
Abstract
Breast cancer (BC) has emerged as the most common malignancy among females. The genomic profile of BC is diverse in nature and complex due to heterogeneity among various geographically different ethnic groups. The primary objective of this study was to carry out a comprehensive mutational analysis of Indian BC cases by performing whole exome sequencing. The cohort included patients with a median age of 48 years. TTN, TP53, MUC16, SYNE1, and OBSCN were the frequently altered genes found in our cohort. The PIK3CA and KLC3 genes are driver genes implicated in various cellular functions and cargo transportation through microtubules, respectively. Except for CCDC168 and PIK3CA, several gene pairings were found to be significantly linked with co-occurrence. Irrespective of their hormonal receptor status, RTK/RAS was observed with frequently altered signaling pathways. Further analysis of the mutational signature revealed that SBS13, SBS6, and SBS29 were mainly observed in our cohort. This study supplements the discovery of diagnostic biomarkers and provides new therapeutic options for the improved management of BC.
Similar content being viewed by others

Introduction
In the recent decade, an increasing burden of cancer has badly impacted every sphere of the human race. A report released by the International Agency for Research on Cancer (IARC) in 2020 suggests that an estimated 19.3 million new cases are diagnosed with a mortality of nearly 10 million cases. Breast cancer surpassed all forms of cancer and accounted for approximately 11.7% of new cases in 2020 worldwide across all ages and sexes, whereas in the case of females, the incidence rate was quite high, at 24.5% (1 in every 4 cases). In India, the projected number of breast cancer patients in 2020 is more than 0.2 million (1 in 29 females) 1.
Breast cancer represents heterogeneity that emerges as a consequence of mutations acquired during the evolution of the disease. There are various risk factors associated with the alteration of genes, which could be somatic or hereditary. It exhibits a disparity in incidence pattern among women aged between 50 and 59 years who are more prone to develop during their lifetime. However, in the present decades, these trends are shifting toward the younger age group 2. Current clinical management focuses mainly on morphological assessment (tumor size, node and metastatic status) as well as molecular markers such as expression of hormone receptors (HR) and amplification of human epidermal growth factor 2 (HER2). However, their clinical behavior shows variations from case to case, even under the same status of HR and HER2 3.
Numerous large-scale sequencing projects 4,5 have been conducted and pinpointed several key genes related to breast cancer. However, these studies were carried out predominantly on breast cancer patients belonging to European ancestry. It is inadequate to assess the susceptibility among non-Western groups based on existing knowledge and can lead to an inaccurate assessment. For instance, EGFR-activating mutations were reported in TNBC patients of Asian ancestry. On the other hand, no such mutation was found in patients of European ancestry 6. This could be due to the heterogeneous nature of mutations and differences in clinical characteristics, risk profiles, and survival across distinct ethnic groups. This makes it necessary to understand the genomic diversity of Indian patients who underlies breast cancer progression.
To explore the diversification of the exome in breast cancer patients, we took advantage of massive parallel sequencing through next-generation sequencing (NGS). NGS has proven to be a potent tool in biomedical and related research. Its application, such as whole exome sequencing (WES), is used to decipher the entire genome's protein-coding region and aid in studying mutational landscapes. The human genome comprises exomes that harbor almost 1–2%, and it is thought to contain relatively 85% of genetic variants of disease 7. To identify genetic abbreviations and driver genes of primary breast cancer in Indian patients, we sequenced the exome region of 97 breast tumors along with matched blood samples. Our results demonstrate the mutational landscapes along with their clinical features. We also analyzed the tumor mutation burden and driver genes that drive cancer progression. Furthermore, we assessed the oncogenic signaling pathway and the mutational signature prevalent in our population.
Result
Overview of cohort characteristics
In this study, we sequenced 97 primary breast tumors and their corresponding leukocytes to determine the landscape of somatic mutations in the AIIMS cohort. The clinical and pathological details of the 97 individuals who participated in this study are outlined in Table 1, primarily including median age, tumor size, lymph node, metastasis, HR, and Her2 neu receptor status. The age at which BC is diagnosed varies from 27 to 75 years, with an average and median age of 50 and 48 years, respectively. Of 97 patients, 49 (50%) presented with T2 stage, i.e., tumor size in the range between 20 and 50 mm. In 56 cases (58%), tumor cells infiltrated one or more internal mammary nodes or axillary lymph nodes, while 16 cases (17%) were positive for distant metastasis. Her2-negative cases (61%) and hormone receptor (either estrogen or progesterone or both)-enriched cases (52%) were predominantly observed in our cohort, with a maximum number of individuals having the HR+/HER2− molecular subtype.
Landscape of somatic mutations across the cohort
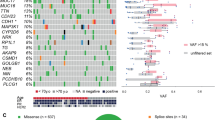
The whole-exome sequencing data revealed that an average of 66.47 million reads per sample were acquired with a 150 bp mean read length. The vast majority of the reads (> 99.34%) were aligned with the human reference genome (GRCh38). We first analyzed the variation in each sample and identified 69,086 mutations, including 65,966 SNVs (60,882 missense, 5010 nonsense, 74 nonstop) and 1,232 small insertions or deletions (766 frameshifts, 466 in-frame). We also detected 1888 variants predicted to affect translation start and splice sites. The above findings showed that missense mutations, SNP variant types, and C > A SNV classes were more prevalent, with 4733 being the highest variant frequency per sample (Fig. 1). We further classified SNPs into transition (ti) and transversion (tv) and found that tv was a more common type of base substitution. The stacked bar plot shows the fraction of six different conversions in each sample (Fig. S1). The summary of the mutation profile of the top 50 mutated genes is described in Fig. 2. TTN (74%), TP53 (59%), MUC16 (55%), SYNE1 (40%), OBSCN (36%), CSMD1 (34%), DST (34%), and PIK3CA (34%) ranked as the top most frequently mutated genes. However, TTN variants are poorly or unlikely associated with cancer. In addition, oncodriveCLUSTL analysis was conducted to investigate the oncogenic driver genes in our cohort. It is based on a positional clustering algorithm and takes advantage of hot spots present across the genomic region. Among the top mutated genes, PIK3CA and KLC3 were discovered to be driver genes that drive tumorigenesis (Fig. 3).

Overview of mutagenesis in AIIMS breast cancer cohort including (A) shows the classification of variants identified in the cohort. The Y-axis represents different variant classifications while the X-axis indicates the number of variants corresponding to each classification. (B) Illustrates the types of variants observed. The Y-axis lists the types of variants including single nucleotide variants (SNVs), insertions, deletions, and indels. (C) The specific classes of SNVs are depicted. The Y-axis includes various SNV classes such as transitions (e.g., T>C & C > T) and transversions (e.g., T>G, T>A, C>G & C>A). (D) The stacked barplot present the number of variants identified in each sample. The Y-axis denotes the number of variants, while the X-axis corresponds to individual samples.

Oncoplot provides a detailed overview of the mutation landscape in the AIIMS breast cancer cohort, focusing on the top 50 highly mutated genes. Each column in the plot represents an individual sample from the AIIMS breast cancer cohort while each row corresponds to one of the top 50 genes. The upper panel displays the mutational burden for each sample and bar graph on the right side of the plot shows the number of samples that have alterations in each gene.

Quantile plot illustrates the driver genes identified by OncodriveCLUSTL in the AIIMS breast cancer. The plot highlights genes predicted to be drivers among the top mutated genes. Among these, PIK3CA and KLC3 were identified as driver genes.
Genomic regions with six or more successive mutations and an average inter-mutation separation of ≤ 1 Kbp were considered to be kataegis. This was observed in the CaBr47 sample, in which hyper-mutations were enriched over chromosomes 1, 3, and 11 (Fig. S2). A detailed distribution of kataegis loci in CaBr47 is provided in Table S1. We further analyzed the tumor mutation burden (TMB) in our patients and compared it with the available datasets of 33 various tumor types in TCGA. It is the ratio of non-inherited mutations per million bases of the genomic sequence. In general, we noted that the mutation burden rate in our patients was higher than that in the other tumor types, followed by skin cutaneous melanoma (SKCM) (Fig. S3).
Association between altered genes
Altered gene‒gene interactions may contribute to the onset or progression of cancer either through synergistic or antagonistic interactions. We next evaluated the mutual exclusivity or co-occurrence of gene variants by performing somatic interaction analysis functions in MAFtools, and Fisher’s exact test was used to examine the significantly associated gene pairs. Mutual exclusivity was observed between mutations in PIK3CA and CCDC168 (OR 0.193704; P value: 0.007106). We also observed co-occurrence of DNAH10 and MUC16 (OR 18.23659, P value: 2.08E−06), IGFN1 and HSPG2 (OR 10.37721, P value: 2.53E−06), TTN and MUC16 (OR 10.88202, P value: 6.68E−06), DNAH10 and MUC16 (OR 18.23659, P value: 0.007106), CSMD1 and HUWE1 (OR 9.481399, P value: 1.10E−05), MUC16 and MDN1 (OR 11.04189, P value: 2.43E−05), AHNAK2 and UTRN (OR 8.190022, P value: 3.01E−05), KMT2D and FAT2 (OR 7.986428, P value: 3.22E−05), HUWE1 and RYR1 (OR 8.078675, P value: 3.95E−05) and HMCN2 and AHNAK2 (OR 7.259601, P value: 5.14E−05) (Fig. 4).

Somatic interaction analysis of the top 50 genes. Co-occurrence and mutual exclusive gene pairs were analyzed with pair-wise Fisher’s exact test. Gene pairs with statistically significant interactions are highlighted by darker color with dots or asterisks depending on whether the p value is < 0.1 or < 0.05.
Oncogenic signaling pathways
Genes were clustered into the pathway, and the OncogenicPathways function in Maftools was employed to obtain the frequency of altered genes involved in the known oncogenic pathway 8. It also calculates the fraction of samples having mutated pathways and ranks the pathway with the highest number of mutant genes. The most enriched oncogenic signaling pathways in breast cancer were TP53 (100%), NRF2 (100%), Hippo (95%), WNT (94%), RTK-RAS (93%), PI3K (90%), TGF-Beta (86%), MYC (85%), NOTCH (83%) and Cell Cycle (80%), and these enrichments were identified in 63, 9, 86, 74, 92, 82, 20, 30, 88 and 24% of samples, respectively (Fig. 5A–B). RTK-RAS was the most commonly altered pathway, encompassing 5 tumor suppressor genes and 33 oncogenes. Of all these genes involved in the RTK-RAS pathway, ROS1 and NF1 were the most immensely mutated in 19 and 12% of samples, respectively (Fig. 5C). The altered genes and their respective samples in the NOTCH, Hippo, and PI3K signaling pathways are shown in Fig. S4.

Pathway enrichment analysis. (A) Bar plot on the left panel shows the fraction of pathways affected while the right panel (B) shows the number of samples affected (C) visualize the complete RTK-RAS pathway. Tumor suppressor genes and oncogenes are indicated in red, and blue font.
Mutational Signatures analysis
Next, we investigated the mutational signature present in our cohort. Generally, mutational processes in tumors can occur through various mechanisms and establish unique patterns known as mutational signatures. Here, we analyze single base substitution (SBS) signatures in which 6 different substitution subtypes (C > A, C > G, C > T, T > A, T > C, and T > G) are used to display each signature's profile. Initially, after generating the trinucleotide matrix, the APOBEC enrichment score was estimated and suggested that APOBEC and non-APOBEC were enriched in 12 and 85 samples, respectively. The tCw motif distribution was found in 13% of APOBEC samples and 3% of non-APOBEC samples. METTL3 and VWCE were the top altered genes in APOBEC enriched sample (Fig. 6).

The plot depicts the overall comparison between APOBEC and non-APOBEC samples. (A) Difference in mutation burden between APOBEC and non-APOBEC enriched samples (B) tCw motifs load in APOBEC samples (C) tCw motifs load in non-APOBEC samples (D) topmost altered genes in non-APOBEC samples.
The cophenetic correlation metric was established to determine the number of signatures. Extracted signatures were compared with the 30 available experimentally validated signatures in the COSMIC database to determine the best match based on their cosine similarity (Fig. S5). With a cosine similarity > 0.68, the top three mutational signatures (SBS13, SBS6, and SBS29) were discovered (Fig. 7). Signature 13 has been linked to the activity of the APOBEC family of cytidine deaminases, which convert cytosine to uracil, while Signature 6 has been attributed to poor DNA mismatch repair and is often seen in microsatellite unstable tumors. Signature 29 has been discovered in tumor samples from individuals exposed to mutagens such as tobacco.

Best match single base substitution (SBS) mutational signatures. The plot highlights the most frequently observed mutational aetiology within the AIIMS breast cancer cohort.
Discussion
Breast cancer (BC) has become a severe public health concern, and its burden impacts the financial, psychological, and physical facets of human life. In women, it is the most frequent cancer, and along with lung and colorectal cancers, it is one of the three most prevalent malignancies worldwide in both sexes. According to a report published by the IARC, BC accounts for over 25% of all new malignancies in women, with 2.3 million women diagnosed globally in 2020. The incidence of BC in 2020 was 0.18 million, with a mortality rate of approximately 51%, and BC is the primary cause of cancer incidence and fatalities among the Indian population 9. Over the past few years, the age-related incidence pattern has significantly shifted. India's BC cases present with a diagnosis at a younger median age (49 years) than the Western population, where the median age at diagnosis is 62 years 10. A similar pattern of median age-related incidence (48 years) was observed in our BC cohort.
According to recent research, ethnicity may significantly influence the genetic propensity to breast cancer. The genetic variants may vary by ethnicity and can be ethnic or race-specific 11,12,13. However, most of today's understanding of the disease's predisposition comes from Western populations. To our knowledge, this is the first study that comprehensively outlines the somatic variants discovered in Indian BC cases. We reported several altered canonical and non-canonical genes. In our cohort, TP53, PIK3CA, KMT2C and ZFHX3 were found to be mutated in 59, 34, 30, and 29% of samples, respectively, compared with 26, 29, 12, and 12% of BC samples in the COSMIC database. Detailed variants of the above mentioned genes are listed in Table S2.
TP53 is a tumor suppressor gene, and its genetic variants (mainly due to frameshift or missense mutations) lead to human malignancies 14. Under normal physiological conditions, it acts as a transcription factor with a pro-apoptotic function, while in diseased conditions, mutational inactivation makes it dysfunctional 15. In our study, the mutation was mainly confined to the kinase region. Mutations in these regions disrupt its configuration and help to gain kinase activity. Thus, in the presence of damaged DNA, it could not arrest the cell cycle and prevent apoptosis, promoting oncogenesis. PIK3CA is an oncogene that plays a significant role in cell signaling, particularly in the phosphoinositide 3-kinase (PI3K) pathway 16. Under normal physiological conditions, PIK3CA regulates various cellular processes, including cell growth, survival, and metabolism. However, when mutated, it can lead to the development of human malignancies 17. PIK3CA mutations are also found in other cancers, such as colorectal cancers (10–15%) and lung cancers (4%) 18,19. Overall, mutations in PIK3CA contribute to the promotion of oncogenesis by driving aberrant cell growth, evading cell death, and enhancing cell survival 20. ZFHX3 is a zinc finger transcription factor-encoding gene that plays a significant role in various cellular processes, including cell differentiation, proliferation, and apoptosis 21. Under normal circumstances, ZFHX3 acts as a tumor suppressor gene, regulates cell growth, prevents the formation of cancerous cells, and modulates the expression of genes involved in key cellular pathways. In diseased conditions, mutational inactivation or loss of ZFHX3 function can occur, disrupting its tumor-suppressive capabilities 22. Mutations in the ZFHX3 gene can lead to increased activity of the protein, which can promote cell growth and proliferation 23. KMT2C is a gene that encodes a histone methyltransferase and plays a crucial role in gene regulation and chromatin remodeling 24. In its normal state, KMT2C helps maintain genomic stability and prevents uncontrolled cell growth by regulating the expression of specific genes involved in cell cycle control and DNA repair 25. Mutations in KMT2C have been identified in various cancers, including gastrointestinal, breast, and ovarian cancers 26. These mutations can lead to functional alterations, affecting the ability of KMT2C to regulate gene expression and chromatin remodeling 27. TMB is the genetic feature of tumorous tissue that varies among patients and tumor types. TMB across 33 different tumor types, including BC (named BRCA), was retrieved from the TCGA database and compared with our cohort. High TMB in the AIIMS cohort correlates with various tumoral immunogenicity and could be a clinical biomarker for immunotherapy response 28,29.
Cancer-causing altered genes show a significant pattern in their co-occurrence and exclusiveness. We performed interaction analysis to test this concurrent and exclusive mutation interaction among the top most altered genes. PIK3CA is one of the driver genes found in our study and was found to be only mutually exclusive with the CCDC168 gene (p < 0.05). CCDC168 (Coiled-Coil Domain Containing 168) is significantly mutated and upregulated in various tumor types, including colorectal cancer and Paget's disease, and is associated with poor prognosis 30,31. The PI3K, NOTCH, and RTK-RAS pathways were the most frequently influenced oncogenic pathways in neuroendocrine breast tumors, while the RTK-RAS, Hippo, and WNT pathways were reported in gastric cancer 32,33. However, in regard to our investigation, the RTK-RAS, WNT, and NOTCH pathways were the most often impacted. Receptor tyrosine kinase (RTK) is one of the most common intracellular signaling pathways that plays an essential role in cell growth, differentiation, and survival 34,35. On the other hand, WNT is a canonical pathway primarily engaged in cell proliferation. In contrast, the Notch pathway is one such juxtacrine signaling pathway that plays a significant role in maintaining progenitor cells, the fate of the cell, differentiation, and cell death 36,37. From a therapeutic perspective, the RTK-RAS and NOTCH pathways are well targeted and can be modulated with FDA-approved drugs 38,39. Conversely, there are no approved chemical compounds that target WNT signaling 40. However, many repurposed drugs or natural compounds are currently in the clinical trial phase 41.
During the tumor formation and development period, multiple mutational events occur, which may contribute to the set of mutational signatures 42. Each signature reveals its mutation etiology, which can be a potential genomic biomarker for personalized therapy. Additionally, it also opens up opportunities for the individual's stratification and for predicting cancer predisposition 43. The vast majority of malignancies are thought to be triggered by somatic mutations 44 acquired either through endogenous (failure of repair mechanism, SBS13 & SBS6) or exogenous (exposure to mutagen, SBS29) mechanisms. Dysregulation of repair machinery or APOBEC activity can lead to hyper-mutation of the DNA that can be linked with the emergence of cancer and represent a major enzymatic source of mutations in cancer. APOBEC3B mRNA shows increased expression in cancers compared to healthy tissue and is associated with disease progression. Additionally, elevated APOBEC3B expression is positively linked with adverse outcomes in ER-positive BC45,46,47. A high rate of mutation is caused by the defect in mismatch repair system, and this can promote the development of cancer. DNA can be damaged by the exposure of mutagen, which can cause cells to grow abnormally. These atypical cells have the potential to develop into cancer. Genomic evidence of SBS13, 6 and 29 was also found in various other malignancies, including BC (Fig. S6). Finally, we could not undertake a survival analysis in this cohort due to the patients' short follow-up duration. Future research will address these issues, and the understanding gained from the present study will assist us in achieving appropriate patient stratification.
Conclusion
In summary, we performed WES on 97 BC cases along with matched blood samples from an Indian population and characterized genes with high somatic mutation frequencies. We also identified several genes with increased mutational frequency in contrast with other BC cases in the COSMIC database. Furthermore, several top mutated genes were enriched in different oncogenic signaling pathways that were primarily responsible for the pathogenesis and development of BC. Mutational signatures SBS13, SBS6 and SBS29 were predominantly observed in our cohort. Moreover, these findings encourage additional investigation to determine the functional significance of altered genes in the development of BC.
Methodology
Sample collection
A total of 97 tumor samples (of a pea size) and 97 matched blood samples (~ 5 ml) were collected in cryovials and EDTA tubes, respectively. Tumor samples and processed blood samples were stored at − 80 °C for further DNA extraction. Clinico-pathological data were retrieved from the patient's records, followed by a questionnaire. All samples were collected after obtaining consent from patients or first-degree relatives after obtaining approval from the institute's ethical committees.
Histological classification
A certified pathologist reviewed all the collected tissue samples for their hormone (estrogen and progesterone) and Her2neu receptor status. Prepared histological slides were confirmed as per Allred scoring (from 0 to 8) of immunohistochemistry (IHC)-stained slides. IHC slides were scored as negative (score 0) or positive (score 1+) for ER or PR. For Her2neu, IHC slides were classified as negative (score 0 or 1), equivocal (score 2), and positive (score 3+). Equivocal IHC slides were confirmed by fluorescence in situ hybridization (FISH).
Library preparation and sequencing
Blood samples (2–5 ml) were collected intravenously from the enrolled patients and then processed as our previously described protocol48. We sequenced 97 tumors and matched 97 peripheral blood samples from the same individuals. After an initial assessment of DNA, a concentration of 200 ng DNA was used for library preparation by using the Agilent SureSelectXT Target Enrichment System. In brief, gDNA was sheared using the Covaris instrument, and the quality assessment was performed using a Bioanalyzer assay. Fragmented DNA of approximately 150–200 bp was used for the end repair reaction, dA-tailing at the 3’ end, adapter ligation, and amplification of the adapter-ligated library, followed by analysis of the amplified library by using a Bioanalyzer assay. Between each successive step, samples were purified using AMPure XP beads, and all these steps were executed as per instructions provided by the manufacturer manual.
Once gDNA libraries were prepared, hybridization was carried out to capture target-specific regions (i.e., exons) by using a probe synthesized by Agilent. Hybridized libraries were separated from non-hybridized libraries by using streptavidin-coated beads. The captured libraries were amplified with indexing primers and purified by AMPure XP beads. Postcapture analysis of the indexed library was evaluated by using a Bioanalyzer and high-sensitivity DNA assay. Finally, the enriched library was paired end sequenced on an Illumina HiSeq 2000, with an average read depth of 100X for blood samples and 200X for tissue samples. It will generates total sequence of more than 35 million and 80 million for blood and tissue samples respectively with a length of 150 bases.
Bioinformatics analysis
The FASTQ raw reads of 97 tumor and corresponding blood samples were quality checked using FastQC v0.11.9, Trimmomatic v0.39 and MultiQC 49,50. Low-quality reads (Phred score < 30), contaminants and adaptors were discarded or trimmed. The quality filtered reads were mapped to the human reference genome (GRCh38) by employing Burrows‒Wheeler Aligner (BWA v0.6) MEM algorithms51,52. The resulting sorted mapped BAM files were mark duplicate by using Picard MarkDuplicates followed by Base Quality Score Recalibration (BQSR) implemented with the Genome Analysis Toolkit (v4.1.9.0) 52. Until this stage, the tools and parameters utilized in the preprocessing analysis have been consistent for both blood and tissue samples. Subsequently, a Panel of Normals (PoN) was generated by employing data from 97 blood samples, and used as a reference in somatic variant calling. The Mutect2 program used tumor bam, its corresponding control normal bam and a panel of normal bam for detecting raw somatic variants54. The raw variants were annotated using SnpEff, Clinvar and ANNOVAR to remove the potential artifacts. We utilize ANNOVAR having more than 20 SNP prediction tools, including SnpEff, SIFT, Polyphen2, Mutation Taster, Mutation Assessor, FATHMM, PROVEAN, and CADD, among others, to predict the adverse effect of variants. Furthermore, we employed InterVar for the clinical interpretation of genetic variants, based on the ACMG/AMP 2015 guideline. In the ANNOVAR program, databases such as gnomAD, ExAC03, avSNP150 and dbnsfp42 have been used to filter germline variant and the variants that are common in the general population are less likely to be pathogenic. This comprehensive approach allows us to identify variants deemed deleterious and pathogenic with increased confidence. The annotated somatic variants of 97 samples were processed using maftools Bioconductor packages to identify highly mutated genes, generate oncoplots, and map TCGA cohorts, driver mutations and COSMIC signatures55,56.
Informed consent
We received informed consent from all of the cases involved in the study.
Institutional review board statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Ethics Committee of All India Institute of Medical Sciences for a study involving humans.
Data availability
The datasets generated during the current study are available in the NCBI repository with BioProject ID- PRJNA935139 & PRJNA1005058.
Code availability
No custom code was used.
Abbreviations
- APOBEC:
-
Apolipoprotein B mRNA-editing enzyme, catalytic polypeptide
- BQSR:
-
Base Quality Score Recalibration
- BC:
-
Breast cancer
- BWA:
-
Burrows‒Wheeler Aligner
- COSMIC:
-
Catalogue of Somatic Mutations in Cancer
- gDNA:
-
Genomics DNA
- HTS:
-
High throughput sequencing
- IARC:
-
International Agency for Research on Cancer
- NGS:
-
Next-generation sequencing
- PoN:
-
Panel of Normals
- SBS:
-
Single base substitution
- SNPs:
-
Single nucleotide polymorphisms
- SNVs:
-
Single nucleotide variants
- TMB:
-
Tumor mutation burden
- WES:
-
Whole exome sequencing (WES)
References
Mathur, P. et al. Cancer statistics, 2020: Report from national cancer registry programme, India. JCO Glob. Oncol. 6, 1063–1075 (2020).
Hirko, K. A. et al. Trends in breast cancer incidence rates by age and stage at diagnosis in Gharbiah, Egypt, over 10 Years (1999–2008). J. Cancer Epidemiol. 2013, 916394 (2013).
Pereira, B. et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 7, 11479 (2016).
Network, C. G. A. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012).
Stephens, P. J. et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 486, 400–404 (2012).
Lamy, P.-J. & Jacot, W. Worldwide variations in EGFR somatic mutations: A challenge for personalized medicine. Diagn. Pathol. 7, 13 (2012).
Choi, M. et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. U. S. A. 106, 19096–19101 (2009).
Sanchez-Vega, F. et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 173, 321–337 (2018).
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Parmar, V. Rising incidence of breast cancer in the young fertile Indian population—a reality check. Indian J. Surg. Oncol. 9, 296–299 (2018).
Whitman, S., Ansell, D., Orsi, J. & Francois, T. The racial disparity in breast cancer mortality. J. Community Health 36, 588–596 (2011).
Henderson, B. E., Lee, N. H., Seewaldt, V. & Shen, H. The influence of race and ethnicity on the biology of cancer. Nat. Rev. Cancer 12, 648–653 (2012).
Ellis, L. et al. Racial and ethnic disparities in cancer survival: The contribution of tumor, sociodemographic, institutional, and neighborhood characteristics. J. Clin. Oncol. 36, 25 (2018).
Ozaki, T. & Nakagawara, A. Role of p53 in cell death and human cancers. Cancers 3, 994–1013 (2011).
Olivier, M., Hollstein, M. & Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2, a001008 (2010).
Hou, P. et al. Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/Akt pathway in thyroid cancer. Clin. Cancer Res. 13, 1161–1170 (2007).
Hoxhaj, G. & Manning, B. D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 20, 74–88 (2020).
Rosty, C. et al. PIK3CA activating mutation in colorectal carcinoma: Associations with molecular features and survival. PloS One 8, e65479 (2013).
Kawano, O. et al. PIK3CA mutation status in Japanese lung cancer patients. Lung Cancer 54, 209–215 (2006).
Shortt, J. & Johnstone, R. W. Oncogenes in cell survival and cell death. Cold Spring Harb. Perspect. Biol. 4, a009829 (2012).
Sun, X. et al. Deletion of atbf1/zfhx3 in mouse prostate causes neoplastic lesions, likely by attenuation of membrane and secretory proteins and multiple signaling pathways. Neoplasia 16, 377–389 (2014).
Hu, Q. et al. ZFHX3 is indispensable for ERβ to inhibit cell proliferation via MYC downregulation in prostate cancer cells. Oncogenesis 8, 28 (2019).
Dong, G. et al. ZFHX3 promotes the proliferation and tumor growth of ER-positive breast cancer cells likely by enhancing stem-like features and MYC and TBX3 transcription. Cancers 12, 3415 (2020).
García-Sanz, P. et al. Chromatin remodelling and DNA repair genes are frequently mutated in endometrioid endometrial carcinoma. Int. J. Cancer 140, 1551–1563 (2017).
Poreba, E., Lesniewicz, K. & Durzynska, J. Histone–lysine N-methyltransferase 2 (KMT2) complexes-a new perspective. Mutat. Res. Rev. Mutat. Res. 790, 108443 (2022).
Zhou, C. et al. Difference of molecular alterations in HER2-positive and HER2-negative gastric cancers by whole-genome sequencing analysis. Cancer Manag. Res. 10, 3945–3954 (2018).
Hoffmann, M. J. & Schulz, W. A. Alterations of chromatin regulators in the pathogenesis of urinary bladder urothelial carcinoma. Cancers 13, 6040 (2021).
O’Meara, T. A. & Tolaney, S. M. Tumor mutational burden as a predictor of immunotherapy response in breast cancer. Oncotarget 12, 394 (2021).
Sammons, S. et al. Genomic evaluation of tumor mutational burden-high (TMB-H) versus TMB-low (TMB-L) metastatic breast cancer to reveal unique mutational features. J. Clin. Oncol. 39, 1091–1091 (2021).
He, Y., Dai, X., Chen, Y. & Huang, S. Comprehensive analysis of genomic and expression data identified potential markers for predicting prognosis and immune response in CRC. Genet. Res. 2022, e45 (2022).
Zhang, G. et al. Whole-exome sequencing reveals frequent mutations in chromatin remodeling genes in mammary and extramammary Paget’s diseases. J. Investig. Dermatol. 139, 789–795 (2019).
Karihtala, P. et al. Comparison of the mutational profiles of neuroendocrine breast tumours, invasive ductal carcinomas and pancreatic neuroendocrine carcinomas. Oncogenesis 11, 53 (2022).
Zagami, C., Papp, D., Daddi, A. A. & Boccellato, F. Morphogen signals shaping the gastric glands in health and disease. Int. J. Mol. Sci. 23, 3632 (2022).
Du, Z. & Lovly, C. M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 17, 1–13 (2018).
Aster, J. C., Pear, W. S. & Blacklow, S. C. The varied roles of notch in cancer. Annu. Rev. Pathol. Mech. Dis. 12, 245–275 (2017).
Dontu, G. et al. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 6, 1–11 (2004).
Sueda, R. & Kageyama, R. Regulation of active and quiescent somatic stem cells by Notch signaling. Devel. Growth Differ. 62, 59–66 (2020).
Yuan, X. et al. Notch signaling: An emerging therapeutic target for cancer treatment. Cancer Lett. 369, 20–27 (2015).
Regad, T. Targeting RTK signaling pathways in cancer. Cancers 7, 1758–1784 (2015).
Kahn, M. Can we safely target the WNT pathway?. Nat. Rev. Drug Discov. 13, 513–532 (2014).
Neiheisel, A., Kaur, M., Ma, N., Havard, P. & Shenoy, A. K. Wnt pathway modulators in cancer therapeutics: An update on completed and ongoing clinical trials. Int. J. Cancer 150, 727–740 (2022).
Alexandrov, L. B. et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020).
Van Hoeck, A., Tjoonk, N. H., van Boxtel, R. & Cuppen, E. Portrait of a cancer: Mutational signature analyses for cancer diagnostics. BMC Cancer 19, 1–14 (2019).
Stratton, M. R., Campbell, P. J. & Futreal, P. A. The cancer genome. Nature 458, 719–724 (2009).
Torgovnick, A. & Schumacher, B. DNA repair mechanisms in cancer development and therapy. Front. Genet. 6, 157 (2015).
Salter, J. D., Bennett, R. P. & Smith, H. C. The APOBEC protein family: United by structure, divergent in function. Trends Biochem. Sci. 41, 578–594 (2016).
Petljak, M. et al. Molecular origins of APOBEC-associated mutations in cancer. DNA repair 94, 102905 (2020).
Kumar, R. et al. Whole exome sequencing identifies novel variants of PIK3CA and validation of hotspot mutation by droplet digital PCR in Breast cancer among Indian Population. Cancer Cell Int. 23, 236 (2023).
Andrews, S. FastQC: A quality control tool for high throughput sequence data (2017).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 43, 1 (2013).
Benjamin, D. et al. Calling somatic SNVs and indels with Mutect2. BioRxiv (2019).
Mayakonda, A., Lin, D.-C., Assenov, Y., Plass, C. & Koeffler, H. P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 28, 1747–1756 (2018).
Tamborero, D., Gonzalez-Perez, A. & Lopez-Bigas, N. OncodriveCLUST: Exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics 29, 2238–2244 (2013).
Acknowledgements
We acknowledge the overall guidance of Dr Rita Mulherkar as chairperson of the review committee of Breast Cancer TF under the grant 5/13/1/TF/AIIMS/2016/NCD-III Indian Council of Medical Research. We appreciate the patients for their consent to participate in this study. We also thanked resident doctors of Dr. B.R.A. Institute Rotary Cancer Hospital and the Department of Surgery, AIIMS for their cooperation and coordination in sample collection.
Funding
This work was supported by the Indian Council of Medical Research through grant 5/13/1/TF/AIIMS/2016/NCD-III. RahK and HG thank the University Grant Commission and Council of Scientific & Industrial Research (CSIR), respectively, for financial support. SA and RakK thank the Indian Council of Medical Research for financial assistance.
Author information
Authors and Affiliations
Contributions
Rahul.K., S.A., H.G., Rakesh.K., J.S., I.H., S.L. did collection of biological samples & conducted wet lab experiments; Rahul.K., S.A. & D.P.: Bioinformatic analysis; S.V.S.D., A.S., A.G. and Atul.B., C.K., Rakesh.K., Amar.B., P.A.: Breast cancer data for cases of genomics and epidemiological studies patient data at AIIMS & Safdarjung Hospital, New Delhi; P.T., S.L., U.A., A.C., A.R.: supervised and helped in wet lab experiments preservation of samples; S.M. & U.A. conducted pathological diagnosis; S.H., U.A., P.T., A.M., G.K.R., T.K., R.S.D.: Critical inputs in overall planning and compilation and analysis of data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kumar, R., Awasthi, S., Pradhan, D. et al. Somatic mutational landscape across Indian breast cancer cases by whole exome sequencing. Sci Rep 14, 18679 (2024). https://doi.org/10.1038/s41598-024-65148-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-65148-4
Keywords
This article is cited by
-
Network pharmacology and AI in cancer research uncovering biomarkers and therapeutic targets for RALGDS mutations
Scientific Reports (2025)
-
Genetic changes that drive breast cancer among Indians
Nature India (2024)
