
Abstract
Callosobruchus maculatus is one of the most competitive stored grain pests, which causes a great loss to agricultural economy. However, due to an inadequacy of high-quality reference genome, the molecular mechanisms for olfactory and hypoxic adaptations to stored environments are unknown and require to be revealed urgently, which will contribute to the detection and prevention of the invasive pests C. maculatus. Here, we presented a high-quality chromosome-level genome of C. maculatus based on Illumina, Nanopore and Hi-C sequencing data. The total size was 1.2 Gb, and 65.17% (797.47 Mb) of it was identified to be repeat sequences. Among assembled chromosomes, chromosome 10 was considered the X chromosome according to the evidence of reads coverage and homologous genes among species. The current version of high-quality genome provides preferable data resources for the adaptive evolution research of C. maculatus.
Similar content being viewed by others


Background & Summary
Callosobruchus maculatus (Coleoptera: Chrysomelidae), commonly known as cowpea weevil, is a kind of universal stored grain insect pest1. C. maculatus feeds on a diverse range of legume seeds, and was originally distributed in the tropical and subtropical areas especially Africa and South Asia2. However, with the global climate change and international communication, C. maculatus were observed in wider regions3 and caused great losses to agricultural grain storage. Each female beetle lays huge amount of eggs on the surface of seeds, then the first instar larvae hatch and tunnel into the seeds where larvae will grow up and pupate by feeding on cotyledon, and complete its lifecycle by emerging as adult beetle1. To control pests and reduce food waste, dozens of approaches have been implemented in the past decades4,5,6,7,8.
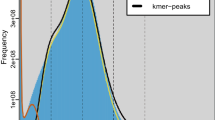
A high-quality chromosome-level genome for C. maculatus (Fig. 1a) supplies a practical and much-needed resource for C. maculatus agricultural pest control due to its immeasurably damage to stored products, which is also valuable for understanding the molecular mechanisms of its physiological activity and evolution relationships in Coleoptera. In the past, a contig-level genome of C. maculatus had been assembled9, but it was smaller than the expected genome size due to unknow reasons10. The C. maculatus genome is highly heterozygous with large proportion of repetitive DNA sequences, rendering substantial challenges for the genome assembly which is also a momentous reason for difference between K-mer analysis and flow cytometry11,12 in genome size13. Compared to the result of K-mer analysis, flow cytometry assay experiment was more credible11,12. The K-mer distribution analysis of the new assembled genome also indicated C. maculatus genome was large with high heterozygosity compared to other known Coleoptera species14,15. To address the challenges posed by high heterozygosity and repetitive DNA sequences, long-read sequencing (Nanopore), next-generation sequencing (Illumina), and high-throughput chromosome conformation capture (Hi-C) mapping have been employed, and these approaches have proven successful in achieving a high level of completeness and continuity in genome assemblies for various plant species16,17 as well as animals14,18.

(a) A picture of male C. maculatus on cowpea. (b) Hi-C heatmap of C. maculatus genome showing interactions among the 10 assembled chromosomes at 1 Mb resolution. Boxes in blue and green colour indicate scaffolds and contigs. (c) The relative gene density, GC content, repeat density, as well as the coverage of second-generation, third-generation, and Hi-C sequencing data in chromosome level are represented from the outside to the center of the circle.
To facilitate C. maculatus control and prevention, a 1.2 Gb novel high-quality chromosome-level genome of C. maculatus was provided in this research stemming from the new whole genome assembly and correct strategies. Here, we sequenced and assembled the high-quality chromosomal-level reference genome of cowpea weevil combining Illumina, Nanopore and Hi-C sequencing technologies. The current version of high-quality genome offers superior data resources study of adaptive evolution in of C. maculatus.
Methods
Sampling and genome sequencing
Adult insects of C. maculatus were intercepted in imported cowpeas originally from Nigeria and transferred via Ethiopia by Beijing Customs, P.R. China, and then inbreed for about 20 generations with full sib-pair mating strategy which were reared on 28 ± 2 °C, 75% relative humidity, 16 h/8 h light/dark photoperiod in ΦA = 90 mm petri dishes and nurtured with cowpeas. Genomic DNA were extracted from 10 male beetles for sequencing using DNeasy Blood & Tissue Kit (QIAGEN, Germany).
Short reads libraries (insert size: 350 bp) were generated using a Next Ultra DNA Library Prep Kit (NEB, USA), and sequenced on Illumina Novaseq6000 platform (PE-150) at Novogene. The raw reads ran through quality control before the next analysis. For short-read data (i.e., Illumina data), the reads which included adapters, the low-quality reads and N bases were removed, a total of 142.48 Gb clean data was obtained (Table 1). The rest of short data were used to estimate genome size based on the K-mer size using Jellyfish19 (2.3.0) with option -s 1.2 G. The genome size was also estimated by flow cytometry (Table 2) using 5 males and 5 female beetles as described before18. Additionally, these short-read data were used to correct the potential base errors in de novo genome assembly. For long-read data (i.e., Nanopore data), genomic DNA was extracted to construct 20-kb libraries and used for Oxford Nanopore sequencing platform at NextOmics and generated 137.33 Gb raw data (Table 1).
The Hi-C sequencing was also conducted at NextOmics, which followed the standard protocol and was sequenced on the Illumina NovaSeq6000 platform. Restriction enzyme DpnII was used to lyse and digest the isolate cells which were from sliced tissues and cross-linked before overnight. The cohesive ends were blunted, reversed, and marked with biotin-14-dATP and purified the DNA by removing biotin from unligated ends. DNA was sheared to 200–300 bp fragments via a Covaris M220 and pulled down the point ligation junctions by Dynabeads MyOneTM Streptavidin C1 after size selection with AMPure XP beads. Finally, a total of 668.22 Gb raw data of Hi-C sequencing was obtained and used to assist genome assemble on chromosome-level.
For transcriptome sequencing of adult female/male C. maculatus, each sample consisted of three beetles, with three replicates for each stage or gender. RNA was extracted using Trizol (Invitrogen, USA), library construction, sequencing on the Illumina NovaSeq6000 platform (PE-150) and quality control was performed in Novogene, and clean data was generated for further analysis.
De novo genome assembly
To combine the advantages of high accuracy of second-generation sequencing and long reads of third-generation sequencing20, this research employed the strategy that both sequencing methods were combinedly used for genome assembly. Genomic DNA was extracted and sequenced via Illumina platform and in prior to the Oxford Nanopore sequencing, the K-mer distribution analysis indicated that the genome size of C. maculatus was 1,358.89 Mb based on 21-mers using the illumina data, which was used to evaluate its size and characteristics, and consistent with the result of flow cytometry assay experiment (1,182.11 Mb, Table 2).
The long-read data was used to assemble the primary genome. NextDenovo21 (2.3.1) package was used to assemble the genome, which had several scripts and seq_stat, a binary script to statistic the information of sequencing data, is one of them. After using seq_stat with option -g = 1,200,000,000 to generate the seed cutoff value, which was required by the main program, run command NextDenovo, the main program, to get the primary genome with default configure. The primary genome was adjusted with option -a = 50 by purge_dups22 from 3 Gb into 1.2 Gb which was confirmed by K-mer analysis and flow cytometry previously23. NextPolish24 (1.2.3) package was used to polish the primary genome with default configure, which integrated the short-read data and the assembled the 1.2 Gb draft genome with an N50 of 1.03 Mb.
The Hi-C paired-end reads were mapped to the above draft genome iteratively using chromap25 (0.2.3) and yahs26 (1.2a1). Finally, juicebox27,28 (2.18) was applied to correct the contig orientation and move the suspicious fragments into unanchored scaffolds via visual exploration of Hi-C heatmaps. After all, 10 assembled chromosomes (Fig. 1b and Table 3), which holds 9 autosomes and one X chromosome (2n = 20, n = 9 + X), consistent with that by karyotype analyses23,29. Totally, 78.5% fragments were anchored to the assembled chromosomes, and the genome length is 1,222.50 Mb (N50 = 117.71 Mb) (Fig. 1b and c).
Identification of X chromosomes
Female and male beetle transcriptomes proved that Chr-10 is X-chromosome since female beetle transcripts showed significant larger coverage on Chr-10 (Table 4). In addition, we also performed integrating collinearity analysis of multiple homologous species genomes to identify X chromosomes. Chromosome X (Chr-X) is a distinctive group, which had been identified in Tribolium castaneum (GCA_000002335.3)30, Harmonia axyridis (GCA_914767665.1)31, and Coccinella septempunctata (GCA_907165205.1)32, and analyzed among the seven Coleoptera species (Fig. 2a). Furthermore, it was reported that several genes, including Trx, Spastin, ARNTH, etc., were located on the Chr-X in T. castaneum, which could be found in all other six Coleoptera species Chr-X (Fig. 2b). Besides, the GO enrichment analysis of all seven Chr-X showed a strong connection to sexual reproduction (Supplementary Fig. 1).

(a) Gene synteny analysis of seven coleopteran species. Homologous genes are linked by grey lines between chromosomes, while X-chromosome homologous genes are linked by blue lines. (b) Gene density of seven coleopteran species X-chromosomes (including predicted chromosomes) is showed from blue (low) to red (high), and white blank meant that no gene are annotated. Nine key genes are located on the chromosomes. Hob: Holotrichia oblita, Psh: Psylliodes chrysocephala, Bra: Brassicogethes aeneus, Cma: C. maculatus, Tca: T. castaneum, Hax: H. axyridis, Cse: C. septempunctata.
Repeat annotation
Repetitive sequence annotation was divided into two types: homologous sequence alignment and ab initio prediction. The homologous sequence alignment was based on the repeat sequence database (RepBase library version: RepeatmaskerEdition-20181026), using Repeatmasker33 (4.1.5) and RepeatProteinMasker to identify the repetitive sequence had known. Ab initio prediction used LTR FINDER34 (0.3.1), RepeatScout35 (1.0.6) and RepeatModeler36 (2.0.4) combined with Repbase nucleotides library and Repbase proteins library. In the beginning, de novo repeat library was established, and then used Repeatmasker to predict them. In addition, in the method of ab initio prediction, tandem repeat finder (TRF)37 (4.0.9) was applied to find tandem repeats (TEs) in the draft genome. Integrated with the result of ab initio prediction, 797.47 Mb (65.17%) repeat elements were identified totally (Fig. 3a).

(a) Taking Repbase as the library, the tandem repeat sequences (TEs) divergence distribution map was obtained by RepeatMasker annotation. The abscissa shows the divergence between the TEs annotated in the C. maculatus genome and the corresponding sequences in Repbase; the ordinate is the percentage of TEs in the genome under a specific divergence, and different TEs are marked with different colours. (b) Venn diagram how the overlap of five databases, Pfam, Swiss-port, KEGG, GO and NR, used in the annotation. 14,458 genes are annotated from five databases and 9,098 genes are in all the databases.
Gene structure prediction
The strategy of Gene structure prediction combined multiple prediction methods including homology prediction (seven species), ab initio prediction and RNA sequences-based prediction. Homology prediction was to compare the coding protein sequence of Drosophila melanogaster (GCA_000001215.4)38, Diabrotica virgifera (GCA_917563875.2)39, T. castaneum, Anoplophora glabripennis (GCA_000390285.2)40, Sitophilus oryzae (GCA_002938485.2)41, Leptinotarsa decemlineata (GCA_000500325.2)42, and Aethina tumida (GCA_024364675.1)43 with the genome sequence of C. maculatus via blast and genewise to predict the gene structure in the genome. Geneid44 (1.4.5), Augustus45 (3.5.0), GlimmerHMM46 (3.0.4), SNAP47 (2006-07-28), and Genscan48 (1.0) were employed with default configure in the ab initio prediction which relied on the statistical characteristics of genome sequence data, codon frequency and exon-intron distribution, to predict gene structure. Program to Assemble Spliced Alignments (PASA)49 (2.5.3) and Cufflinks50 (2.2.1) were applied in the RNA sequences-based prediction method with default settings. Based on the above prediction results, combined with the transcriptome comparison data, the EVidenceModeler (EVM)49 (2.1.0) and Liftoff51 (1.6.3) was used to integrate the gene sets predicted by various methods into a non-redundant and more complete gene set with different weights (Supplementary Fig. 2). Finally, used PASA to correct the EVM annotation results combined with the transcriptome assembly results, added untranslated region (UTR) and variable splicing and other information to get the final gene set. Using homology prediction, ab initio prediction and RNA sequences-based prediction, totally 14,458 genes were predicted (Table 5, Fig. 3b), and 91.9% proteins were conserved in BUSCO52 (5.4.1) analysis with protein mode based on Insecta odb10 (Table 6). And using the same method for previous version of the genome showed that annotated gene-sets have completeness value was 84.2% with 63.6% single copy, while the original completeness value was 75% which based on arthropod datasets10.
Gene function prediction and Non-coding RNA annotation
The gene functions were identified by aligning to Swiss‐prot, the nonredundant sequence databases: Nucleotide collection (NR and NT), eukaryotic orthologous groups of proteins (KOG), KEGG, TrEMBL and using BLAST53 (2.15.0 + ) with an E-value cutoff of 1e-5. The Blast2GO54 (6.0) was employed to annotate gene functions in the GO database based on the aligned results from the NR database. The molecular pathways of predicted genes, which might be involved, were detected through search and annotation for the KEGG database. Using Interproscan55 (5.62–94.0) to search in the Pfam (35.0), PRINTS (42.0), SMART (9.0) databases, known motifs and domains in the C. maculatus genome were found. The ___domain boundaries of interesting proteins were searched on the Pfam website. In all, 14,013 of 14,458 genes were supported by functional annotation from the databases (Fig. 3b). It is worth noting that several genes related to hypoxia, odorant, and immunity were not well annotated in the previous genome, such as olfactory receptors and clip-___domain serine proteases which had been spot checked.
According to the structural characteristics, tRNAscan-SE56 (1.3.1) was used to identify the tRNA in the genome. Meanwhile, because of the highly conservative of it, the rRNA sequences of related species were used as a reference sequence and aligned with C. maculatus genome via blast. Additionally, the covariance model of the Rfam family was used, and the INFERNAL57 (1.1.4) that came with Rfam to predict the information of miRNA and snRNA on the genome. There were 493,139 bp non-coding RNA, and most of them is tRNA (2,827 genes for 206,000 bp), while it was 6,948 tRNA genes in the previous genome10.
Data Records
The whole raw data has been deposited at the NCBI Sequence Read Archive under BioProject number PRJNA1048654 and BioSample ID SAMN38657795 for C. maculatus. Raw sequencing data (Illumina, Nanopore, Hi-C and RNA-seq data) have been deposited in the Sequence Read Archive database as SRP47724758. The final genome assembly and gene annotation results have been deposited in GenBank59 and Figshare60.
Technical Validation
To evaluate the completeness of the assembly, BWA61 (0.7.17) was used to align the short-reads data with genome while Minimap262 (2.17) aligned the long-reads data and the coverage depth for assembled chromosomes were calculated via SAMtools63 (1.16.1) (Table 7). The chromosome-level genome was also evaluated via BUSCO52 (5.4.1) which was compared with Insecta odb10 with 1367 genes, and the results showed that 98.4% and 0.3% conserved core genes were identified as completed and fragmented (Table 6). These results showed that the assembled C. maculatus chromosome-level genome has an elevated level of completeness.
Code availability
All commands and pipelines utilized in the data processing were executed in accordance with the manuals and protocols of the respective bioinformatics software. In instances where detailed parameters were absent, default parameters were employed. The version of the software used is delineated in the Methods section. Notably, no custom programming or coding was incorporated.
References
Kalpna, Hajam, Y. A. & Kumar, R. Management of stored grain pest with special reference to Callosobruchus maculatus, a major pest of cowpea: A review. Heliyon 8, e08703 (2022).
Naseri, B., Ebadollahi, A. & Hamzavi, F. Oviposition preference and life-history parameters of Callosobruchus maculatus (Coleoptera: Chrysomelidae) on different soybean (Glycine max) cultivars. Pest Management Science 78, 4882–4891 (2022).
Global Biodiversity Information Facility Secretariat. GBIF Backbone Taxonomy, https://www.gbif.org/species/1047343 (2023).
Ranabhat, S., Zhu, K. Y., Bingham, G. V. & Morrison, W. R. III Mobility of phosphine-susceptible and -resistant Rhyzopertha dominica (Coleoptera: Bostrichidae) and Tribolium castaneum (Coleoptera: Tenebrionidae) after exposure to controlled release materials with existing and novel active ingredients. Journal of Economic Entomology 115, 888–903 (2022).
Caswell, G. H. The storage of cowpeas in the northern states of Nigeria. Proceedings of the agricultural society of Nigeria 5, 4–6 (1970).
Pimbert, M. A model of host plant change of Zabrotes Subfasciatus Boh. (Coleoptera: Bruchidae) in a traditional bean cropping system in Costa Rica. Biological Agriculture & Horticulture 3, 39–54 (1985).
Keever, D. W. & Daniel Cline, L. Effect of light trap height and light source on the capture of Cathartus quadricollis (Guérin-Méneville) (Coleoptera: Cucujidae) and Callosobruchus maculatus (F.) (Coleoptera: Bruchidae) in a warehouse. Journal of Economic Entomology 76, 1080–1082 (1983).
New, J. H. & Rees, D. P. Laboratory studies on vacuum and inert gas packing for the control of stored-product insects in foodstuffs. Journal of the Science of Food and Agriculture 43, 235–244 (1988).
Sayadi, A. Callosobruchus maculatus, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:CAACVG000000000 (2019).
Sayadi, A. et al. The genomic footprint of sexual conflict. Nature Ecology & Evolution 3, 1725–1730 (2019).
Adan, A., Alizada, G., Kiraz, Y., Baran, Y. & Nalbant, A. Flow cytometry: basic principles and applications. Critical Reviews in Biotechnology 37, 163–176 (2017).
Kron, P., Suda, J. & Husband, B. C. Applications of flow cytometry to evolutionary and population biology. Annual Review of Ecology, Evolution, and Systematics 38, 847–876 (2007).
Blommaert, J. Genome size evolution: towards new model systems for old questions. Proceedings of the Royal Society B: Biological Sciences 287, 20201441 (2020).
Zhang, L. et al. Chromosome-level genome assembly of the predator Propylea japonica to understand its tolerance to insecticides and high temperatures. Molecular Ecology Resources 20, 292–307 (2020).
Fu, X. et al. Long-read sequence assembly of the firefly Pyrocoelia pectoralis genome. GigaScience 6, gix112 (2017).
Wang, P. et al. The genome evolution and domestication of tropical fruit mango. Genome Biology 21, 60 (2020).
Shang, J. et al. The chromosome-level wintersweet (Chimonanthus praecox) genome provides insights into floral scent biosynthesis and flowering in winter. Genome Biology 21, 200 (2020).
Yang, J. et al. Chromosome-level reference genome assembly and gene editing of the dead-leaf butterfly Kallima inachus. Molecular Ecology Resources 20, 1080–1092 (2020).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Shendure, J. et al. DNA sequencing at 40: past, present and future. Nature 550, 345–353 (2017).
Nextomics. NextDenovo https://github.com/Nextomics/NextDenovo (2020).
Guan, D. Purge Dups https://github.com/dfguan/purge_dups (2020).
Arnqvist, G. et al. Genome size correlates with reproductive fitness in seed beetles. Proc Biol Sci 282, 20151421 (2015).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2019).
Zhang, H. et al. Fast alignment and preprocessing of chromatin profiles with Chromap. Nature Communications 12, 6566 (2021).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics 39, btac808 (2023).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Systems 3, 99–101 (2016).
Neva, C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Systems 3, 95–98 (2016).
Yadav, J. S. Karyological studies on the three species of Bruchidae (Coleoptera). Caryologia 24, 157–166 (1971).
Liu, Y. et al. Tribolium castaneum strain Georgia GA2, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:AAJJ00000000 (2016).
Wellcome Sanger Institute. Harmonia axyridis, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:CAJZBN000000000 (2021).
Wellcome Sanger Institute. Coccinella septempunctata, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:CAJRAZ000000000 (2021).
Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics 5, 4.10.11–14.10.14 (2004).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research 35, W265–268 (2007).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl 1), i351–358 (2005).
Smit, A. & Hubley, R. RepeatModeler Open-1.0 www.repeatmasker.org/ (2015).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research 27, 573–580 (1999).
Adams, M. D. et al. Release 6 of the Drosophila melanogaster genome. GenBank https://identifiers.org/insdc.gca:GCA_000001215.4 (2017).
National Center for Biotechnology Information. Genome assembly PGI_DIABVI_V3a. GenBank https://identifiers.org/ncbi/insdc.gca:GCA_917563875.2 (2022).
Murali, S. et al. Anoplophora glabripennis isolate ALB-LARVAE, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:AQHT00000000 (2017).
Parisot, N. et al. Sitophilus oryzae breed Bouriz, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:PPTJ00000000 (2019).
Murali, S. et al. Leptinotarsa decemlineata strain Imidocloprid resistant, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:AYNB00000000 (2017).
Evans, J. et al. Aethina tumida isolate Nest 87, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JALKMD000000000 (2022).
Alioto, T., Blanco, E., Parra, G. & Guigó, R. Using geneid to identify genes. Current Protocols in Bioinformatics 64, e56 (2018).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve De novo gene finding. Bioinformatics 24, 637–644 (2008).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. Journal of Molecular Biology 268, 78–94 (1997).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biology 9, R7 (2008).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols 7, 562–578 (2012).
Shumate, A. & Salzberg, S. L. Liftoff: accurate mapping of gene annotations. Bioinformatics 37, 1639–1643 (2021).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Chan, P. P. T. M. L. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods in Molecular Biology 1962, 1–14 (2019).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP477247 (2024).
Lu, H.-R., Ge, S.-Q., Li, X.-Y. & Zou, Z. Callosobruchus maculatus breed cowpea beetle isolate GSQ-2024a, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JBDIZO000000000 (2024).
Lu, H.-R., Ge, S.-Q., Li, X.-Y. & Zou, Z. Genome data of Callosobruchus maculatus. Figshare https://doi.org/10.6084/m9.figshare.24893025 (2023).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 37, 4572–4574 (2021).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008 (2021).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 32270460), the Third Xinjiang Scientific Expedition Program (No. 2021xjkk0605), the National Natural Science Foundation of China (No. 32370522), the National Key Plan for Scientific Research and Development of China (No. 2022YFC2602500 and No. 2021YFC2600100), and the Science and Technology fund from GACC (No. 2023HK051). Sincere thanks to Doctor Jie Yang (Northwestern Polytechnical University) for her help.
Author information
Authors and Affiliations
Contributions
S.Q.G., Z.Z. and X.Y.L. designed the research. H.R.L. and J.W.H. planned the project. S.Q.G., L.J.Z., X.S.W., X.Y.Z., W.L.F. and C.H.C. prepared the material for sequencing. H.R.L. and C.Y.M. designed the genome assembly, annotation, and analyses. H.R.L. performed evolutionary analysis and gene identification. Z.Z.H. provided the photograph. H.R.L. drafted the manuscript. H.R.L., J.W.H., L.Z., Z.Z.H., X.Y.L., Z.Z. and S.Q.G. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lu, HR., Mao, CY., Zhang, LJ. et al. High-quality reference genome of cowpea beetle Callosobruchus maculatus. Sci Data 11, 799 (2024). https://doi.org/10.1038/s41597-024-03638-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03638-w
