
Abstract
Thermal injuries represent one of the most severe forms of trauma to the human body, with a high annual incidence of burn victims globally. Skin regeneration and wound healing following thermal injury constitute a complex process involving various cell types and cytokine interactions. By Single-cell ATAC sequencing (scATAC-seq), we elucidated the molecular mechanisms underlying the dermal regeneration and healing processes following thermal injury in a rat model. Tissue samples were harvested for sequencing at predetermined intervals (0 h, 12 h, 24 h, 3 d, 7 d, 11 d, 15 d, and 19 d post-injury), yielding 28,179 high-quality single cells. Our comprehensive analysis revealed 28 distinct cell populations throughout the regenerative process, encompassing various subsets of keratinocytes, fibroblasts, and immune cells, exhibiting temporal heterogeneity across samples. Furthermore, we investigated the chromatin accessibility landscape of individual cell types and identified enriched transcription factor binding motifs, corroborating the robustness and validity of our data. Our dataset provides a valuable resource for further elucidation of burnt skin regeneration and healing processes.
Similar content being viewed by others

Background & Summary
The skin represents the largest and one of the most complex organ systems in mammals. It serves as a protective barrier lining the external surfaces of the mammalian body, shielding organisms from environmental insults1. Mammalian skin is a double-layered structure consisting of the epidermis and dermis and is characterized by a heterogeneous cellular composition that reflects its multifaceted functions and intricate organization2. Globally, many individuals sustain thermal injuries to the skin annually. The underlying cellular and molecular mechanisms of this process remain to be fully elucidated. Following burn injury, the healing process of damaged skin can be divided into three primary phases: (1) inflammatory, (2) proliferative, and (3) remodelling3,4. During these stages of burn wound healing, multiple cell types and cytokines interact in a coordinated manner to promote tissue regeneration and repair3,5. However, the transcription factors and epigenetic regulatory mechanisms underlying this process remain incompletely understood.
Single-cell assay for transposase-accessible chromatin sequencing (scATAC-seq) is a cutting-edge technology that has garnered substantial attention in recent years6. Compared to single-cell RNA sequencing (scRNA-seq), scATAC-seq can better capture subtle epigenetic regulatory variations among individual cells. This technique is particularly efficacious in elucidating transcriptional regulatory networks within cellular populations7. While numerous studies have employed scRNA-seq technology to investigate skin regeneration and wound healing processes8,9,10,11,12, research utilizing single-cell ATAC sequencing to decipher relevant DNA regulatory elements remains comparatively limited. While transcriptomic profiling enables the elucidation of functionally distinct cell populations within complex tissues, the integration of epigenetic data offers a more comprehensive understanding of the regulatory mechanisms governing these expression profiles and their maintenance13,14. The latter approach is critical for a comprehensive analysis of burn wound healing mechanisms, offering unique insights into the epigenetic landscape of the cellular populations involved in tissue repair.
In this study, we utilized scATAC-seq technology to elucidate the regeneration and healing process of rat skin following thermal injury. Tissue samples were collected for sequencing at eight time points post-injury: 0 h, 12 h, 24 h, 3 days, 7 days, 11 days, 15 days, and 19 days. Our findings reveal dynamic alterations in the cellular composition of the injured rat dermis and epidermis, as well as changes in the chromatin accessibility of specific cell types. In summary, our research provides a valuable resource for deciphering epigenetic regulation during the skin regeneration process following burn injuries.
Methods
Animal experiments and tissue collection
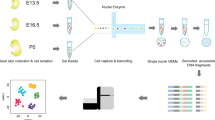
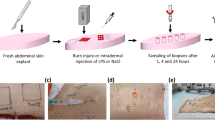
All animal experiments were conducted in accordance with protocols approved by the Department of Burn Surgery at the First Affiliated Hospital of Naval Medical University, Shanghai, China. Eight-week-old male Sprague‒Dawley rats (250–300 g) were utilized. Prior to the procedure, the rats were depilated to ensure complete exposure of the dorsal skin. On the day of the experiment, the rats were anaesthetized with isoflurane and placed on a heated pad with their head and limbs secured to facilitate dorsal skin exposure. A sponge pad was preheated to approximately 100 °C in boiling water for more than 10 minutes and then applied to the dorsal skin for 10 seconds to induce a burn. After the burn, the area was covered with oil gauze and secured with a pressure bandage. Tissue samples from the burn sites were collected at the following intervals: 0 hours, 12 hours, 24 hours, 3 days, 7 days, 11 days, 15 days, and 19 days post-burn. The samples were excised and prepared for single-cell ATAC-seq to assess local and systemic physiological, pathological, and inflammatory responses.
Nuclear isolation from rat skin tissue
Nuclei were isolated from rat skin tissue for single-cell ATAC-seq using a refined enzymatic digestion and mechanical extraction approach15. Fresh skin samples, with a total area of approximately 1 cm², were first rinsed in PBS to remove visible fat and muscle debris. The cleaned tissue was then minced into small fragments using sterile surgical scissors and transferred to a 2 mL centrifuge tube. The tissue fragments were enzymatically digested by adding 1 mL of enzyme solution 1 (0.1% collagenase and 0.01% DNase) and incubating at 37 °C with gentle agitation (130 rpm) for 25 minutes. After digestion, the sample was centrifuged at 500 × g for 5 minutes, and the supernatant was discarded. The resulting pellet was resuspended in 10 mL of enzyme solution 2 (0.1% collagenase) and further incubated at 37 °C with agitation for an additional 60 minutes. The digested tissue suspension was then filtered through a 70 µm cell strainer to remove undigested fragments and washed with 2% FBS in DPBS. The suspension was further filtered through a 30 µm cell strainer. The filtered cell suspension was centrifuged at 300 × g for 5 minutes at 4 °C, and the pellet was resuspended in 1 mL of PBS containing 0.04% BSA. The cell viability and concentration were determined using trypan blue exclusion or an automated cell counter. The prepared nuclei were then utilized for scATAC-seq library preparation, ensuring high-quality samples for accurate chromatin accessibility analysis.
scATAC-seq library preparation and sequencing
For the preparation of single-cell ATAC-seq libraries, we employed the DNBelab C Series Single-Cell ATAC Library Prep Set (MGI, H-020-000747-00)16. To investigate chromatin accessibility changes following burn injury in rats, we generated a total of 19 libraries. Specifically, 3 libraries were prepared from samples collected at 0 hours post-burn; 2 libraries were generated for each of the time points at 12 hours, 24 hours, 3 days, 7 days, 11 days, and 19 days post-burn; and 4 libraries were prepared for samples collected at 15 days post-burn. The barcoded scATAC-seq libraries were generated from transposed single-cell suspensions. In summary, the protocol involved droplet encapsulation, preamplification, emulsion breakage, collection of capture beads, DNA amplification, and purification. Afterwards, indexed sequencing libraries were prepared according to the manufacturer’s instructions. The concentrations of the sequencing libraries were measured via a Qubit ssDNA Assay Kit (Thermo Fisher Scientific). Sequencing was performed on the MGI DNBSEQ-T1 platform, with read lengths of 115 bp for read 1, 69 bp for read 2, and 10 bp for the sample index, following the sequencing scheme of the China National GenBank (CNGB)17.
Preprocessing of scATAC-seq data
The preprocessing of scATAC-seq data followed a series of well-defined steps. First, raw sequencing reads were demultiplexed to separate insertions and barcodes. The reads were then filtered using PISA (version 1.1)18, with a minimum sequencing quality threshold of 20. PISA is available at https://github.com/shiquan/PISA. The sequencing reports for the scATAC-seq datasets are detailed in Table 1. After filtering, the reads were aligned to the rat reference genome (mRatBN7.2) using BWA (version 0.7.17-r1188)19. The aligned BAM files were subsequently processed using bap2 (version 0.6.2) to group barcodes belonging to the same cell for downstream analysis20. This preprocessing workflow ensured high-quality data for further chromatin accessibility analysis.
Quality control for scATAC-seq data
We used the AchR (version 1.0.2)21 package to create arrow files and ArchR projects via the ‘createArrowFiles’ and ‘ArchRProject’ functions. The doublet scores of the cells were calculated by the ‘addDoubletScores’ function. In this way, low-quality cells were filtered with the same criteria for all scATAC-seq libraries: unique nuclear fragments (nFrags) >1000 and an enrichment score of the transcriptional start site (TSS) >4. We also filtered doublets with parameters of filterRatio = 1 by the ‘filterDoublets’ function.
Dimension reduction and scATAC cell clustering
The ‘addIterativeLSI’ function in ArchR21 was used to implement the iterative latent semantic indexing (LSI) dimensionality reduction. The ‘addClusters’ function was subsequently used to cluster cells via the default Seurat22 graph clustering approach in this reduced-dimensional subspace. We also implemented uniform manifold approximation and projection (UMAP) in this subspace to visualize all the cells via the ‘addUMAP’ function.
Batch effect correction and library integration
We tried a batch effect correction tool called Harmony with the ‘addHarmony’ function on our scATAC-seq data to correct for batch effects. However, no significant batch effects were detected. Therefore, we directly merged all the libraries into one ArchR21 object for downstream analysis.
Cell type annotation and subpopulation identification
Using the ‘getMarkerFeatures’ function and the ‘plotEmbedding’ function with default parameters, we obtained and visualized the marker genes for each cell cluster. We accurately annotated each cell type manually on the basis of the cell markers summarized in previous studies. To demonstrate the high quality of the data, we separately filter some of the fibroblasts and keratinocytes to find subclusters via the ‘addClusters’ function, with ‘resolution = 0.15’.
Integration of scRNA-seq data and scATAC-seq data
We first obtained single-cell RNA-seq data from radiation-induced skin injury (GSE193564)23. We then converted our burnt skin scATAC-seq ArchR objects into Seurat objects using gene scores. We subsequently integrated the publicly available scRNA-seq datasets and our scATAC-seq data using Seurat, employing the anchor-based CCA integration approach for multimodal integration.
Cell type-specific peak calling
Before calling the cell type-specific peak, we made pseudobulk replicates by using the ‘addGroupCoverages’ function with the key parameter ‘groupBy = celltype’. With cell type pseudobulk replicates created, we used MASC224 to call peaks for each cell type via the ‘addReproduciblePeakSet’ function with the following parameters: groupBy = cluster_name, pathToMacs2 = pathToMacs2, genomeAnnotation = genomeAnnotation, geneAnnotation = geneAnnotation, genomeSize = 2647899415. The marker peaks of each cell type were identified via the ‘addMarkerFeatures’ function with useMatrix = “PeakMatrix”.
Motif enrichment for cell types
We obtained the mouse motif annotation database “mouse_pwms_v1” from the chromVARmotifs package (https://github.com/GreenleafLab/chromVARmotifs). Then, we add motif annotation to the ArchR object via the ‘addMotifAnnotations’ function. Next, motif enrichment analysis was executed by using the ‘peakAnnoEnrichment’ function on the marker peaks of each cell type obtained previously.
Temporal profiling of chromatin accessibility dynamics
To elucidate the temporal changes in chromatin accessibility, we extracted the accessibility matrices of peaks from monocytes and vascular endothelial cells. For each peak, we calculated the average accessibility matrix across different time points. We employed the Mfuzz25 R package (version 2.58.0) to perform C-means clustering on the accessible peaks in both monocytes and vascular endothelial cells. We subsequently utilized the clusterProfiler26 R package (version 4.6.0) to perform Gene Ontology (GO) functional enrichment analysis for different peak clusters produced by Mfuzz.
TF footprint analysis
We used the ‘getPositions’ function to obtain the genome positions of the cell type-specific motifs. With group coverages calculated previously, we computed footprints for marker motifs via the ‘getFootprints’ function and visualized them via the ‘plotFootprints’ function.
Data Records
The chromatin accessibility landscapes of burnt rat skin at different healing times were profiled via the scATAC-seq technique. The panoramic landscape consisting of 28179 high-quality single cells provides a valuable resource for exploring the epigenetic regulation of the burnt skin healing process at the single-cell level. All related raw fastq files have been submitted to the CNGB Nucleotide Sequence Archive (CNSA) under accession number CNP000614427. Moreover, the cell type-peak matrices and other metadata have been stored in Figshare (https://figshare.com/s/f21a969a6e598f8d3551)28.
Technical Validation
Data quality control for scATAC-seq
In this study, single-cell nuclei were extracted from rat skin at 0 h, 12 h, 24 h, and 3, 7, 11, 15, and 19 days after burn injury (see Methods). H&E-stained sections of burn skin samples at each time point revealed distinct skin structure statuses. Round-shaped cells (indicated by dashed arrows at 12 h and 24 h and dashed boxes at 3D) were identified as myeloid cells. These cells first emerged at 12 h post-injury and then progressively increased, peaking at 3D. Between 7D and 15D, elongated spindle-shaped cells demarcated by solid boxes and solid arrows were recognized as fibroblasts, which exhibited extensive proliferation to repopulate the wound. Although wound closure was observed from 15D to 19D, histological analysis revealed incomplete restoration of native skin architecture. (Figs. 1a, 2b). At each time point, two single-cell ATAC-seq libraries, both derived from the same individual rat, were generated. However, at the Day 15 time point, four distinct libraries were constructed using biological replicates from two rats: libraries 15D-1 and 15D-2 originated from one rat, whereas libraries 15D-3 and 15D-4 were prepared from a second rat. The total number of raw reads from all the libraries was 11,660,674,859. In addition, 10,244,835,427 passed quality control (Table 1). Next, the raw fastq data were processed via a standardized pipeline including alignment to the genome and ArchR21 downstream analysis (Fig. 1b).

Overview of the experimental design and data analysis workflow for scATAC-seq of burnt rat skin. (a) Schematic representation of the experimental timeline and sample collection. Burn injury was induced, and skin samples were harvested at 0 h, 12 h, 24 h, 3 days, 7 days, 11 days, 15 days, and 19 days post-injury for single-cell assays for transposase-accessible chromatin sequencing (scATAC-seq). (b) scATAC-seq analysis workflow.

Quality control and preprocessing of scATAC-seq data from burnt rat skin. (a) Violin plot showing the distribution of log10(nFragments) and TSS enrichment scores for each library, with libraries grouped by burn injury status and time points. (b) H&E-stained sections of rat skin under normal conditions and at various time points post-burn (0–19 days) (×20). Immune cell infiltration is observed in the early burn stage (≤3D), followed by extensive proliferation of fibroblasts and parenchymal cells (7D–11D) and extracellular matrix remodelling (15D–19D). (c) Heatmap showing the correlations between different libraries. (d) Scatter plot comparing log10(nFragments) and TSS enrichment scores, revealing the quality of the data. (e) UMAP plot visualizing the distribution of cells across different time points post-burn injury, with each dot representing a single cell and the colour indicating the time point. (f) Transcription start site (TSS) enrichment profiles for each library. (g) Bar chart showing the number of cells captured at each time point.
To ensure that each cell was of high quality, we calculated transcriptional start site (TSS) enrichment scores, unique fragments, and the number of fragments of each cell. After filtering low-quality cells with the criterion (TSS > 4, nFrags > 1000, Doublet filterRatio = 1) (see Methods), we reserved a total of 28179 high-quality cells with chromatin accessibility profiles. The number of cells retained at each time point is shown in a bar plot (Fig. 2g). Among these remaining cells, the mean number of fragments per cell and the mean TSS enrichment were 11,539.45 and 9.60672, respectively (Table 1) (Fig. 2a,d,f). We also calculated the correlation of each scATAC-seq library data expression profile on the basis of the peak accessibility matrix (Fig. 2c). The UMAP colour at different time points during skin wound healing revealed diverse cell type distributions during the healing process (Fig. 2e).
Cell type identification by chromatin accessibility
After that, all cells collected from the skin at all time points of burn healing were subjected to unsupervised clustering on the basis of gene score values. On the basis of the differential chromatin accessibility within some clusters, we implemented cell-subpopulation clustering in one keratinocyte cluster and two fibroblast clusters. Finally, we defined 28 cell clusters, including endothelial cells, fibroblasts, muscle cells, immune cells, epidermal keratinocytes, and hair follicle keratinocytes. Among all keratinocyte subtypes, three subtypes of epidermal keratinocytes (IFE_basel1 (Krt5, Krt15), IFE_basel2 (Prss27, S100a8), and IFE_spinous (Krt1, Krt10)) and eight subtypes of cells in hair follicles (HFSC (Lgr5, Lhx2), IRS (Krt73, Shh, Krt27), cortex (Krt36, Krt27, Krt81), ORS (Fst, Sdc1, Krt17, Gata6), melanocytes (Mitf, Mlana), TAC (Shh, Wnt10b, Krt27), HFSC_activate (Lgr5, Tp63), and sebaceous glands (Thrsp, Acsbg1, Elovl5)) were identified. Among all fibroblast subtypes, the dermal papilla expresses the most unique marker genes (Enpp2, Crabp1, Hhip, Rspo3, and Gldn). Our data revealed that myofibroblasts expressed the following markers: Postn, Runx2, Sfrap2 and Tnn. Other fibroblasts were divided into FB_COL, which more highly expresses Col1a1 and Col1a2; FB_pro_inf, which more highly expresses some inflammation-related genes (Il6, Cxcl12, and Ccl2); FB_papi, which more highly expresses Enpp2, Crabp1, Cspg4 and Colec12; and FB_Tnc, which more highly expresses Tnc and other myofibroblast marker genes. We also detected 4 immune cell populations, namely, neutrophils (S100a8, S100a9, and Cxcr2), monocytes (Cd14, Ctss, Ptprc, and Cd68), dendritic cells (DCs) (Cd74, Irf8, Irf4, and Irf7) and innate lymphoid cells (Xcl1, Klrd1, Cpa3, and Cd69). The muscle cells consisted of two cell types: pericytes (Acta2, Tagln) and smooth muscle cells (Notch3, Rgs5). The endothelial cells were divided into 4 groups: lymphatic endothelial cells (Lyve1, Pdpn) and 3 types of vascular endothelial cells. Interestingly, we also detected a neural cell type (Scn7a, Plp1) (Fig. 3d). The chromatin accessibility of these genes is also shown (Fig. 3g). These cells constitute the microenvironment during the healing process of rat skin burn injuries.

Identification of major cell types through scATAC-seq analysis during the rat burnt skin healing process. (a) UMAP plot displaying the separation of six primary cell types on the basis of their chromatin accessibility profiles. Each cluster is labelled with its corresponding cell type. (b) Stacked bar graph showing the proportion of each cell type across all samples. (c) UMAP visualization of the cell type-specific gene activity score. (d) Heatmap of cell type-specific marker gene activity scores. (e) Genome browser view of the aggregated scATAC-seq chromatin accessibility profiles of housekeeping genes and cell type-specific gene loci.
By comparing the proportions of cell types at different healing time points, we determined that keratinocytes, including epithelial keratinocytes and hair follicle keratinocytes, were the most abundant in normal samples (Fig. 3b). The number of keratinocytes, especially epidermal keratinocytes, was significantly decreased after burn injury, indicating keratinocyte apoptosis. Moreover, the proportions of immune cells, such as neutrophils, monocytes, and innate lymphoid cells, increased significantly at 0 h, 12 h, and 24 h after burn injury, indicating immune infiltration in burn wounds (Fig. 3b).
Integrative analysis of public rat injured skin scRNA-seq datasets
To validate the reliability of our scATAC-seq data and reveal the deeper biological importance, we performed integrative analysis between our scATAC-seq dataset and publicly available scRNA-seq data of radiation-induced skin injuries (spanning 0, 7, 14, and 28 days post-injury). We reanalyzed and annotated the scRNA-seq data on the basis of the marker genes reported in the original publications. (Supplementary Fig. 1c). The integration of multi-omics data revealed a high degree of correlation between cell types in our scATAC-seq and public scRNA-seq data. (Supplementary Fig. 1a,b). Notably, our chromatin accessibility profiles enabled higher-resolution cellular clustering (Supplementary Fig. 1d), exemplified by the identification of six fibroblast subtypes, including the presence of myofibroblasts in the postrepair period of burned skin and distinct subpopulations of keratinocytes that constitute the epidermis and hair follicles, respectively. These findings underscore both the superior discriminative power of scATAC-seq in cell type delineation and the comprehensive nature of our dataset.
Dynamic changes in chromatin accessibility hinder the skin healing process
Given that our time series data comprehensively captured the early immune response and the vascularization process during the skin healing process, we aimed to describe the changes in gene accessibility and the key molecular mechanisms involved in these processes over time. Consequently, we extracted accessible peaks from monocytes and vascular endothelial cells and analysed the temporal trends of gene expression accessibility using the Mfuzz, which employs a fuzzy c-means clustering approach. In the analysis of monocytes, we identified three key peak clusters, Mono1 (n = 251), Mono2 (n = 180), and Mono3 (n = 321), which exhibited high accessibility at 12 hours, 12–24 hours, and 24 hours post-injury, respectively. In vascular endothelial cells, VE1 (n = 267), VE2 (n = 122), and VE3 (n = 271) were highly accessible at 12 hours, 24 hours, and 3–19 days post-injury, respectively (Fig. 4a).

Identification of temporal chromatin accessibility through the burnt skin healing process. (a) Six key peak clusters identified by Mfuzz: Mono1/2/3 (monocytes) and VE1/2/3 (vascular endothelial cells). (b) GO functional enrichment for peak clusters in monocytes and vascular endothelial cells.
We performed GO functional enrichment analysis on the nearest genes corresponding to these accessible peaks. The results revealed that the functions influenced by the accessible peaks in Mono1 are associated primarily with immune response activation, chemokine production, and monocyte migration. These findings suggest that at 12 hours post-injury, monocytes are in a state of immune activation and migrate towards the wound. The accessible peaks in Mono2 mainly affect functions related to interleukin-2 production, as well as the activation of B cells, T cells, and monocyte differentiation. This finding indicates that between 12 and 24 hours post-injury, monocytes begin to further differentiate into mature monocyte subtypes and release cytokines such as interleukin-2 to influence the activation of other lymphoid cells. The accessible peaks in Mono3 primarily influence the differentiation of leukocytes and myeloid cells, as well as the accessibility of genes related to chemotaxis and cell adhesion. They regulate the activity of immune cells and promote signalling interactions with other cells (Fig. 4b).
The accessible peaks in VE1 primarily influenced genes associated with hypoxia, as well as the regulation of leukocyte differentiation. These findings indicate that at 12 hours post-injury, vascular endothelial cells adapt to environmental changes and secrete cytokines that influence immune regulation. The peaks in VE2 mainly affected genes related to angiogenesis regulation and development, suggesting that by 24 hours post-injury, vascular endothelial cells have already opened up chromatin regions associated with vascularization to promote tissue repair. The peaks in VE3, in addition to influencing genes related to endothelial cell differentiation, predominantly affected genes associated with cell-matrix adhesion and the response to biotic stimuli. This finding suggests that from 3 to 19 days post-injury, vascular endothelial cells enter a state of endothelial differentiation and interact with the external environment through cell adhesion and lipopolysaccharides (Fig. 4b).
Cell type-specific peak calling and motif enrichment
For each cell type, we performed pseudobulk replicates and peak calling, ultimately obtaining a total of 748906 peak sets. We also identified cell type markers and implemented motif enrichment on these peaks to identify potential transcription factors (TFs) associated with these open chromatin regions. Interestingly, Kruppel family29,30 (Klf4, Klf7) and AP-2 family31,32 (Tcfap2a, Tcfap2b, and Tcfap2c) genes, such as IRS and IFE_Spinous, were enriched in keratinocytes. En133, Hoxc534 and Alx335 were specifically enriched in hair follicle keratinocytes, such as those in the cortex and TAC. In addition, the p5336 family (Trp53, Trp63, Trp73) was more highly enriched in epidermal keratinocytes. Dendritic cells were enriched for motifs of the Bcl1137,38 family (Bcl11a, Bcl11b) and the Runx37 family (Runx1, Runx2, Runx3). Sebaceous glands were enriched for the Ar39, Pgr and Nr3c1 motifs (Fig. 5a). To further confirm the dependability of these motifs, transcription factor (TF) footprinting was performed, which confirmed that the motif bound to the TFs. Indeed, the results were consistent with those of the motif enrichment analysis (Fig. 5b).

Transcription factor-binding site enrichment and motif activity profiling in the rat burnt skin healing process. (a) Heatmap depicting the enrichment of transcription factors (TFs) across various cell types. (b) Footprint profiles for selected TFs in different cell types.
In summary, the results provide high-quality data that we generated to profile the epigenetic process of burn wound healing.
Usage Notes
The pipeline of scATAC-seq data analysis, including read quality control, read mapping, read counting, low-quality cell removal, dimension reduction, unsupervised clustering, peak calling and motif enrichment, was run on the Linux operating system. All source R codes used for data analysis are provided online.
Code availability
All the codes for scATAC-seq downstream analysis in this paper were shared online. (https://figshare.com/s/f21a969a6e598f8d3551) and (https://github.com/BGI-TaoWang/Rat-burnt-skin-scATAC-seq-atlas).
References
Joost, S. et al. Single-Cell Transcriptomics of Traced Epidermal and Hair Follicle Stem Cells Reveals Rapid Adaptations during Wound Healing. Cell Rep 25, 585–597.e587 (2018).
Watt, F. M. Mammalian skin cell biology: at the interface between laboratory and clinic. Science 346, 937–940 (2014).
Oka, T., Ohta, K., Kanazawa, T. & Nakamura, K. Interaction between Macrophages and Fibroblasts during Wound Healing of Burn Injuries in Rats. Kurume Med J 62, 59–66 (2016).
Wilkinson, H. N. & Hardman, M. J. Wound healing: cellular mechanisms and pathological outcomes. Open Biol 10, 200223 (2020).
Waasdorp, M. et al. The Bigger Picture: Why Oral Mucosa Heals Better Than Skin. Biomolecules 11 (2021).
Lu, C. et al. Application of Single-Cell Assay for Transposase-Accessible Chromatin with High Throughput Sequencing in Plant Science: Advances, Technical Challenges, and Prospects. Int J Mol Sci 25 (2024).
Lin, L. & Zhang, L. Joint analysis of scATAC-seq datasets using epiConv. BMC Bioinformatics 23, 309 (2022).
Guerrero-Juarez, C. F. et al. Single-cell analysis reveals fibroblast heterogeneity and myeloid-derived adipocyte progenitors in murine skin wounds. Nat Commun 10, 650 (2019).
Mascharak, S. et al. Multi-omic analysis reveals divergent molecular events in scarring and regenerative wound healing. Cell Stem Cell 29, 315–327.e316 (2022).
Wasko, R. et al. Langerhans cells are essential components of the angiogenic niche during murine skin repair. Dev Cell 57, 2699–2713.e2695 (2022).
Pal, D. et al. Identification of a physiologic vasculogenic fibroblast state to achieve tissue repair. Nat Commun 14, 1129 (2023).
Correa-Gallegos, D. et al. CD201(+) fascia progenitors choreograph injury repair. Nature 623, 792–802 (2023).
Buenrostro, J. D. et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015).
Lake, B. B. et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol 36, 70–80 (2018).
Corces, M. R. et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14, 959–962 (2017).
Yu, Y. et al. Single-Nucleus Chromatin Accessibility Landscape Reveals Diversity in Regulatory Regions Across Distinct Adult Rat Cortex. Front Mol Neurosci 14, 651355 (2021).
Huang, J. et al. A reference human genome dataset of the BGISEQ-500 sequencer. Gigascience 6, 1–9 (2017).
Shi, Q., Liu, S., Kristiansen, K. & Liu, L. The FASTQ+ format and PISA. Bioinformatics 38, 4639–4642 (2022).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Lareau, C. A. et al. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat Biotechnol 37, 916–924 (2019).
Granja, J. M. et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat Genet 53, 403–411 (2021).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e3529 (2021).
Yan, T. et al. Single-cell RNA-Seq analysis of molecular changes during radiation-induced skin injury: the involvement of Nur77. Theranostics 14, 5809–5825 (2024).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008).
Kumar, L. & M, E. F. Mfuzz: a software package for soft clustering of microarray data. Bioinformation 2, 5–7 (2007).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb) 2, 100141 (2021).
CNGB Nucleotide Sequence Archive https://doi.org/10.26036/CNP0006144 (2023).
Wang, T. Single-cell chromatin landscapes of Rat burnt skin healing process. figshare https://doi.org/10.6084/m9.figshare.26934424 (2024).
Lyu, Y. et al. KLF5 governs sphingolipid metabolism and barrier function of the skin. Genes Dev 36, 822–842 (2022).
McConnell, B. B., Ghaleb, A. M., Nandan, M. O. & Yang, V. W. The diverse functions of Krüppel-like factors 4 and 5 in epithelial biology and pathobiology. Bioessays 29, 549–557 (2007).
Sahlén, P. et al. Chromatin interactions in differentiating keratinocytes reveal novel atopic dermatitis- and psoriasis-associated genes. J Allergy Clin Immunol 147, 1742–1752 (2021).
Smits, J. P. H. et al. The aryl hydrocarbon receptor regulates epidermal differentiation through transient activation of TFAP2A. bioRxiv (2023).
Aldea, D. et al. Differential modularity of the mammalian Engrailed 1 enhancer network directs sweat gland development. PLoS Genet 19, e1010614 (2023).
Awgulewitsch, A. Hox in hair growth and development. Naturwissenschaften 90, 193–211 (2003).
Mallarino, R. et al. Developmental mechanisms of stripe patterns in rodents. Nature 539, 518–523 (2016).
Viganò, M. A. et al. New p63 targets in keratinocytes identified by a genome-wide approach. Embo j 25, 5105–5116 (2006).
Mann-Nüttel, R. et al. The transcription factor reservoir and chromatin landscape in activated plasmacytoid dendritic cells. BMC Genom Data 22, 37 (2021).
Ippolito, G. C. et al. Dendritic cell fate is determined by BCL11A. Proc Natl Acad Sci USA 111, E998–1006 (2014).
Bläuer, M. et al. Location of androgen receptor in human skin. J Invest Dermatol 97, 264–268 (1991).
Acknowledgements
This project was supported by the CAMS Innovation Fund for Medical Sciences (2019-I2M-5-076), the Clinical Key Discipline Project of Shanghai, the Shanghai Top Priority Research Center Project (2023ZZ02013), the National Natural Science Foundation of China (81971836), the National Natural Science Foundation of China (81930057,81772076), the Achievements Supportive Fund (2018-CGPZ-B03), the Excellent Academic Leader Project of the Shanghai Science and Technology Committee (23XD1425000), the Deep Blue Talent Project of Naval Medical University, the 234 Academic Climbing Programme of Changhai Hospital and Achievements Supportive Fund (2018-CGPZ-B03), the Postdoctoral Fellowship Program of CPSF, and the Shanghai Rising-Star Program (Sailing Special Program) (No. 23YF1458400). We thank the China National GeneBank for offering the sequencing services for this project.
Author information
Authors and Affiliations
Contributions
T.W. and S.J. conceived the idea. J.L. collected the samples with the guidance of R.L. J.C. generated the data with the assistance of Y.Y. T.W. analysed the data. T.W. and B.Z. wrote the manuscript. B.Z. drew a flowchart. X.W. oversaw the study and revised the manuscript. A final manuscript review and approval were completed by all the authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, T., Zhu, B., Lu, J. et al. Single-cell chromatin landscapes associated with the burnt skin healing process in rats. Sci Data 12, 639 (2025). https://doi.org/10.1038/s41597-025-04928-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-04928-7
