
Abstract
As an important economic and ecological fish, Amur minnow (Phoxinus lagowskii) plays a significant role in food products as well as evolutionary, ecological research. However, a high-quality chromosome-level genome of P. lagowskii is not currently available. In this study, we report a T2T (Telomere-to-telomere) genome for P. lagowskii with chromosome-level. The finally assembled genome size is 1.04 G, with a contig N50 of 41.7 Mb, comprising 25 chromosomes. The transposable elements constituted 512.40 Mb (49.22%) of the assembled P. lagowskii genome, with DNA transposons 25.02% being the predominant repeat type. A total of 2,4610 protein-coding genes were predicted in P. lagowskii genome, with 99.96% of these genes being functionally annotated. The identification of telomeres, BUSCO assessment, mapping coverage, and sequencing depth collectively demonstrated the high quality of the genome assembly. The T2T genomic information serves as an invaluable resource for studies in evolution, comparative genomics, fish breeding applications, and ecological research.
Similar content being viewed by others


Background & Summary
The Amur minnow (Phoxinus lagowskii) is a commercially important cold-water fish species native to Northeast China, the Korean Peninsula, and Far Eastern Russia1,2,3. P. lagowskii belongs to the family Cyprinidae or Leuciscidae and the genus Phoxinus which owns high species diversity due to Its wide latitudinal distribution. This species could be confused with the Larcophyllum genus4 due to morphological similarities, the small size of individuals in certain species, and the overlapping of habitats5,6. It was previously known as Phoxinus lagowskii variegatus, Phoxinus lagowskii Dybowskii7. It is an omnivorous fish that feeds mainly on aquatic insects, molluscs, zooplankton, cladopods, and aquatic plants8,9. P. lagowskii primarily inhabits the upper reaches of river tributaries or the middle to upper layers of reservoirs and lakes10. They prefer to inhabit cool, flowing water and tend to live in groups in mountainous ditches and streams characterized by rapid, clear water with high dissolved oxygen and low temperature9,11,12. In China, this species is primarily distributed in the main stem and tributaries of the Heilongjiang River, as well as the Rehe River, Liaohe River, and the middle and upper reaches of the Yellow River, extending to the Yangtze River Basin13. It can serve as an indicator species for evaluating environmental changes. The aquaculture production of P. lagowskii, as a food source, has increased rapidly14,15.
As an economically and ecologically important species, P. lagowskii had been subject to artificial seedling breeding programs in various regions of China. However, investigations into P. lagowskii had been limited due to the lack of genomic resources, particularly a reference genome. Currently, only a limited number of studies have focused on the transcriptomic responses to low temperature stress and recovery1, cold stress response9,16, oxygen deficiency14 and chronic saline-alkaline stress10 without reference genome. To date, only a single Phoxinus genome has been sequenced. For genomic-level studies in the Phoxinus genus, the Eurasian minnow Phoxinus phoxinus (L.) genome, sized at 950.50 Mb with a Contig N50 of 2.0 Mb, had been reported17. The advantages and importance of T2T genome assembly lie in its ability to resolve gaps in previously inaccessible regions, such as telomeres, centromeres, and repetitive elements, thereby enhancing fish research18. Additionally, novel genes associated with centromere/telomeres regions can be identified, further enriching our understanding of genomic structures19. In this study, we present a completely T2T (Telomere-to-telomere) genome assembly for P. lagowskii, integrating Nanopore ultralong reads, PacBio HiFi long reads, DNBSeq short reads, and Hi-C data. The assembled T2T P. lagowskii genome had a size of approximately 1.04 Gb, with a contig N50 of 41.7 Mb and 48 telomeres. The assembled genome was gap-free, consisting of a single contig for each chromosome. A total number of 24,610 protein-coding genes were annotated in the P. lagowskii genome, among which 99.96% were functionally annotated. This T2T genome assembly and annotation provide an invaluable foundational resource for breeding applications, ecological studies, and comparative genomics and evolution.
Methods
Sample collection
The sample, an individual P. lagowskii fish (bogy length ~14.5 cm, weight ~35 g) of unknown gender, was collected on June 5, 2024, from Liaohe River (Taizi River), China (Fig. 1a). Due to P. lagowskii lacks secondary sexual characteristics. The fish was captured during the non-breeding period, making it impossible to determine the sex information. The live individual was promptly transported to the laboratory at the Freshwater Fisheries Center of the Chinese Academy of Fishery Sciences in Wuxi for subsequent sequencing. The muscle tissue samples were harvested and rapidly cryopreserved in liquid nitrogen, then stored at −80 °C for subsequent extraction of genomic DNA and Hi-C analysis. High molecular weight (HMW) nuclear genomic DNA of P. lagowskii was extracted using the QIAGEN Blood & Cell Culture DNA Midi Kit (QIAGEN, Germany). The quality of the extracted DNA was assessed through measurements of DNA concentration, OD260/280 ratio, OD260/230 ratio, and DNA integrity. Five muscle tissue samples were utilized for RNA extraction using the TRIZOL Reagent (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer’s protocol.

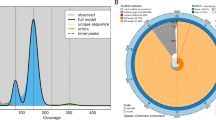
The characteristics of P. lagowskii. (a)The picture of the P. lagowskii. (b) The genomescope plot of the P. lagowskii genome survey. (c) The circos plot of the P. lagowskii T2T genome. a: gene density in 1-Mb sliding windows; b: GC content in non-overlapping 1 Mb windows; c: percentage of tandem repeats in 1 Mb sliding windows; d: percentage of interspersed repeats in 1 Mb sliding windows; e: the heatmap of telomeric locations; f: the position of centromeres; g: the length of chromosomes in the size of Mb.
Short-read sequencing and genome survey
The genome size of P. lagowskii was estimated through short-read sequencing prior to long-read sequencing. The library with a short insert size (350–500 bp) was prepared according to the MGI Library Prep Reagents protocol. A total of 157.83 Gb raw data were generated using the PE 150 sequencing strategy on the DNBSEQ-T7 platform. The adapter and low-quality reads were removed and filtered using Fastp v0.20.0 pipeline with the parameter (--average_qual 15 -l 150)20. The high-quality clean short-read data totaling 148.16 Gb were obtained, with a Q20 value of 98.53% and a Q30 value of 95.11%. A 17-mer was utilized to determine the k-mer frequency, and the distribution profile was generated using Jellyfish v2.2.621. The genome size was estimated to be 989.83 Mb with formula: Genome size = K-mer/Peak depth (131,647,395,869/133). The genomic characteristics of P. lagowskii, including an estimated genome size (~966.57 Mb), heterozygosity (0.949%), and repeat content (60.4%), were determined using Genomescope 1.022 (Fig. 1b).
Long-read sequencing and Hi-C sequencing
With the estimated genome size of approximately 1 Gb, the PacBio and Nanopore long-read sequencing were employed for genome assembly. A PacBio library (insert size ~20 kb) was constructed for P. lagowskii following the SMRTbell Express Template Prep Kit 2.0 protocol (Pacific Biosciences, USA). A total number of 1,520,742 highly accurate long reads were sequenced using the PacBio Revio SMRT cell with HiFi model. A total of 27.18 Gb HiFi long reads, with an N50 length of 18,182 bp, were generated for the genome assembly (Table 1). An ultra-long nanopore library was constructed following the Ligation sequencing 1D kit (SQK-LSK109, Oxford Nanopore Technologies, Oxford, UK) protocol. A single Nanopore cell was sequenced on the PromethION platform (Oxford Nanopore Technologies), yielding 4,826,740 reads and generating 154.91 Gb of raw data. The longest reads and N50 length of ONT data were 538,660 bp and 49,841 bp, respectively. After excluding the reads which below 50 kb, a total of 1,089,739 reads (76.97 Gb) of clean data were used for genome assembly (Table 1). The average read length and N50 length, excluding reads shorter than 50 kb, were 70,630 bp and 69,302 bp, respectively. The Hi-C data can be useful for anchoring the contigs to chromosomes. In this study, approximately 1.5 g of muscle tissue from P. lagowskii was fixed in 1% formaldehyde at room temperature for a duration of 10 to 30 minutes. The cross-linked DNA of P. lagowskii was digested using MboI (New England Biolabs, Ipswich, USA). The resulting cohesive ends were labeled with biotin through incubation with biotin-14-dATP and Klenow fragment. Following the completion of proximity ligation, crosslinking reversal, and DNA purification, the Hi-C products were subsequently enriched and utilized for library construction. Then, a total of 147.2 Gb of Hi-C raw data were sequenced with DNBSEQ-T7 platform. The raw reads were processed using Fastp v0.20.0 pipeline with the parameter (--average_qual 15 -l 150)20. A total of 146.2 Gb of clean data, with a Q20 value of 97.46%, was utilized to anchor the contigs to chromosomes (Table 1).
Genome assembly
To obtain the T2T genome assembly of P. lagowskii, PacBio HiFi reads were initially employed to assemble the primary contigs using Hifiasm (v 0.19.6) with default parameters23. The redundant sequences were subsequently eliminated using the purge_dups v1.2.6 program with parameters “-a 70 -b 500 −l 10000 -m 300 -f 0.8”24. The assembled genome size was approximately 1.16 Gb, with a contig N50 value of 7.28 Mb with Hifiasm. Then, the assembled P. lagowskii contigs were anchored to chromosomes using Hi-C data. The total of 146.2 Gb Hi-C data were aligned to the assembled contigs using BWA v 0.7.1225. After eliminating duplicate and erroneous mapping reads, the valid pairs reflecting contact interactions were retained. The inter-/intra-chromosomal interactions were calculated. The Hi-C contacts were processed using juicer pipeline v1.526. The assembled contigs were clustered, sorted and oriented into groups/chromosomes utilizing the 3D-DNA pipeline, version 18011426. The manual verification was involved and refined using JUICEBOX Assembly Tools v2.15.0727. The assembled contigs of P. lagowskii were finally anchored and oriented onto 25 chromosomes (Fig. 1c, 2), resulting in a total length of 1.05 Gb with scaffold N50 of 41.6 Mb. The maximum scaffold and contig size were 60.46 Mb and 28.83 Mb, respectively (Table 2). A total of 511 gaps remained across 25 chromosomes. To address this, ultra-long reads were employed for gap closing, and NECAT v 20200119 was utilized with default parameters for error correction28. The assembled chromosome-level genome of P. lagowskii was used as a reference to close those gaps using consensus ONT ultra-long reads, ultimately yielding a completely gap-free genome. The gap-free P. lagowskii genome size was 1.04 Gb and the contig/scaffold N50 was 41.7 Mb, with each chromosome represented by a single contig (Tables 2, 3).

Hi-C intra-chromosomal interaction map in P. lagowskii with 500 Kb.
Genome annotation
The assembled P. lagowskii T2T genome was annotated by identifying repetitive elements and protein-coding genes. For repeat identification, the homolog and Ab initio annotation were integrated to annotate the repeat sequences. The homologous sequences of P. lagowskii genome were detected and classified using RepeatProteinMask v4.0.729 and RepeatMasker v4.0.730 based on the Repbase library31. For Ab initio annotation, the de novo repetitive element database was constructed using RepeatModeler v1.0.432 and LTRharvest33. The Tandem repeats and long terminal repeats were identified using Tandem Repeat Finder v4.10.034 and LTR finder v1.0.735, respectively. Finally, a total of 49.22% of assembled P. lagowskii genome was classified as repetitive elements (Table 4). The proportions of DNA, long interspersed nuclear elements (LINEs), long terminal repeats (LTRs) and among the repeat sequences were 25.02%, 14.53% and 18.94%, respectively, while short interspersed nuclear elements (SINEs) accounted for only 0.92% of the P. lagowskii genome (Table 4). The annotation of protein-coding genes was performed using three strategies including RNA-Seq assisted annotation, de novo prediction and homology-based prediction. The transcriptome data were sequenced form five different tissues of P. lagowskii. A total of 32.14 Gb of clean data was generated, with individual data ranging from 5.68 Gb to 6.62 Gb. The transcriptome data were assembled using Trinity (v2.15.1)36 with parameters “--min_contig_length 200 --min_kmer_cov 4 --min_glue 4 --bfly_opts ‘-V 5 --edge-thr = 0.1 --stderr’ --genome_guided_max_intron 10000”. The transcripts were generated and coding region were predicted using PASA (v2.4.1)37 and TransDecoder v5.5.0 (https://github.com/TransDecoder/TransDecoder), respectively. The sequenced transcriptome data were mapped onto the P. lagowskii genome using Hisat2 v2.1.038 with parameters “--sensitive --no-discordant --no-mixed -I 1 -X 1000 --max-intronlen 1000000”. The mapping rate of the five transcriptomes ranged from 84.86% to 88.5%. Then, the mapped reads of five samples were assembled using Stringtie v1.3.539 with parameters (-j 3 -c 5 -f 0.3 -g 100 -s 10000). The coding sequence was predicted using TransDecoder v5.5.0 (https://github.com/TransDecoder/TransDecoder) with default parameters. For de novo prediction, the gene models of the P. lagowskii genome were predicted using Augustus v3.2.140 with default parameters. For homology-based prediction, protein sequences of six representative fish species, namely Danio rerio (GCF_000002035.6), Carassius auratus (GCF_003368295.1), Carassius carassius (GCF_963082965.1), Cyprinus carpio (GCF_018340385.1), Megalobrama amblycephala (GCF_018812025.1) and Rhinichthys klamathensis (GCF_029890125.1) were retrieved from NCBI (National Center for Biotechnology Information). Combined these three strategies results, coding structures were annotated using GeMoMa version 1.941, and PASA (v2.4.1)37 was employed for the refined the gene structure. Finally, a total of 24,610 genes were obtained for P. lagowskii genome (Table 5). The average gene length, intron length, and CDS length of the annotated P. lagowskii genome were 17,934 bp, 1,785 bp, and 1,700 bp, respectively (Table 5).
Functional annotation
The gene set of 24,610 genes were functionally annotated using diamond v0.8.23 with an E-value threshold of 1E-5 based on the five databases including NR (NCBI nonredundant protein), TrEMB (http://www.uniprot.org), KOG42, KEEG (Kyoto Encyclopedia of Genes and Genomes, http://www.genome.jp/kegg/), Swiss-Prot (http://www.gpmaw.com/html/swiss-prot.html). The protein motifs and domains were identified using the InterProScan with InterPro 93.043. The GO Ontology (GO) was classified from the results of InterProScan44. The annotation of 24,599 predicted genes (99.96%) out of the total 24,610 genes can be found by at least one database (Fig. 3b and Table 6). Of these functional proteins, 19,367 genes (~78.7%) were supported by five databases (Fig. 3a).

Functional annotation of P. lagowskii protein-coding genes. (a) Venn diagram of functional annotation of the P. lagowskii genome. (b) The functional annotation statistics in seven databases.
Data Records
The sequencing data of P. lagowskii have been deposited in the NCBI SRA database under BioProject accession PRJNA123158145, including DNA short-read survey data (SRA: SRR32570940), DNA short-read Hi-C data (SRA: SRR32570943), DNA long-read Nanopore data (SRA: SRR32570942), DNA long-read PacBio HiFi data (SRA: SRR32570941), and RNA short-read data (SRR32570944–SRR32570948).
The Whole Genome Shotgun project has been deposited at GenBank under accession JBNYWR00000000046.
The files of genome assembly and gene structure annotation have been deposited at Figshare47.
Technical Validation
Evaluation of the P. lagowskii genome quality and gene set
To evaluate the quality of the P. lagowskii genome, the short-reads were mapped to the T2T assembled reference genome with a mapping rate of 99.67% using BWA v 0.7.1225. Additionally, the HiFi long-reads and Nanopore long-reads were also mapped to the P. lagowskii reference genome using Minimap248 with the mapping rate of 100% and 99.99%, respectively. The mapped coverage of HiFi reads was 99.39%. The analysis of sequencing depth and GC content based on HiFi reads revealed that the GC values were tightly clustered around 40% with a pronounced peak, suggesting minimal contamination (Fig. 4a). The BUSCO evaluation of P. lagowskii genome demonstrated that 97.1% of the BUSCOs were classified as Complete, with 95.5% being single-copy and complete BUSCOs, while 1.6% were identified as duplicated and complete BUSCOs (Fig. 4b). The BUSCO evaluation of the gene set demonstrated that 97.45% of the BUSCOs were classified as Complete, with 95.99% being single-copy and complete BUSCOs, while 1.46% were identified as duplicated and complete BUSCOs (Table 7). As T2T genome assembly in the genus of Phoxinus, both the genome and gene set quality are higher than that previously published Eurasian minnow (Phoxinus Phoxinus) genome (Table 7). The accuracy of the P. lagowskii genome assembly was assessed via Merqury using 21-mer based on HiFi reads, resulting in a quality value (QV) score of 32.58, and derived 99.95% accuracy. To confirm the T2T genome quality, telomeres were identified using quarTeT pipeline49 to search for telomere repeat sequences and centromere regions in P. lagowskii genome. A total of 48 telomeres were assembled and identified for P. lagowskii genome, except one end of chromosome 16 and 23, suggesting the new genome was assembled nearly telomere-to-telomere (Table 8). The maximum total number of telomeres repeats at the left and right ends were 825 and 905, respectively (Table 8). The newly T2T P. lagowskii genome were compared with the previous published Phoxinus Phoxinus genome using MUMmer4 with nucmer model (-c 100 -l 20 and delta-filter -l 20000). The mummer analysis of genome revealed one chromosome to one chromosome clearly and high homology (>80%), and suggested chromosome anchoring was accurate (Fig. 5). Furthermore, gene synteny analysis was conducted by comparing with P. phoxinus. The results revealed highly conserved gene synteny, demonstrating a one-to-one correspondence that confirmed the accuracy of chromosome anchoring (Fig. 6).

Genome evaluation of P. lagowskii. (a) GC content and depth distribution of HiFi reads in P. lagowskii genome. (a) BUSCO assessment of P. lagowskii and P. Phoxinus genomes.

The sequence comparison between P. lagowskii and P. Phoxinus genomes.

The synteny comparison of P. lagowskii to Phoxinus Phoxinus using protein-coding gene synteny.
Code availability
No specific software or code was developed for this study. The experiments and bioinformatics were performed accordance with the protocols detailed in the Methods section.
References
Sun, M. et al. Transcriptome, histology, and enzyme activities analysis of liver in Phoxinus lagowskii to the low temperature stress and recovery. Comparative Biochemistry and Physiology Part D: Genomics and Proteomics 52, 101317, https://doi.org/10.1016/j.cbd.2024.101317 (2024).
Xue, Z. et al. Effects of habitat fragmentation on the population genetic diversity of the Amur minnow (Phoxinus lagowskii). Mitochondrial DNA. Part B, Resources 2, 331–336, https://doi.org/10.1080/23802359.2017.1331319 (2017).
Yu, T. et al. Effect of dietary L‐carnitine supplementation on growth performance and lipid metabolism in Rhynchocypris lagowski Dybowski fed oxidized fish oil. Aquaculture Research 51, 3698–3710 (2020).
Fujita, T. & Hosoya, K. Biochemical and morphological comparison between two Japanese daces, phoxinus lagowskii steindachneri and P. oxycephalus jouyi in the sympatric sites. Japanese Journal of Ichthyology 50, 55–62 (2003).
Sakai, H. et al. Phylogenetic and taxonomic relationships of northern Far Eastern phoxinin minnows, Phoxinus and Rhynchocypris (Pisces, Cyprinidae), as inferred from allozyme and mitochondrial 16S rRNA sequence analyses. Zoological science 23, 323–331, https://doi.org/10.2108/zsj.23.323 (2006).
Chen, T., Jiao, L. & Ni, L. The phylogeographical pattern of the Amur minnow Rhynchocypris lagowskii (Cypriniformes: Cyprinidae) in the Qinling Mountains. Ecology and Evolution 12, e8924, https://doi.org/10.1002/ece3.8924 (2022).
Ergens, R. Gyrodactylus konovalovi sp. n. (Gyrodactylidae: Monogenoidea) from Phoxinus lagowskii Dybowski. Folia parasitologica 23, 87–89 (1976).
Gou, N., Ji, T., Zhang, J., Bian, K. & Wang, K. Population Characteristics and Seasonal Variation of Brachymystax lenok tsinlingensis’s Main Bait Organism Lagowskiella lagowskii in National Nature Reserves of Rare Aquatic Wildlife of Heihe River. Northwest Journal of Agricultural Sciences 27, 7 (2018).
Chen, Y. et al. Histology, fatty acid composition, antioxidant and glycolipid metabolism, and transcriptome analyses of the acute cold stress response in Phoxinus lagowskii. Comparative Biochemistry and Physiology Part D: Genomics and Proteomics 50, 101242, https://doi.org/10.1016/j.cbd.2024.101242 (2024).
Zhou, H. et al. Effects of chronic saline-alkaline stress on gill, liver and intestinal histology, biochemical, and immune indexes in Amur minnow (Phoxinus lagowskii). Aquaculture 579, 740153, https://doi.org/10.1016/j.aquaculture.2023.740153 (2024).
Ding, F. & Mao, Z. Observation and Analysis of Water Temperature in Ice-Covered Shallow Lake: Case Study in Qinghuahu Lake. Water 13, 3139 (2021).
Yang, Y. et al. Histopathological, hematological, and biochemical changes in high-latitude fish Phoxinus lagowskii exposed to hypoxia. Fish Physiology and Biochemistry 47, 919–938, https://doi.org/10.1007/s10695-021-00947-4 (2021).
Kang, X. et al. Assessment of Habitat Suitability of Juvenile Phoxinus lagowskii in Taizi River. Journal of Ecotoxicology 6, 310–320 (2011).
Wang, J. et al. Comparison of effects in sustained and diel-cycling hypoxia on hypoxia tolerance, histology, physiology and expression of clock genes in high latitude fish Phoxinus lagowskii. Comparative Biochemistry and Physiology Part A: Molecular & Integrative Physiology 260, 111020 (2021).
Hu, T., Ye, C., Ning, Z., Liu, T. & Mu, W. Effect of Toxicity of Chromium (VI) Stressors Alone and Combined to High Temperature on the Histopathological, Antioxidation, Immunity, and Energy Metabolism in Fish Phoxinus lagowskii. Fishes 9, 168 (2024).
Zhang, T. et al. Identification, characterization, and function of GRP94 and HSP90β in cold stress response in cold water fish Phoxinus lagowskii. Aquaculture Reports 28, 101443 (2023).
Nunn, A. D., Moccetti, P. & Hänfling, B. The genome sequence of the Eurasian minnow, Phoxinus phoxinus (Linnaeus, 1758). Wellcome open research 9, 504, https://doi.org/10.12688/wellcomeopenres.22867.1 (2024).
Zhou, Y. et al. Telomere-to-telomere genome and resequencing of 254 individuals reveal evolution, genomic footprints in Asian icefish, Protosalanx chinensis. GigaScience 14, https://doi.org/10.1093/gigascience/giae115 (2025).
Yin, D. et al. Telomere-to-telomere gap-free genome assembly of the endangered Yangtze finless porpoise and East Asian finless porpoise. GigaScience 13, https://doi.org/10.1093/gigascience/giae067 (2024).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204, https://doi.org/10.1093/bioinformatics/btx153 (2017).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898, https://doi.org/10.1093/bioinformatics/btaa025 (2020).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell systems 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Chen, Y. et al. Efficient assembly of nanopore reads via highly accurate and intact error correction. Nature Communications 12, 60, https://doi.org/10.1038/s41467-020-20236-7 (2021).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11, https://doi.org/10.1186/s13100-015-0041-9 (2015).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl 1), i351–358, https://doi.org/10.1093/bioinformatics/bti1018 (2005).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenetic and genome research 110, 462–467, https://doi.org/10.1159/000084979 (2005).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics Chapter 4, 4.10.11–14.10.14, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18, https://doi.org/10.1186/1471-2105-9-18 (2008).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 35, W265–268, https://doi.org/10.1093/nar/gkm286 (2007).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature biotechnology 29, 644–652, https://doi.org/10.1038/nbt.1883 (2011).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 31, 5654–5666, https://doi.org/10.1093/nar/gkg770 (2003).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature biotechnology 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol 20, 278, https://doi.org/10.1186/s13059-019-1910-1 (2019).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644, https://doi.org/10.1093/bioinformatics/btn013 (2008).
Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. Methods in molecular biology (Clifton, N.J.) 1962, 161–177, https://doi.org/10.1007/978-1-4939-9173-0_9 (2019).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59, https://doi.org/10.1186/1471-2105-5-59 (2004).
Mulder, N. & Apweiler, R. InterPro and InterProScan: tools for protein sequence classification and comparison. Methods in molecular biology (Clifton, N.J.) 396, 59–70, https://doi.org/10.1007/978-1-59745-515-2_5 (2007).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29, https://doi.org/10.1038/75556 (2000).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP567941 (2025).
Zhou, Y. whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JBNYWR000000000.1 (2025).
Zhou, Y. Telomere-to-telomere genome assembly of Phoxinus lagowskii. figshare. Dataset. https://doi.org/10.6084/m9.figshare.28537136.v1 (2025).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Lin, Y. et al. quarTeT: a telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Hortic Res 10, uhad127, https://doi.org/10.1093/hr/uhad127 (2023).
Acknowledgements
This work was financially supported by the earmarked fund for the National Key Research and Development Program of China (NO. 2023YFD2400900), the Central Public-interest Scientific Institution Basal Research Fund, CAFS(NO.2023TD65).
Author information
Authors and Affiliations
Contributions
D. Xu, J. Jian designed and conceived the study. Y. Zhou, D. Fang, C. Wang, M. Zhang, Y. You, Y. Liu and G. Deng collected the samples, conducted experiments, J. Jian, Y. Zhou, C. Chen, Y. Peng and B. Wang performed bioinformatics analysis. Y. Zhou, J. Jian and D. Xu wrote and revised the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, Y., Chen, C., Fang, D. et al. Telomere-to-telomere genome assembly of Phoxinus lagowskii. Sci Data 12, 1025 (2025). https://doi.org/10.1038/s41597-025-05367-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-05367-0
