
Abstract
The COVID-19 pandemic has underscored the critical need for precise diagnostic methods to distinguish between similar respiratory infections, such as COVID-19 and Mycoplasma pneumoniae (MP). Identifying key biomarkers and utilizing machine learning techniques, such as random forest analysis, can significantly improve diagnostic accuracy. We conducted a retrospective analysis of clinical and laboratory data from 214 patients with acute respiratory infections, collected between October 2022 and October 2023 at the Second Hospital of Nanping. The study population was categorized into three groups: COVID-19 positive (n = 52), MP positive (n = 140), and co-infected (n = 22). Key biomarkers, including C-reactive protein (CRP), procalcitonin (PCT), interleukin- 6 (IL-6), and white blood cell (WBC) counts, were evaluated. Correlation analyses were conducted to assess relationships between biomarkers within each group. The random forest analysis was applied to evaluate the discriminative power of these biomarkers. The random forest model demonstrated high classification performance, with area under the ROC curve (AUC) scores of 0.86 (95% CI: 0.70–0.97) for COVID-19, 0.79 (95% CI: 0.64–0.92) for MP, 0.69 (95% CI: 0.50–0.87) for co-infections, and 0.90 (95% CI: 0.83–0.95) for the micro-average ROC. Additionally, the precision-recall curve for the random forest classifier showed a micro-average AUC of 0.80 (95% CI: 0.69–0.91). Confusion matrices highlighted the model’s accuracy (0.77) and biomarker relationships. The SHAP feature importance analysis indicated that age (0.27), CRP (0.25), IL6 (0.14), and PCT (0.14) were the most significant predictors. The integration of computational methods, particularly random forest analysis, in evaluating clinical and biomarker data presents a promising approach for enhancing diagnostic processes for infectious diseases. Our findings support the use of specific biomarkers in differentiating between COVID-19 and MP, potentially leading to more targeted and effective diagnostic strategies. This study underscores the potential of machine learning techniques in improving disease classification in the era of precision medicine.
Similar content being viewed by others


Introduction
Acute respiratory infections (ARIs) significantly contribute to global morbidity and mortality, particularly in developing nations1. Lower respiratory infections were the fourth leading cause of death in 2019, causing approximately 2.5 million deaths, mainly affecting children and older adults2,3,4,5. The COVID-19 pandemic has changed the epidemiology of respiratory viruses and Mycoplasma pneumoniae (MP)6. MP, a major pathogen, contributed significantly to pneumonia epidemics in Beijing between 2015 and 20207. The pandemic highlighted the need for precise diagnostics to differentiate diseases with overlapping symptoms like pneumonia. Differentiating COVID-19 from pneumonia, especially co-infections, is vital for proper diagnosis and improved outcomes. Biomarkers are key in identifying disease pathways and guiding therapies.
Biomarkers, including clinical and molecular indicators, are crucial for diagnosing ARIs, assessing disease severity, and monitoring treatment responses8. For COVID-19 and MP, biomarkers like C-reactive protein (CRP), procalcitonin (PCT), and interleukin 6 (IL-6) are key to understanding disease progression and outcomes9. Del Valle et al. found an inflammatory cytokine signature, including IL-6, predicting COVID-19 severity and survival10. The varying expression of these biomarkers among patient groups offers insights into underlying mechanisms, forming the basis for tailored diagnostics11.
Studies show machine learning’s effectiveness in medical diagnostics. Wynants et al. reviewed COVID-19 prediction models, highlighting machine learning’s potential12. Recent advancements in computational biology and machine learning have revolutionized medical research13. Machine learning, a subset of artificial intelligence, develops algorithms enabling computers to learn from and make predictions or decisions based on data14. These techniques identify patterns in large datasets not apparent through traditional methods15. High-dimensional data refers to datasets with numerous features, complicating analysis and model building due to the curse of dimensionality. Random forest is an ensemble learning method that constructs multiple decision trees during training and outputs the mode of the classes for classification or the mean prediction for regression16. It excels in handling high-dimensional data and managing overfitting via the ensemble approach, making it ideal for exploring relationships between clinical features and disease states17,18. Compared to other algorithms: support vector machines are effective but sensitive to kernel choice and computationally intensive; neural networks require extensive tuning and large datasets; logistic regression struggles with complex relationships; K-nearest neighbors is computationally expensive and less effective in high-dimensional spaces19. Random forest highlights feature importance and handles variable interactions, making it suitable for our study.
Studies emphasize the importance of biomarkers in managing COVID-19; however, their role in ARIs involving COVID-19 and MP in China remains underexplored. Moreover, there is a gap in using machine learning to differentiate between COVID-19, MP, and co-infections. We used the random forest algorithm to analyze a cohort of patients with COVID-19, MP, and co-infections, focusing on key biomarkers and clinical features. Utilizing a multidimensional dataset of demographic and clinical parameters, our analysis incorporated statistical methods, machine learning, and feature importance analysis to identify key biomarkers in ARI patients. This study aims to enhance infectious diagnostics by integrating computational methods with clinical data, ultimately advancing precision medicine strategies.
Results
Descriptive statistics and group characteristics
Shapiro-Wilk normality tests for each group yielded P-values less than 0.05, indicating non-normal distributions and justifying the use of non-parametric methods in subsequent analyses.
The study population consisted of 214 patients categorized into three groups: COVID-19 positive (n = 52), MP positive (n = 140), and co-infected with both (n = 22). The baseline characteristics of the participants are summarized in Tables 1 and 2, with statistical analysis using Python 3.7. Differences in demographic and clinical characteristics between the groups were analyzed using the Kruskal-Wallis H test for continuous variables and the Chi-square test for categorical variables.
The median age differed significantly among the groups: COVID-19 (74.0 years), MP (6.0 years), and co-infected (34.5 years) (P < 0.001). No significant difference was observed in the sex distribution among the three groups (P = 0.861).
Significant differences were noted in the levels of CRP, PCT, and IL-6 across the groups, with P-values of < 0.001, 0.003, and 0.017, respectively as determined by the Kruskal-Wallis H test. WBC counts did not show significant variation (P = 0.886) (Table 1).
Biomarker correlation analysis
The correlation analysis (Figs. 1, 2 and 3) illustrates the relationships between key biomarkers and demographic variables for each group: COVID-19, MP, and co-infected.

Correlation heatmap of continuous variables in COVID-19 positive group. WBC, white blood cell count; CRP, C-reactive Protein; PCT, procalcitonin; IL6, interleukin-6. A positive correlation between CRP and WBC (0.46). A moderate correlation between CRP and Age (0.29). Other correlations were relatively low, indicating weak or no linear relationship among those variables in this group.

Correlation heatmap of continuous variables in MP positive group. MP, Mycoplasma pneumoniae; WBC, white blood cell count; CRP, C-reactive protein; PCT, procalcitonin; IL6, interleukin-6. A positive correlation between CRP and WBC (0.46). A positive correlation between CRP and WBC (0.46), CRP and Age (0.29).

Correlation heatmap of continuous variables in co-infected group. WBC, white blood cell count; CRP, C-reactive protein; PCT, procalcitonin; IL6, interleukin-6. A positive correlation between CRP and Age (0.46), CRP and WBC (0.48).
In the COVID-19 cohort (Fig. 1): CRP showed a moderate positive correlation with WBC (r = 0.46) and age (r = 0.29).
In the MP cohort (Fig. 2): IL-6 correlated with WBC counts (r = 0.29) and PCT (r = 0.21). Additionally, there were correlations between CRP and IL-6 (r = 0.19) and CRP and PCT (r = 0.11).
In the Co-infected Cohort (Fig. 3): CRP correlated positively with WBC (r = 0.48) and age (r = 0.46).
Random forest model performance
In this study, the random forest classifier categorized patients into three distinct classes based on their disease status: Class 1, COVID-19 positive; Class 2, MP positive; and Class 3, co-infected with both. The ROC curves for each class demonstrated the model’s classification efficacy. The area under the ROC curve (AUC), which ranges from 0 to 1 with 1 indicating perfect classification performance, was 0.86 (95% CI: 0.70–0.97) for COVID-19, 0.79 (95% CI: 0.64–0.92) for MP, 0.69 (95% CI: 0.50–0.87) for co-infections, and 0.90 (95% CI: 0.83–0.95) for micro-average ROC curve (Fig. 4).

Receiver operating characteristic (ROC) curves for the multi-class classification performance of the random forest model. 95% CI, 95% confidence interval; class 1 (COVID-19 positive), AUC = 0.86 (95% CI: 0.70–0.97); class 2 (Mycoplasma pneumoniae positive), AUC = 0.79 (95% CI: 0.64–0.92); class 3 (co-infected), AUC = 0.69 (95% CI: 0.50–0.87); micro-average ROC curve: AUC = 0.90 (95% CI: 0.83–0.95).
Precision-recall curve analysis: Fig. 5 displays the precision-recall curves for the random forest classifier, including the micro-average curve, which summarizes the classifier’s overall performance with an AUC of 0.80 (95% CI: 0.69–0.91). The individual curves for each class reveal the classifier’s ability to distinguish between them. Class 2 (MP) achieved the highest AUC of 0.87 (95% CI: 0.75–0.97), indicating strong performance. Class 1 (COVID-19) had an AUC of 0.71 (95% CI: 0.45–0.92), while Class 3 (co-infected) had the lowest AUC of 0.40 (95% CI: 0.08–0.73), highlighting difficulties in accurately identifying instances of Class 3.

Precision-recall curve for the random forest classifier. 95% CI, 95% confidence interval; class 1 (COVID-19 positive), AUC = 0.71 (95% CI: 0.45–0.92); class 2 (Mycoplasma pneumoniae positive), AUC = 0.87 (95% CI: 0.75–0.97); class 3 (Co-infected), AUC = 0.40 (95% CI: 0.08–0.73); micro-average ROC curve, AUC = 0.80 (95% CI: 0.69–0.91).
Confusion matrix: The confusion matrix (Fig. 6) illustrates the model’s predictive accuracy, with the highest correct classification rates for the MP group (precision = 0.85, recall = 0.89). The overall accuracy was 0.77, with the F1-score as follows: COVID-19 (0.62), MP (0.87), and co-infections (0.00). These metrics indicate robust performance in classifying COVID-19 and MP patients, with improvement needed for the co-infection group (Table 3).

Confusion matrix for the random forest classifier’s performance in predicting patient groups. MP, Mycoplasma pneumoniae; COVID-19 positive group shows 9 correctly classified, 3 misclassified as MP positive, and none as co-infected. MP positive group shows 41 correctly classified, 5 misclassified as COVID-19 positive, and none as co-infected. Co-infected group shows 0 correctly classified, 3 misclassified as COVID-19 positive, and 4 as MP positive. Overall accuracy of the model is 0.77, demonstrating moderate classification accuracy for COVID-19 and MP groups but significant misclassification in co-infected cases.
Feature importance (Table 4): Age, CRP, IL-6, and PCT were the most significant predictors in distinguishing patient groups using random forest analysis (Fig. 7). SHAP analysis confirmed these findings, indicating that age and CRP had the highest impact, while IL-6 and PCT also played significant roles in certain contexts (Fig. 8).

Feature importance in random forest classifier. PCT, procalcitonin; WBC, white blood cell count; CRP, C-reactive protein; IL-6, interleukin-6; ADV, adenovirus; PIV, human parainfluenza viruses; FLUA, influenza A virus; RSV, respiratory syncytial virus. This bar chart displays the importance of each feature in the random forest classifier. The height of each bar represents the significance of the feature.

Feature importance in SHAP analysis. SHAP, Shapley additive explanations; PCT, procalcitonin; CRP, C-reactive protein; WBC, white blood cell count; IL-6, interleukin-6; ADV, adenovirus; PIV, human parainfluenza viruses; FLUA, influenza A virus; RSV, respiratory syncytial virus. The height of each bar represents the mean SHAP value, indicating the contribution of each feature to the model’s predictions.
SHAP summary plot
The SHAP summary plot (Fig. 8) visualizes feature contributions. Age had the highest impact on predictions (SHAP value: 0.27), followed by CRP (0.25), PCT (0.14), and IL-6 (0.14). This analysis elucidates the intricate relationships between the features and their combined effect on the classification.
Discussion
In this study, elevated levels of CRP, PCT, and IL-6 in COVID-19 patients indicate a more intense inflammatory response compared to MP and co-infected groups. CRP signifies severe inflammation, PCT suggests bacterial co-infection, and elevated IL-6 indicates a cytokine storm common in severe COVID-19 cases. These biomarkers help differentiate between COVID-19, MP, and co-infections, providing insights into infection severity and guiding clinical diagnostics and management strategies.
Our findings indicated significant differences in key biomarkers (age, CRP, IL-6, PCT) across different patients with ARIs. Elevated CRP and IL-6 in COVID-19 and co-infected groups highlight their role in inflammation, aligning with previous studies that link these markers to severe COVID-19 cases10,20,21. For instance, Del Valle et al. identified an inflammatory cytokine signature, including elevated IL-6, which predicted COVID-19 severity and survival10. Moore and June also reported that cytokine release syndrome plays a significant role in severe COVID-19 cases, further emphasizing the relevance of IL-620. Distinguishing COVID-19 from MP using these biomarkers is crucial for accurate diagnostics and improved patient outcomes22. Age was a significant differentiator among groups, with the highest median age in the COVID-19 group, followed by co-infected and MP groups. This finding aligns with literature highlighting the severe impact of COVID-19 on older populations, emphasizing the need for targeted interventions13,23.
The random forest algorithm showed robust classification performance24 with high AUC scores for COVID-19 and MP groups. The model’s capability to manage high-dimensional data and highlight influential factors underscores the utility of machine learning in biomedical research12,25. Wynants et al. reviewed various prediction models for COVID-19 outcomes, highlighting the potential of machine learning models in clinical settings12. Integrating age, CRP, IL-6, and PCT into diagnostic models can enhance disease classification precision and diagnostic strategies12,25.
Precision-recall curves demonstrate the random forest classifier’s overall performance with a micro-average AUC of 0.80. However, the low AUC for co-infections (0.40) highlights the model’s limited effectiveness in this subgroup. This could be due to the small sample size of the co-infected group (n = 22), which restricts the model’s ability to learn distinctive patterns. Additionally, overlapping clinical presentations of co-infected patients with those of COVID-19 or MP alone contribute to the challenge. To improve the model, future research should consider increasing the sample size of co-infected cases and incorporating additional biomarkers. Advanced techniques, such as ensemble learning, could also enhance classification accuracy. Accurate identification of co-infections is vital for effective clinical diagnosis and treatment.
Random forest feature importance analysis identified age, CRP, IL-6, and PCT as key predictors. SHAP analysis confirmed these findings, emphasizing age and CRP, and the context-specific importance of IL-6 and PCT. Random forest feature importance can overestimate the significance of features higher in the trees due to its method of averaging impurity decreases, not accounting for feature interactions. In contrast, SHAP values consider each feature’s impact on predictions, including interactions, leading to a more accurate ranking. This often results in a different ranking compared to random forest importance scores.
Limitations and future directions
While our study offers valuable insights, it has limitations. First, the sample size, though substantial, represents a single-center cohort, limiting the generalizability. Future studies should use multi-center data to validate these findings across diverse populations26. Beam and Kohane stressed the importance of multi-center studies and large datasets in validating machine learning models in healthcare26.
Second, the study focused on a limited set of biomarkers. We identified significant biomarkers like CRP, IL-6, and PCT, but may have overlooked others. Future research should explore more biomarkers, including genomic and proteomic data, to better understand the pathophysiology of COVID-19 and MP.
Third, the random forest model, despite its robustness, struggles with high-dimensional data when there are few variables. The limited diversity of decision trees may impact performance. Future research should expand the variable set and compare results with other machine learning algorithms to improve robustness.
Fourth, the retrospective design of this study introduces biases like incomplete or inconsistent data. We reviewed and validated the data using robust statistical methods to mitigate these biases. However, residual bias may still exist. Future prospective studies are recommended to confirm our findings and provide a more comprehensive understanding.
Finally, addressing missing data is crucial. In this study, missing values for continuous variables were imputed using median values, and categorical data were imputed using mode values. Sensitivity analyses assessed the impact of these imputations on the study’s findings.
Methods and materials
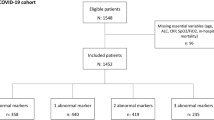
This study aims to distinguish between COVID-19 and MP infections using clinical and biomarker data analyzed through random forest analysis. Our research focuses on identifying significant biomarkers and clinical features that can accurately classify patients into COVID-19 positive, MP positive, and co-infected groups. The process flow of this study is illustrated in Fig. 9.

Study design and patient selection flowchart.
Study design and participants
This retrospective study was conducted at the Department of Clinical Laboratory, Second Hospital of Nanping in Fujian Province, China. We reviewed medical records of 214 patients with ARIs diagnosed between October 2022 and October 2023. The study population was divided into three groups: COVID-19 positive (n = 52), MP positive (n = 140), and co-infected with both COVID-19 and MP (n = 22). Diagnosis of MP followed the Chinese Medical Association guidelines, which included clinical symptoms, chest X-ray abnormalities, and positive seroconversion results for MP antibodies. Diagnosis of COVID-19 was based on a positive reverse transcriptase-polymerase chain reaction (RT-PCR) test for SARS-CoV-2 (severe acute respiratory syndrome coronavirus)27.
Inclusion criteria
-
(1)
Diagnosis of ARIs: Patients diagnosed with ARIs between October 2022 and October 2023.
-
(2)
Specific pathogen groups: Patients must belong to one of the following groups:
COVID-19 positive (confirmed by RT-PCR test for SARS-CoV-2); MP positive (confirmed by clinical symptoms, chest X-ray abnormalities, and positive seroconversion results for MP antibodies); Co-infected with both COVID-19 and MP;
-
(3)
Community-acquired pneumonia (CAP): Patients must meet the criteria for CAP as per the guidelines by the Respiratory Society of the Chinese Medical Association for adults (2016)28 and children (2013)29;
-
(4)
Hospitalization: Patients who were admitted to the hospital for acute respiratory tract infection, including COVID-19 and/or MP infections, within the last three months from the date of diagnosis.
Exclusion criteria
-
(1)
Underlying conditions: Patients with underlying conditions that could confound the study results, including: immunodeficiency disorders, asthma, allergic diseases, infections other than respiratory infections;
-
(2)
Special populations: Pregnant or lactating women;
-
(3)
Mental health: Individuals with severe mental disorders;
-
(4)
Other conditions: Patients with other significant medical conditions that could interfere with the study, such as chronic obstructive pulmonary disease (COPD) or other chronic respiratory conditions.
Biomarkers and nucleic acid measurements
Blood samples were taken from peripheral veins before the initiation of antibiotic therapy. This approach ensures that the measured biomarkers and pathogens are not influenced by prior treatment, providing accurate baseline values. Biomarkers such as CRP, PCT, IL-6, WBC, and IgM antibodies against MP were measured within 24 h of admission.
These measurements included PCT, CRP, IL-6, and WBC. Serum PCT levels were measured using the Getein MAGICL 6000 Chemiluminescence analyzer with accompanying reagents (Jiangsu, China). CRP levels were measured using the Genrui PA300 fully automatic Specific Protein Analyzer with accompanying reagents (Shenzhen, China). IL-6 levels were measured using the Beijing Hotgen Automatic Chemiluminescence Immunoassay Analyzer C2000 with accompanying reagents (Beijing, China). Furthermore, the Sysmex XN-9000 hematology analyzer (Sysmex Corporation, Kobe, Japan) was utilized for conducting a complete blood cell count, including measurements of WBC. The direct chemiluminescence method was employed to detect IgM antibodies against MP in accordance with the instructions of the kit. Antibody levels were measured using a chemiluminescence immunoassay analyzer (iFlash 3000, YHLO, China). These measurements were performed in accordance with standard procedures. According to the manufacturer’s instructions, CRP values above 8.0 mg/L, WBC counts above 10.0 × 109/L, PCT levels above 0.5 ng/mL, and IL-6 levels above 0.7 pg/mL were considered positive.
Before initiating anti-infective therapy upon admission, a nasopharyngeal swab was collected for the detection of coronavirus and respiratory virus nucleic acids. The respiratory viruses included human respiratory syncytial virus (RSV), adenovirus (ADV), human metapneumovirus (HMPV), human parainfluenza virus types 1, 2, and 3 (PIV-1,PIV-2, PIV-3), and influenza virus A (FLUA) and B (FLUB). The nucleic acids of these viruses were detected using the Shanghai Hongshi SLAN-96P Real-Time Quantitative Polymerase Chain Reaction Instrument (Shanghai, China) with reagents from the BioGerm nucleic acid detection kit (fluorescent PCR method) (Shanghai, China). The SARS-CoV-2 nucleic acid was detected using the Applied Biosystems 7500 Real-Time PCR System (USA) with reagents from the Chongqing Zhongyuan SARS-CoV-2 Nucleic Acid Detection Kit (PCR-Fluorescent Probe Method) (Chongqing, China). Results for SARS-CoV-2 and the respiratory viruses were interpreted as positive when the cycle threshold (Ct) values of the N gene (nucleocapsid) and ORF1ab gene (open reading frame 1a and 1b) were below 40, in accordance with the latest guideline in China (Trial 9th version)30 .
To ensure the accuracy and reliability of our measurements, we used validated measurement techniques and regularly calibrated the equipment. All equipment was calibrated according to the manufacturer’s instructions, and regular maintenance checks were performed to ensure consistent performance. Validation studies confirmed the accuracy and precision of the assays used.
Statistical analysis
Statistical analysis was conducted using the Python3.7 programming environment (Python Software Foundation). Continuous variables were initially tested for normal distribution using the Shapiro-Wilk test, which is particularly effective for detecting deviations from normality in small sample sizes. Categorical variables are presented as numbers and percentages. For non-normally distributed measurements, continuous variables are presented as median values and interquartile ranges (IQR). Non‑parametric tests were used to analyze continuous variables across the three groups, specifically the Kruskal‑Wallis H test. Categorical data were analyzed using the Chi-square test or Fisher’s exact test, as appropriate. The Chi-square test was used to assess the association between categorical variables when the sample sizes were sufficiently large, while Fisher’s exact test was applied when sample sizes were small or expected frequencies were less than 5. A p-value < 0.05 was considered to indicate a statistically significant difference.
Data handling
To ensure the accuracy and completeness of our analysis, we carefully addressed missing data. Missing values for continuous variables were imputed using median values, while missing categorical data were imputed using the mode. This approach helps to reduce bias and maintain the integrity of the dataset.
Correlation analysis
The correlation analysis was conducted separately for the COVID-19 cohort, the MP cohort, and the co-infected cohort. Spearman correlation coefficients were calculated to assess the relationships between key biomarkers and demographic variables within each group. The results were visualized using correlation heatmaps to clearly display the distinct relationships in each cohort.
Random forest model
Python libraries and environment
The random forest model was implemented in the Python 3.7.
NumPy (version 1.21.0) for numerical operations31, Pandas (version 1.3.0) for data manipulation32, Scikit-learn (version 0.24.2) for machine learning algorithms and model evaluation33, Matplotlib (version 3.4.2) for data visualization34, SHAP (version 0.39.0) for Shapley additive explanations analysis35. The analysis was conducted in a local computing environment without using a GPU for analysis.
Dataset and features
The dataset used for the random forest analysis included the following features: Age, Sex, RSV, ADV, PIV, FLUA, PCT, IL-6, WBC, and CRP. The target variable was the patient group classification: COVID-19 positive, MP positive, and co-infected.
Standardization
The dataset was standardized using the “StandardScaler” to ensure uniformity and improve the model’s performance. “StandardScaler” is a preprocessing technique that standardizes features by removing the mean and scaling to unit variance. This process is important because it ensures that each feature contributes equally to the model, preventing any single feature from disproportionately influencing the results due to differing scales.
Data splitting and cross-validation
The dataset was split into training (70%) and testing (30%) sets using the “train_test_split” function to ensure an unbiased evaluation of the model. Additionally, within the training set, five-fold cross-validation was performed to optimize the hyperparameters and prevent overfitting.
Model training and hyperparameter tuning
Initially, “GridSearchCV” was used to determine the optimal hyperparameters for the random forest classifier, including the number of estimators, maximum depth, and minimum samples split. “GridSearchCV” is a method for systematically working through multiple combinations of parameter tunes, cross-validating as it goes to determine which tune gives the best performance. The cross-validation process involved five-fold cross-validation, where the data were divided into five subsets, and the model was trained on four subsets while the remaining subset was used for validation. This process was repeated five times, with each subset used exactly once as the validation set. Based on the results of this five-fold cross-validation, the final model was trained with the parameters: “max_depth = None”, “min_samples_split = 5”, and “n_estimators = 200”, based on performance considerations. The random forest classifier was trained using the “OneVsRestClassifier” strategy to handle the multi-class classification problem. “OneVsRestClassifier” is a strategy that involves training a single classifier per class, with the samples of that class as positive samples and all other samples as negatives, effectively converting a multi-class problem into several binary classification problems.
ROC curve and precision-recall curve analysis
The ROC curve and AUC were computed for each class to evaluate the model’s performance. The ROC curve is a graphical plot that illustrates the diagnostic ability of a binary classifier system as its discrimination threshold is varied. The ROC curve is important for evaluating the trade-offs between sensitivity and specificity in classifiers36. The “roc_curve” and “auc” functions from “sklearn.metrics” were utilized for this purpose. The micro-average ROC curve was also computed to provide an overall performance metric.
To evaluate the performance of the random forest classifier, we used the precision-recall curve, which is particularly informative for imbalanced datasets. An imbalanced dataset is one where the classes are not represented equally, leading to challenges in model training as the classifier may become biased towards the majority class. The precision-recall curve is a plot that shows the trade-off between precision and recall for different threshold settings. The area under the precision-recall curve (“PR AUC”) is a useful metric for assessing model performance in these scenarios37. The dataset was split into training and testing sets with a ratio of 7:3. The random forest classifier was trained using the training set, and its predictions were evaluated using the testing set.
Confusion matrix
A confusion matrix was constructed to evaluate the classification accuracy and visualize the performance of the random forest model. It is a table used to describe the performance of a classification model by displaying the true positives, true negatives, false positives, and false negatives. This matrix provides a comprehensive overview of how well the model distinguishes across three classes.
Random forest feature importance and SHAP analysis
Random forest feature importance scores were computed to understand the contribution of each feature to the classification model. These scores indicate how much each feature contributes to the model’s predictions and were visualized using a bar plot to easily identify the most influential features.
To further explain the feature importance and enhance the transparency of the random forest model, we applied SHAP (Shapley additive explanations) analysis. SHAP values provide a unified measure of feature importance by calculating the contribution of each feature to the prediction. Based on cooperative game theory, SHAP explains individual predictions by computing the contribution of each feature. We used the Python SHAP library to compute SHAP values for each feature and visualize their impact on the model’s predictions. This approach helps in understanding the model’s decision-making process by highlighting how each feature contributes to the prediction of each class.
The study protocol was approved by the Medical Ethics Committee of the Second Hospital of Nanping (Approval No. 202409). All methods were performed in accordance with relevant guidelines and regulations. Due to its retrospective nature, the study was exempted from requiring written informed consent by the Second Hospital of Nanping.
Conclusion
In conclusion, our study demonstrates the potential of computational methods in distinguishing among COVID-19 positive, MP positive, and co-infected groups based on clinical and biomarker data. The significant differences in age, CRP, IL-6, and PCT among these groups highlight their clinical relevance, offering a deeper understanding of the disease mechanisms and providing a foundation for developing targeted interventions. Specifically, elevated CRP and IL-6 levels were strongly associated with COVID-19 severity, while age and PCT levels played crucial roles in distinguishing between MP and co-infected cases. As we continue to confront the challenges posed by infectious diseases, integrating computational approaches in clinical research will be indispensable in advancing patient care and therapeutic development. Integrating SHAP values into our analysis enhanced the interpretability of the random forest model, providing a detailed understanding of feature contributions and interactions. This approach underscores the importance of using advanced machine learning techniques to improve diagnostic precision and patient outcomes in the era of precision medicine.
Data availability
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the global burden of Disease Study 2013. Lancet 385, 117–171. https://doi.org/10.1016/s0140-6736(14)61682-2 (2015).
Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 396, 1204–1222 (2020). https://doi.org/10.1016/s0140-6736(20)30925-9
Li, Z. J. et al. Etiological and epidemiological features of acute respiratory infections in China. Nat. Commun. 12, 5026. https://doi.org/10.1038/s41467-021-25120-6 (2021).
Zhang, S. Y., Shi, J., Zhuo, Y. & Wang, T. Q. Impact of COVID-19 on the distribution of pathogenic bacteria in the lower respiratory tract of the elderly. Immun. Inflamm. Dis. 11, e931. https://doi.org/10.1002/iid3.931 (2023).
Yang, F. F. et al. Global morbidity and mortality of lower respiratory infections: a population -based study. Respir. Med. 205, 107042. https://doi.org/10.1016/j.rmed.2022.107042 (2022). https://doi.org:.
Chow, E. J., Uyeki, T. M. & Chu, H. Y. The effects of the COVID-19 pandemic on community respiratory virus activity. Nat. Rev. Microbiol. 21, 195–210. https://doi.org/10.1038/s41579-022-00807-9 (2023).
Wang, X. et al. Mycoplasma pneumoniae triggers pneumonia epidemic in autumn and winter in Beijing: a multicentre, population-based epidemiological study between 2015 and 2020. Emerg. Microbes Infect. 11, 1508–1517. https://doi.org/10.1080/22221751.2022.2078228 (2022).
Smedemark, S. A. et al. Biomarkers as point-of-care tests to guide prescription of antibiotics in people with acute respiratory infections in primary care. Cochrane Database Syst. Rev. 10, CD010130 (2022). https://doi.org/10.1002/14651858.CD010130.pub3
Li, P., Wang, W., Zhang, X., Pan, J. & Gong, L. Observational retrospective clinical study on clinical features of macrolide-resistant Mycoplasma pneumoniae pneumonia in Chinese pediatric cases. Sci. Rep. 14, 5632. https://doi.org/10.1038/s41598-024-55311-2 (2024).
Del Valle, D. M. et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 26, 1636–1643. https://doi.org/10.1038/s41591-020-1051-9 (2020).
Li, Y., Min, L. & Zhang, X. Usefulness of procalcitonin (PCT), C-reactive protein (CRP), and white blood cell (WBC) levels in the differential diagnosis of acute bacterial, viral, and mycoplasmal respiratory tract infections in children. BMC Pulm Med. 21, 386. https://doi.org/10.1186/s12890-021-01756-4 (2021).
Wynants, L. et al. Prediction models for diagnosis and prognosis of covid-19: systematic review and critical appraisal. BMJ 369, m1328. https://doi.org/10.1136/bmj.m1328 (2020).
Obermeyer, Z. & Emanuel, E. J. Predicting the future—big data, machine learning, and clinical medicine. N. Engl. J. Med. 375, 1216–1219. https://doi.org/10.1056/NEJMp1606181 (2016).
Qiu, J., Wu, Q., Ding, G., Xu, Y. & Feng, S. A survey of machine learning for big data processing. EURASIP J. Adv. Signal Process. 67 (2016). https://doi.org/10.1186/s13634-016-0355-x (2016).
Sarker, I. H. Data science and analytics: An overview from data-driven smart computing, decision-making and applications perspective. SN Comput. Sci. 2, 377. https://doi.org/10.1007/s42979-021-00765-8 (2021).
Breiman, L. Random forests. Mach. Learn. 45, 5–32 https://doi.org/10.1023/A:1010933404324 (2001).
Shamsolmoali, P. et al. Image synthesis with adversarial networks: a comprehensive survey and case studies. Inform. Fusion 72, 126–146. https://doi.org/10.1016/j.inffus.2021.02.014 (2021). https://doi.org
Li, B. R., Zhuo, Y., Jiang, Y. Y. & Zhang, S. Y. Random forest differentiation of Escherichia coli in elderly sepsis using biomarkers and infectious sites. Sci. Rep. 14, 12973. https://doi.org/10.1038/s41598-024-63944-6 (2024).
Itoo, F., Singh, S. & Meenakshi & Comparison and analysis of logistic regression, Naïve Bayes and KNN machine learning algorithms for credit card fraud detection. Int. J. Inform. Technol. 13, 1503–1511 (2021).
Moore, J. B. & June, C. H. Cytokine release syndrome in severe COVID-19. Science 368, 473–474. https://doi.org/10.1126/science.abb8925 (2020).
Chen, G. et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Investig. 130, 2620–2629. https://doi.org/10.1172/JCI137244 (2020).
Li, X. et al. Risk factors for severity and mortality in adult COVID-19 inpatients in Wuhan. J. Allergy Clin. Immunol. 146, 110–118. https://doi.org/10.1016/j.jaci.2020.04.006 (2020).
Zhou, F. et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395, 1054–1062. https://doi.org/10.1016/S0140-6736(20)30566-3 (2020).
Sakagianni, A. et al. Prediction of COVID-19 mortality in the Intensive Care Unit using machine learning. Stud. Health Technol. Inf. 302, 536–540. https://doi.org/10.3233/shti230200 (2023).
Topol, E. J. High-performance medicine: the convergence of human and artificial intelligence. Nat. Med. 25, 44–56. https://doi.org/10.1038/s41591-018-0300-7 (2019).
Beam, A. L. & Kohane, I. S. Big data and machine learning in health care. JAMA 319, 1317–1318. https://doi.org/10.1001/jama.2017.18391 (2018).
Escandón, K. et al. COVID-19 false dichotomies and a comprehensive review of the evidence regarding public health, COVID-19 symptomatology, SARS-CoV-2 transmission, mask wearing, and reinfection. BMC Infect. Dis. 21, 710. https://doi.org/10.1186/s12879-021-06357-4 (2021).
Cao, B. et al. Diagnosis and treatment of community-acquired pneumonia in adults: 2016 clinical practice guidelines by the Chinese Thoracic Society, Chinese Medical Association. Clin. Respir. J. 12, 1320–1360 (2018).
Alemayheu, G. et al. Children hospitalized with community-acquired pneumonia complicated by effusion: a single-centre retrospective cohort study. BMC Pediatr. 23, 181. https://doi.org/10.1186/s12887-023-04004-2 (2023).
Diagnosis and Treatment Protocol for Novel Coronavirus Pneumonia (Trial Version 7). Chin. Med. J. (Engl) 133, 1087–1095. https://doi.org/10.1097/cm9.0000000000000819 (2020).
Harris, C. R. et al. Array programming with NumPy. Nature 585, 357–362. https://doi.org/10.1038/s41586-020-2649-2 (2020).
Reback, J. et al. pandas-dev/pandas: pandas 1.3.0. Zenodo. https://doi.org/10.5281/zenodo.3509134 (2020).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011). https://jmlr.org/papers/volume12/pedregosa11a/pedregosa11a.pdf
Hunter, J. D. Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 9, 90–95. https://doi.org/10.1109/mcse.2007.55 (2007).
Lundberg, S. M. & Lee, S. I. A unified approach to interpreting model predictions. Adv. Neural Inf. Process. Syst. 30, 4765–4774. https://proceedings.neurips.cc/paper/2017/file/8a20a8621978632d76c43dfd28b67767-Paper.pdf (2017).
Fawcett, T. An introduction to ROC analysis. Pattern Recognit. Lett. 27, 861–874. https://doi.org/10.1016/j.patrec.2005.10.010 (2006).
Saito, T. & Rehmsmeier, M. The precision-recall plot is more informative than the ROC plot when evaluating binary classifiers on imbalanced datasets. PLoS One 10, e0118432. https://doi.org/10.1371/journal.pone.0118432 (2015).
Acknowledgements
We would like to thank the staff of the Department of Clinical Laboratory, The Second Hospital of Nanping, for their assistance in data collection and analysis.
Funding
Financial support from Natural Science Foundation Joint Project of Ningde, China, Grant/Award Number: 2022J54.
Author information
Authors and Affiliations
Contributions
X. Z., J. Z. and X-M. D. Conceptualization, Methodology, Formal analysis, Investigation, Data Curation, Writing—Original Draft Preparation, Visualization. Contributed equally to this work. F-M. F. and J-M. W. Conceptualization, Methodology, Software, Validation, Formal analysis, Investigation, Resources, Data Curation, Writing—Review & Editing, Visualization. Contributed equally to this work. Z-Y. Z. and X-Q. Z. Software, Validation, Formal analysis, Data Curation, Writing—Review & Editing, Visualization. Y-X. L. and S-Y. Z. Conceptualization, Resources, Data Curation, Supervision, Project Administration, Funding Acquisition, Writing—Review & Editing, Correspondence.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, X., Zhang, J., Deng, XM. et al. Using random forest and biomarkers for differentiating COVID-19 and Mycoplasma pneumoniae infections. Sci Rep 14, 22673 (2024). https://doi.org/10.1038/s41598-024-74057-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-74057-5
