
Abstract
The rumen microbiome plays an important role in providing energy and protein to the host. Manipulation of rumen microbiome during early life may have a long-term beneficial effect on the health, growth performance, and feed efficiency of ruminants. To better understand the profiles and functional potentials of rumen microbiome in young ruminants, metagenomic binning was performed to investigate the rumen microbiome of goat kids from one to 84 days of age. A total of 797 metagenome-assembled genomes (MAGs) were recovered from the rumen of 42 Laiwu black goat kids. Our findings provide fundamental knowledge of the rumen microbiome during early life based on metagenomic binning, which may provide insights into effective strategies to achieve long-term beneficial effects on animal health and production.
Similar content being viewed by others
Background & Summary
Ruminants can utilize a variety of dietary components that are not digestible by the mammals. This is achieved through microbial fermentation taking place primarily in the rumen, the fore-stomach of ruminants that hosts a diverse microbial community. As the key microbial fermentation products, volatile fatty acids (VFA) can provide up to 75% of total metabolizable energy requirement in ruminants1. In addition, microbial protein contributes 60% to 85% of amino acids reaching the small intestine of ruminants2. It is therefore crucial to promote rumen fermentation in order to enhance the productivity of ruminants. Effective manipulation of rumen fermentation requires a deep understanding of the rumen microbiome, consisting of both the microbiota (community of microorganisms) and their “theater of activity” (structural elements, metabolites/signal molecules, and the surrounding environmental conditions)3. It is generally accepted that the gut microbiome in infancy is more flexible and less resilient compared with mature gut microbiome4, and thus external disturbance in early life such as the use of antibiotics or food/feed additive may have a long-term impact on the health, growth, and development of human/animals5,6,7. Therefore, the knowledge of early life rumen microbiome may provide opportunities to effectively engineer microbiome for an enhanced fermentation capacity as well as growth performance in the future.
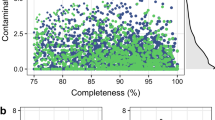
In recent years, the recovery of draft genome sequences also known as metagenome-assembled genomes (MAGs) achieved by metagenomic binning along with bioinformatic analyses allows insights into ecological adaptation and metabolic diversity of uncultured rumen microbiome8,9,10. However, studies of the rumen microbial communities in ruminants during early life rarely go beyond the characterization of community composition based on 16 S rRNA metataxonomic analysis11,12. In our previous study, we investigated the temporal changes in gut microbiota in goat kids from birth to postweaning based on metagenome and metatranscriptome13. However, binning was not involved in that study. Here, we present a description of draft genomes (MAGs) in the rumen of goat kids at 1, 7, 14, 28, 42, 56, 70 and 84 days of age using the samples as we previously reported13. A total of 797 MAGs with completeness >80% (91.7% ± 5.3%) and contamination <10% (1.9% ± 2.0%) were recovered, among which 475 (60.8%) could be considered as high-quality draft MAGs (completeness >90%, contamination <5%) (Fig. 1).

Assessment of the percentage of completeness and contamination of the 797 metagenome-assembled genomes (MAGs) recovered from the rumen of goat kids based on metagenomic binning.
Methods
Animals and phenotypes
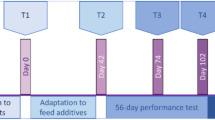
The datasets used in the current study originated from our previous experiment conducted at a goat farm in Laiwu city, Shandong province, China (36.3 N, 117.6 E)14 in accordance with the guidance of the Animal Ethics Committee of the Chinese Academy of Agricultural Sciences (Protocol Number: AEC-CAAS-20191105). A total of 48 healthy female Laiwu goat kids were used in the study and slaughtered at 1, 7, 14, 28, 42, 56, 70, and 84 days of age (n = 6 each age) prior to morning feeding. Ruminal digesta samples were collected, snap-frozen in liquid nitrogen, and subsequently stored in a freezer at −80 °C for sequencing analysis. During the experiment, all goat kids were fed with colostrum at 1 day of age and dam’s milk until 60 days of age. A solid concentrate was introduced at 30 days of age and all goats had ad libitum access to water and starter. The ingredients of the starter included corn, bran, soybean, DDGS, and detoxified cotton meal, containing 18.03% crude and 29.62% NDF, respectively.
DNA extraction and metagenome sequencing
Total genomic DNA from 0.1 g ruminal digesta samples was extracted using a DNeasy PowerSoil Kit (Cat. No.12888, Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The purity and integrity of the genomic DNA was evaluated using 1.0% agarose gel electrophoresis and a Nanodrop ND-1000 (Thermo Fisher Scientific, Wilmington, DE). The DNA quality of 6 samples (two samples from 1 day of age, and one sample from 7, 28, 42, and 84 days of age) was low and thus were not used for sequencing. After quality and quantity checks, metagenomic libraries were constructed using TruSeq DNA PCR-Free Library Preparation Kit (Illumina, San Diego, CA). Each metagenomic library was initially quantified using a Qubit 2.0 fluorimeter (Invitrogen, Carlsbad, CA). Metagenomic sequencing was performed using the Illumina HiSeq 4000 (150 bp paired-end) platform at the Realbio Technology Center (Shanghai, China).
Metagenomic assembly and binning
Trimmomatic (v0.39)15 was used to trim artificial sequences (adapters), cut low quality bases (quality scores < 20), and remove short reads (<50 bp). Bowtie2 (v2.5.2)16 was then used to remove potential host DNA contamination by mapping non-rDNA reads to the latest goat (Capra hircus) genome (ARS1.2). After quality control, MEGAHIT (v1.2.9)17 was used to assemble the contigs for each sample individually with ‘--min-contig-len 1000’ options. A total of 1,776,085,267 paired-end reads were assembled into 4,783,675 contigs. The genomic bins were then reconstructed using MetaBAT2 (v2.15)18, MaxBin2 (v2.2.7)19, CONCOCT (v1.1.0)20, and COMEBin (v1.0.0)21, respectively. The bins were subjected to dRep (v2.5.4)22 with ‘-p 16 -comp 80 -con 10 -str 100 -strW 0’ in order to remove potential replicate bins and only those bins with completeness >80% and contamination <10% were considered as MAGs.
Technical Validation
The information regarding the number of raw, trimmed, host-removed reads is presented in Supplementary Table 1, and the quality information regarding the MAGs is presented in Supplementary Table 2. Considering that the biomass in rumen digesta collected from neonatal goat kids during the first weeks of life could be potentially low, a negative control without sample DNA was sequenced to make sure there was no contamination from laboratory agents or sequencing platform, and no read was assigned to the negative control. MEGAHIT (v1.2.9)17 was used to assemble the contigs for each sample individually with ‘–min-contig-len 1000’ options to remove contigs shorter than 1,000 bp. After assembly, the clean reads were then mapped against their own assembly using Bowtie2 (v2.5.2)16. Samtools (v0.1.19)25 was then used to convert the output to BAM file format and ‘jgi_summarize_bam_contig_depths’ script18 was used to calculate the coverage for each of the bam files. The coverage files were combined for multi-coverage binning with the option -minContig 150026. Four binning methods, including MetaBAT2 (v2.15)18, MaxBin2 (v2.2.7)19, CONCOCT (v1.1.0)20, and COMEBin (v1.0.0)21 were in order to minimize biases and to maximize the certainty of the binning results. These binners were reported to show excellent overall performance and generate high-quality scaffold cluster21,27. The draft bins were subjected to dRep (v3.4.2)22 and only those with completeness >80% and contamination <10% were kept, which is a generally accepted criterion for constructing MAGs in the rumen8,28.
Code availability
No custom code was used in this study.
References
Bergman, E. N. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 70, 567–590 (1990).
Storm, E., Orskov, E. R. & Smart, R. The nutritive value of rumen micro-organisms in ruminants. 2. The apparent digestibility and net utilization of microbial N for growing lambs. Br. J. Nutr. 50, 471–478 (1983).
Berg, G. et al. Microbiome definition re-visited: old concepts and new challenges. Microbiome 8, 103 (2020).
Lozupone, C. A., Stombaugh, J. I., Gordon, J. I., Jansson, J. K. & Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230 (2013).
Guo et al. How early-life gut microbiota alteration sets trajectories for health and inflammatory bowel disease? Front. Nutr. 8, 690073 (2021).
Sommer, F., Anderson, J. M., Bharti, R., Raes, J. & Rosenstiel, P. The resilience of the intestinal microbiota influences health and disease. Nat. Rev. Microbiol. 15, 630–638 (2017).
Belanche, A. et al. Inoculation with rumen fluid in early life accelerates the rumen microbial development and favours the weaning process in goats. Anim. Microbiome 3, 11 (2021).
Stewart, R. D. et al. Compendium of 4,941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat. Biotechnol. 37, 953–961 (2019).
Xie, F. et al. An integrated gene catalog and over 10,000 metagenome-assembled genomes from the gastrointestinal microbiome of ruminants. Microbiome 9, 137 (2021).
Xue, M. et al. Investigation of fiber utilization in the rumen of dairy cows based on metagenome- assembled genomes and single-cell RNA sequencing. Microbiome 10, 11 (2022).
O’Hara, E. et al. Investigating temporal microbial dynamics in the rumen of beef calves raised on two farms during early life. FEMS Microbiol. Ecol. 96, fiz203 (2020).
Ma, T. et al. Linking perturbations to temporal changes in diversity, stability, and compositions of neonatal calf gut microbiota: prediction of diarrhea. ISME J. 14, 2223–2235 (2020).
Chai, J. et al. Metagenomics reveals the temporal dynamics of the rumen resistome and microbiome in goat kids. Microbiome. 12, 14 (2024).
Zhuang, Y. et al. Longitudinal investigation of the gut microbiota in goat kids from birth to postweaning. Microorganisms 8, 1111 (2020).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Langmead, B. & Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, D., Liu, C. M., Luo, R., Sadakane, K. & Lam, T. W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019).
Wu, Y.-W., Simmons, B. A. & Singer, S. W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607 (2016).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146 (2014).
Wang, Z. et al. Effective binning of metagenomic contigs using contrastive multi-view representation learning. Nat. Comm. 15, 585 (2024).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868 (2017).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP326656 (2021).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP517025 (2024).
Li, H., Handsaker, B., Wysoker, A., Fennell, T. & Ruan, J. The sequence alignment/map format and SAMtools. Bioinformatic 25, 2078–2079 (2009).
Mattock, J. & Watson, M. A comparison of single-coverage and multi-coverage metagenomic binning reveals extensive hidden contamination. Nat. Methods 20, 1170–1173 (2023).
Yang, C. et al. A review of computational tools for generating metagenome-assembled genomes from metagenomic sequencing data. Comput. Struct. Biotechnol. J. 19, 6301–6314 (2021).
Wilkinson, T. et al. 1200 high-quality metagenome-assembled genomes from the rumen of African cattle and their relevance in the context of sub-optimal feeding. Genome Biol. 21, 229 (2020).
Acknowledgements
The authors are grateful to Drs. Jianmin Wang, Zhibin Ji, Guizhi Wang, Tianle Chao, Xunhe Yin and Meng Li from Shandong Agricultural University for sample collection. This work was funded by the Central Public-Interest Scientific Institution Basal Research Fund of Chinese Academy of Agricultural Sciences (Y2022QC10; 1610382024009), Agricultural Science and Technology Innovation Program of Chinese Academy of Agricultural Sciences (CAAS-ASTIP-2023-IFR-04; CAAS-IFR-ZDRW202404), and China Agriculture Research System of MOF and MARA (CARS-38).
Author information
Authors and Affiliations
Contributions
T.M., Y.Z., Y.T., X.F., Q.D., and N.Z. conceived the study. Y.Z., K.C., and Y.B. performed the animal trial, sample collection, and the laboratory processing of samples. T.M., Y.Z., and W.L. completed the bioinformatic analyses and led the writing of the paper, with all authors reviewing draft versions of the manuscript. Y.T., X.F., and N.Z. provided funding and resources to perform the analysis.
Corresponding authors
Ethics declarations
Competing interests
The authors declared no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, T., Zhuang, Y., Lu, W. et al. Seven hundred and ninety-seven metagenome-assembled genomes from the goat rumen during early life. Sci Data 11, 897 (2024). https://doi.org/10.1038/s41597-024-03703-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03703-4
