), suggesting that ATP6V1C could be part of the DMXL1–V1-ATPase complex; thus, whether this interaction is important for V-ATPase assembly in mammals warrants further studies.
DMXL2A1712V has been previously identified as a causative mutation in developmental and epileptic encephalopathy 81 (DEE81)70. The corresponding residue in DMXL1 (A1685) is located within the ATP6V1A-binding interface (Fig. 7a). Replacement of Ala with Val could alter the packing of the helix containing F1701 in DMXL1 (F1728 in DMXL2), a residue that is conserved in the Rav1–Vma1 interaction interface (Fig. 7a). A recent report indicated that DMXL2–ROGDI–WDR7 is present in synaptic vesicles71, and therefore it is conceivable that p.V1712A in DMXL2 could negatively affect V-ATPase assembly on synaptic vesicles, which could contribute to the pathogenesis of DEE81.
The simplest model for CASM-dependent DMXL1 recruitment to lysosomes would involve interaction with ATG8 proteins conjugated on the membrane. Substituting a candidate LIR motif near the C-terminal WD40 ___domain or deleting this region in DMXL1 disrupts lysosomal recruitment (Fig. 8). However, we have yet to detect DMXL1–GABARAP or LC3B interaction post-MLSA5 treatment, possibly due to: (1) low affinity, (2) membrane dependence lost during detergent extraction or (3) intermediate proteins or modifications. Notably, the DMXL17A mutant, with reduced ATP6V1A interaction, is poorly recruited to membranes (Fig. 7c), suggesting that V1-ATPase components are needed for efficient lysosomal targeting. A similar ‘coincidence detector’ mechanism was proposed for LRRK2 recruitment to lysosomes, which also relies on CASM and interactions with Rab GTPases30. Although understanding the precise signals and interactions that facilitate DMXL1–V1-ATPase recruitment to lysosomes will require further study, our results provide a framework for the pathway and reveal a mechanism that directly links TRPML1-dependent cation efflux with maintenance of lysosomal pH.
Methods
Materials
Antibodies
The following antibodies were used in this study: rabbit polyclonal DMXL1, Bethyl Laboratories, cat. no. A304-685A, RRID: AB_2620880 (western blot dilution (WB): 1:500); rabbit monoclonal LC3B (D11), Cell Signaling Technology, cat. no. 3836, RRID: AB_2137707 (WB, 1:1,000); rabbit monoclonal LAMP1 (D2D11), Cell Signaling Technology, cat. no. 9091, RRID: AB_2687579 (WB, 1:1,000; immunofluorescence dilution (IF): 1:200); rabbit polyclonal YIPF4, Proteintech, cat. no. 15473-1-AP, RRID: AB_2217206 (WB, 1:1,000); mouse monoclonal Golgin-97 (CDF4), Thermo Fisher Scientific, cat. no. A-21270, RRID: AB_221447 (WB, 1:1,000); rabbit monoclonal calreticulin (D3E6), Cell Signaling Technology, cat. no. 12238, RRID: AB_2688013 (WB, 1:1,000); rabbit monoclonal COX IV (3E11), Cell Signaling Technology, cat. no. 4850, RRID: AB_2085424 (WB, 1:1,000); rabbit monoclonal GPNMB (E4D7P), Cell Signaling Technology, cat. no. 38313, RRID: AB_2799131 (WB, 1:1,000); mouse monoclonal β-actin (AC-15), Santa Cruz, cat. no. sc-69879, RRID: AB_1119529 (WB, 1:5,000); mouse monoclonal TRPML1 (F-10), Santa Cruz, cat. no. sc-398868 (WB, 1:500); rabbit monoclonal ATG16L1 (D6D5), Cell Signaling Technology, cat. no. 8089, RRID: AB_10950320 (WB, 1:1,000); mouse monoclonal Hsp90 (4F10), Santa Cruz, cat. no. sc-69703, RRID: AB_2121191 (WB, 1:2,000); rabbit monoclonal HA-tag (C29F4), Cell Signaling Technology, cat. no. 3724, RRID: AB_1549585 (WB, 1:1,000); rabbit monoclonal ATP6V1B2 (D2F9R), Cell Signaling Technology, cat. no. 14617, RRID: AB_2798541 (WB, 1:1,000); rabbit monoclonal ATP6V1D (EPR11326(B)), Abcam, cat. no. ab15748, RRID: AB_2732041 (WB, 1:1,000); rabbit monoclonal ATP6V1A (EPR19270), Abcam, cat. no. ab199326, RRID: AB_2802119 (WB, 1:1,000); rabbit monoclonal ATP6V0D1 (EPR18320), Abcam, cat. no. ab202897, RRID: AB_2802121 (WB, 1:1,000); rabbit polyclonal DMXL2, Proteintech, cat. no. 24415-1-AP, RRID: AB_2879534 (WB, 1:500); rabbit monoclonal Legumain (EPR14718), Abcam, cat. no. ab183028, RRID: AB_3291610 (WB, 1:1,000); rabbit monoclonal Cathepsin B (D1C7Y), Cell Signaling Technology, cat. no. 31718, RRID: AB_2687580 (WB, 1:500); rabbit polyclonal TGN46, Proteintech, cat. no. 13573-1-AP, RRID: AB_10597396 (IF: 1:200); rabbit polyclonal GM130, Proteintech, cat. no. 11308-1-AP, RRID: AB_2115327 (IF: 1:200); mouse monoclonal giantin (9B6), Abcam, cat. no. ab37266, RRID: AB_880195 (IF: 1:200); mouse monoclonal HA-tag (6E2), Cell Signaling Technology, cat. no. 2367, RRID: AB_10691311 (IF: 1:200); rabbit monoclonal α-tubulin (11H10), Cell Signaling Technology, cat. no. 2125, RRID: AB_2619646 (WB, 1:1,000); rabbit monoclonal GAPDH (D16H11), Cell Signaling Technology, cat. no. 5174, RRID: AB_10622025 (WB, 1:1,000); mouse monoclonal LAMP1 (H4A3), BD Biosciences, cat. no. 555798, RRID: AB_396132 (IF: 1:400); rabbit polyclonal TFEB, Cell Signaling Technology, cat. no. 4240, RRID: AB_11220225 (WB, 1:1,000); mouse monoclonal mNeonGreen (32F6), ChromoTek, cat. no. 32f6, RRID: AB_2827566 (WB, 1:1,000); goat anti-rabbit-IgG heavy and light conjugates linked to horseradish peroxidase ((H+L)–HRP conjugate), Bio-Rad, cat. no. 1706515, RRID: AB_11125142 (WB, 1:1,000); goat anti-mouse-IgG (H+L)–HRP conjugate, Bio-Rad, cat. no. 1706516, RRID: AB_2921252 (WB, 1:1,000); goat anti-rabbit IgG (H+L) Alexa-Fluor-488-conjugated highly cross-adsorbed secondary antibody Thermo Fisher Scientific, cat. no. A-11034, RRID: AB_2576217 (IF: 1:200); and goat anti-mouse-IgG (H+L) Alexa-Fluor-647-conjugated highly cross-adsorbed secondary antibody Thermo Fisher Scientific, cat. no. A-21235, RRID: AB_2535804 (IF: 1:200).
Reagents
Reagents used in the experiments included: MLSA5, MedChemExpress, cat. no. HY-152182; bafilomycin A1, Cayman Chemicals, cat. no. 11308; torin1, Sellekchem, cat. no. S2827; brefeldin A, Cayman Chemicals, cat. no. 11861; pierce anti-HA magnetic beads, Thermo Fisher Scientific, cat. no. 88837; TMTpro 18-plex label reagent set, Thermo Fisher Scientific, cat. no. A52045; MS-grade trypsin protease, Thermo Fisher Scientific, cat. no. 90305; MS-grade lysyl endopeptidase, Thermo Fisher Scientific, cat. no. NC9242798; ChromoTek mNeonGreen-trap magnetic agarose, ChromoTek, cat. no. ntma; ChromoTek HA-trap magnetic agarose, ChromoTek, cat. no. atma; DSSO, Thermo Fisher Scientific, cat. no. A33545; Sep-Pak C8 1 cc vac cartridge, 50 mg Sorbent, Waters, cat. no. WAT054965; pHLys red, Dojindo, cat. no. L265; SiR-lysosome, Cytoskeleton, cat. no. CY-SC012; DQ-BSA red, Thermo Fisher Scientific, cat. no. D12051; HiFi DNA Assembly Master Mix, NEB, cat. no. 2621; Hoechst 33342, trihydrochloride trihydrate, Thermo Fisher Scientific, cat. no. H1399; and EM-grade paraformaldehyde 16% aqueous solution, Electron Microscopy Sciences, cat. no. 15710.
MLSA5 (MedChemExpress, HY152182) was dissolved in DMSO to a stock concentration of 10 mM; all experiments were performed with a working concentration of 5 µM. Bafilomycin A1 (Cayman Chemicals, cat. no. 11308) was dissolved in DMSO to a stock concentration of 1 mM; the working concentration was 1 µM. Torin1 (Selleckchem, cat. no. S2827) was dissolved in DMSO to a stock concentration of 1 mM; the working concentration was 1 µM. Brefeldin A (Cayman Chemicals, cat. no. 11861) was dissolved in DMSO to a stock concentration of 5 mg ml–1; the working concentration was 5 µg ml–1. Nocodazole (Sigma, cat. no. M1404) was dissolved in DMSO to a stock concentration of 5 mg ml–1; the working concentration was 5 µg ml–1. DQ-BSA red (Thermo, cat. no. D12051) was dissolved in DPBS to a stock concentration of 2 mg ml–1; the working concentration was 20 µg ml–1.
Human cell lines
U2OS (ATCC, cat. no. HTB-96), A101D (ATCC, cat. no. CRL-7898) and 293T (ATCC, cat. no. CRL-3216) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Thermo, cat. no. 11995065) supplemented with 10% fetal bovine serum (FBS) and maintained in an 37 °C incubator with 5% CO2. For imaging experiments, cells were grown in DMEM medium without phenol red (Thermo, cat. no. 21063029) supplemented with 10% FBS.
CRISPR–Cas9 gene editing
Generation of knockout cell lines
U2OS-KO cells were generated by introducing Cas9 and single guide RNA (sgRNA) through lentiviral particles packaged using lenticrisprv2-opti (Addgene, cat. no. 163126) and lenticrisprv2-bsd (Addgene, cat. no. 98293). Stably infected cells were selected with 2 µg ml–1 puromycin for 4–5 days, at which uninfected control cells were completely killed. Puromycin-selected cells were then single-cell sorted into 96-well plates on a SONY SH800S sorter. Next-generation sequencing with Illumina MiSeq was used to sequence the gene-edited locus for each single cell clone, and OutKnocker analysis72 was utilized to identify clones with out-of-frame indels. Clones were further verified by immunoblotting.
The following sgRNA sequences were used to target the indicated gene: MCOLN1, 5′-CTTGGCTCGAAACTTGTCGC-3′; ATG16L1, 5′-CACTGGAGGCATGGACCGCA-3′; DMXL1, 5′-GAAAATCCATTTACTGCTCG-3′; and DMXL2, 5′-AGTGCCAGTGGCTTAAAACT-3′.
Generation of HA–dTAG–DMXL1 knock-in cell line
The precise integration into target chromosome (PITCh) system73 was implemented to N-terminally tag the endogenous DMXL1 locus in U2OS cells with an HA epitope fused to dTAG74. The microhomology arms of the targeting vector, pCRIS–PITChv2–Puro–dTAG (Addgene, cat. no. 91793), were modified for the DMXL1 locus: left microhomology sequence, 5′-GCAGGACTAGGGCGCCGACA-3′; right microhomology sequence, 5′- TGAACCTGCACCAGGTGCTG-3′. The following sgRNA was cloned into px330–BbsI–PITCh (Addgene, cat. no. 127875): 5′-ACATGAACCTGCACCAGGTG-3′. U2OS cells were transfected simultaneously with the targeting vector and the px330-PITCh vector; 72 h after transfection, cells were subjected to selection with 2 µg ml–1 puromycin until puromycin-resistant colonies formed. Puromycin-resistant cells were single-cell sorted into 96-well plates on a SONY SH800S sorter, and correct knock-in of the targeting vector was confirmed by PCR using the following primers: forward, 5′-ACCCGTGGCATGAGCTGGAT-3′; reverse, 5′-AAGCAGTGGTCGCCAGGGTT-3′. Successful knock-in was further confirmed by immunoblotting.
Lysosome immunoprecipitation
Cells were made suitable for affinity-based isolation of lysosomes37 by stably expressing TMEM192–mRFP–3×HA (Addgene, cat. no. 134631). For experiments comparing multiple genotypes, cell lines were bulk sorted using a SONY SH800S sorter with the RFP filter set to achieve similar expression levels of TMEM192–mRFP–3×HA. Lyso-IP was conducted on confluent cells in 15-cm plates, and all steps were performed on ice or at 4 °C. Cells were rinsed with ice-cold PBS and gently scraped into ice-cold PBS. Cells were pelleted by centrifugation at 1,000g for 5 min at 4 °C. Cells were resuspended in ice-cold KPBS buffer (136 mM KCl, 10 mM KH2PO4, pH 7.25, supplemented with protease inhibitors) and lysed either by a Potter-Elvehjem homogenizer (30 strokes) or passing through a 25-G needle (10 strokes). The lysate was centrifuged at 1,500g for 10 min at 4 °C to obtain the postnuclear supernatant, which was incubated with prewashed magnetic anti-HA beads (Thermo, cat. no. 88837) for 30 min at 4 °C with end-to-end rotation. The lysosome-bound beads were washed twice with KPBS with 150 mM NaCl and once with KPBS. Lysosomes were eluted from beads at 37 °C for 30 min in KPBS with 0.5% NP-40. The eluates were subsequently processed for immunoblotting or quantitative proteomics.
Preparing Lyso-IP samples for TMT-proteomics
Lyso-IP samples were reduced with TCEP (final concentration of 5 mM) for 30 min at 25 °C with shaking at 1,000 r.p.m., followed by alkylation with iodoacetamide (final concentration of 15 mM) for 30 min at 25 °C in the dark. DTT (final concentration of 15 mM) was added to quench the reaction at 25 °C for 15 min with shaking in the dark. Proteins were precipitated by adding trichloroacetic acid (TCA) to a final concentration of 20% and were incubated on ice for 1 h. Precipitates were pelleted by centrifugation at 20,000g for 20 min at 4 °C, and pellets were washed three times with ice-cold acetone. Pellets were dried in a vacuum centrifuge and then reconstituted in 200 mM EPPS pH 8.0 for digestion. Samples were digested with LysC at 37 °C for 2 h, followed by overnight digestion with trypsin at 37 °C. Digested peptides were labeled with TMT reagents in a solution with 28% (vol/vol) acetonitrile for 1.5 h at 25 °C. The labelling reaction was quenched with 0.5% (wt/vol) hydroxylamine for 15 min at room temperature. To ensure equal peptide representation in each channel, the labeled samples were pooled in a 1:1 ratio and then dried in a vacuum centrifuge. The pooled sample was then fractionated into six final fractions using the high-pH reverse-phase peptide fractionation kit (Thermo, cat. no. 84868). Fractions were desalted by C18 StageTip before being reconstituted in a solution of 5% acetonitrile and 5% formic acid for liquid chromatography—tandem MS (LC–MS/MS) processing.
Preparation of crude lysosome fraction
A crude lysosome fraction was prepared using a modified version of a previously reported method for analysis of V-ATPase enrichment on lysosomes53. In brief, cells were resuspended in ice-cold lysis buffer (20 mM HEPES pH 7.5, 250 mM sucrose, 1 mM EDTA, protease inhibitor cocktail) and passed through a 25-G needle for 20 strokes. The cell lysate was centrifuged at 2,500g for 10 min at 4 °C, and in a new tube, the post-nuclear supernatant was centrifuged at 20,000g for 20 min at 4 °C to pellet the crude lysosome fraction. This pellet was resuspended in buffer containing 2% SDS (50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 2% SDS) and analyzed by immunoblotting.
Membrane and cytosol fractionation
Cells were washed once in ice-cold PBS, scraped from the plate into ice-cold PBS, and pelleted by centrifugation at 500g for 5 min at 4 °C. The cell pellet was resuspended in ice-cold hypotonic lysis buffer (20 mM HEPES pH 7.5, 10 mM KCl, 2 mM MgCl2, 1 mM DTT, protease inhibitor cocktail), incubated on ice for 10 min, passed through a 25-G needle for 20 strokes and then incubated on ice again for 10 min. The cell lysate was then centrifuged at 1,000g for 10 min at 4 °C, and the postnuclear supernatant was transferred to a prechilled 1.5-ml ultracentrifuge tube (Beckman, cat. no. 357448). The postnuclear supernatant was centrifuged at 150,000g for 1 h at 4 °C using a TLA-55 fixed-angle rotor (Beckman, cat. no. 366725) in the Optima MAX-XP Ultracentrifuge. The supernatant was collected as the cytosol fraction and the pellet, the membrane fraction, was resuspended in 2% SDS buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 2% SDS).
Stable expression of DMXL1 constructs
DMXL1–mNG
Stable expression of DMXL1 C-terminally tagged with mNG was achieved through piggyBac transposon-mediated transgene delivery75. DMXL1 complementary DNA (Horizon Discovery, cat. no. MHS6278-213246318), followed by a SGLRS linker and mNG, was cloned into a piggyBac transposon vector (cDNA expression driven by EF1ɑ promoter) using HiFi DNA Assembly (NEB, cat. no. E2621). U2OS cells were simultaneously transfected with the transposon vector and the piggyBac transposase expression vector (System Biosciences, cat. no. PB210PA-1) using PEI MAX (Polysciences, cat. no. 24765); 72 h post-transfection, cells were either bulk-sorted or single-cell sorted on a SONY SH800S sorter using the GFP filter set. Expression of DMXL1–mNG was confirmed by confocal microscopy.
DMXL1–mSG2–HA
Constructs used to stably express DMXL1 C-terminally tagged with mSG2 (monomeric StayGold)60 and 3×HA were generated similarly to DMXL1–mNG using HiFi DNA Assembly (NEB, cat. no. E2621). DNA encoding mSG2–3×HA was obtained as a gene fragment from Twist Bioscience. To generate the DMXL1 interface mutant (DMXL17A), a gene fragment with the corresponding mutations (Fig. 7a) was purchased from Twist Bioscience. The DMXL1 ΔLIR and ΔWD40_3 mutants were generated through HiFi DNA Assembly-based mutagenesis. Open reading frames (ORF) were cloned into the PB-CMV-MCS-puro piggyBac transposon vector (Addgene, cat. no. 219794). U2OS cells were simultaneously transfected with the transposon vector and the piggyBac transposase expression vector (System Biosciences, cat. no. PB210PA-1) using PEI MAX (Polysciences, cat. no. 24765); 72 h after transfection, cells were either subjected to puromycin selection (2 µg ml–1) or single-cell sorted on a SONY SH800S sorter using the GFP filter set. Expression was verified by immunoblot analysis and confocal microscopy.
Stable expression of mScarlet–LAMP1, Halo–GFP–LC3B and TFEB–GFP
Lentiviral particles were used to infect cells and introduce stable expression of transgenes. Lentiviral particles were generated in 293T cells by cotransfecting lentiviral packaging plasmids, VSV-G (Addgene, cat. no. 8454) and psPAX2 (Addgene, cat. no. 12260), with lentiviral transfer plasmids. mScarlet–LAMP1 was cloned from pcDNA3_mScarlet–LAMP1 (Addgene, cat. no. 185138) into pLX304 (Addgene, cat. no. 25890). Halo–GFP–LC3B was cloned from pMRX–No–HaloTag7–mGFP–LC3B (Addgene, cat. no. 184901) into plenti–UBC-gate–3×HA–pGK–PUR (Addgene, cat. no. 107393). TFEB–GFP was cloned from pEGFP–N1–TFEB (Addgene, cat. no. 38119) into pLX304 (Addgene, cat. no. 25890). All recombinant DNA cloning was done using HiFi DNA Assembly (NEB, cat. no. E2621).
Rescue of ATG16L1 in ATG16L1-KO cells
Lentiviral particles were used to infect ATG16L1-KO cells, facilitating stable expression of the beta-isoform of ATG16L1WT and ATG16L1K490A. Lentiviral particles were produced in 293T cells by cotransfecting lentiviral packaging plasmids, VSV-G (Addgene, 8454) and psPAX2 (Addgene, 12260), with lentiviral transfer plasmids. The ORF of ATG16L1 was obtained from the ORFeome library43. A Q5 site-directed mutagenesis kit (NEB, cat. no. E0554) was used to generate ATG16L1K490A. ATG16L1WT and ATG16L1K490A were subsequently cloned into pLX304 (Addgene, cat. no. 25890) using HiFi DNA Assembly (NEB, cat. no. E2621).
Expression and mCherry and mCherry–SidK in U2OS DMXL1–mNG cells
Expression of mCherry and SidK–mCherry was achieved using lentiviral delivery of transgenes into U2OS cells stably expressing DMXL1–mNG. ORFs encoding mCherry and SidK–mCherry were cloned into pLX304 (Addgene, cat. no. 25890) using HiFi DNA Assembly (NEB, cat. no. E2621). The ORF encoding SidK was obtained from pSAB35 (Addgene, cat. no. 175787). Lentiviral particles were generated in 293T cells by cotransfecting the lentiviral packaging plasmids, VSV-G (Addgene, cat. no. 8454) and psPAX2 (Addgene, cat. no. 12260), with lentiviral transfer plasmids. U2OS DMXL1–mNG cells were infected with supernatant containing lentivirus in the presence of 8 µg ml–1 polybrene. The medium was changed 24 h after infection and incubated for an additional 24 h before samples were collected for immunoprecipitation and proteomic analysis. More than 95% of cells expressed mCherry or SidK–mCherry, as confirmed using a ZOE fluorescent cell imager (Bio-Rad).
DMXL1 interaction proteomics
DMXL1 interaction proteomics were performed from cells stably overexpressing DMXL1–mNG and cells endogenously HA-tagged at the DMXL1 gene locus; mNeongreen-Trap (Proteintech, cat. no. ntma-20) and HA-Trap (Proteintech, cat. no. atma-20) magnetic agarose beads, respectively, were used to immunoprecipitate DMXL1, following the manufacturer’s protocol with modifications. Cells were rinsed with ice-cold PBS and scraped with ice-cold lysis buffer (50 mM EPPS pH 8.0, 150 mM NaCl, 0.5 mM EDTA, 1% NP-40, protease inhibitors) and incubated on ice for 30 min with occasional vortexing. The lysate was centrifuged at 21,000g for 20 min at 4 °C, and the supernatant was incubated with prewashed magnetic beads for 2 h at 4 °C with end-over-end rotation. After binding, beads were washed twice with wash buffer (50 mM EPPS pH 8.0, 150 mM NaCl).
Preparing DMXL1-IP samples for TMT-proteomics
Immunoprecipitated beads were resuspended in reducing buffer (50 mM EPPS pH 8.0, 2 M urea, 5 mM TCEP) and incubated at 25 °C for 30 min with shaking at 1,000 r.p.m. on a thermomixer. Iodoacetamide was added (final concentration of 15 mM) to alkylate samples at 25 °C for 30 min with shaking at 1,000 r.p.m. in the dark. To quench the reaction, DTT was added to a final concentration of 15 mM and incubated at 25 °C for 15 min with shaking in the dark. On-bead digestion was initiated with the addition of LysC for 2 h at 37 °C. The samples were then diluted with 50 mM EPPS pH 8.0 to decrease the urea concentration to 1 M; trypsin was then added for further digestion overnight at 37 °C. The digestion was quenched by adding formic acid to a final concentration of 1.4%. Peptides were separated from magnetic beads and desalted using a C18 StageTip. Peptides were then labeled with TMT reagents in a solution with 28% (vol/vol) acetonitrile for 1.5 h at 25 °C. The labeling reaction was quenched with 0.5% (wt/vol) hydroxylamine for 15 min at room temperature. Labeled samples were pooled in a 1:1 ratio to ensure that the same amount of peptide was represented in each channel. The pooled sample was dried in a vacuum centrifuge, and the resulting pellet was resuspended in 5% formic acid solution and the pH was adjusted to 2–3. The pooled sample was desalted using a C18 StageTip and dried again in a vacuum centrifuge. The dried sample was reconstituted in a solution of 5% acetonitrile and 5% formic acid for LC–MS/MS processing.
Quantitative proteomics
Mass spectrometry data acquisition
Mass spectrometry data were collected using an Orbitrap Lumos, Orbitrap Eclipse or Orbitrap Ascend mass spectrometer (Thermo) coupled with an nLC-1200 or Vanquish Neo liquid chromatograph. Peptides were separated on a 100-µm-inner-diameter microcapillary column packed with approximately 35 cm of Accucore C18 resin (2.6 µm, 150 Å, Thermo). MS data were acquired with parameters summarized in each Supplementary Table.
TMT data analysis
Spectra were converted to mzXML using MSconvert76. Database searching included all entries from the human UniProt reference Database (downloaded February 2020). The database was concatenated with one composed of all protein sequences for that database in reversed order. Searches were performed using a 50-ppm precursor ion tolerance for total protein level profiling. The product ion tolerance was set to 0.03 Da. These wide mass tolerance windows were chosen to maximize sensitivity in conjunction with Comet searches and linear discriminant analysis77,78. For TMT experiments, TMTpro labels on lysine residues and peptide N termini (+304.207 Da), as well as carbamidomethylation of cysteine residues (+57.021 Da), were set as static modifications, and oxidation of methionine residues (+15.995 Da) was set as a variable modification. Peptide-spectrum matches (PSMs) were adjusted to a 2% false discovery rate (FDR)79,80. PSM filtering was performed using a linear discriminant analysis, as described previously78, culminating in a final protein-level FDR of 2% (ref. 79). PSMs were filtered on the basis of a summed signal-to-noise ratio (SNR) greater than 200 across the TMT plex and for precursor signals with an isolation purity exceeding 0.5 of the MS1 isolation window. Quantification of the differential abundance of proteins was done using statistical models included in the MSstats package81. To normalize protein input across TMT channels, the raw intensities of all PSMs were summed, and the total intensity per channel was normalized to the median summed intensity across the TMTpro plex. Protein intensities were calculated by summing the input-normalized TMT intensities for the constituent peptide PSMs82, serving as a weighted average quantification. Statistical significance between experimental conditions was calculated using the Student’s t-test on normalized log2-transformed protein TMT intensities, and the resulting P values were adjusted for multiple hypothesis testing using the Benjamini–Hochberg method.
DMXL1-IP cross-link proteomics
DMXL1-IP cross-link proteomics was performed in cells stably overexpressing DMXL1–mNG, and DMXL1–mNG IP was performed as described above. DMXL1–mNG was immunoprecipitated from 10 confluent 15-cm plates, and on-bead cross-linking was done using DSSO (Thermo, cat. no. A33545) at a final concentration of 1 mM for 40 min at room temperature with end-to-end rotation. The cross-linking reaction was quenched by adding 1 M Tris pH 7.5 to a final concentration of 50 mM and incubating for 30 min at room temperature. The on-bead cross-linked sample was subjected to on-bead digestion. Digested cross-linked peptides were desalted using a 50 mg C8 Sep-Pak column (Waters, cat. no. WAT054965). Desalted peptides were fractionated by strong cation exchange (SCX) chromatography using a PolyLC PolySulfoethyl A 100-mm column (diameter 2.1 mm, 3 μm particle size). A 70-min linear gradient of mobile phase (0.5 M NaCl in 20% acetonitrile (ACN), 0.05% formic acid) was used at a flow rate of 0.18 ml min–1. Fractions were collected between 30 min and 60 min, dried in a SpeedVac and desalted using a C8 StageTip. Unfractionated and 11 pooled fractionated samples were reconstituted in 5% ACN, 5% formic acid and analyzed by LC–MS/MS83.
Samples were analyzed on an Orbitrap Fusion Lumos mass spectrometer with FAIMSpro. Chromatography was performed on a Proxeon EASY-nLC pump 1000 with a 15-cm-long 100-μm capillary column packed with Accucore 150 C18 resin (2.6 μm, 150 Å). A 90-min linear gradient was used, varying from 5% to 30% ACN in 0.125% formic acid at a flow rate of 0.3 µl min–1. Data were acquired using an HCD-MS2 strategy84,85 with the FAIMS Pro set to −50, −60 and −75 CV86. MS1 was acquired on the Orbitrap (120,000 resolution, 400–1,600 m/z, standard AGC target). Peptide fragmentation was performed by HCD at 21%, 27% and 33% normalized collision energy for peptide with 4–8 charge. MS2 was acquired on the Orbitrap (60,000 resolution, 1.6 m/z isolation window, 200% normalized AGC, maximum injection time 120 ms).
Cross-links in purified DMXL1–mNG pull-downs were identified with Scout v.1.4.14 (https://github.com/diogobor/Scout/releases/tag/1.4.14) at a FDR of 1%, as previously described58, and with the XlinkX module in Thermo Proteome Discoverer (version 2.5.0.400) with a FDR of 5% (refs. 84,87). A search was performed against the human canonical proteome (Uniprot Swiss-Prot 2022-05). The parameters were set to 10 ppm precursor mass tolerance, 20 ppm FTMS fragment mass tolerance, a maximum of 3 miscleavages and a minimum peptide length of 5. Carbamidomethylation and N-terminal acetylation were included as fixed and variable modification, respectively.
Immunoblot analysis of DMXL1–mSG2–HA immunoprecipitates
Cells from confluent 15-cm plates were rinsed once with ice-cold PBS, scraped into ice-cold PBS and centrifuged at 500g for 5 min at 4 °C to pellet the cells. The cell pellet was resuspended in ice-cold lysis buffer (50 mM EPPS pH 8.0, 10 mM KCl, 0.1% NP-40, protease inhibitor cocktail) and incubated on ice for 10 min, then passed through a 25-G needle for 12 strokes, and then incubated on ice for an additional 10 min. Buffer without NP-40 was added to dilute lysate to 0.05% NP-40. The cell lysate was then clarified by centrifugation at 8,000g for 10 min at 4 °C. The supernatant was transferred to a new tube, and the protein concentration was measured using BCA assay (Thermo, cat. no. 23225). Samples were normalized to a total of 1 mg of protein and incubated with anti-HA magnetic beads (Thermo, cat. no. 88836) for 2 h in 4 °C with end-over-end rotation. Ten percent of normalized sample was taken as input. Before incubation with lysate, anti-HA magnetic beads were equilibrated by washing the beads three times with wash buffer (50 mM EPPS pH 8.0, 10 mM KCl, 150 mM NaCl, 0.02% Tween-20). After incubation, beads were washed three times with wash buffer and samples were eluted by incubating the beads in elution buffer (50 mM TEAB pH 8.5, 5% SDS) at 37 °C for 30 min and then at 98 °C for 4 min. Input and immunoprecipitates samples were then analyzed by immunoblot.
Spinning disk confocal microscopy
Live-cell imaging of DMXL1-mNG recruitment to lysosomes
Cells stably expressing DMXL1–mNG and mScarlet–LAMP1 were seeded in 24-well no. 1.5 high-performance glass-bottom plates (Cellvis, cat. no. P24-1.5H-N) and cultured in DMEM without phenol red medium supplemented with 10% FBS. The plate was mounted in an Okolab stage top incubator warmed to 37 °C with 5% CO2 for live-cell image acquisition. Cells were treated directly on the stage with MLSA5 and imaged immediately. Images were collected with a Yokogawa CSU-X1 spinning disk confocal on a Nikon Ti-E motorized inverted microscope equipped with a Nikon Plan Fluor ×40/1.3-NA oil objective, and the Perfect Focus System was used to maintain focus over time. mScarlet–LAMP1 (561 nm) and DMXL1–mNeongreen (488 nm) were excited sequentially with a Nikon LUN-F XL solid state laser combiner (405 nm (80 mW), 488 nm (80 mW), 561 nm (65 mW), 640 (60 mW)) using a Semrock Di01–T405/488/568/647 dichroic mirror. Fluorescence emission spectra were collected through a Chroma ET455/50m (for 405 nm), Chroma ET525/36m (for 488 nm), Chroma ET605/52m (for 561 nm) and Chroma ET700/75m (for 640 nm) filter set (Chroma Technologies). Images were acquired with a Hamamatsu ORCA-Fusion BT sCMOS camera (6.5 µm2 photodiode) and NIS-Elements image acquisition software. Images were collected every 1 min using an exposure time of 100 ms (for both 561 nm and 488 nm). At each timepoint, 11 z-series optical sections were collected with a step-size of 0.3 µm, using the NIDAQ Piezo Z focus motor. Z-series are displayed as maximum z-projections, and intensity measurements were performed using Fiji (ImageJ). For intensity measurements, background subtraction was processed using the rolling ball background subtraction method for each channel. Subsequently, Otsu’s method was used to threshold the mScarlet–LAMP1 signal to select regions of interest (ROIs) corresponding to lysosomes. The selected ROI was applied to the mNeongreen channel, and the mNeongreen fluorescence intensity was measured for each cell for every timepoint.
Fixed-cell immunofluorescence imaging
Fixed-cell immunofluorescence imaging was performed on cells seeded in 24-well no. 1.5 high-performance glass bottom plates (Cellvis, P24-1.5H-N). Cells were rinsed once with PBS and fixed with 4% paraformaldehyde (Electron Microscopy Sciences) at room temperature for 15 min. For staining endogenously tagged DMXL1, cells were permeabilized with 0.5% Triton X-100 for 10 min at room temperature, and for staining LAMP1 in Salmonella-infected cells, cells were permeabilized with methanol for 5 min at −20 °C. After permeabilization, cells were rinsed three times with PBS for 5 min each, and were then blocked for 1 h at room temperature with a solution of 5% BSA and 100 mM glycine. Samples were then incubated overnight at 4 °C with primary antibodies diluted in blocking buffer. After this, cells were washed three times with PBS and incubated for 1 h at room temperature with secondary antibodies diluted in blocking buffer. Nuclei were stained with Hoechst33342, and samples were imaged in PBS. Images were acquired at room temperature with a Yokogawa CSU-X1 spinning disk confocal on a Nikon Ti-E motorized inverted microscope equipped with a Nikon Plan Fluor ×40/1.3-NA oil objective and a Nikon Plan Apo λD ×60/1.42-NA oil objective. Signals of 405/488/561/640 fluorophores were excited sequentially with a Nikon LUN-F XL solid state laser combiner (405 nm (80 mW), 488 nm (80 mW), 561 nm (65 mW), 640 (60 mW)) using a Semrock Di01–T405/488/568/647 dichroic mirror. Fluorescence emissions were collected through a Chroma ET455/50 m (for 405 nm), Chroma ET525/36 m (for 488 nm), Chroma ET605/52 m (for 561 nm), and Chroma ET700/75 m (for 640 nm) filter set (Chroma Technologies). Images were acquired with a Hamamatsu ORCA-Fusion BT sCMOS camera (6.5 µm2 photodiode) and NIS-Elements image acquisition software. For colocalization analysis, 30-pixel × 30-pixel regions in the cytosol were drawn, and Otsu’s method was used to threshold signal on both channels. Pearson’s coefficient was measured using the BIOP JACoP plugin in Fiji (ImageJ).
Live-cell lysosome pH measurements
The day before measurements were taken, 100,000 cells were seeded in 24-well no. 1.5 high-performance glass-bottom plate (Cellvis, cat. no. P24-1.5H-N). On the day that measurements were taken, cells were incubated with SiR-Lysosome (1:1,000, Cytoskeleton) and pHLys Red (1:1,000, Dojindo) in DMEM and 10% FBS for 1 h. The stains were then washed out and chased with phenol-red free DMEM with 10% FBS for 3 h before imaging. For BafA1 treatment, 1 µM BafA1 was treated 2.5 h into the chase for 30 min before live-cell imaging on a spinning disk confocal microscope equipped with a Nikon Plan Apo λD ×20/0.75-NA air objective. To establish the pH calibration curve, WT cells were bathed in calibration buffers, with the pH adjusted to 3, 4, 5, 6 and 7, supplemented with 10 µM monensin88. For both the experimental and pH-calibration conditions, five to six fields of view were imaged and analyzed in their entirety. This process was repeated for each independent experiment. Image analysis was performed using Fiji (ImageJ). For each field of view, background subtraction was processed using the rolling ball background subtraction method for each channel. Subsequently, Otsu’s method was used to threshold the SiR-Lysosome signal to select ROIs corresponding to lysosomes. The selected ROI was applied to the pHLys Red channel and then fluorescence intensity was measured. The fluorescence intensity of the pHLys Red channel was then fitted to the calibration curve to calculate the pH.
DQ-BSA
DQ-BSA Red (Thermo, cat. no. D12051) was prepared in PBS at a stock concentration of 2 mg ml–1 and used at a working concentration of 20 µg ml–1. Cells were seeded in 24-well plates (glass-bottom for imaging) and incubated with DQ-BSA Red for 4 h with or without cotreatment before analysis by spinning disk confocal microscopy or flow cytometry. For microscopy analysis, images were acquired with cells incubated at 37 °C with 5% CO2 using an Okolab stage top incubator. Images were collected with a Yokogawa CSU-X1 spinning disk confocal on a Nikon Ti-E motorized inverted microscope, equipped with a Nikon Plan Apo λD ×20/0.75-NA air objective. DQ-BSA Red signal (561 nm) was excited with a Nikon LUN-F XL solid-state laser combiner (405 nm (80 mW), 488 nm (80 mW), 561 nm (65 mW), 640 (60 mW)) using a Semrock Di01–T405/488/568/647 dichroic mirror. Fluorescence emissions were collected through a Chroma ET455/50m (for 405 nm), Chroma ET525/36m (for 488 nm), Chroma ET605/52m (for 561 nm) and Chroma ET700/75m (for 640 nm) filter set (Chroma Technologies). Images were acquired with a Hamamatsu ORCA-Fusion BT sCMOS camera (6.5 µm2 photodiode) and NIS-Elements image acquisition software. DQ-BSA Red intensity measurements were performed in Fiji (ImageJ). Otsu’s method was used to threshold the DQ-BSA Red signal in each cell and measure signal intensity. For flow cytometry analysis, cells were detached and separated into single cells by passing through a cell strainer with a mesh size of 35 µm. The DQ-BSA Red signal intensity was measured on a BD LSR II flow cytometer (BD Biosciences), and 10,000 cells were analyzed for each condition per experiment. Data were analyzed using FlowJo.
Immunoblot analysis
Cell lysates for immunoblot analysis were prepared by directly lysing cells on plates in a buffer containing 50 mM Tris-Cl pH 7.5, 150 mM NaCl, 1 mM EDTA and 2% SDS. Protein concentrations were determined by BCA assay (Thermo, cat. no. 23225), and all samples were adjusted to ensure equal protein amounts. Protein samples were resolved on either 7.5% or 4–20% gradient pre-cast polyacrylamide Tris-Glycine gels (Bio-Rad) and transferred to PVDF membranes (Millipore) using a wet-transfer system (Bio-Rad). Membranes were blocked with 5% skim-milk in TBST (0.05% Tween-20). Membranes were probed with primary antibodies overnight at 4 °C and secondary antibodies at room temperature for 1 h. Chemiluminescence was visualized using the Bio-Rad Chemidoc MP Imaging System. Immunoblot densitometry quantification was performed on unsaturated images with ImageStudio software (LICOR).
Structural analysis
Structural predictions were performed using AlphaFold 3 (https://golgi.sandbox.google.com), as described46, and models were visualized using Pymol. LIR searches64 were performed at https://ilir.warwick.ac.uk/search.php. Prediction of LIR interactions with GABARAP was performed as described65 using the AF3 server46 (https://golgi.sandbox.google.com). ipTM scores for LIR predictions are provided in Supplementary Table 4.
Endolysosomal electrophysiology
Enlarged endolysosomes were generated by treating U2OS cells (WT and ATG16L1-KO) overnight with 1 µM vacuolin. A patch pipette was used to rupture the plasma membrane and release the enlarged endolysosomes into the bath solution, which was used to mimic the cytoplasm and contained (in mM): 144 K-Gluconate, 4 NaCl, 10 HEPES, 1 EGTA, 2 MgCl2 and 0.39 CaCl2, adjusted to pH 7.2 with KOH. Patch–clamp experiments were performed using EPC-10 amplifiers controlled by Patchmaster software (HEKA). Currents were sampled at 10 kHz and filtered at 2.9 KHz. Following seal formation and break-in to whole-endolysosome mode, 240-ms voltage ramps from −40 mV to +100 mV were applied every 4 s. Current amplitudes were assessed at +100 mV. Patch pipettes were pulled from borosilicate glass tubing (Warner Instruments) and filled with pipette solution containing (in mM): 145 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 MES, 10 HEPES and 10 Glucose, adjusted to pH 4.6 with NaOH. Resistance of the microelectrodes was typically in the range between 4 and 8 MΩ. TRPML1 channels were activated by the addition of 5 µM MLSA5 to the preparation until a stable current was achieved.
Salmonella infection
Bacterial culture conditions
The Salmonella strains used in this study are described in Supplementary Table 7. These strains were grown overnight at 37 °C without shaking in LB (Invitrogen) containing 300 mM NaCl to induce high expression of the SPI-1 secretion system. The following day, overnight cultures were diluted 1:20 in fresh, prewarmed LB-salt medium and grown with shaking until an optical density at 600 nm of 0.9–1.2 was reached. Bacteria were then washed once in sterile PBS and resuspended in infection medium before infection.
Infection
Eighty-five thousand cells were seeded in a 24-well no. 1.5 high-performance glass-bottom plate (Cellvis, P24-1.5H-N) and infected with Salmonella at a multiplicity of infection of 50. For infection, plates were centrifuged at 750g for 10 min in a 37 °C prewarmed centrifuge. Plates were then stored in an incubator at 37 °C with 5% CO2 for an additional hour. After incubation, cells were rinsed with PBS three times and fixed with pre-warmed 4% paraformaldehyde (Electron Microscopy Sciences) at 37 °C for 10 min. Subsequently, samples were processed for immunofluorescence as described in ‘Fixed-cell immunofluorescence imaging.’
Quantification
Salmonella-containing vacuoles (SCVs) were defined by LAMP1-positive compartments containing mCherry-positive Salmonella. GFP–LC3B and DMXL1–mNG-positive SCVs were identified by colocalization of GFP or mNG signal with LAMP1 (imaged using the 640-nm channel). The percentage of GFP–LC3B or DMXL1–mNG-positive SCVs was calculated by dividing the number of GFP–LC3B or DMXL1–mNG-positive SCVs to the total number of SCVs. The calculation was performed in Microsoft Excel version 16.81.
Statistics and reproducibility
All statistical tests, except proteomics analysis, were performed using Prism 9 (GraphPad). Normality and equal variances were tested and the statistical conditions and tests used are indicated in each figure legend. Quantification plots show the mean, and error bars represent the s.d.
No statistical method was used to predetermine sample size, but our sample sizes are similar to those reported in previous publications33,89,90. Experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment.
In Extended Data Figure 1f, TMT channel 130N, which corresponds to a replicate for MLSA5 0 min, was excluded from analysis because of poor TMT labelling.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository91 with the following dataset identifiers: PXD054968, PXD054967, PXD054973 and PXD061791.
The following structural data form the Protein Data Bank (PDB) were used as part of our structural modelling efforts: 6XBW, 9COP and 7VEC.
Unprocessed immunoblots and numerical source data are available in source data. Microscopy data are available at https://doi.org/10.5281/zenodo.15270572. All other data supporting the findings of this study are available from the corresponding author on reasonable request. Source data are provided with this paper.
Code availability
This paper does not report original software or code.
References
Lawrence, R. E. & Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 21, 133–142 (2019).
Mindell, J. A. Lysosomal acidification mechanisms. Annu. Rev. Physiol. 74, 69–86 (2012).
Li, P., Gu, M. & Xu, H. Lysosomal ion channels as decoders of cellular signals. Trends Biochem. Sci. 44, 110–124 (2019).
Xu, H. & Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 77, 57–80 (2015).
Dong, X. P. et al. Activating mutations of the TRPML1 channel revealed by proline-scanning mutagenesis. J. Biol. Chem. 284, 32040–32052 (2009).
LaPlante, J. M. et al. Identification and characterization of the single channel function of human mucolipin-1 implicated in mucolipidosis type IV, a disorder affecting the lysosomal pathway. FEBS Lett. 532, 183–187 (2002).
Xu, H., Delling, M., Li, L., Dong, X. & Clapham, D. E. Activating mutation in a mucolipin transient receptor potential channel leads to melanocyte loss in varitint-waddler mice. Proc. Natl Acad. Sci. USA 104, 18321–18326 (2007).
Nakamura, S. et al. LC3 lipidation is essential for TFEB activation during the lysosomal damage response to kidney injury. Nat. Cell Biol. 22, 1252–1263 (2020).
Scotto Rosato, A. et al. TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKKβ/VPS34 pathway. Nat. Commun. 10, 5630 (2019).
Venkatachalam, K., Wong, C. O. & Zhu, M. X. The role of TRPMLs in endolysosomal trafficking and function. Cell Calcium 58, 48–56 (2015).
Alavi, M. S., Soheili, V. & Roohbakhsh, A. The role of transient receptor potential (TRP) channels in phagocytosis: a comprehensive review. Eur. J. Pharmacol. 964, 176302 (2024).
Lloyd-Evans, E. & Platt, F. M. Lysosomal Ca2+ homeostasis: role in pathogenesis of lysosomal storage diseases. Cell Calcium 50, 200–205 (2011).
Chen, Q. et al. Structure of mammalian endolysosomal TRPML1 channel in nanodiscs. Nature 550, 415–418 (2017).
Schmiege, P., Fine, M., Blobel, G. & Li, X. Human TRPML1 channel structures in open and closed conformations. Nature 550, 366–370 (2017).
Hirschi, M. et al. Cryo-electron microscopy structure of the lysosomal calcium-permeable channel TRPML3. Nature 550, 411–414 (2017).
Dong, X. P. et al. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca2+ release channels in the endolysosome. Nat. Commun. 1, 38 (2010).
Feng, X. et al. Drosophila TRPML forms PI(3,5)P2-activated cation channels in both endolysosomes and plasma membrane. J. Biol. Chem. 289, 4262–4272 (2014).
Fine, M., Schmiege, P. & Li, X. Structural basis for PtdInsP(2)-mediated human TRPML1 regulation. Nat. Commun. 9, 4192 (2018).
McCartney, A. J., Zhang, Y. & Weisman, L. S. Phosphatidylinositol 3,5-bisphosphate: low abundance, high significance. Bioessays 36, 52–64 (2014).
Durgan, J. & Florey, O. Many roads lead to CASM: diverse stimuli of noncanonical autophagy share a unifying molecular mechanism. Sci. Adv. 8, eabo1274 (2022).
Durgan, J. et al. Non-canonical autophagy drives alternative ATG8 conjugation to phosphatidylserine. Mol. Cell 81, 2031–2040 (2021).
Goodwin, J. M. et al. GABARAP sequesters the FLCN-FNIP tumor suppressor complex to couple autophagy with lysosomal biogenesis. Sci. Adv. 7, eabj2485 (2021).
Gui, X. et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567, 262–266 (2019).
Fischer, T. D., Wang, C., Padman, B. S., Lazarou, M. & Youle, R. J. STING induces LC3B lipidation onto single-membrane vesicles via the V-ATPase and ATG16L1-WD40 ___domain. J. Cell Biol. 219, e202009128 (2020).
Inomata, M. et al. Macrophage LC3-associated phagocytosis is an immune defense against Streptococcus pneumoniae that diminishes with host aging. Proc. Natl Acad. Sci. USA 117, 33561–33569 (2020).
Meyer-Morse, N. et al. Listeriolysin O is necessary and sufficient to induce autophagy during Listeria monocytogenes infection. PLoS ONE 5, e8610 (2010).
Lopez de Armentia, M. M., Gauron, M. C. & Colombo, M. I. Staphylococcus aureus alpha-toxin induces the formation of dynamic tubules labeled with LC3 within host cells in a Rab7 and Rab1b-dependent manner. Front. Cell Infect. Microbiol 7, 431 (2017).
Bentley-DeSousa, A., Roczniak-Ferguson, A. & Ferguson, S. M. A STING–CASM–GABARAP pathway activates LRRK2 at lysosomes. J. Cell Biol. 224, e202310150 (2025).
Beale, R. et al. A LC3-interacting motif in the influenza A virus M2 protein is required to subvert autophagy and maintain virion stability. Cell Host Microbe 15, 239–247 (2014).
Fletcher, K. et al. The WD40 ___domain of ATG16L1 is required for its non-canonical role in lipidation of LC3 at single membranes. EMBO J. 37, e97840 (2018).
Florey, O., Gammoh, N., Kim, S. E., Jiang, X. & Overholtzer, M. V-ATPase and osmotic imbalances activate endolysosomal LC3 lipidation. Autophagy 11, 88–99 (2015).
Fracchiolla, D. & Martens, S. Sorting out ‘non-canonical’ autophagy. EMBO J. 37, e98895 (2018).
Hooper, K. M. et al. V-ATPase is a universal regulator of LC3-associated phagocytosis and non-canonical autophagy. J. Cell Biol. 221, e202105112 (2022).
Timimi, L. et al. The V-ATPase/ATG16L1 axis is controlled by the V(1)H subunit. Mol. Cell 84, 2966–2983 (2024).
Ulferts, R. et al. Subtractive CRISPR screen identifies the ATG16L1/vacuolar ATPase axis as required for non-canonical LC3 lipidation. Cell Rep. 37, 109899 (2021).
Xu, Y. et al. A bacterial effector reveals the V-ATPase-ATG16L1 axis that initiates xenophagy. Cell 178, 552–566 (2019).
Abu-Remaileh, M. et al. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 358, 807–813 (2017).
Smardon, A. M., Tarsio, M. & Kane, P. M. The RAVE complex is essential for stable assembly of the yeast V-ATPase. J. Biol. Chem. 277, 13831–13839 (2002).
Wang, H. & Rubinstein, J. L. CryoEM of V-ATPases: assembly, disassembly, and inhibition. Curr. Opin. Struct. Biol. 80, 102592 (2023).
Jaskolka, M. C., Winkley, S. R. & Kane, P. M. RAVE and Rabconnectin-3 complexes as signal dependent regulators of organelle acidification. Front. Cell Dev. Biol. 9, 698190 (2021).
Smardon, A. M., Nasab, N. D., Tarsio, M., Diakov, T. T. & Kane, P. M. Molecular interactions and cellular itinerary of the yeast RAVE (regulator of the H+-ATPase of vacuolar and endosomal membranes) complex. J. Biol. Chem. 290, 27511–27523 (2015).
Merkulova, M. et al. Mapping the H+ (V)-ATPase interactome: identification of proteins involved in trafficking, folding, assembly and phosphorylation. Sci. Rep. 5, 14827 (2015).
Huttlin, E. L. et al. The BioPlex Network: a systematic exploration of the human interactome. Cell 162, 425–440 (2015).
Cho, N. H. et al. OpenCell: endogenous tagging for the cartography of human cellular organization. Science 375, eabi6983 (2022).
Eaton, A. F., Danielson, E. C., Capen, D., Merkulova, M. & Brown, D. Dmxl1 Is an essential mammalian gene that is required for V-ATPase assembly and function in vivo. Function 5, zqae025 (2024).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Nusinow, D. P. et al. Quantitative proteomics of the Cancer Cell Line Encyclopedia. Cell 180, 387–402 (2020).
Carey, K. L. et al. TFEB transcriptional responses reveal negative feedback by BHLHE40 and BHLHE41. Cell Rep. 33, 108371 (2020).
Vasanthakumar, T. & Rubinstein, J. L. Structure and roles of V-type ATPases. Trends Biochem. Sci. 45, 295–307 (2020).
Yan, Y., Denef, N. & Schupbach, T. The vacuolar proton pump, V-ATPase, is required for notch signaling and endosomal trafficking in Drosophila. Dev. Cell 17, 387–402 (2009).
Beck, M. et al. The quantitative proteome of a human cell line. Mol. Syst. Biol. 7, 549 (2011).
Itzhak, D. N., Tyanova, S., Cox, J. & Borner, G. H. Global, quantitative and dynamic mapping of protein subcellular localization. eLife 5, e16950 (2016).
Ratto, E. et al. Direct control of lysosomal catabolic activity by mTORC1 through regulation of V-ATPase assembly. Nat. Commun. 13, 4848 (2022).
Boyle, K. B. et al. TECPR1 conjugates LC3 to damaged endomembranes upon detection of sphingomyelin exposure. EMBO J. 42, e113012 (2023).
Huttlin, E. L. et al. Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell 184, 3022–3040 (2021).
Wang, H., Tarsio, M., Kane, P. M. & Rubinstein, J. L. Structure of yeast RAVE bound to a partial V(1) complex. Proc. Natl Acad. Sci. USA 121, e2414511121 (2024).
Merkley, E. D. et al. Distance restraints from crosslinking mass spectrometry: mining a molecular dynamics simulation database to evaluate lysine–lysine distances. Protein Sci. 23, 747–759 (2014).
Clasen, M. A. et al. Proteome-scale recombinant standards and a robust high-speed search engine to advance cross-linking MS-based interactomics. Nat. Methods 21, 2327–2335 (2024).
Zhu, Y. et al. Cross-link assisted spatial proteomics to map sub-organelle proteomes and membrane protein topologies. Nat. Commun. 15, 3290 (2024).
Ando, R. et al. StayGold variants for molecular fusion and membrane-targeting applications. Nat. Methods 21, 648–656 (2024).
Abbas, Y. M., Wu, D., Bueler, S. A., Robinson, C. V. & Rubinstein, J. L. Structure of V-ATPase from the mammalian brain. Science 367, 1240–1246 (2020).
Zhao, J. et al. Molecular basis for the binding and modulation of V-ATPase by a bacterial effector protein. PLoS Pathog. 13, e1006394 (2017).
Xu, L. et al. Inhibition of host vacuolar H+-ATPase activity by a Legionella pneumophila effector. PLoS Pathog. 6, e1000822 (2010).
Jacomin, A. C., Samavedam, S., Promponas, V. & Nezis, I. P. iLIR database: a web resource for LIR motif-containing proteins in eukaryotes. Autophagy 12, 1945–1953 (2016).
Stuke, J. F. M. & Hummer, G. AlphaFold2 SLiM screen for LC3-LIR interactions in autophagy. Autophagy https://doi.org/10.1080/15548627.2025.2493999 (2025).
Chino, H. et al. Phosphorylation by casein kinase 2 enhances the interaction between ER-phagy receptor TEX264 and ATG8 proteins. EMBO Rep. 23, e54801 (2022).
Platt, F. M., d’Azzo, A., Davidson, B. L., Neufeld, E. F. & Tifft, C. J. Lysosomal storage diseases. Nat. Rev. Dis. Prim. 4, 27 (2018).
Martina, J. A., Raben, N. & Puertollano, R. SnapShot: lysosomal storage diseases. Cell 180, 602–602 e601 (2020).
Somogyi, A. et al. The synthetic TRPML1 agonist ML-SA1 rescues Alzheimer-related alterations of the endosomal-autophagic-lysosomal system. J. Cell Sci. 136, jcs259875 (2023).
Esposito, A. et al. Biallelic DMXL2 mutations impair autophagy and cause Ohtahara syndrome with progressive course. Brain 142, 3876–3891 (2019).
Coupland, C. E. et al. High-resolution electron cryomicroscopy of V-ATPase in native synaptic vesicles. Science 385, 168–174 (2024).
Schmid-Burgk, J. L. et al. OutKnocker: a web tool for rapid and simple genotyping of designer nuclease edited cell lines. Genome Res. 24, 1719–1723 (2014).
Sakuma, T., Nakade, S., Sakane, Y., Suzuki, K. T. & Yamamoto, T. MMEJ-assisted gene knock-in using TALENs and CRISPR–Cas9 with the PITCh systems. Nat. Protoc. 11, 118–133 (2016).
Nabet, B. et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 14, 431–441 (2018).
Woodard, L. E. & Wilson, M. H. piggyBac-ing models and new therapeutic strategies. Trends Biotechnol. 33, 525–533 (2015).
Chambers, M. C. et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 (2012).
Beausoleil, S. A., Villen, J., Gerber, S. A., Rush, J. & Gygi, S. P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 (2006).
Huttlin, E. L. et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 (2010).
Elias, J. E. & Gygi, S. P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 (2007).
Elias, J. E. & Gygi, S. P. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol. Biol. 604, 55–71 (2010).
Kohler, D. et al. MSstats Version 4.0: statistical analyses of quantitative mass spectrometry-based proteomic experiments with chromatography-based quantification at scale. J. Proteome Res. 22, 1466–1482 (2023).
Plubell, D. L. et al. Extended multiplexing of tandem mass tags (TMT) labeling reveals age and high fat diet specific proteome changes in mouse epididymal adipose tissue. Mol. Cell Proteom. 16, 873–890 (2017).
Gonzalez-Lozano, M. A. et al. Stitching the synapse: cross-linking mass spectrometry into resolving synaptic protein interactions. Sci. Adv. 6, eaax5783 (2020).
Liu, F., Lossl, P., Scheltema, R., Viner, R. & Heck, A. J. R. Optimized fragmentation schemes and data analysis strategies for proteome-wide cross-link identification. Nat. Commun. 8, 15473 (2017).
Liu, F., Rijkers, D. T., Post, H. & Heck, A. J. Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nat. Methods 12, 1179–1184 (2015).
Schnirch, L. et al. Expanding the depth and sensitivity of cross-link identification by differential ion mobility using high-field asymmetric waveform ion mobility spectrometry. Anal. Chem. 92, 10495–10503 (2020).
Klykov, O. et al. Efficient and robust proteome-wide approaches for cross-linking mass spectrometry. Nat. Protoc. 13, 2964–2990 (2018).
Ma, L., Ouyang, Q., Werthmann, G. C., Thompson, H. M. & Morrow, E. M. Live-cell microscopy and fluorescence-based measurement of luminal pH in Intracellular Organelles. Front. Cell Dev. Biol. 5, 71 (2017).
Florey, O., Kim, S. E., Sandoval, C. P., Haynes, C. M. & Overholtzer, M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat. Cell Biol. 13, 1335–1343 (2011).
Hundley, F. V. et al. Endo-IP and lyso-IP toolkit for endolysosomal profiling of human-induced neurons. Proc. Natl Acad. Sci. USA 121, e2419079121 (2024).
Deutsch, E. W. et al. The ProteomeXchange consortium at 10 years: 2023 update. Nucleic Acids Res. 51, D1539–D1548 (2023).
Acknowledgements
We thank members of the Harper lab for feedback and I. R. Smith for implementation of statistical analysis. We also thank the Center for Imaging Technology and Education (CITE) and the Flow Cytometry Core at Harvard Medical School for microscopy and flow cytometry support, respectively. This work was supported by Warren Alpert Foundation (to J.W.H.), NIH (R01NS083524, R01NS110395 to J.W.H.; R01GM132129 to J.A.P.) and a Rubicon Postdoctoral Fellowship (M.A.G.-L.).
Author information
Authors and Affiliations
Contributions
Conceptualization: C.L., J.W.H.; cell biology, proteomics, biochemistry: C.L.; cross-linking proteomics: M.A.G.-L and C.L.; mass spectrometry: J.A.P.; structural prediction/visualization: C.L., J.W.H., T.F.; electrophysiology: T.B., Z.N., D.D.C. under supervision of M.M.M.; Salmonella experiments: M.J.G.E. and C.L. under supervision of S.H. The manuscript was written by J.W.H. and C.L. with input from all authors.
Corresponding author
Ethics declarations
Competing interests
J.W.H. is a co-founder of Caraway Therapeutics, a subsidiary of Merck, and is a member of the scientific advisory board for Lyterian Therapeutics. T.B., Z.N., D.D.C. and M.M.M. participated in this study as employees of Caraway Therapeutics. The other authors declare no competing interests.
Peer review
Peer review information
Nature Structural & Molecular Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Melina Casadio, in collaboration with the Nature Structural & Molecular Biology team. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Quantitative proteomics captures recruitment of proteins to lysosomes upon TRPML1 activation, related to Fig. 1.
a, Gene editing of MCOLN1 in U2OS cells was performed using CRISPR-Cas9, generating out of frame deletions in exon 2, as based on MiSeq analysis of the locus in B7 and C7 clones. Yellow highlighted sequence indicates the position of the gRNA target. b, The indicated WT and MCOLN1 KO U2OS cell lines were treated with MLSA5 (24 h) or left untreated and whole cell extracts were analyzed by immunoblotting with the indicated antibodies. Blot is representative of three independent experiments. c, Volcano plot [log2FC versus -log10(q-value)] for Lyso-IP from U2OS cells treated with MLSA5 for 1, 2 or 4 h compared with untreated cells. d, TMT reporter ion intensity for DMXL1 in Lyso-IP samples from cells treated with MLSA5 for 0, 0.5, 1, 2, or 4 h. Each point indicates the intensity of DMXL1 in one of three biological replicate experiments, with the average shown by the bars. e, Validation of Lyso-IP in A101D melanoma cell line. Cell extracts from A101D cells stably expressing TMEM192-mRFP-3xHA were used for Lyso-IP (α-HA immunoprecipitation). Input and Lyso-IP samples were analyzed by immunoblotting with the indicated antibodies. Blot is representative of three independent experiments. f, Volcano plot [log2FC versus -log10(p-value)] for Lyso-IP from A101D cells treated with MLSA5 for 20 min (left) and 4 h (right) compared with untreated cells, showing enrichment of DMXL1 and WDR7 (blue points), ATG8 proteins (red points) and ATG8ylation machinery (yellow points). P values were calculated using a two-sided Student’s t-test and was not adjusted for multiple comparisons.
Extended Data Fig. 2 Analysis of ATG16L1 KO U2OS cells and the role of ATG16L1 in DMXL1 recruitment during TRPML1 activation, related to Fig. 1.
a, Gene editing to create ATG16L1 KO U2OS cells. MiSeq reads are shown for clone 1C10, showing single nucleotide insertion or deletion. Location of gRNA target is highlighted in yellow. b, Validation of ATG16L1 KO cells for defects in CASM. U2OS cells of the indicated genotypes were left untreated or treated with MLSA5 for 2 h prior to immunoblotting of cell extracts for the indicated proteins. Blot is representative of three independent experiments. c, Whole-endolysosome patch clamp recordings measured from endolysosomes isolated from U2OS WT or ATG16L1 KO cells. The holding potential was 0 mV and the blue line indicates 0 current level. Ringer represents the basal endolysosomal current prior to the addition of 5 µM MLSA5. Perfusion of the TRPML1 agonist resulted in the activation of an outward current (from inside of the endolysosome to the bath) in both types of endolysosomes. d, Current density (pA/pF at +100 mV) recorded from U2OS WT (n=8) and U2OS ATG16L1 KO (n=9) endolysosomes in response to 5 µM MLSA5. Every individual measurement as well as mean ± SD for each group are plotted (Supplementary Table 3). Unpaired two-sided Student’s t-test with Welch’s correction was used to determine statistical significance. e, Correlation plot displaying the relationship between lysosomal enrichment and ATG16L1-dependence. f, TMT reporter ion intensity for DMXL1 in Lyso-IP samples from WT or ATG16L1 KO cells treated with MLSA5 for 2 h. Each point indicates the intensity of DMXL1 in one of three biological replicate experiments, with the average shown by the bars.
Extended Data Fig. 3 MCOLN1/TRPML1 is required for DMXL1 recruitment to lysosomes in response to MLSA5, related to Fig. 2.
a, Live-cell imaging of WT (top) or MCOLN1 KO (bottom) U2OS cells stably expressing DMXL1-mNG upon treatment with MLSA5. Lysosomes were marked using a lysosomal-specific dye, SiR-Lyso. Images at 2 min interval, up to 10 min, are shown. Scale bar is 10 microns; inset scale bar is 3 microns. b, Fold change in mean fluorescence intensity over 20 min. Quantification was performed on the following total number cells from three biological replicates: WT, n=20; MCOLN1 KO, n=24. Data are presented as mean values ± SD. Two-way repeated measures ANOVA with the Geisser-Greenhouse correction followed by Dunnett’s multiple comparison test was used to assess significance. Indicated statistical significance is for the 20 min timepoint. ****, p-value ≤ 0.0001.
Extended Data Fig. 4 Analysis of HA-dTAG-DMXL1 knock-in cells and characterization of cellular pools of DMXL1, related to Fig. 2.
a, Strategy for knocking in an HA-dTAG epitope into the N-terminus of DMXL1. b,c, PCR validation of knock-in clones (panel b) and by immunoblotting of extracts from cells using α-HA or α-DMXL1 antibodies (panel c). PCR and immunoblot was performed once. d, Immunofluorescence of HA-dTAG-DMXL1 in edited U2OS cells, with and without MLSA5 treatment (2 h). Cells were stained with either of two Golgi markers (GM130 or TGN46), LAMP1 to mark endolysosomes, or α-HA to detect HA-dTAG-DMXL1. Scale bar is 10 microns. e, Quantification of colocalization between LAMP1 and HA-dTAG-DMXL1 by Pearson correlation coefficient (PCC). Each point represents the PCC value from individual cells; control, n=30 cells; MLSA5, n=30 cells. Data are presented as mean values ± SD. Unpaired two-sided Student’s t-test was used for statistical comparison. *, p-value ≤ 0.05; **, p-value ≤ 0.01; ***, p-value ≤ 0.001; ****, p-value ≤ 0.0001; ns, not significant. f, Confocal images of fixed WT U2OS cells stably expressing DMXL1-mNG and immunostained with an α-Giantin antibody. Scale bar is 10 microns. Micrographs are from a single experiment. g, WT U2OS cells stably expressing DMXL1-mNG and mScarlet-LAMP1, to mark lysosomes, were pre-treated with BFA to disrupt the Golgi and then subjected to live-cell imaging after co-treatment with MLSA5. Images at 2 min interval, up to 10 min, are shown. Scale bar is 10 microns; inset scale bar is 3 microns. Fold change in mean fluorescence intensity over 20 min is shown in Fig. 2e. h, Confocal images of fixed WT U2OS cells stably expressing DMXL1-mNG treated with nocodazole (top) or pre-treated with nocodazole (5 ug/ml; 1 h) and co-treated with MLSA5 (5 μM, 2 h) (bottom). Immunostaining of LAMP1 was used to mark lysosomes (magenta). Scale bar is 10 microns; inset scale bar is 5 microns. Micrographs are from a single experiment. i, Immunoblot of cytosol and membrane fractions prepared from WT U2OS cells and U2OS cells stably overexpressing DMXL1-mNG, probed with the indicated antibodies. Blot is representative of three independent experiments.
Extended Data Fig. 5 Structural predictions of DMXL1/2-ROGDI-WDR7 and the relationship between Rav1-C domains in DMXL1 and Rav1, related to Fig. 4.
a, Overlay of AF3 model of yeast Rav1-Rav2 with Rav1-Rav2 from 9COP56. Residues 1–21, 60–77, 227–293, and 349–351 are not present in Rav2 from 9COP. b, Overlay of the major helical regions of the Rav1-C ___domain for Rav1 (residues 756-1190) and the corresponding region in DMXL1 (residues 1318-1878). Green arrow indicates the ___location of the Rav1-C ___domain within the larger helical ___domain of DMXL1. c, Dmxl2-Rogdi-Wdr7 prediction by AF3. Lys-Lys crosslinks for Dmxl2-Rogdi-Wdr7 complex identified by Zhu et al.59 are shown by red lines (see Supplementary Table 4).
Extended Data Fig. 6 Analysis of DMXL1/2-deficient cells, related to Fig. 5.
a, DNA sequence analysis of U2OS cells gene edited to create homozygous knockout of DMXL1 (clone 2B4). b, DNA sequence analysis of U2OS cells gene edited to create homozygous knockout of DMXL1 and heterozygous knockout of DMXL2 (clone C2). Locations of gRNA target sequences is shown in yellow. c, Immunoblot of the indicated U2OS cells with α-DMXL1 or α-DMXL2 antibodies, using α-Actin as a loading control. Blot is representative of three independent experiments. d, Immunoblotting of WT, DMXL1−/− and DMXL1−/−;DMXL2+/− cell extracts with α-TFEB or α-LC3B after treatment of cells with MLSA5 or Torin1 (2 h). Blots were probed with α-Hsp90 as loading control. Blot is representative of three independent experiments. e, Live-cell confocal images showing nuclear translocation of TFEB-GFP in WT, DMXL1−/− and DMXL1−/−;DMXL2+/− cells after treatment with MLSA5 or Torin1 (6 h). Scatter plot shows fold change of nuclear to cytoplasmic ratios of TFEB-GFP intensity. Quantification was performed on the following total number of cells from three biological replicates: WT: Control n=43, MLSA5 n=43, Torin1 n=41; DMXL1−/−: Control n=41, MLSA5 n=45, Torin1 n=42; DMXL1−/−;DMXL2+/−: Control n=43, MLSA5 n=40; Torin1 n=43. Fold change is normalized to the average ratio from control condition for each genotype. Each dot represents measurement from an individual cell and the line represents the mean. Kruskal-Wallis test followed by Dunn’s multiple comparisons test was used for statistical comparison. ***, p-value ≤ 0.001; ****, p-value ≤ 0.0001; ns, not significant. f, Immunoblotting of WT, DMXL1−/− and DMXL1−/−;DMXL2+/− cell extracts with α-GPNMB after treatment of cells with MLSA5 or Torin1 (24 h). Blots were probed with α-Hsp90 as loading control. Blot is representative of three independent experiments. g, Confocal images showing fluorescence intensity of lysosomal pH sensor, pseudo-colored to scale, in WT U2OS cells after treatment with BafA1 (2 h). Quantification shown in Fig. 5b. Scale bar is 20 microns.
Extended Data Fig. 7 Analysis of lysosomal hydrolytic activity in U2OS cells deficient for DMXL1/2, related to Figs. 6 and 7.
a, Confocal images showing fluorescence intensity of DQ-BSA Red in WT U2OS cells after treatment with BafA1 (4 h). Scale bar is 20 microns. Quantification shown in Fig. 6d. b, Mean fluorescence intensity of DQ-BSA Red in the indicated U2OS cells measured by flow cytometry (10,000 cells/replicate; experiment was performed in biological quadruplicate). Quantification represents n=4 independent experiments. Data are presented as mean values ± SD. One-way ANOVA followed by Dunnett’s multiple comparison test was used for statistical comparison within control group and MLSA5 treated group. Unpaired two-sided Student’s t-test was used to assess statistical significance between WT control and WT MLSA5 treated conditions, **, p-value ≤ 0.01; ***, p-value ≤ 0.001; ****, p-value ≤ 0.0001; ns, not significant. c, DQ-BSA Red flow cytometry assays, with or without MLSA5 treatment (4 h), in WT or DMXL1−/−;DMXL2+/− U2OS cells and in two independent clones of DMXL1−/−;DMXL2+/− cells with expression of DMXL1-mNG, which rescue the defect in DQ-BSA Red cleavage. d, Fold change in DQ-BSA Red intensity (MLSA5/control) from two independent experiments with the indicated cell types.
Extended Data Fig. 8 Analysis and prediction of DMXL1-V-ATPase interaction interfaces.
a, Summary of pairwise AF3 scores for full-length DMXL1 or Rav1-C helical ___domain with all V1-ATPase subunits or selected V1-ATPase subunits, respectively. The three-way AF3 score for the helical portion the DMXL1 Rav1-C ___domain together with both ATP6V1A and ATP6V1C is also included. b, AF3 prediction of the DMXL1 helical ___domain of Rav1-C (green) with ATP6V1C (cyan) and ATP6V1A (yellow). Two distinct binding sites are observed. c, AF3 prediction of the Rav1 helical ___domain of Rav1-C (blue) with Vma5 (light blue) and Vma1 (lime). Predicted binding sites are analogous to that seen with the human counterparts. d, AF3 models for interaction of Vma5 (cyan) with Rav1 (blue) (top panel) and ATP6V1C (cyan) with DMXL1 (green) (bottom panel) showing conserved structural features at the interaction interface.
Supplementary information
Supplementary Tables
Supplementary Tables 1–7.
Supplementary Video 1
Recruitment of DMXL1–mNG to lysosomes in response to MLSA5 treatment. Lysosomes are marked with mScarlet–LAMP1. Top, WT U2OS cells. Bottom, ATG16L1-KO U2OS cells. Scale bar, 10 µm. Relevant to Figure 2. In .avi format.
Supplementary Video 2
Movie of the DMXL1–ROGDI–WDR7 structural model, including cross-links, allowing the ___location of crosslinks to be readily observed. In .avi format.
Source data
Source Data Fig. 1
Unprocessed immunoblots.
Source Data Fig. 2
Unprocessed immunoblots and numerical source data.
Source Data Fig. 3
Numerical source data.
Source Data Fig. 5
Unprocessed immunoblots and numerical source data.
Source Data Fig. 6
Unprocessed immunoblots and numerical source data.
Source Data Fig. 7
Unprocessed immunoblots and numerical source data.
Source Data Fig. 8
Unprocessed immunoblots and numerical source data.
Source Data Extended Data Fig. 1
Unprocessed immunoblots.
Source Data Extended Data Fig. 2
Unprocessed immunoblots.
Source Data Extended Data Fig. 3
Numerical source data.
Source Data Extended Data Fig. 4
Unprocessed immunoblots and numerical source data.
Source Data Extended Data Fig. 6
Unprocessed immunoblots and numerical source data.
Source Data Extended Data Fig. 7
Numerical source data and example of gating strategy for flow cytometry.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lee, C., Eldridge, M.J.G., Gonzalez-Lozano, M.A. et al. DMXL1 promotes recruitment of V1-ATPase to lysosomes upon TRPML1 activation. Nat Struct Mol Biol (2025). https://doi.org/10.1038/s41594-025-01581-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41594-025-01581-x
