
Abstract
Microbes living inside or around sugarcane (Saccharum spp.) are crucial for their resistance to abiotic and biotic stress, growth, and development. Sequences of microbial genomes and genes are helpful to understand the function of these microbes. However, there is currently a lack of such knowledge in sugarcane. Here, we combined Nanopore and Illumina sequencing technologies to successfully construct the first high-quality metagenome-assembled genomes (MAGs) and gene catalogues of sugarcane culturable microbes (GCSCMs), which contained 175 species-level genome bins (SGBs), and 7,771,501 non-redundant genes. The SGBs included 79 novel culturable bacteria genomes, and 3 bacterial genomes with nitrogen-fixing gene clusters. Four single scaffold near-complete circular MAGs (cMAGs) with 0% contamination were obtained from Nanopore sequencing data. In conclusion, we have filled a research gap in the genomes and gene catalogues of culturable microbes of sugarcane, providing a vital data resource for further understanding the genetic basis and functions of these microbes. In addition, our methodology and results can provide guidance and reference for other plant microbial genome and gene catalogue studies.
Similar content being viewed by others

Background & Summary
Sugarcane (Saccharum spp.) is an important cash crop, whose stalks are rich in sugar, and are widely used in food, energy production, and industrial raw materials1. Globally, sugarcane has been planted over 27 million hectares, and is commonly grown in 120 countries and regions2. In recent years, the increase in sugarcane yields has been stagnant due to the excessive use of chemical fertilizers3, soil acidification4,5, pests6 and diseases7 in sugarcane cultivation. How to improve the yield has become a current research hotspot in the field of sugarcane agricultural research. Endophytes and rhizosphere soil microbes, the two primary forms of microbiota, are crucial for fostering plant growth, development and tolerance to stresses8,9. Hence, it is imperative to investigate the community composition and potential functions of sugarcane endophytes and its rhizosphere soil microbes, to screen functional strains that are beneficial to sugarcane yield enhancement, and to develop and utilize applicable bacterial resources for the sustainable development of the sugarcane industry.
Endophytes are microbes that live inside plant tissues (leaves, stems, roots), and may form a symbiotic relationship with plant10. These microbes can promote plant growth and development by fixing nitrogen and providing growth-regulating substances11. Since the initial isolation of endophytes from sugarcane by Dobereiner in 196112,13, several endophytic flora from 21 genera, including Bacillus, Burkholderia, Enterobacteriaceae, and Pantoea, have been characterized from sugarcane tissues (stems and leaves)14,15,16,17. Many culturable bacteria have been isolated from sugarcane’s rhizosphere soil and roots, including Azospirillum, Burkholderia, Klebsiella, Enterobacter and Erwinia18,19,20,21. Due to limits of culture conditions, in the context of traditional colony culture and single bacteria isolation procedures, it is typical to result in a significant reduction in the number of microbial species detected in the sample22,23,24. Furthermore, isolating and identifying individual microbe necessitates a substantial allocation of both human and material resources. Thus, in this research, we employed a technique to culture microbes from sugarcane’s inner tissues and rhizosphere soil by using multiple plant genotypes and media, which is able to enhance the cultivation of microbes.
In recent years, microbial genomics research has experienced significant advancement due to the ongoing progress in high-throughput sequencing technology25,26. These technologies, such as Nanopore and Illumina sequencing, have proven effective in acquiring comprehensive genomic and gene sequence data of microbial populations27,28, and have greatly facilitated the investigation of microbes’ genetic makeup and functional characteristics. The construction of reference genomes and gene catalogues of microbes for the global oceans29, human25,30, soil31,32, and animal gut33,34,35 has been completed. However, there are few applications and reports on the construction of genomes and gene catalogues using plant microbiota metagenomes, and there need to be more research on sugarcane microbiota. Therefore, constructing a complete genome and gene catalogue of culturable microbial species of sugarcane is necessary for studying sugarcane microbiota.
To cover this void, we sequenced 48 samples (mixtures of culturable microbes) from multiple plant compartments, genotypes, and sugarcane species by Nanopore sequencing and Illumina sequencing (Fig. 1A). Through this study, we have the following findings: (1) Constructed a non-redundant gene catalogue (GCSCMs)36 of culturable microbes in sugarcane containing 7,771,501 genes; (2) Assembled 175 species-level genome bins (SGBs)37,38 at the species level by the metagenome assembly technique, which included 77 potentially novel culturable bacterial species; (3) Successfully assembled single scaffold circular genomes37,38 from Nanopore Long Reads (LRs) with 0% contamination and near-complete. Thus, the utilization of Nanopore and Illumina sequencing technologies in constructing the genome and gene catalogue of the sugarcane culturable microbiome held the potential to enhance our comprehension of the characteristics of this microbiome. In summary, the results provide substantial genomes and gene resources of endophytes and rhizosphere soil microbial resources in sugarcane to explore sugarcane-microbe interaction and microbial functions.

Metagenome-assembled genomes (MAGs) construction and assembly process. (A) Schematic diagram of constructing MAGs using Nanopore and Illumina sequencing data. S. spon, S. robu, S. off, and S. hybrid stood for S. spontaneum, S. robustum, S. officinarum and Saccharum hybrid cultivar, respectively. Three genotypes for each species were included for culture of microbes (see Supplementary Data 1 for detail). Five media were used, including NA (Nutrient Agar), AM (Ashby’s Medium), BM (Burke’s Medium), R2A (R2A Medium), and PDA (Potato Dextrose Agar). C-Bins1 represented bins generated in the first round of correction, and C-Bins2 represented bins generated in the second round of correction. (B) Distribution of genome integrity and distribution of quality classes of MAGs. Horizontal coordinates denoted MAG length, vertical coordinates denoted MAG integrity, and n represented the number of MAGs. (C) Distribution of integrity and contamination of high-quality MAGs. Horizontal coordinates indicated MAG completeness, and vertical coordinates indicated MAG contamination. (D–F) Comparison of three critical metrics of assembly results using Nanopore and Illumina sequence data, with vertical coordinates indicating the length of the most extended overlapping group, MAG length, and N50 value, respectively.
Methods
Sample collection, isolation and culture of microbes
In order to ensure the diversity of samples, three species and one hybrid of sugarcane (S. spontaneum, S. robustum, S. officinarum, and S. hybrid) with three genotypes from each were chosen for this study (Fig. 1A, Supplementary Data 1). These materials included 48 samples for microbial isolation and cultivation, including leaves (the first leaf of the sugarcane plant that is fully green from the bottom to the top), stems (taken from the second node above the ground), and roots (the soil still adhering to the roots was collected as rhizosphere soil samples by vigorous shaking). The samples were immediately maintained in sterile bags and appropriately kept in an incubator set at 4 °C. Within 24 hours of sample collection, culturable microbes from sugarcane were isolated and cultured.
For the purpose of isolating rhizosphere soil microbes, 5 g of roots retaining small particles of soil attached need to be taken, and 100 ml of sterile water added. Then, it was washed using ultrasonic oscillation (Model: KQ-600E, Frequency: 28KHZ) for 1 min, and the step was repeated 2 times. After the oscillatory washing, the sample was left to stand for 10 min in order to isolate the rhizosphere soil microbes. The treatment of leaves, stems, and roots required surface sterilization. Leaves, stems, and roots (5 g after oscillatory washing as stated above) were soaked in 75% alcohol for 3 min and rinsed with sterile water 5 times after soaking. Then, the leaves, stems, and roots were immersed in 3% sodium hypochlorite for 7, 5, and 3 min, respectively, before being rinsed five times with sterile water. For the isolation of endophytes from leaves, stems, and roots, the processed samples were clamped into the sterile mortar with tweezers and fully ground by adding 100 ml of sterile water. After grinding, it was left to stand for 10 min in horizontal flow clean bench (SW-CJ-1CU). The four supernatants obtained above were the original bacterial suspension. Except for the 10-fold dilution of the original bacterial solution needed for rhizosphere soil, the original bacterial suspension was used for the cultivation of culturable microbes. Next, 100 μl was aspirated and spread separately on five kinds of solid media, namely Nutrient Agar, Ashby’s Medium, Burke’s Medium, R2A Medium, and Potato Dextrose Agar (Table 1). After spreading evenly by the rollerball method, the plates were inverted and incubated at 28 °C for 72 h.
DNA extraction and quality control
Following a 72-hour incubation to ensure the growth of the culturable microbes (Fig. 2), we conducted whole-genome metagenome sequencing on the genomic DNA extracted.1 ml of sterile water was aspirated as a rinse solution using a sterile pipette in horizontal flow clean bench. Rinsing was repeated 4 times, and the rinsed solution was aspirated into a 50 ml centrifuge tube. In order to collect all the colonies on the five solid media, rinses were repeated four times, and the rinses from the same bacterial suspension were aspirated into a 50 ml centrifuge tube. A total of 48 rinses of culturable microbes from different plant compartments and different genotypes of sugarcane were obtained. After shaking and mixing, 2 ml of the rinse suspension was pipetted into 2 ml centrifuge tubes and centrifuged separately, the supernatant was discarded, and the precipitate was kept at −80 °C immediately until the microbial DNA extraction for Illumina data sequencing. Meanwhile, 3 ml of the rinses from the same sugarcane species (3 genotypes, 4 plant compartments) were aspirated into the same 50 ml centrifuge tube and centrifuged. The supernatant was discarded, and the precipitate was immediately kept at −80 °C until the microbial DNA extraction for sequencing.

Growth of sugarcane culturable microbes after 72 h. Horizontal coordinates indicated the three sugarcane species and a hybrid (each with three genotypes), and vertical coordinated indicate the plant compartments for the isolates. PDA (Potato Dextrose Agar), R2A (R2A Medium), NA (Nutrient Agar), AM (Ashby’s Medium), and BM (Burke’s Medium) denoted five different media.
To isolate high-quality DNA of culturable culturable microbes from sugarcane, we used the CTAB/NaCl method for DNA extraction39. The integrity of the DNA was tested by capillary electrophoresis using a fragment analyser (AATI) with the use of a Qubit® 2.0 fluorometer and a Nanodrop kit for precise quantification and purity determination. The quality of the extracted DNA was assessed using 1% agarose gel electrophoresis.
Library construction and sequencing
To create Illumina sequencing libraries, we combined the NEBNext® UltraTM DNA Library Preparation Kit (New England Biolabs, USA) with 1 μg of DNA. Index codes were appended to the sequencing primers. The ends of the isolated DNA were repaired after being sonicated into 350 bp fragments. Following end repair, an adenine was added to the 3’ ends of the DNA fragments, and then adaptor sequences were ligated to both ends of the A-tailed DNA. These libraries were cleaned using Beckman Coulter’s AMPure XP technology in Brea, California, USA. With the use of an Agilent 2100 Bioanalyzer and real-time fluorescence quantitative PCR, the purified products were examined for size distribution and quantity. All samples were subjected to paired-end sequencing utilizing an Illumina NovaSeq 6000 platform with a read length of 150 base pairs (PE150) once the library’s quality had been confirmed.
According to the manufacturer’s instructions, we used 2.5 μg of extracted DNA for library preparation using the SQK-LSK110 Ligation Sequencing Kit (Oxford Nanopore Technologies, Oxford, UK) to create PromethION libraries. A Megaruptor (Diagenode, NJ, USA) was used to process the DNA, and BluePippin was used to screen for DNA fragments longer than 10 Kb. The lengths of the repaired pieces were verified, and specific barcodes and poly(A)tail were inserted. After mixing samples with various barcodes in equimolar proportions and purifying them, the DNA libraries were ready. A Qubit fluorometer was used to measure the DNA concentration. Utilizing Nanopore sequencing technology (Oxford Nanopore PromethION Sequencer: PromethION P48), sequencing was carried out after ensuring the libraries’ quality.
Data quality control
Low-quality base pairs, and reads shorter than 150 bp of raw data obtained from the Illumina sequencing platform were removed using fastp (v0.19.4 https://github.com/OpenGene/fastp)40. Parameters were set to “--cut_by_quality3 -W 4 -M 20 -n 5 -c -l 50 -w 3”. The Nanopore reads fast5 data were converted to fastq format using Guppy (v.3.03 https://community.nanoporetech.com) for Nanopore sequencing data. Then, NanoPlot (ver.1.18.2 https://github.com/wdecoster/NanoPlot)41 was used to perform quality control on the fastq format data by filtering low-quality sequences, splice sequences, and etc. The threshold value of the filtering criterion was set to meanQ > 7 to remove low-quality sequences, and the parameters were set to ‘-t 20,--loglength,--N50’. After quality control, the clean and high-quality reads obtained from both sequencing methods were used for further in-depth analyses.
Illumina sequencing yielded a total of 1.1 Tb of Illumina sequencing data42. After quality control of the sequencing data, 1.08 Tb of clean, high-quality pair-ended data were retained for further analysis, and an average of 22.50 Gb/sample was attained (Supplementary Data 1). Nanopore sequencing is commonly employed to generate long reads, which are then utilized to assemble circular metagenome-assembled genomes (cMAGs). To improve the quality of metagenome assembly, we mixed 48 samples into four pooled samples by sugarcane species (Supplementary Data 2) for Nanopore sequencing. We obtained 61.35 Gb of clean long reads sequencing data42 (65.63 Gb of raw data, with data efficiency > 93.48%) with an average read length of 8.01 Kb (Supplementary Data 2).
Metagenomics assembly and binning
To obtain comprehensive genomic data, a single-sample de novo assembly of quality-controlled Illumina sequencing data was performed using MetaSPAdes (v.3.13.0 https://github.com/ablab/spades)43 with default K-mer parameters. Only contigs of ≥ 1000 bp were retained. A total of 13,481,007 contigs were obtained with a minimum length of 1000 bp. The quality-controlled Illumina sequencing data were mapped to the contigs using BWA MEM (v.0.7.17 https://github.com/lh3/bwa)44 to produce Sam format files containing the comparison information. Samtools (v.1.10 https://github.com/samtools/samtools/releases)45 was used to convert the Sam files to Bam format. Sequencing depths of contigs were generated from Bam files using the script jgi_summarise_BAM_contig_depth that comes with MetaBAT2 (v.2.12.1 https://github.com/bioboxes/metaBAT)46 Based on the sequence characteristics and sequencing depth of these contigs, a total of 1485 bins were generated from 13,481,007 contigs using MetaBAT2.
For Nanopore sequencing data, the four Nanopore datasets were individually de novo assembled using metaFlye (v.2.8.3 https://github.com/fenderglass/Flye)47 after quality control. The Nanopore reads were mapped onto the contigs generated from the Nanopore sequencing data by minimap2 (v.2.22 https://github.com/lh3/minimap2)48 to produce Sam format files containing comparison information. The next binning steps were consistent with the processing of Illumina sequencing data. In total, 62 bins were generated from the Nanopore sequencing data. To improve the reliability of binning, we performed two rounds of correction. The first round of correction was a self-correction, where Nanopore reads were rearranged onto contigs by Medaka (v0.6.5 https://github.com/nanoporetech/medaka) to obtain consensus sequences. Then, the second round of correction was performed, using Pilon (v.1.12 https://github.com/broadinstitute/pilon)49 to map Illumina reads onto the consensus sequence based on the Pilon correction of BWA-MEM (v.0.7.17) to correct Indel errors.
A total of 1547 bins were generated from Illumina reads and Nanopore reads.The Illumina sequencing data produced bins with a maximum length of 23.79 Mb and a maximum N50 value of 2.97 Mb, and the Nanopore sequencing data produced bins with a maximum length of 11.78 Mb and a maximum N50 value of 2.30 Mb (Supplementary Data 3). 717 MAGs were generated by removing duplications of all bins usingdRep (v.1.1.2 https://github.com/MrOlm/drep)50 at 99% ANI (equivalent to strain level), with 681 and 36 bins from Illumina sequencing and Nanopore sequencing data, respectively (Supplementary Data 4). The completeness and contamination of the above 717 MAGs were estimated based on the lineage_wf workflow using CheckM (v.1.0.7 https://github.com/Ecogenomics/CheckM)51. After quality assessment, no complete eukaryotic and viral genomes were found in the obtained non-redundant metagenome-assembled genomes (MAGs). Subsequently, we focused on the analysis of prokaryotic MAGs, and obtained a total of 185 metagenome-assembled genomes (SMAGs) of culturable bacteria from sugarcane (Supplementary Data 5). According to the “Minimum Information about Metagenome Assembled Genomes (MIMAG)” standard52, all of these assembled SMAGs met or exceeded the standard of medium quality (defined as > 50% completeness and < 10% contamination)53 (Fig. 1B). Of these, 171 SMAGs were from Illumina sequencing, and 14 SMAGs were from Nanopore sequencing. Among the 185 SMAGs, 139 had high-quality genomes (defined as > 80% completeness and 10% contamination) (Fig. 1C), including 95 with > 95% completeness and < 5% contamination, 12 with > 95% completeness and 0% contamination, and 4 with 100% completeness and 0% contamination (Supplementary Data 5). The Nanopore data outperformed Illumina data in assembly length, N50 value, and overlap cluster length for high-quality SMAGs (Fig. 1D).
Phylogenetic analysis and annotation of SMAGs
To determine the phylogenetic affiliation and diversity of the 185 SMAGs,we used the “classify_wf” workflow in GTDB-TK (v.0.3.0; default settings http://gtdb.ecogenomic.org/)54 to identify 120 bacterial marker genes and constructed multiple sequence pairs based on these marker genes. The generated multiple sequence comparison FASTA files were subjected to maximum likelihood phylogenetic tree inference using IQ-TREE. Final visualization was performed using the ChiPlot web tool (https://www.chiplot.online/).
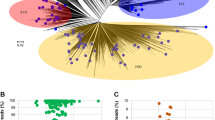
These SMAGs consisted of four phylum levels, namely Proteobacteria (n = 137), Bacteroidota (n = 38), Firmicutes (n = 8), and Bdellovibrionota (n = 1). The phylogenetic tree constructed using SMAGs also confirmed this finding (Fig. 3A). Further analysis showed that all 105 of the 185 SMAGs were identified at the species level (Fig. 3B), and 80 (43.22%) did not match the reference genome in the GTDB, and thus represented species or strains without genome sequenced before (Fig. 3C).

Bacterial community distribution of assembled SMAGs. (A) Phylogenetic tree of culturable microbes from sugarcane. Background colours in the outer circles represented microbial phyla. (B) Mulberry diagram showing the distribution of 185 SMAGs at different taxonomic levels. (C) Number of MAGs at each taxonomic level by GTDB-TK classification.
Complete or near complete genomes for sugarcane culturable microbes
From high-quality species-level genome bins (SGBs, >90% completeness and <5% contamination), we obtained 112 complete or near-complete species-level genomes bins (N-SGBs), of which 108 were assembled using Illumina sequencing data, and four using Nanopore sequencing data (Supplementary Data 6). Bacteroidota (21.4%) and Proteobacteria (75.0%) accounted for 96.4% of the 112 N-SGBs (Fig. 4A). For the 73 N-SGBs with reference genomes in the GTDB database, significant similarity was observed in terms of GC content between our assembly and the reference genomes (Fig. 4B). These N-SGBs showed improved genome quality in terms of completeness and contamination (Fig. 4C). Notably, the completeness of 8 genomes we assembled was 0.02% to 26.92% higher, and 31 of our assembled genomes had contamination rates 0.02% to 4.05% lower than their respective reference genomes from GTDB database (Supplementary Data 7). For the 39 unknown N-SGBs (uN-SGBs), they were widely distributed in 26 different genera (Fig. 4A, Supplementary Data 8). Two uN-SGBs contained a cluster of conserved nitrogen-fixing genes in typical nitrogen-fixing bacteria (Supplementary Data 8).

Overview of complete or near-complete genomes. (A) Number of complete or nearly complete species-level genomes bins (N-SGBs) and percentage of unknown N-SGBs (uN-SGBs) in each phylum. (B) Distribution of GC content in N-SGBs and corresponding reference genomes, with the horizontal coordinates indicating the cumulative number of genomes and the vertical coordinates showing the GC content, the yellow solid line marking N-SGBs with reference genomes, and the blue dashed line indicating reference genomes. (C) Distribution of completeness and contamination N-SGBs and corresponding reference genomes, with the horizontal coordinates indicating the cumulative number of genomes and the vertical coordinates indicating the completeness and contamination values, respectively, and the yellow solid line marking N-SMAGs with reference genomes and the blue solid line indicating the reference genomes.
Illumina sequencing data typically results in fragmented assembled contigs in the metagenome assembly of microbial genomes. In contrast, Nanopore sequencing of long reads can be used to assemble near-complete cyclic MAGs (cMAGs), and our results confirmed this conclusion. Four Nanopore sequencing data genomes were assembled into single scaffold cyclic MAGs (cMAGs) with an average completeness of 97.50% and contamination of 0.82% (Supplementary Data 9). Among the four cMAGs, the contamination of SMAGNO90 and SMAGNO131 was 0. All the four cMAGs contained complete bacterial genome information, including 16 S, 5 S, and 23SrRNA genes and 18 tRNAs (Supplementary Data 9), and fulfilled the MINMAG criteria for “high-quality” MAGs set by the Genome Standards Consortium. 16 S rRNA genes were predicted using RNAmmer (v.1.2 http://www.cbs.dtu.dk/services/RNAmmer/)55. The cMAGs were annotated using the Bakta web tool (https://bakta.computational.bio), including the prediction of coding sequences (CDS), tRNAs and rRNAs. To understand the function of cMAGs, the predicted CDS was compared with the eggNOG database56 and KEGG database57 using DIAMOND (v.0.9.9.110 http://github.com/ bbuchfink/ diamond)58.
Gene catalogue construction, taxonomic annotation and abundance analysis
Gene prediction by Prodigal (v.2.6 https://github.com/hyattpd/Prodigal)59 on contigs assembled from Illumina reads and Nanopore reads yielded 22.17 M open reading frames (ORFs). ORFs less than 100 bp in length were discarded, and then clustered to construct an initial non-redundant gene catalogue using CD-HIT-EST (v.4.5.8 https://github.com/weizhongli/cdhit)60. The longest ORF from each group was selected as a representative of that group. After clustering at 95% nucleotide sequence identity, we obtained a non-redundant gene catalogue of sugarcane culturable microbes (GCSCMs) containing 7,771,501 non-redundant genes. High-quality reads from Illumina sequencing of each sample were aligned to the gene catalogue using BWA-MEM (v.0.7.17), and gene abundance was calculated in transcripts per million (TPM)61 and corrected for variations in gene lengths and mapped read segments for each sample.
ORFs were translated into protein sequences, and compared with the NR_euk database62 to determine the species classification information of the genes using Kaiju (https://github.com/bioinformatics-centre/kaiju). 82.4% of the genes in the GCSCMs were derived from bacteria, archaea, eukaryotes, and viruses (Fig. 5A, Supplementary Data 10), and these genes were annotated to 151 phyla, 3465 genera, and 24,084 species (Supplementary Data 11). The dominant phyla were Proteobacteria (65%), Bacteroidetes (14%), Firmicutes (8%), Ascomycota (6%), and Actinobacteria (3%) (Fig. 5B). Among the 3465 identified genera, the top 10 in terms of number of genes were Pseudomonas, Burkholderia, Paraburkholderia, Rhizobium, Chitinophaga, Bacillus, Trichoderma, Paenibacillus, Dyella, and Massilia (Fig. 5C).

Species classification and functional annotation of the GCSCMs. (A) Species Taxonomic classification of the GCSCMs. (B,C) Categorization of gene catalogues at the phylum and genus level. Horizontal coordinates indicated the different phylum (genus) levels, and vertical coordinates indicated the total number of genes annotated to each phylum (genus) level.
Data Records
The microbial genes of this study were submitted to the National Center for Biological Information (CNCB https://www.cncb.ac.cn/), China, with the BioProject accession (PRJCA019660). Gene catalogues (GCSCMs) of sugarcane culturable microbes were provided herein (https://ngdc.cncb.ac.cn/omix/release/OMIX004891)36. The metagenome sequencing data, and assembled genomes of this study were submitted to the National Center for Biotechnology Information, USA, with the BioProject accession (PRJNA1096928). Raw data were provided herein (NCBI Sequence Arch https://identifiers.org/ncbi/insdc.sra:SRP500217)41; Metagenome-assembled genomes of sugarcane culturable microbes (SMAGs) were provided herein (NCBI GenBank https://identifiers.org/ncbi/insdc:JBCNJQ000000000-JBCNNK000000000)37,38. Culturable bacterial single scaffold cyclic MAGs (cMAGs) from sugarcane were provided herein (NCBI GenBank https://identifiers.org/ncbi/insdc:JBCNJQ000-000000-JBCNNK000000000)37,38.
Technical Validation
The completeness and contamination of the above 718 MAGs were estimated based on the lineage_wf workflow using CheckM (v.1.0.7 https://github.com/Ecogenomics/CheckM), which generated 185 SMAGs that met or exceeded moderate quality thresholds (≥50% completeness and ≤10% contamination), with quality scores for each MAG calculated based on completeness - 5*contamination. Gene prediction by Prodiga on contigs assembled from Illumina reads and Nanopore reads yielded 22.17 M open reading frames (ORFs).
Code availability
All the tools mentioned in the data analysis used in this study were publicly available and the sources and versions of the analytical programs and codes were indicated in the Materials and Methods. No custom code or pipelines were generated in this manuscript.
References
Verma, K. K. et al. Impact of agroclimatic variables on proteogenomics in sugar cane (Saccharum spp.) Plant Productivity. Acs Omega 7, 22997 (2022).
Aslam, U., Tabassum, B., Nasir, I. A., Khan, A. & Husnain, T. A virus-derived short hairpin RNA confers resistance against sugarcane mosaic virus in transgenic sugarcane. Transgenic Res 27, 203 (2018).
Li, Y. & Yang, L. Sugarcane agriculture and sugar industry in China. Sugar tech 17, 1 (2015).
Pang, Z. et al., Continuous sugarcane planting negatively impacts soil microbial community structure, soil fertility, and sugarcane agronomic parameters. Microorganisms 9, (2021).
Tayyab, M. et al. Sugarcane monoculture drives microbial community composition, activity and abundance of agricultural-related microorganisms. Environ Sci Pollut R 28, 48080 (2021).
Geetha, M. V. et al. in Pests and Their Management, edited by Omkar, pp. 241 (Springer Singapore, Singapore, 2018).
Viswanathan, R. & Rao, G. P. Disease scenario and management of major sugarcane diseases in India. Sugar Tech 13, 336 (2011).
Da, S. A. et al. Exploitation of new endophytic bacteria and their ability to promote sugarcane growth and nitrogen nutrition. Anton Leeuw Int J G 112, 283 (2019).
Liu, Q. et al. Response of sugarcane rhizosphere bacterial community to drought stress. Front Microbiol 12, 716196 (2021).
Jha, P., Panwar, J. & Jha, P. N. Mechanistic insights on plant root colonization by bacterial endophytes: a symbiotic relationship for sustainable agriculture. Environmental Sustainability 1, 25 (2018).
Gupta, A., Gopal, M. & Tilak, K. V. Mechanism of plant growth promotion by rhizobacteria. Indian J Exp Biol 38, 856 (2000).
Dobereiner, J. Nitrogen-fixing bacteria of the genus Beijerinckia Derx in the rhizosphere of sugar cane. Plant Soil 15, 211 (1961).
Cavalcante, V. A. & Dobereiner, J. A new acid-tolerant nitrogen-fixing bacterium associated with sugarcane. Plant Soil 108, 23 (1988).
Malviya, M. K. et al. Beneficial linkages of endophytic Burkholderia anthina MYSP113 towards sugarcane growth promotion. Sugar Tech 21, 737 (2019).
Pereira, L. B., Andrade, G. S., Meneghin, S. P., Vicentini, R. & Ottoboni, L. Prospecting plant growth-promoting bacteria isolated from the rhizosphere of sugarcane under drought stress. Curr Microbiol 76, 1345 (2019).
Dong, Z. et al. A nitrogen-fixing endophyte of sugarcane stems (a new role for the apoplast). Plant Physiol 105, 1139 (1994).
Lacava, P. T. & Azevedo, J. L. in Bacteria in Agrobiology: Crop Productivity, edited by D. K, Maheshwari, M., Saraf and A., Aeron, pp. 1 (Springer Berlin Heidelberg, Berlin, Heidelberg, 2013).
Ahmad, F., Ahmad, I., Aqil, F., Ahmed, W. A. & Sousche, Y. S. Plant growth promoting potential of free-living diazotrophs and other rhizobacteria isolated from Northern Indian soil. Biotechnol J 1, 1112 (2006).
Sajjad Mirza, M. et al. Isolation, partial characterization, and the effect of plant growth-promoting bacteria (PGPB) on micro-propagated sugarcane in vitro. Plant Soil 237, 47 (2001).
Teixeira, L. C. et al. Bacterial diversity in rhizosphere soil from Antarctic vascular plants of Admiralty Bay, maritime Antarctica. ISME J 4, 989 (2010).
Prashar, P., Kapoor, N. & Sachdeva, S. Rhizosphere: its structure, bacterial diversity and significance. Reviews in Environmental Science and Bio/Technology 13, 63 (2014).
Yang, C. H., Crowley, D. E., Borneman, J. & Keen, N. T. Microbial phyllosphere populations are more complex than previously realized. P Natl Acad Sci USA 98, 3889 (2001).
Gewin, V. Genomics: Discovery in the dirt. Nature 439, 384 (2006).
Gupta, R., Anand, G., Gaur, R. & Yadav, D. Plant–microbiome interactions for sustainable agriculture: a review. Physiol Mol Biol Pla 27, 165 (2021).
Qin, J. et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59 (2010).
Brown, C. T. et al. Unusual biology across a group comprising more than15% of ___domain Bacteria. Nature 523, 208 (2015).
Jain, M., Olsen, H. E., Paten, B. & Akeson, M. The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol 17, 239 (2016).
Lu, H., Giordano, F. & Ning, Z. Oxford nanopore minion sequencing and genome assembly. Genom Proteom Bioinf 14, 265 (2016).
Tully, B. J., Graham, E. D. & Heidelberg, J. F. The reconstruction of 2,631 draft metagenome-assembled genomes from the global oceans. Sci Data 5, 170203 (2018).
Zeng, S. et al. A compendium of 32,277 metagenome-assembled genomes and over 80 million genes from the early-life human gut microbiome. Nat Commun 13, 5139 (2022).
Lou, J. et al. Metagenomic sequencing reveals microbial gene catalogue of phosphinothricin-utilized soils in South China. Gene 711, 143942 (2019).
Lemos, L. N., Mendes, L. W., Baldrian, P. & Pylro, V. S. Genome-resolved metagenomics is essential for unlocking the microbial black box of the soil. Trends Microbiol 29, 279 (2021).
Xie, F. et al. An integrated gene catalog and over 10,000 metagenome-assembled genomes from the gastrointestinal microbiome of ruminants. Microbiome 9, 137 (2021).
Feng, Y. et al. Metagenome-assembled genomes and gene catalog from the chicken gut microbiome aid in deciphering antibiotic resistomes. Commun Biol 4, 1305 (2021).
Li, C. et al. Expanded catalogue of metagenome-assembled genomes reveals resistome characteristics and athletic performance-associated microbes in horse. Microbiome 11, 7 (2023).
CNCB https://ngdc.cncb.ac.cn/omix/release/OMIX004891 (2023).
NCBI GenBank https://identifiers.org/ncbi/insdc:JBCNJQ000000000 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc:JBCNNK000000000 (2024).
Griffith, G. W., Ozkose, E., Theodorou, M. K. & Davies, D. R. Diversity of anaerobic fungal populations in cattle revealed by selective enrichment culture using different carbon sources. Fungal Ecol 2, 87 (2009).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884 (2018).
De Coster, W., D’Hert, S., Schultz, D. T., Cruts, M. & Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666 (2018).
NCBI Sequence Arch https://identifiers.org/ncbi/insdc.sra:SRP500217 (2024).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19, 455 (2012).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589 (2010).
Etherington, G. J., Ramirez-Gonzalez, R. H. & MacLean, D. bio-samtools 2: a package for analysis and visualization of sequence and alignment data with SAMtools in Ruby. Bioinformatics 31, 2565 (2015).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. Peerj 7, e7359 (2019).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol 37, 540 (2019).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094 (2018).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. Plos One 9, e112963 (2014).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool forfast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J 11, 2864 (2017).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25, 1043 (2015).
Bowers, R. M. et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol 35, 725 (2017).
Pan, S., Zhu, C., Zhao, X. M. & Coelho, L. P. A deep siamese neural network improves metagenome-assembled genomes in microbiome datasets across different environments. Nat Commun 13, 2326 (2022).
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk:a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925 (2019).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomalRNA genes. Nucleic Acids Res 35, 3100 (2007).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res 47, D309 (2019).
Kanehisa, M. et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 42, D199 (2014).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59 (2015).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010).
Huang, Y., Niu, B., Gao, Y., Fu, L. & Li, W. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26, 680 (2010).
Wagner, G. P., Kin, K. & Lynch, V. J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theor Biosci 131, 281 (2012).
Tovo, A., Menzel, P., Krogh, A., Cosentino Lagomarsino, M. & Suweis, S. Taxonomic classification method for metagenomics based on core protein families with Core-Kaiju. Nucleic Acids Res 48, e93 (2020).
Acknowledgements
This work was financially supported by the National Key Research and Development Program (2021YFD1200204), the Guangxi Natural Science Foundation (GK AA22117002, 2020GXNSFAA297039 and GK AD21075011), the Yunnan Seeds and Seed Industry Joint Laboratory Project (202205AR070001-09), Chongzuo science and technology project (CK 20220619), and the ‘One Hundred Person’ Project of Guangxi Province, Science.
Author information
Authors and Affiliations
Contributions
X.Y. and W.D. conceived and designed the experiments. L.W. and H.L. performed the experiments and analyzed the data. L.Z., T.Q., P.L., J.S., Y.D., C.H. and H.Y. performed some of the experiments. L.W., H.L. and X.Y. wrote the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, L., Lin, H., zhang, L. et al. Construction of high-quality genomes and gene catalogue for culturable microbes of sugarcane (Saccharum spp.). Sci Data 11, 534 (2024). https://doi.org/10.1038/s41597-024-03379-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03379-w
