
Abstract
The ratmouth barbel (Ptychidio jordani) is a critically endangered freshwater fish from the Cyprinidae family, primarily due to overfishing and habitat disruption. To address the challenges of its shrinking wild populations and the difficulties in artificial reproduction, we sequenced, assembled, and annotated a high-quality chromosome-level genome of P. jordani using next-generation short-read sequencing, third-generation long-read sequencing, and Hi-C sequencing. The final genome assembly was 1.14 Gb, consisting of 25 chromosomes with a contig N50 of 25.14 Mb and a scaffold N50 of 42.91 Mb. We identified 25,183 protein-coding genes, 751.75 Mb of repeats, and 19,373 ncRNAs. Methylation loci on most chromosomes ranged from 1,000 to 3,000 per 100 kb window. Gene expression levels across various tissues were analyzed, revealing 12,135 (caudal fin), 11,465 (liver), 14,438 (gill), 12,413 (heart), 8,301 (spleen), and 3,578 (kidney) differentially expressed genes compared to muscle. The comprehensive genomic and transcriptomic resources generated here will aid in understanding the ecology, adaptation, and environmental responses of P. jordani, supporting future research and conservation efforts.
Similar content being viewed by others

Background & Summary
The ratmouth barbel (Ptychidio jordani Myers, 1930) is a freshwater ray-finned fish in the Cyprinidae family, native to the Pearl River drainage basin in China1. It prefers fast-flowing, clear streams and rivers with gravel substrates, occupying a bottom-dwelling ecological niche and primarily feeding on mollusks and algae2. It is known to reach a maximum length of 37 cm and is easily identified by its notable barbels, which are critical for its foraging behavior3. Due to its high muscle fat content, tender and delicate flesh, and excellent taste, P. jordani has high economic value and is considered one of the four most renowned fish in the Pearl River region. Historically an important species for local fisheries, P. jordani has experienced a drastic population decline due to overfishing and habitat disruption, primarily caused by dam construction4,5. In 2011, the IUCN classified P. jordani as critically endangered6. Subsequently, in 2016, P. jordani (wild populations) was recorded in the “Red List of China’s Vertebrates” and classified as a critically endangered species in China7. Additionally, P. jordani presents significant challenges for artificial breeding, as it has proven difficult to reproduce in controlled environments8. Given the sharp decline in wild populations and the difficulties in achieving artificial reproduction, urgent conservation measures are needed for P. jordani.
However, developing effective conservation strategies is challenging due to the current lack of detailed research on the biology and ecological characteristics of P. jordani. Gaining insight into the genetic composition of P. jordani is vital for identifying the evolutionary adaptations that have enabled its persistence in specific freshwater environments. While the mitochondrial DNA and microsatellite marker studies have provided preliminary insights into its genetic diversity and population structure8,9,10, the lack of a chromosome-level genome sequence significantly limits the depth of current research. A chromosome-level genome sequence provides a high-resolution resource to uncover key genetic and evolutionary traits, including population-specific adaptations, genetic diversity, and sex determination mechanisms11,12,13. This information is fundamental for designing targeted conservation strategies, such as identifying at-risk populations, preserving genetic diversity, and implementing effective restocking and habitat restoration programs. Furthermore, it can facilitate advancements in artificial breeding techniques by identifying genes linked to reproduction, growth, and adaptive traits, thereby enhancing breeding efficiency and promoting aquaculture sustainability.
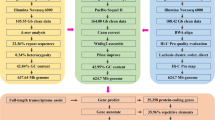
In this study, we present the chromosome-level genome assembly of P. jordani (Fig. 1), utilizing multiple sequencing technologies, including next-generation short-read sequencing and third-generation long-read sequencing, supplemented by high-throughput chromosome conformation capture (Hi-C) techniques. Ultimately, we sequenced and assembled a high-quality chromosome-level genome of P. jordani, with the final genome size of 1.14 Gb, comprising 25 chromosomes with a contig N50 of 25.14 Mb and a scaffold N50 of 42.91 Mb. We also evaluated the methylation level of the assembled genome and identified the distribution of methylation loci across the chromosomes. Additionally, we conducted transcriptomic analyses across muscle, caudal fin, liver, gill, heart, spleen, and kidney. These analyses provided a deep understanding of gene expression patterns in P. jordani. Thus, we constructed a detailed and comprehensive genomic map of P. jordani, facilitating the identification of critical genes and regulatory elements related to the species’ growth, reproduction, and adaptation to the environmental conditions. Our research substantially enriches the genomic resources available for P. jordani, providing valuable data for comparative genomic studies within the Cyprinidae family and supporting both conservation efforts and the development of artificial breeding techniques.

The genome structure of Ptychidio jordani. From outer to inner circles: (a) chromosomes (Chr1-Chr25), (b) gene density, (c) repeats coverage, (d) distribution of non-coding RNAs, including rRNA (black), snRNA (red), tRNA (green), miRNA (blue), and (e) GC ratio.
Methods
Ethics statement
This study was performed in compliance with ethical standards and approved by the Laboratory Animal Ethics Committee of the Pearl River Fisheries Research Institute under License No. LAEC-PRFRI-2024-04-01.
Sample collection
A cultured specimen of P. jordani was collected from Guangdong Hanyu Ecological Technology Co., LTD, Guangzhou city, Guangdong Province, China (113°30′45″N, 22°55′54″E). Following lethal anesthesia of the specimen, multiple tissue samples, including muscle, caudal fin, liver, gill, heart, spleen, and kidney, were collected, frozen in liquid nitrogen, and stored at −80 °C for subsequent DNA and RNA extraction.
Genomic DNA extraction, species identification and sequencing
Muscle tissue was utilized for next-generation sequencing, PacBio HiFi sequencing, and Hi-C sequencing. Genomic DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen, Germany) following the manufacturer’s instructions. The concentration and purity of the DNA were measured using a NanoDrop 2000 spectrophotometer (NanoDrop Technologies, USA).
Due to the ambiguous morphology of the Ptychidio genus, we initially identified the species using the COI mitochondrial marker. COI primers and PCR protocols were adopted from14. We then blasted the target COI sequence against the other species in the genus Ptychidio and conducted a phylogenetic tree using all available COI sequences in the CoSFISH website and its online analysis tools15. The COI phylogenetic result confirmed that our specimen is P. jordani (Figure S1).
For the genome survey, next-generation sequencing was conducted using a DNA library prepared with the MGIEasy Universal DNA Preparation Reagent. The library was constructed through a series of steps, including fragmentation, end-repair, adapter ligation, and subsequent amplification. Sequencing was performed on the BGISEQ DNBSEQ-T7 platform, producing 150 bp paired-end reads with an insert size of 350 bp. SOAPnuke (version 2.1.0)16 was used to filter the raw reads, removing low-quality sequences and adapter contaminants, resulting in 55.57 Gb of clean data.
The PacBio HiFi library was prepared using SMRTbell prep kit 3.0 (PacBio 102-182-700) following the manufacturer’s protocols. High-quality genomic DNA was sheared to a target size range of 15–18 kb using the Megaruptor 3 system (Diagenode), followed by size selection with the AMPure PB bead kit (PacBio 102-182-500). The enriched fragments were then end-repaired, A-tailed, and ligated to SMRTbell adapters. The prepared library was then sequenced on the PacBio Revio platform, generating 42.95 Gb of HiFi reads.
The Hi-C library was constructed according to the Hi-C library protocol by Rao et al.17, utilizing frasergen Hi-C kit. Sequencing was carried out on the BGISEQ DNBSEQ-T7 platform, resulting in 123.62 Gb of raw reads. The sequencing depth for Hi-C was 108.6X, ensuring comprehensive coverage for chromosomal interaction mapping. After trimming with Trimmomatic (version 0.40)18, we obtained 110.55 Gb of polished Hi-C reads.
Transcriptome sequencing
Tissue samples of muscle, caudal fin, liver, gill, heart, spleen, and kidney were used for RNA-seq analysis. RNA was extracted from each tissue sample using the TRIzol reagent (Invitrogen, MA, USA) according to the manufacturer’s instructions. The concentration and purity of the extracted RNA were measured using Nanodrop 2000 spectrophotometer (NanoDrop Technologies, USA). RNA integrity was evaluated through agarose gel electrophoresis and Agilent 2100 bioanalyzer (Agilent Technologies, Palo Alto, CA). Subsequently, RNA-seq libraries were individually constructed for each tissue sample using the qualified RNA. The mRNA was purified from the total RNA using poly‐T oligo‐attached magnetic beads. Sequencing libraries were then generated from the purified mRNA using the VAHTS Universal V6 RNA-seq Library Kit for MGI (Vazyme, Nanjing, China) following the manufacturer’s recommendations. Finally, the libraries were sequenced on the BGISEQ DNBSEQ-T7 platform, producing 150 bp paired-end reads for each tissue sample. Detailed RNA-seq data information for each tissue sample is provided in Table 1.
Genome size estimation
Based on next-generation short reads, the k-mer frequency with k = 17 was used to estimate the genome size, heterozygosity, and repeat content of P. jordani using GCE software (version 1.0.2)19 (Figure S2). The analysis revealed that the estimated genome size was approximately 1.08 Gb, with a heterozygosity rate of 0.43% and a repeat content of 57.88%.
Genome assembly
The draft genome was assembled using PacBio Hi-Fi reads processed with Hifiasm (version 0.19.5)20 under default parameters, resulting in 251 contigs with a contig N50 of 25.14 Mb and an estimated genome size of approximately 1.15 Gb. Polished Hi-C reads were then mapped to the draft contig-level genome assembly using Juicer (version 1.6)21 to generate interaction matrices. These matrices enabled the anchoring and scaffolding of 100 contigs, accounting for 97.48% of the total sequences, into 25 chromosomes using 3D-DNA (version 180922)22 under default parameters (Fig. 2). The remaining 132 unanchored contigs were analyzed using BLAST (version 2.11.0+)23 against the NCBI NT (Nucleotide Sequence Database) to identify and remove potential contaminants, including bacterial and organelle-derived sequences. The final genome assembly consisted of 232 contigs, with a contig N50 of 25.14 Mb, a scaffold N50 of 42.91 Mb, and a genome size of 1.14 Gb. The number of chromosomes is consistent with karyotypic findings from a previous study24. Detailed chromosome information is provided in Table S1. The summarized assembly information is provided in Table 2.

Hi-C interaction heatmap of the P. jordani genome. The x- and y-axes represent genomic positions (in Mb), with darker red indicating stronger chromosomal interactions. The color scale (right) reflects interaction frequencies.
Repeat identification
Identification of repetitive sequences was performed using a combination of de novo and homology-based approaches. RepeatModeler (version 2.0.1, default parameters) and LTR-FINDER (version 1.0.7, parameter -w 2 -C)25 were utilized to construct a de novo library of repetitive sequences. This library was subsequently employed to identify repetitive regions using RepeatMasker (version 4.1.2)26 with the parameter -nolow -no_is -norna -parallel 2. Additionally, tandem repeats were detected using the Tandem Repeats Finder (Trf) software (version 4.09.1)27 with the parameter 2 7 7 80 10 50 2000 -d -h. Homologous repeats were identified by aligning the assembled genome sequences against the RepBase database (http://www.girinst.org/repbase) using RepeatMasker (version 4.1.2) and RepeatProteinMask (version 1.36)26. By merging the results from both methods, we identified a total of 751.75 Mb repeats, accounting for 66.04% of the genome (Table 3). Among these repetitive sequences, transposable elements (TEs) accounted for 56% of the genome. Among TEs, the most abundant type was DNA transposons (Table 4).
Gene prediction and functional annotation
Gene prediction was conducted using a combination of ab initio prediction, homology-based annotation, and transcriptome-based prediction. The ab initio prediction was performed using Augustus (version 3.4.0)28 with the parameter –noInFrameStop = true -strand = both and Genscan (version 1.0)29 with default parameters. For homology searching, the protein sequences of the four phylogenetically closely related species, including Cirrhinus molitorella, Onychostoma macrolepis30, Labeo rohita31 and Puntigrus tetrazona12, were downloaded from NCBI and aligned against the assembled genome using BLAST (version 2.11.0+)23 with an e-value ≤ 1e–05. Subsequently, Exonerate (version 2.4.0)32 with the parameter –model protein2genome was used to predict gene structures. For transcriptome-based prediction, we first used SOAPnuke (version 2.1.0)16 to filter the raw RNA-seq data, with parameters -lowQual = 20, -nRate = 0.005 and -qualRate = 0.5. The clean RNA-seq reads, combined from all tissue samples, were assembled using Trinity (version 2.8.5)33 and then aligned to the assembled genome using HISAT2 (version 2.2.1)34. The aligned reads were subsequently assembled into transcripts with StringTie (version 2.1.7)35. The non-redundant reference gene set was compiled by integrating gene predictions from three different methods using MAKER (version 3.01.03)36 and subsequently annotated with PASA (version 2.4.1)37. Finally, a total of 25,183 protein-coding genes were identified (Table 5). This gene set was functionally annotated by aligning it against the InterPro38, SwissProt39, TrEMBL39, NR (ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz), GO40, and KEGG41 databases using Diamond (version 2.0.7)42. Consequently, 98.28% (24,750) of the predicted protein-coding genes obtained functional annotations (Table 6).
Non-coding RNA annotation
We also annotated the non-coding RNAs using various tools: tRNAs were identified with tRNAscan-SE (version 2.0.9)43, rRNAs were predicted using RNAmmer (version 1.2)44 with the parameter -S euk -multi -m lsu,ssu,tsu. Additionally, miRNAs and snRNAs were detected with infernal (version 1.1.4)45 as implemented in the Rfam database46. Finally, 19,373 ncRNAs were annotated, including 458 miRNAs, 9,692 tRNAs, 8,533 rRNAs and 690 snRNAs (Table 7).
Methylation profiling
HiFi reads were aligned to the assembled genome using pbmm2 (version 1.13.1) (https://github.com/PacificBiosciences/pbmm2), an implementation within minimap2 (version 2.21)47, with the parameters–preset HIFI–sort -j 16 -J 16. The resulting sorted BAM file was then used as input for pb-CpG-tools (version 2.2.0-x86_64) (https://github.com/PacificBiosciences/pb-CpG-tools/) to determine the site-level modification proportions, producing probabilistic estimates of CpG methylation patterns. Based on the coverage of C sites, we used loci with sequencing depth greater than 5 to calculate DNA methylation levels and to count the number of methylation loci in different methylation regions. Ultimately, the overall 5mC methylation proportion of the sample was 79.41%. The distribution of methylation loci was analyzed at the assembled chromosome level. Chromosomes were divided into bins, and the average methylation loci per bin was calculated using a 100 kb window (Fig. 3). The majority of chromosomes had methylation loci ranging from approximately 1,000 to 3,000 per bin. Outliers were observed across all chromosomes, with some bins showing exceptionally high numbers of methylation loci, reaching up to 8,000, while the lowest values were close to zero. These outliers may highlight potential hotspots or areas of differential regulation within the genome.

The distribution of methylation loci across chromosomes in the genome of P. jordani.
Gene expression analysis
Gene expression levels were quantified using the fragments per kilobase of transcript per million mapped reads (FPKM) method with RSEM (version 1.3.3)48 under default parameters. Differentially expressed genes (DEGs) were identified with a false discovery rate (FDR) <0.05 and a log2 fold change (log2FC) >1 or <−1 using edgeR (version 3.22.5)49. The differentially expressed genes (DEGs) among all tissue samples were displayed in Fig. 4. Hierarchical clustering was performed on both genes and tissues to highlight similarities and differences in gene expression profiles. In the comparative analysis with muscle tissue, 12,135 DEGs were identified in the caudal fin, 11,465 in the liver, 14,438 in the gill, 12,413 in the heart, 8,301 in the spleen, and 3,578 in the kidney. Detailed information on the up-regulated and down-regulated DEGs between each pair of tissues is provided in Table S2.

Heatmap of differentially expressed genes (DEGs) across different tissues. The color key indicates the expression levels, with blue representing lower expression and red representing higher expression. The colored bars on the vertical axis represent hierarchical clustering of DEGs, with each color corresponding to a distinct gene cluster based on similar expression patterns across tissues.
Data records
All raw sequencing data from this study have been deposited in the NCBI SRA database. Specifically, next-generation short reads (SRR29932654)50, PacBio third-generation reads (SRR29932653)51, Hi-C reads (SRR29932652)52, and RNA-seq reads (SRR29932645-SRR29932651)53,54,55,56,57,58,59 are available under BioProject accession number PRJNA1137526. The final chromosome-level assembled genome file is accessible in the GenBank database under accession number JBGFUG00000000060, and the genome annotation files are available on Figshare61. The gene expression matrix for all tissues has been deposited in the GEO database under accession number GSE28230662, and the methylation profiling data are available under accession number GSE28231163.
Technical Validation
The completeness of the genome assembly was evaluated using BUSCO (version 5.2.2)64, which indicated a high level of completeness with a score of 97.3%. Additionally, we mapped the next-generation reads to the assembled genome using BWA (version 0.7.17)65 with the parameter mem -t 50, achieving a mapping rate of 99.47% and an overall coverage of 99.75%. Similarly, for the HiFi reads, mapped using minimap2 (version 2.21)47 with the parameter -ax map-hifi, the mapping rate was 99.93% with an overall coverage of 100%. We also evaluated the assembly using Merqury (version 1.3)66 to assess the consistency of the genome. Merqury k-mer-based analysis showed a QV (Quality Value) score of 45.40 (Table 2). To evaluate gene prediction accuracy and reliability, we examined the distribution of exon number, intron number, and GC content in the genes and coding sequences (CDS) of P. jordani and four phylogenetically related species (C. molitorella, O. macrolepis, L. rohita and P. tetrazona). The consistent distribution patterns observed among these species further validated the annotated gene dataset for P. jordani (Fig. 5). All the aforementioned results indicate that the assembled genome is complete and highly reliable. The quality of RNA-seq clean reads across all tissues was assessed using SOAPnuke (version 2.1.0)16, yielding Q20 and Q30 values ranging from 98.70% to 99.99% and 89.32% to 97.52%, respectively (Table S3). These results underscore the high reliability and robustness of the RNA-seq data.

Comparative analysis of genomic elements, including exon number, intro number, GC rate of genes and coding sequences (CDS), among P. jordani and the other four phylogenetically closely related species.
Code availability
We used standard bioinformatics tools and followed instructions of the manuals and protocols. Details about the software versions and settings we used were fully described in the Methods section. No custom code was developed for this study.
References
Li, S. Z. China freshwater fishes distribution (in Chinese). (Science Press, 1981).
Liao, G. et al. Age, growth, feeding habits and reproduction of the mouse fish Ptychidio jordani on the Pearl River. China. J. Fish. China. 10, 71–86 (1986).
He, J. et al. Length‐Weight Relationships of Twenty‐Nine Native Fish Species from Hongshui River, Southern China. J. Appl. Ichthyol. 2023, 1744641 (2023).
Shuai, F. M. et al. Fish diversity and distribution pattern of the Pearl River system in Guangxi. Acta Hydrobiol. Sin. 44 (2020).
He, J. et al. Age, growth, reproduction and status of resource development of Ptychidio jordani, a critically endangered freshwater fish in the Hongshui River, China. J. Fish Biol. 104, 399–409 (2024).
Zhao, H. H. & Chen, X. Y. Ptychidio jordani. The IUCN Red List of Threatened Species 2011: e.T166166A6185765. Accessed on 09 August 2024.
Jiang, Z. et al. Red list of China’s vertebrates. Biodiversity Science 24, 500 (2016).
Zhu, X. P. et al. Genetic analysis of ratmouth barbell (Ptychidio jordani Myers) from different geographic sub‐populations in the Pearl River Basin using microsatellite markers. J. Appl. Ichthyol. 25, 148–152 (2009).
Zhao, N., Chen, F. & Liao, X. The complete mitochondrial genome of the ratmouth barbell, Ptychidio jordani (Cypriniformes Cyprnidae). Mitochondrial DNA A DNA Mapp Seq Anal 27, 692–693 (2014).
Peng, M. et al. Genetic diversity analysis of Ptychidio jordani in Xijiang River flowing through Guangxi Province based on mitochondrial Cytb gene sequence. South China Fisheries Science 16, 10–18 (2020).
Xu, P. et al. The allotetraploid origin and asymmetrical genome evolution of the common carp Cyprinus carpio. Nat. Commun. 10, 4625 (2019).
Li, J. T. et al. Parallel subgenome structure and divergent expression evolution of allo-tetraploid common carp and goldfish. Nat. Genet. 53, 1493–1503 (2021).
Gong, G. et al. Origin and chromatin remodeling of young X/Y sex chromosomes in catfish with sexual plasticity. Natl. Sci. Rev. 10, nwac239 (2023).
Peng, J., Wang, X., Wang, D. & He, S. Application of DNA barcoding based on the mitochondrial COI gene sequences in classification of Culter (Pisces: Cyprinidae). Acta Hydrobiol. Sin. 33, 271–276 (2009).
Wang, Y. et al. CoSFISH: a comprehensive reference database of COI and 18S rRNA barcodes for fish. Database-Oxford 2024, baae038 (2024).
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7, gix120 (2018).
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Preprint at https://arxiv.org/abs/1308.2012 (2013).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods. 18, 170–175 (2021).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Liu, Y. H. et al. Morphological character and karyotype of Ptychidio jordani Myers in Pearl River. J. Fish. China. 31, 721–725 (2007).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 35, W265–W268 (2007).
Tarailo‐Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics 25, 4–10 (2009).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580 (1999).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34, W435–W439 (2006).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94 (1997).
Sun, L. et al. Chromosome‐level genome assembly of a cyprinid fish Onychostoma macrolepis by integration of nanopore sequencing, Bionano and Hi‐C technology. Mol. Ecol. Resour. 20, 1361–1371 (2020).
Arick, M. A. et al. A high-quality chromosome-level genome assembly of rohu carp, Labeo rohita, and its utilization in SNP-based exploration of gene flow and sex determination. G3-GENES GENOM GENET 13, jkad009 (2023).
Slater, G. S. C. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform 6, 1–11 (2005).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 12, 357–360 (2015).
Pertea, M. et al. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667 (2016).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform 12, 1–14 (2011).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 31, 5654–5666 (2003).
Finn, R. D. et al. InterPro in 2017—beyond protein family and ___domain annotations. Nucleic Acids Res 45, D190–D199 (2017).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence data bank and its supplement TrEMBL in 1999. Nucleic Acids Res 27, 49–54 (1999).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 44, D457–D462 (2016).
Buchfink, B., Reuter, K. & Drost, H. G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods. 18, 366–368 (2021).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res 49, 9077–9096 (2021).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35, 3100–3108 (2007).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res 33, D121–D124 (2005).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform 12, 1–16 (2011).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29932654 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29932653 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29932652 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29932651 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29932650 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29932649 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29932648 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29932647 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29932646 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29932645 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc:JBGFUG000000000 (2024).
Mu, X. D. The genome annotation of Ptychidio jordani. figshare https://doi.org/10.6084/m9.figshare.26394937 (2024).
NCBI GEO https://identifiers.org/geo/GSE282306 (2024).
NCBI GEO https://identifiers.org/geo/GSE282311 (2024).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Phillippy Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 21, 1–27 (2020).
Acknowledgements
This work was supported by the Guangdong Agricultural Technology Service - Major Agricultural Technology Rural Promotion Project (2130106), China-ASEAN Maritime Cooperation Fund (CAMC-2018F), the Rural Revitalization Strategy Special Provincial Organization and Implementation Project Funds (2023SBH00001), National Freshwater Genetic Resource Center (FGRC18537).
Author information
Authors and Affiliations
Contributions
Yuanyuan Wang: Conceptualization; data curation and analysis; investigation; writing (original draft, review and editing). Yexin Yang: Data curation; methodology; writing (review and editing). Yuli Wu: Data curation; funding acquisition; writing (review and editing). Yi Liu, Qingyong Li, Chao Liu, Zhiyong Jiang, Wanying Jiang: Sample collection, data curation; resources, writing (review and editing). Fangcan Chen: Sample collection, resources, writing (review and editing). Xidong Mu: Conceptualization; data curation; funding acquisition; resources; supervision; writing (review and editing).
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Y., Yang, Y., Wu, Y. et al. Chromosome-level genome assembly of the ratmouth barbel, Ptychidio jordani. Sci Data 11, 1435 (2024). https://doi.org/10.1038/s41597-024-04331-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-04331-8
