
Abstract
Fruit size is an important quality trait in different market classes of Cucumis sativus L., an economically important vegetable cultivated worldwide, but the genetic and molecular mechanisms that control fruit size are largely unknown. In this study, we isolated a natural cucumber mutant, short fruit 1 (sf1), caused by a single recessive Mendelian factor, from the North China-type inbred line CNS2. In addition to significantly decreased fruit length, other fruit-related phenotypic variations were also observed in sf1 compared to the wild-type (WT) phenotype, indicating that sf1 might have pleiotropic effects. Microscopic imaging showed that fruit cell size in sf1 was much larger than that in WT, suggesting that the short fruit phenotype in sf1 is caused by decreased cell number. Fine mapping revealed that sf1 was localized to a 174.3 kb region on chromosome 6. Similarly, SNP association analysis of bulked segregant RNA-Seq data showed increased SNP frequency in the same region of chromosome 6. In addition, transcriptomic analysis revealed that sf1 might control fruit length through the fine-tuning of cytokinin and auxin signalling, gibberellin biosynthesis and signal transduction in cucumber fruits. Overall, our results provide important information for further study of fruit length and other fruit-related features in cucumber.
Similar content being viewed by others

Introduction
Cucumber (Cucumis sativus L., 2n = 14), a member of the family Cucurbitaceae, is one of the most economically important vegetable crops cultivated throughout the world. As one of the most significant fruit-related traits, fruit length is a key consideration in cucumber breeding. Previous studies have demonstrated that fruit length is quantitatively inherited, and several quantitative trait loci (QTLs) have been identified and analysed. Wenzel et al. were the first to investigate the QTLs that affect fruit quality traits in F2:3 families from a cross between the inbred lines Gyl4 and PI432860; in their studies, three QTLs were identified and shown to be responsible for cucumber fruit length1. Subsequently, two QTLs associated with fruit length were identified by Serquen et al. in F2:3 populations developed from G421 × H-19 cucumber inbred lines2. By analysing recombinant inbred lines (RILs) from the same cross between G421 and H-19, Fazio et al. identified twelve QTLs that contribute to cucumber fruit length3. Yuan et al.4 constructed F2 and F2:3 families from a narrow cross between inbred lines S94 and S06 and identified one QTL for fruit length in the F2 population and six QTLs in F2:3 families, three of which affected fruit length in autumn and three of which affected fruit length in spring. Additionally, five QTLs were identified and shown to be responsible for mature fruit length in four environments using an RIL population developed from CC3 × SWCC8 cucumber inbred lines5. With the aid of QTL models, 12 consensus QTLs were also shown to determine cucumber fruit length6. In addition, transcriptomic analysis revealed that cucumber fruit length might be regulated by microtubules, CDKs-cyclins, and expansin-mediated cell division and cell expansion and that some transcription factors might function as key upstream players7.
Fruit growth and development are closely associated with cell division and expansion, and cell division and expansion can be modulated by plant hormones such as cytokinins, gibberellins and auxin. For example, it has been reported that cytokinin concentration and the expression of genes associated with cytokinin biosynthesis are maintained at high levels during early tomato fruit development, and different temporal patterns of expression of different classes of cytokinins and genes associated with their biosynthetic pathways have been observed, providing evidence for a significant role of cytokinin signalling in the cell division phase of tomato fruit development8. A positive relationship between endogenous cytokinin level and cucumber fruit size has also been demonstrated9. Gibberellins control both cell proliferation and expansion rates, processes that rely on the destruction of DELLAs10. Significant roles for gibberellin biosynthesis and signalling in both cell division and cell expansion have also been observed during fruit growth and development11, 12. In general, the results of studies showing that auxin functions significantly in plant cell division and expansion are well accepted13, whereas evidence that the coordination of plant hormones is involved in the regulation of fruit length is still ambiguous.
In this study, we obtained a spontaneous mutant, short fruit 1 (sf1), from a cucumber inbred line CNS2 with long fruits (wild type, WT) and characterized the sf1 mutant through comprehensive methodologies, including genetics, physiology, histology and comparative transcriptomics. Based on the results, we proposed a new mechanism by which sf1 might control fruit length in cucumber by fine-tuning cytokinin and auxin signalling and by regulating gibberellin biosynthesis and signal transduction.
Results
Isolation and phenotypic analysis of the sf1 mutant
We isolated a natural mutant, sf1, with short fruits from the cucumber inbred line CNS2, a North China type with long fruits (Fig. 1A). The fruit length of sf1 was approximately half that of WT at the commodity maturity stage (Fig. 1A), but plant height, stem diameter, leaf area and the first female and male flower nodes of this mutant line were similar to those of the WT (Supplementary Figure 1).

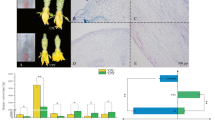
Phenotypes of WT and sf1. (A) Fruit of WT, F1 and sf1 at 11 days after anthesis (DAA). (B) Fruit cracking in sf1 at 20 DAA. (C,D) Distribution of bloom trichomes on the pericarp of WT and sf1 at 9 DAA. (E,F) Fruit spines and tubercules of WT and sf1 at 11 DAA. (G,H) Seed cavities of WT and sf1 fruits at 9 DAA. (I,J) Stigmas from WT and sf1 at 0 DAA. Fruit hardness (K) and crispness (L) in WT and sf1 at 0, 9 and 23 DAA. Acid-soluble lignin content (M) in WT and sf1 cucumbers at 9 DAA. Scale bars represent 2 cm in (A),(B),(G) and (H), 2 mm in (E),(F),(I) and (J), and 500 μm in (C) and (D). ‘**’and ‘*’ indicate significant differences from WT at the 0.01 and 0.05 probability levels, respectively. Vertical bars represent standard deviation (n = 3).
In addition to decreased fruit length, many other fruit-related traits and stigma size were affected in sf1 (Fig. 1). The fruits of sf1 had a light green peel, no tubercle and cracked easily under moist conditions, whereas the WT fruits had a dark green peel and tubercles and did not crack at all (Fig. 1A–D,K and L). Significantly fewer bloom trichomes on the fruit surface resulted in more glossy fruits in sf1 than in WT (Fig. 1A,C and D). There were also differences in the fruit spines of sf1 and WT (Fig. 1E and F). A cucumber fruit spine has an upper portion with a single-cell arrangement and a warty base (Fig. 1E and F). The height of the upper portion of the sf1 spine was 43% greater than that of WT due to an increase in the cell number in this portion of the spine, but the height of the spine base was 63% that of WT; as a result, the height of the whole spine of sf1 was 15% greater than that of WT (Supplementary Figure 2A–D). We also found that the diameters of the stigma and seed cavity in sf1 were 28% and 30% greater, respectively, than those in WT (Supplementary Figure 2E and G). Accordingly, the ratio of seed cavity diameter to fruit diameter increased by 53% in sf1 relative to that in WT (Supplementary Figure 2F).
Higher chlorophyll and carotenoid content was observed in the pericarp, but more carotenoids were present in the sarcocarp of sf1 than of WT (Supplementary Figure 2H and I). The cell wall is a protective structure of plant cells and is primarily composed of cellulose, hemicellulose and lignin14. Fruit hardness and crispness, two important quality parameters in cucumber, are closely related to the composition of the cell wall15,16,17. Fruit hardness refers to the capability of fruit to resist the penetration of outside objects into it. Fruit crispness refers to the capability of fruit to resist cracking when an outside force is exerted on it. In this study, sf1 fruits displayed increased hardness and crispness (Fig. 1K and L), indicating possible involvement of sf1 in the regulation of cell wall components. To investigate this hypothesis, we analysed the content of cell wall components in sf1 and WT fruits at 9 days after anthesis (DAA), when obvious cracking of sf1 fruits began to occur. Strikingly increased lignin content was observed in the sf1 pericarp compared to that in the WT pericarp (Fig. 1M).
These significantly influenced traits of sf1 plants were closely associated with the short-fruit phenotype, and no segregation was observed in the progenies, indicating that sf1 might have pleiotropic effects on fruit-related traits.
Growth and histological features of sf1 fruit
Kinematic analysis of fruit growth revealed that sf1 fruits display a growth trend similar to that of WT (Fig. 2A) but undergo much slower growth throughout the entire growing phase (Fig. 2B). As a result, the difference in fruit length between sf1 and WT continuously increased; at the end of the investigation, the fruit length of sf1 was only approximately half of the WT fruit length (Fig. 1A).

Kinematic analysis of fruit growth and pericarp cell size and number in WT and sf1. Fruit length (A) and daily increase in fruit length (B) were determined in WT and sf1 from 0 to 22 DAA. Pericarp cell size of WT (C–E) and sf1 (F–H) at 0 (C and F), 9 (D and G) and 23 DAA (E and H). Cell number per square millimetre (I) of WT and sf1 pericarp was measured at three time points. ‘**’ indicates significant difference from WT at the 0.01 probability level. Scale bars represent 50 μm in (C) and 10 μm in (D–H). Vertical bars represent standard deviation (n = 18 for A and B, and n = 6–70 for I).
To illuminate the cellular mechanism underlying the short fruit of sf1, we measured the cell number and cell size in the pericarp of WT and sf1 fruits at 0, 9 and 23 DAA. We found significantly larger cells and fewer cells per square millimetre in sf1 than in WT (Fig. 2C–I). Similar differences were observed in the sarcocarp of WT and sf1 fruits (Supplementary Figure 3). These results indicate that the decrease in fruit length associated with sf1 is due to a decrease in cell number.
Fine mapping of sf1
To genetically map the sf1 locus, an F2 population was first constructed by crossing sf1 with ZG, a European greenhouse-type inbred line with short, light green, non-tuberculate fruits. Thirty-five polymorphic simple sequence repeat (SSR) markers were used in 24 F2 plants for rough mapping. The sf1 locus was localized to a 4,456 kb region between SSR16451 and SSR22801 on chromosome 6 (Fig. 3). Because of the similarity in fruit size between sf1 and ZG, it was difficult to distinguish mutant-phenotype plants from ZG plants in the F2 population. Therefore, we developed another F2 population from a cross between sf1 and ‘Chinese long’ 9930 that has fruits similar to those of the WT parental line of sf1. All F1 progeny showed a long-fruit phenotype similar to that of WT. In the F2 population, long- and short-fruit plants segregated in a 3:1 ratio (79 long vs. 19 short; x 2 = 1.65 < x 2 0.05 = 3.84; p > 0.05), demonstrating that the short-fruit phenotype of the sf1 mutant is controlled by a single recessive nuclear gene. Two markers, STS1 (sequence-tagged site 1) and SSR21886, were then used to screen the F2 population derived from the cross between sf1 and ‘Chinese long’ 9930, and 33 recombinant individuals were selected from 6,720 F2 plants (Supplementary Table 1). These recombinant individuals were transplanted into a solar greenhouse for fine mapping of sf1 using three additional markers (SNP1, SNP2 and SNP3). The sf1 locus was finally localized to an approximately 174.3 kb region (from 11,584,292 to 11,758,559) between markers SNP1 and SNP2 (Fig. 3). In this region, 15 annotated genes (Csa6G176930, Csa6G176940, Csa6G177440, Csa6G178440, Csa6G178940, Csa6G178950, Csa6G179450, Csa6G179460, Csa6G179470, Csa6G179480, Csa6G180980, Csa6G180990, Csa6G181000, Csa6G181500 and Csa6G181510) were identified. The total genomic size of these 15 annotated genes was only 37.4 kb, and the remaining 136.9 kb was intergenic. The 15 annotated genes together with most of the non-coding region in the 174.3 kb region were analysed by sequencing, but no variation between sf1 and WT (CNS2) was detected. Some partial segments (approximately 5.8 kb) in the 174.3 kb region were not sequenced because they could not be successfully amplified by PCR. We further detected the expression levels of these 15 annotated genes by semi-quantitative RT-PCR, and 12 of the 15 genes displayed no difference in expression level compared with that in WT (Supplementary Figure 4). The expression of the remaining three genes (Csa6G180980, Csa6G180990 and Csa6G181500) was not detected, possibly due to low or no expression. This evidence indicates that there might be an unannotated gene or unknown regulatory element in this region. At the same time, we also identified two STS markers (STS2 and STS3) closely associated with the sf1 locus (Fig. 3) that can be used in future cucumber breeding.

Mapping of the sf1 gene. The sf1 gene was fine-mapped to a 174.3 kb region between markers SNP1 and SNP2 on chromosome 6 in an F2 population containing 6,720 individuals from a cross between sf1 and ‘Chinese long’ 9930.
Gene expression analysis in sf1 fruits at different developmental stages
To investigate the molecular basis underlying the changes in fruit traits in sf1, comparative transcriptomic analysis of cucumber fruits was performed at three time points representing key developmental stages of cucumber fruit [cell proliferation (6 DBA), fruits sampled from sf1 and WT; transition from slow to fast growth of cucumber fruit (2 DBA) and fastest growth (9 DAA), fruits sampled from the F2 population of a cross between sf1 × ‘Chinese long’ 9930],which was based on our preliminary investigation and a previous study18. High-throughput RNA sequencing (RNA-Seq) produced 51, 167 and 128 million reads at these time points, respectively, for the mutant pools and 50, 117 and 66 million reads for the WT pools (Supplementary Table 2). After trimming low-quality reads and adapter sequences with Cutadapt (v.1.6) using a Cutoff Quality Score of 30, 47, 155 and 116 million reads from the mutant pools and 46, 109 and 55 million reads from the WT pools were successfully mapped to the cucumber genome (http://www.icugi.org/cgi-bin/ICuGI/genome/home.cgi?ver=2&organism=cucumber&cultivar=Chinese-long) and combined with known gene annotations (cucumber_ ChineseLong_v2.gff3) using the improved version of Tophat2 (Supplementary Table 2). All transcripts longer than 200 nt were used to predict the coding potential with the UniRef90 database; the genes with absolute values for logFC > 1 and q-value < 0.05 were defined as differentially expressed genes (DEGs) between the WT groups and the mutant groups using the R (v.3.1.0) language with the edgeR package. We found 266 DEGs, of which 173 were up-regulated and 93 were down-regulated in the mutant compared to WT at 6 DBA (Supplementary Figure 5). At 2 DBA, 263 DEGs were obtained; of these, 82 were significantly up-regulated and 181 were significantly down-regulated (Supplementary Figure 5). At 9 DAA, 478 DEGs were identified, 200 of which were up-regulated and 278 of which were down-regulated (Supplementary Figure 5). A Venn diagram (Supplementary Figure 5) shows that there was less DEG commonality between the 6 and 2 DBA and the 9 DAA age groups. To further confirm the DEGs identified by RNA-Seq analysis, quantitative PCR (qPCR) was performed with total RNA from sf1 and WT fruits to test the variation in expression of 35 randomly selected genes (Supplementary Table 3). The genes whose variations in expression were tested by qPCR displayed the same expression patterns as in RNA-Seq analysis (Supplementary Table 3), indicating that the RNA-Seq results were reliable.
Furthermore, SNPs were identified using the SAM tools, and variants with QUAL values higher than 20 were filtered as SNPs with high confidence. SNPs that were closely associated with genotype were further identified through Fisher’s exact test with p-value < 0.01. SNP-rich regions were identified on chromosomes 1 and 6 (from 10,053,234 to 11,631,603) at 2 DBA and on chromosome 6 (from 11,603,968 to 11,631,493) at 9 DAA in fruits of the mutant groups compared to the WT groups (Fig. 4). The SNP-enriched region on chromosome 6 was detected at both of the investigated time points and overlapped (from 11,603,968 to 11,631,493) with the region to which the sf1 gene was fine-mapped (Fig. 4).

Chromosomal distributions of SNPs and DEGs in the cucumber genome. The SNPs and DEGs of all genes expressed in the fruit samples of the mutant vs. WT groups at 2 DBA (A) and 9 DAA (B), which came from the F2 population from a cross between sf1 and ‘Chinese long’ 9930, are shown as the up direction (○) and the down direction (○) from the zero point on the same y-axis, respectively. The purple rectangles indicate the overlapping region (11,603,968 to 11,631,493) between the fine-mapped 174.3 kb region and the SNP-enriched region on chromosome 6. The number of SNPs and DEGs was calculated based on non-overlapping 50-kb bins.
To determine the functions of these DEGs, we performed gene ontology (GO) term enrichment analysis (p-value < 0.01) for the significantly influenced genes in fruits of the mutant groups at 6 DBA, 2 DBA and 9 DAA compared to the WT groups. The most significantly enriched GO terms were ‘Regulation of biosynthetic process’, ‘Regulation of gene expression’ and ‘Regulation of transcription, DNA-templated’ in the biological process group (Fig. 5). Only ‘DNA binding’ in the molecular function group was observed in mutant fruits at all three investigation time points (Fig. 5). KEGG analysis of these DEGs using a hyper-geometric distribution test revealed pathways that were closely associated with fruit phenotype and showed significant differences in fruits of mutant vs. WT groups. At 6 DBA, the significantly influenced genes included AUX1, which is involved in auxin signalling (Fig. 6A1), and HP, which is involved in cytokinin signalling (Fig. 6B1). At 2 DBA, the significantly influenced genes included AUX1, Aux/IAA, GH3 and SAUR, which are involved in auxin signalling (Fig. 6A2), and HP, which is involved in cytokinin signalling (Fig. 6B2). At 9 DAA, the significantly influenced genes included AUX1, Aux/IAA and SAUR (Fig. 6A3), HP (Fig. 6B3), and KAO, GA20 ox , GA2 ox and GID1, which is involved in gibberellin biosynthesis and signal transduction (Fig. 6C). At the same time, significant differences in the content of IAA, zeatin and GA3 were observed in sf1 fruits compared with WT fruits (Supplementary Table 4). In addition, BGLU, COMT, 4CL, REF1, UGT72E and Peroxidase, which are known to be in the phenylpropanoid biosynthesis pathway, and CrtR-b, β-Carotene-isomerase, CrtZ, CCD7 and NCED in the carotenoid biosynthesis pathway, were significantly influenced at 9 DAA (Supplementary Figure 6; Supplementary Figure 7). These results provide molecular evidence for the phenotypic variations in sf1 fruits.

Significantly enriched GO terms (p-value < 0.01) in the cucumber fruits of the mutant vs. WT groups at 6 DBA (A), 2 DBA (B) and 9 DAA (C). GO terms were categorized into biological processes, cellular components and molecular functions based on their p-values.

DEG-enriched pathways from the KEGG analysis. (A) DEG-enriched auxin signalling in the cucumber fruits of the mutant vs. WT groups at 6 DBA (A1), 2 DBA (A2) and 9 DAA (A3). (B) DEG-enriched cytokinin signalling in the cucumber fruits of the mutant vs. WT groups at 6 DBA (B1), 2 DBA (B2) and 9 DAA (B3). (C) DEG-enriched gibberellin biosynthesis and signalling in the cucumber fruits of the mutant vs. WT group at 9 DAA. The red closed rectangles represent the genes that were up-regulated, and the green closed rectangles represent the down-regulated genes. The number of rectangles indicates the fold gene expression difference between the mutant and WT fruits. Aux/IAA, Auxin/indole-3-acetic acid protein; ARF, auxin-response factor; SAUR, small auxin up RNA; CRE1, cytokinin response 1; HP, histidine phosphotransfer protein; B-RR, type-B response regulator; A-RR, type-A response regulator; KAO, ent-kaurenoic acid oxidase; GA, gibberellin; GA20ox, GA 20-oxidase; GA2ox, GA 2-oxidase; GID1, gibberellin insensitive dwarf 1.
We also identified significantly differentially expressed transcription factors in fruits of the mutant vs. the WT groups. At 6 DBA, the expression of 29 transcription factors belonging to the following gene families was up-regulated: AP2/ERF, GRAS, HSF, LFY, MADS, NAC, WRKY, YABBY and Zinc finger protein; by contrast, the expression of 7 transcription factors belonging to the following gene families was down-regulated: bZIP, MYB, NAC, TCP, WD40, WRKY and Zinc finger protein (Fig. 7; Table 1). At 2 DBA, the expression of 12 transcription factors belonging to the following gene families was up-regulated: bHLH, bZIP, MADS, MYB, WRKY, YABBY and Zinc finger protein; by contrast, the expression of 13 transcription factors belonging to the gene families AP2/ERF, bZIP, MADS, MYB, SBP, WRKY, YABBY and Zinc finger protein was down-regulated (Fig. 7; Table 1). At 9 DAA, there were 10 up-regulated transcription factors belonging to the gene families AP2/ERF, HSF, MYB, NAC, YABBY, and Zinc finger protein and 39 down-regulated transcription factors belonging to the gene families AP2/ERF, bHLH, bZIP, HSF, MYB, NAC, NF-YA, WRKY, and Zinc finger protein (Fig. 7; Table 1). These results suggest that transcription factors might play a critical role in determining fruit length.

Heatmap showing the differential expression of selected transcriptional factors in the mutant vs. WT group fruits at 6 DBA, 2 DBA and 9 DAA. Genes highly or weakly expressed in the mutant vs. WT group fruits are shown in red and green, respectively. Genes without expression in the mutant vs. WT group fruits are shown in black. The heatmap was generated using cluster 3.0 software.
Discussion
Fruit size, one of the most important appearance quality parameters for vegetables such as cucumber and tomato, commonly displays high variation among cultivars. Many QTLs that control cucumber fruit length have been detected in different populations, developmental stages and growing seasons, and 14 were mapped on chromosome 65,6,7, 19,20,21,22,23,24. Twelve of the 14 QTLs on chromosome 6 were named FL6.1 (or MFL6.1or Sfl6.1), but their positions differed. In addition to five QTLs, qFL6.1 and qMFL6.1 detected by Weng et al., Sfl6.1 and Sfl6.2 detected by Miao et al., and fl6.1 detected by Yuan et al. (only their genetic positions are available: from 43.5 to 77.3 cM, from 43.5 to 54.3 cM, from 45.7 to 49.7 cM, from 49.7 to 52.4 cM, and from 183.2 to 186.9 cM, respectively)6, 19, 20, the remaining nine QTLs’ physical positions have been detailed as follows: from 9.245 to 10.817 Mb and from 10.816 to 11.520 Mb for two fl6.1s detected by Bo et al. under two different growing conditions, respectively5; from 12.488 to 12.711 Mb for fl6.1 detected by Wei et al.21; from 12.931 to 19.653 Mb for fl6.1 detected by Wang et al.22; from 15.630 to 22.271 Mb for MFL6.1detected by Weng et al.6; from 23.205 to 23.755 Mb for fl6.1 detected by Qi et al.23; from 23.544 to 24.578 Mb for fl6.1 detected by Cheng et al.24; and from 24.289 to 27.896 Mb for fl6.1 and fl6.2 detected by Miao et al.19. In addition, Jiang et al. identified a fruit length-related locus in the region from 18.1 Mb to 19.0 Mb on chromosome 6 through SNP association analysis based on transcriptomic data7. However, none of the regions in which these reported QTLs/genes were localized overlaps with the region to which the sf1 gene was mapped (from 11.584 to 11.759 Mb) on chromosome 6, indicating that sf1 might be an uncharacterized locus.
We further analysed the 174.3 kb region in which sf1 was mapped using PCR-based sequencing and found no difference in the 168.5 kb sequenced region between sf1 and WT. In addition, there were no differences in expression among the genes in this region (Supplementary Figure 4). Therefore, the responsible mutation is still unknown. This discrepancy might be explained with the following two possibilities. First, in the candidate region, 5.8 kb segments (if no gap) have not yet been sequenced due to technical difficulties, thus, we do not know whether there are mutations or gaps in the un-sequenced region. In fact, there are still many un-assembled scaffolds in the cucumber genome. In this study, we also found that there are 9 DGEs at 6 DBA, 64 DGEs at 2 DBA and 10 DGEs at 9 DAA mapped on un-assembled scaffolds. Second, epigenetic differences might be responsible for the mutation. Variations such as DNA methylation might occur at the epigenetic level, which could influence gene expression either inside or outside the mapped regions because of the complexity of cis-acting regulatory DNA elements25,26,27. Further studies should address this problem in the future.
It has been well demonstrated that cell division and cell enlargement play critical roles in final fruit length, diameter and yield. To illuminate how sf1 regulates fruit length, we analysed sf1 fruits using histological, physiological and comparative transcriptomic approaches. We found that the cell number of sf1 fruits was significantly lower than that of WT fruits (Fig. 2I). Comparative transcriptomic analysis revealed that a cucumber orthologue of Arabidopsis histidine phosphotransfer proteins (AHPs), which are a critical component of plant cytokinin signalling, was significantly repressed at three investigated time points in the mutant group fruits compared to its expression in the WT group fruits (Fig. 6B). AHPs belong to a protein family that contains five members, AHP1-AHP5; these proteins are required for transfer of a phosphoryl group from the Arabidopsis histidine kinase (AHKs) to Arabidopsis response regulators (ARRs)28. To date, all reported results on AHPs suggest that, with the exception of AHP428, they function positively in cytokinin signalling. Mutations in these positive AHPs can cause a series of developmental abnormalities, such as shortened primary roots, reduced vascular development, decreased fertility, increased numbers of embryos and seeds, and adventitious root development29. It has been demonstrated that cucumber fruit cell division primarily occurs prior to anthesis and that the rate of cell division then gradually decreases until 5 days post pollination (DPP)30, 31. The significantly decreased expression of histidine phosphotransfer protein (HP) in sf1 fruits during cucumber fruit development provides a possible molecular explanation for the dramatic reduction in cell number, the main reason for the short-fruit phenotype of sf1.
Although some genes responsible for gibberellin biosynthesis and signalling were stimulated, the expression of GID1 (gibberellin insensitive dwarf 1) was significantly lower in sf1 fruits at 9 DAA (Fig. 6C). GID1 is a gibberellin receptor that can stimulate the degradation of DELLA in plants through a ubiquitin-proteasome pathway32. DELLA proteins are repressors of GA signalling and can restrict GA-mediated plant growth and development, including fruit development33. Binding of GA to GID1 can facilitate GID1-DELLA interaction and thus stimulate the degradation of DELLAs34. It has been reported that cucumber fruit elongation occurs most rapidly between 4 DPP and 12 DPP and that the peak rate of elongation occurs at approximately 8 DPP18, 35. The significant reduction in GID1 expression at 9 DAA (Fig. 6C) might block fruit elongation at a key stage and thus exacerbate the shortening of fruit length in sf1.
Previous studies have proposed a positive relationship between cell size and fruit length in cucumber35. However, in this study, we observed that although sf1 fruits displayed dramatically decreased fruit length and decreased daily growth (Fig. 2A and B), cell size in sf1 fruits was significantly increased relative to that in WT fruits, possibly due to abnormalities in auxin signalling in sf1 fruits. In particular, at 2 DBA the expression of key auxin-responsive genes was significantly higher in the mutant group fruits than in the WT group fruits from the F2 population (Figs 2C–H and 6A). The significantly larger cell size in sf1 fruits might be a compensatory response to the decrease in cell number. Similar effects have been reported in cucumber, in which a decrease in cell number is compensated for by an increase in cell size when assimilated supply is limited36; in Arabidopsis mutants that affect leaf size by producing decreased cell numbers but increased cell size37; in Arabidopsis mutants that affect integument development by causing increased cell division and reduced cell size38; and in the Antirrhinum majus floral mutant formosa (fo), which has increased petal size but smaller cells39. The abnormal cell size might also be an important reason for the increased cracking of sf1 fruits (Fig. 1B and 2C–H). At the same time, we observed significant effects of the sf1 mutation on the expression of transcription factors that play critical roles in plant hormone signalling (Fig. 7; Table 1). These up- or down-regulated transcription factors might bridge the three signalling pathways of cytokinin, gibberellins and auxin to co-regulate fruit length in the sf1 mutant. Tan et al. also reported that the transient balance of endogenous hormones might play a key role in the regulation of cucumber fruit length40.
In addition to the significant decrease in fruit length (Fig. 1A), significant changes in other fruit-related traits also occurred in the sf1 mutant (Fig. 1B–J), indicating that sf1 has pleiotropic effects. Genes with pleiotropic effects have been widely identified and well characterized in previous studies. For example, a rice mutant losing FISH BONE (FIB) gene function, an orthologue of TRYPTOPHAN AMINOTRANSFERASE OF ARABIDOPSIS (TAA) genes, has pleiotropic abnormal phenotypes, including decreased leaf size with larger lamina joint angles, smaller panicles, abnormal vascular development and organ identity, defects in root development and reduced endogenous IAA levels41. In Populus trichocarpa, an orthologue of the class III homeodomain-leucine zipper transcription factor gene REVOLUTA has been demonstrated to be significantly associated with poplar fungal and rust resistance, leaf abscission, cellulose content, and auxin signalling42. In cucumber, a putative R2R3-MYB transcription factor gene has been shown to control both black spine and orange mature fruit colours of cultivated cucumber, thus displaying pleiotropic effects43.
Comparative transcriptomic analysis provided some molecular evidence for the pleiotropic effects of the sf1 gene as well. For example, fruit hardness and crispness are mainly dependent on the composition of the fruit’s cell walls15,16,17; thus, changes in the content of cell wall components might result in variations in fruit hardness and crispness. In sf1 fruits, significantly higher lignin content was observed (Fig. 1M), consistent with the observed significant stimulation of the lignin biosynthesis pathway (Supplementary Figure 6; Supplementary Table 5). This might be the reason sf1 fruits display greater fruit hardness and crispness. Additionally, sf1 fruits were shown to have significant differences in carotenoid biosynthesis (Supplementary Figure 7; Supplementary Table 5), providing molecular evidence for sf1-mediated regulation of carotenoid content in cucumber fruits (Supplementary Figure 2I). However, full explanation of other altered traits such as spine morphology, stigma and seed cavity size in sf1 requires further study.
In summary, the results of the present study demonstrated that sf1 is a pleiotropic effector of the external and internal qualities of cucumber fruits. Altered expression of genes in the auxin, cytokinin signalling and GA biosynthesis and signalling pathways might be the reason for the decrease in cell number in the sf1 mutant, which led to a decrease in fruit length. Future studies, such as cloning and functional analysis of sf1, are needed to better understand the molecular mechanisms involved in the regulation of cucumber fruit growth and development.
Materials and Methods
Plant materials and growth conditions
Cucumber (Cucumis sativus L., 2n = 14) inbred line CNS2 (WT, North China type), ZG (North European type), ‘Chinese long’ 9930 (North China type), and the sf1 mutant were used. Plants were grown for two generations each year from 2011 to 2016 in the greenhouse of the experimental field at Shandong Agricultural University. Standard management was performed during the cultivation period.
Phenotype investigation
Fruit length (FL) was measured from 0 DAA to 22 DAA. Daily fruit growth (DFG) was calculated based on the following formula: DFG = FLn − FL(n−1), where FLn and FL(n−1) represent fruit length at n and n−1 DAA (1 ≤ n ≤ 22). Each parameter was determined from 18 biological repeats.
Fruit quality analysis
Cucumber fruits from WT and sf1 were sampled at 0, 9 and 23 DAA, and their hardness and crispness were measured using a texture analyser (TA.XT Plus, Stable Micro Systems Ltd) with a P/2N and the following settings: 3 mm s−1 pre-test speed, 1 mm s−1 test speed, 0.5 mm s−1 post-test speed, 5 mm penetration depth and 10 g minimum trigger force.
WT and sf1 fruits were sampled at 9 DAA and dried at 55 °C for 48 h. The dried samples were weighed and ground to pass a 1-mm sieve. Subsequent analysis of acid-soluble lignin content in the samples was performed according to the previously described method44 using a modification for amylase45.
Three biological repeats were used for the determination of each parameter.
Microscopic investigation of sf1 fruits
Pericarp and sarcocarp cell size in WT and sf1 fruits were investigated using a scanning electron microscope (SEM) at 0, 9 and 23 DAA according to the method previously described46. After 24-h fixation in 2.5% (w/v) glutaraldehyde at 4 °C, the pericarp and sarcocarp samples were washed three times with full-strength phosphate- buffered saline (PBS) and further processed with 1% (v/v) OsO4. The OsO4-processed samples were dehydrated through six volume concentrations of ethanol (30%, 50%, 70%, 80%, 90% and 100%) three times, critical- point-dried with liquid CO2 and coated with gold palladium (EIKO IB-3). The resulting samples were viewed under a SEM (JSM-6610LV). Cell number per unit area of pericarp was then calculated using Image J software (v 2.1.4.7, NIH).
Stigmas from WT and sf1 at 0 DAA and spines of WT and sf1 fruits at 11 DAA were investigated under a dissection microscope (LEICA M165 FC).
Fine mapping of the sf1 gene
For mapping the sf1 locus, the sf1 mutant was crossed with ZG and ‘Chinese long’ 9930 to construct F2 segregation populations. Thirty-five SSR markers were used to perform preliminary mapping of sf1 with the F2 population from the cross between sf1 and ZG. In total, 6,720 F2 plants from the cross between sf1 and ‘Chinese long’ 9930 were further analysed with 22 SNP markers for fine mapping of sf1. The PCR products were checked by ethidium bromide (EB) staining under UV light following separation on agarose gels. All primers used for mapping are listed in Supplementary Table 6.
Construction of RNA libraries for comparative transcriptomic analysis
Transcriptomic profiling was analysed using cucumber fruits from WT and sf1 at 6 DBA and from the F2 population from sf1 × ‘Chinese long’ 9930 at 2 DBA and 9 DAA. The middle part of the fruits at all three stages was used to extract total RNA. The F2 population from sf1 × ‘Chinese long’ 9930 was used at the two later time points to test the fine mapping results obtained through SNP association analysis. Thirty individual plants were used for fruit sampling; these fruits were then pooled as one biological sample for WT and mutant phenotyping. mRNA was isolated from total RNA using oligo-dT magnetic beads and fragmented with fragmentation buffer for cDNA synthesis. After the addition of adenine, the resulting cDNAs were linked to adapters and purified by gel electrophoresis; finally, cDNAs approximately 250 bp in length were collected for PCR amplification. After quality checking and quantification with an Agilent 2100 Bioanalyzer and the ABI StepOnePlus Real-Time PCR System, all libraries were sequenced on the Illumina HiSeqTM 2000 platform at Honortech (Beijing, China).
Bioinformatics analysis
Removal of low-quality and adaptor sequences from the raw data was performed with Cutadapt (v1.6)47. Clean sequencing data for each sample were aligned to the reference genome (http://www.icugi.org/cgi-bin/ICuGI/genome/home.cgi?ver=2&organism=cucumber&cultivar=Chinese-long) and combined with known gene annotations (cucumber_ChineseLong_v2.gff3) using the improved version of Tophat2 software (v 2.1.0)48 with the following parameters: read-mismatches 2, mate-inner-dist 50, mate-std-dev 80 and library-type fr-unstranded. Four procedures in Cufflinks49, including cufflinks, cuffmerge, cuffquant and cuffnorm, were used to reconstruct transcripts, identify novel transcripts, quantify transcripts and normalize expression values as FPKM (fragments per kilobase of transcript per million mapped reads).
CPC software with the UniRef90 database was used to predict the coding potential of all transcripts longer than 200 nt50. Transcripts scored greater than 0 were identified as encoding genes, and those scored less than 0 were considered non-coding genes. Functional annotation for expressed transcripts was performed using the BLASTX program (v 2.2.29) based on the NR, eggNOG and Swiss-Port databases with an e-value cutoff of <1e-5. Identification of domains and gene families and GO analysis were performed using InterProScan (v. 5.8–49.0). GO plant slim level 2 was used to obtain GO functional classifications for all transcripts. Mapping of the transcripts to KEGG pathways was performed using KOBAS 2.051 with default parameters.
The R (v.3.1.0) language with the edgeR package was used to identify DEGs. The fold change (FC) between the two groups was calculated based on the following formula: logFC = log2 (mutant group/WT group). Genes that differed in expression between the mutant and WT groups were defined as DEGs in this study if the absolute value of the logFC was greater than 1 and the q-value was less than 0.05. A hypergeometric distribution test was carried out to identify GO functions and KEGG pathways in which the DEGs were significantly enriched with p-values < 0.01 in comparison to the total expressed transcripts. Python 2.7.5 with numpy (1.9.2), scipy (0.15.1) and matplotlib (1.4.3) was used to perform cluster analysis of the DEGs based on Ward’s method with Euclidean distance as a measurement of similarity.
Based on alignments by Tophat2, SNPs were identified using SAM tools (v. 0.1.19) with the set parameters of -Q13 and -Q20. Variants with QUAL values higher than 20 were filtered as SNPs with high confidence. Fisher’s exact test was used to further identity the SNPs associated with the genotype through the cutoff of p-value < 0.01, which was based on the principle of MutMap52.
References
Wenzel, G., Kennard, W. C. & Havey, M. J. Quantitative trait analysis of fruit quality in cucumber: QTL detection, confirmation, and comparison with mating-design variation. Theor Appl Genet 91, 53–61, doi:10.1007/BF00220858 (1995).
Serquen, F. C., Bacher, J. & Staub, J. E. Mapping and QTL analysis of horticultural traits in a narrow cross in cucumber (Cucumis sativas L.) using random amplified polymorphic DNA markers. Mol Breed 3, 257–268, doi:10.1023/A:1009689002015 (1997).
Fazio, G., Staub, J. E. & Stevens, M. R. Genetic mapping and QTL analysis of horticultural traits in cucumber (Cucumis sativus L.) using recombinant inbred lines. Theor Appl Genet 107, 864–874, doi:10.1007/s00122-003-1277-1 (2003).
Yuan, X. J. et al. Genetic linkage map construction and ___location of QTLs for fruit-related traits in cucumber. Plant Breeding 127, 180–188, doi:10.1111/j.1439-0523.2007.01426.x (2008).
Bo, K., Ma, Z., Chen, J. & Weng, Y. Molecular mapping reveals structural rearrangements and quantitative trait loci underlying traits with local adaptation in semi-wild Xishuangbanna cucumber (Cucumis sativus L. var. xishuangbannanesis Qi et Yuan). Theor Appl Genet 128, 25–39, doi:10.1007/s00122-014-2410-z (2015).
Weng, Y. et al. QTL mapping in multiple populations and development stages reveals dynamic quantitative trait loci for fruit size in cucumbers of different market classes. Theor Appl Genet 128, 1747–1763, doi:10.1007/s00122-015-2544-7 (2015).
Jiang, L. et al. Transcriptomic analysis reveals the roles of microtubule-related genes and transcription factors in fruit length regulation in cucumber (Cucumis sativus L.). Sci Rep 5, 8031, doi:10.1038/srep08031 (2015).
Matsuo, S., Kikuchi, K., Fukuda, M., Honda, I. & Imanishi, S. Roles and regulation of cytokinins in tomato fruit development. J Exp Bot 63, 5569–5579, doi:10.1093/jxb/ers207 (2012).
Ogawa, Y., Nishikawa, S., Inoue, N. & Aoki, S. Promotive effects of different cytokinins on the fruit growth in Cucumis sativus L. J Jpn Soc Hortic Sci 59, 597–601, doi:10.2503/jjshs.59.597 (1990).
Achard, P. et al. Gibberellin signaling controls cell proliferation rate in Arabidopsis. Curr Biol 19, 1188–1193, doi:10.1016/j.cub.2009.05.059 (2009).
Asahina, M. et al. Gibberellin produced in the cotyledon is required for cell division during tissue reunion in the cortex of cut cucumber and tomato hypocotyls. Plant Physiol 129, 201–210, doi:10.1104/pp.010886 (2002).
Zhang, C. et al. Biologically active gibberellins and abscisic acid in fruit of two late-maturing Japanese pear cultivars with contrasting fruit size. J Amer Soc Hort Sci 132, 452–458 (2007).
Wang, L. & Ruan, Y. L. Regulation of cell division and expansion by sugar and auxin signaling. Front Plant Sci 4, 163, doi:10.3389/fpls.2013.00163 (2013).
Keegstra, K. Plant cell walls. Plant Physiol 154, 483–486, doi:10.1104/pp.110.161240 (2010).
Boerjan, W., Ralph, J. & Baucher, M. Lignin biosynthesis. Annu Rev Plant Biol 54, 519–546, doi:10.1146/annurev.arplant.54.031902.134938 (2003).
Cai, C., Xu, C. J., Li, X., Ferguson, I. & Chen, K. S. Accumulation of lignin in relation to change in activities of lignification enzymes in loquat fruit flesh after harvest. Postharvest Biol Technol 40, 163–169, doi:10.1016/j.postharvbio.2005.12.009 (2006).
Salentijn, E. M. J., Aharoni, A., Schaart, J. G., Boone, M. J. & Krens, F. A. Differential gene expression analysis of strawberry cultivars that differ in fruit-firmness. Physiol Plant 118, 571–578, doi:10.1034/j.1399-3054.2003.00138.x (2003).
Ando, K., Carr, K. M. & Grumet, R. Transcriptome analyses of early cucumber fruit growth identifies distinct gene modules associated with phases of development. BMC Genomics 13, 518, doi:10.1186/1471-2164-13-518 (2012).
Miao, H. et al. Mapping QTLs for fruit-associated traits in Cucumis sativus L. Scientia Agricultura Sinica 44, 5031–5040 (2011).
Yuan, X. et al. Genetic mapping and QTL analysis of fruit and flower related traits in cucumber (Cucumis sativus L.) using recombinant inbred lines. Euphytica 164, 473–491, doi:10.1007/s10681-008-9722-5 (2008).
Wei, Q. et al. An SNP-based saturated genetic map and QTL analysis of fruit-related traits in cucumber using specific-length amplified fragment (SLAF) sequencing. BMC Genomics 15, 1158, doi:10.1186/1471-2164-15-1158 (2014).
Wang, M. et al. Quantitative trait loci associated with fruit length and stalk length in cucumber using RIL population. Acta Bot Boreal–Occident Sin 34, 1764–1770 (2014).
Qi, J. et al. A genomic variation map provides insights into the genetic basis of cucumber domestication and diversity. Nat Genet 45, 1510–1515, doi:10.1038/ng.2801 (2013).
Cheng, Z. et al. QTL mapping of fruit length in cucumber. China Vegetables 12, 20–25 (2010).
Stam, M. et al. The regulatory regions required for B’ paramutation and expression are located far upstream of the maize b1 transcribed sequences. Genetics 162, 917–930 (2002).
Dickel, D. E. et al. Function-based identification of mammalian enhancers using site-specific integration. Nat Methods 11, 566–571, doi:10.1038/nmeth.2886 (2014).
Lindholm, M. E. et al. An integrative analysis reveals coordinated reprogramming of the epigenome and the transcriptome in human skeletal muscle after training. Epigenetics 9, 1557–1569, doi:10.4161/15592294.2014.982445 (2014).
Nishiyama, R. et al. Arabidopsis AHP2, AHP3, and AHP5 histidine phosphotransfer proteins function as redundant negative regulators of drought stress response. Proc Natl Acad Sci USA 110, 4840–4845, doi:10.1073/pnas.1302265110 (2013).
Hutchison, C. E. et al. The Arabidopsis histidine phosphotransfer proteins are redundant positive regulators of cytokinin signaling. Plant Cell 18, 3073–3087, doi:10.1105/tpc.106.045674 (2006).
Marcelis, L. F. M. & Hofman-Eijer, L. R. B. Cell division and expansion in the cucumber fruit. J Hortic Sci 68, 665–671, doi:10.1080/00221589.1993.11516398 (1993).
Fu, F. Q. et al. A role of brassinosteroids in early fruit development in cucumber. J Exp Bot 59, 2299–2308, doi:10.1093/jxb/ern093 (2008).
Ueguchi-Tanaka, M., Nakajima, M., Motoyuki, A. & Matsuoka, M. Gibberellin receptor and its role in gibberellin signaling in plants. Annu Rev Plant Biol 58, 183–198, doi:10.1146/annurev.arplant.58.032806.103830 (2007).
Sun, T. Gibberellin-GID1-DELLA: A pivotal regulatory module for plant growth and development. Plant Physiol 154, 567–570, doi:10.1104/pp.110.161554 (2010).
Murase, K., Hirano, Y., Sun, T. P. & Hakoshima, T. Gibberellin-induced DELLA recognition by the gibberellin receptor GID1. Nature 456, 459–463, doi:10.1038/nature07519 (2008).
Ando, K. & Grumet, R. Transcriptional profiling of rapidly growing cucumber fruit by 454-pyrosequencing analysis. J Amer Soc Hortic Sci 135, 291–302 (2010).
Marcelis, L. F. M. Effect of assimilate supply on the growth of individual cucumber fruits. Physiol Plant 87, 313–320, doi:10.1111/j.1399-3054.1993.tb01736.x (1993).
Ferjani, A., Horiguchi, G., Yano, S. & Tsukaya, H. Analysis of leaf development in fugu mutants of Arabidopsis reveals three compensation modes that modulate cell expansion in determinate organs. Plant Physiol 144, 988–999, doi:10.1104/pp.107.099325 (2007).
Truernit, E. & Haseloff, J. Arabidopsis thaliana outer ovule integument morphogenesis: ectopic expression of KNAT1 reveals a compensation mechanism. BMC Plant Biol 8, 35, doi:10.1186/1471-2229-8-35 (2008).
Delgado-Benarroch, L., Weiss, J. & Egea-Cortines, M. The mutants compacta ähnlich, Nitida and Grandiflora define developmental compartments and a compensation mechanism in floral development in Antirrhinum majus. J Plant Res 122, 559–569, doi:10.1007/s10265-009-0236-6 (2009).
Tan, J. et al. A novel allele of monoecious (m) locus is responsible for elongated fruit shape and perfect flowers in cucumber (Cucumis sativus L.). Theor Appl Genet 128, 2483–2493, doi:10.1007/s00122-015-2603-0 (2015).
Yoshikawa, T. et al. The rice FISH BONE gene encodes a tryptophan aminotransferase, which affects pleiotropic auxin-related processes. Plant J 78, 927–936, doi:10.1111/tpj.12517 (2014).
Porth, I. et al. Extensive functional pleiotropy of REVOLUTA substantiated through forward genetics. Plant Physiol 164, 548–554, doi:10.1104/pp.113.228783 (2014).
Li, Y., Wen, C. & Weng, Y. Fine mapping of the pleiotropic locus B for black spine and orange mature fruit color in cucumber identifies a 50 kb region containing a R2R3-MYB transcription factor. Theor Appl Genet 126, 2187–2196, doi:10.1007/s00122-013-2128-3 (2013).
van Soest, P. J. & Robertson, J. B. Systems of analysis for evaluating fibrous feeds. In: Proc Int Workshop on Standardization Analytical Methodology Feeds (eds Pigden, W. J. et al.) 49–60 (IDRC, 1980).
Bidlackr, J. E. & Buxton, D. R. Content and deposition rates of cellulose, hemicellulose, and lignin during regrowth of forage grasses and legume. Can J Plant Sci 72, 809–818, doi:10.4141/cjps92-097 (1992).
Chen, C. et al. Transcriptome profiling reveals roles of meristem regulators and polarity genes during fruit trichome development in cucumber (Cucumis sativus L.). J Exp Bot 65, 4943–4958, doi:10.1093/jxb/eru258 (2014).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17, 10–12, doi:10.14806/ej.17.1.200 (2011).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36, doi:10.1186/gb-2013-14-4-r36 (2013).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Prot 7, 562–578, doi:10.1038/nprot.2012.016 (2012).
Kong, L. et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res 35, W345–W349, doi:10.1093/nar/gkm391 (2007).
Xie, C. et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res 39, W316–W322, doi:10.1093/nar/gkr483 (2011).
Abe, A. et al. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol 30, 174–178, doi:10.1038/nbt.2095 (2012).
Acknowledgements
This work was supported by funding from the National Natural Science Foundation of China (31171977, 31222048 and 31401894), the Natural Science Foundation of Shandong Province (JQ201309), the ‘Taishan Scholar’ Foundation of the People’s Government of Shandong Province, the Program for Changjiang Scholars and Innovative Research Team in University (IRT1155), and the Henry Fok Education Foundation (131032).
Author information
Authors and Affiliations
Contributions
L.W. and Z.R. conceived and designed the experiments. C.C. provided sf1 mutant. L.W., S.Z., H.Z., P.L., Q.G. and J.L. performed the experiments. L.W., S.Z. and Z.R. analyzed the data. L.W. and R.Z. wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, L., Cao, C., Zheng, S. et al. Transcriptomic analysis of short-fruit 1 (sf1) reveals new insights into the variation of fruit-related traits in Cucumis sativus . Sci Rep 7, 2950 (2017). https://doi.org/10.1038/s41598-017-02932-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-02932-5
This article is cited by
-
Fine mapping and identification of regulatory genes for fruit neck length in bottle gourd (Lagenaria siceraria)
Euphytica (2024)
-
Mapping and identification of CsSF4, a gene encoding a UDP-N-acetyl glucosamine-peptide N-acetylglucosaminyltransferase required for fruit elongation in cucumber (Cucumis sativus L.)
Theoretical and Applied Genetics (2023)
-
Genome-wide association study of the candidate genes for grape berry shape-related traits
BMC Plant Biology (2022)
-
Localization of quantitative trait loci for cucumber fruit shape by a population of chromosome segment substitution lines
Scientific Reports (2020)
-
Genetic architecture of fruit size and shape variation in cucurbits: a comparative perspective
Theoretical and Applied Genetics (2020)
