
Abstract
Diabetic retinopathy (DR) is the most common microvascular complication in diabetic patients, and recent studies have shown that immune regulatory mechanisms are closely associated with retinal damage in DR. Therefore, this study focused on exploring immune cells and immune-related genes (IRGs) in DR and gaining insight into the ceRNA mechanisms by which IRGs regulate DR progression. Four datasets from human DR model retinal tissues were obtained from the Gene Expression Omnibus (GEO) database. R software was first used to identify differentially expressed mRNAs (DE-mRNAs) in the dataset GSE160306-mRNAs, then the distribution of immune cells in the gene matrix was analyzed by xCell and ImmuCellAI, ImmPort and InnateDB database were used to obtain immune-related hub genes (IRHGs) in the DR, and finally the STRING online tool and Cytoscape to construct the immune-related ceRNA network. The datasets GSE102485, GSE160308 and GSE160306-lncRNAs were used to validate the results of the ceRNA network further. The results of immune cell infiltration analysis showed that macrophages are important immune cells in DR; immune-related gene screening showed that FCGR2B is an IRHG in DR, and 2 immune-related ceRNA networks of IRHG were obtained: DDN-AS1/miR-10a-5p/FCGR2B and LINC01515/miR-10a-5p/FCGR2B. Our study suggests that infiltration of immune cells, especially the immune role of macrophages, is an important component of DR progression; the immune-related hub gene FCGR2B and its ceRNA network may be a key regulatory network for DR progression. The discovery of key immune cells, IRHG and ceRNA networks in this study may provide new prospects for early intervention and targeted treatment of DR.
Similar content being viewed by others


Introduction
Diabetic retinopathy (DR) is the most common microvascular complication of diabetes, and the risk of blindness in diabetic patients is 25 times that of non-diabetic patients1,2. Since the development of diabetes mellitus (DM) is progressive. It causes severe visual impairment, resulting in treatment difficulty if DR is not promptly diagnosed and treated3. Therefore, how to diagnose DR at an early stage and control its progression has become the focus of attention4. However, the current diagnosis of DR based on comprehensive ophthalmic examination is not only costly but also not conducive to rapid diagnosis5. Therefore, it is particularly important to find biomarkers that are more specific for DR and can provide a more rapid diagnosis for elucidating the molecular mechanism of DR occurrence and assisting DR treatment.
Competitive endogenous RNA (ceRNA) networks with the components of ncRNA and mRNA are novel mechanisms with a regulatory role at the transcriptional level. Studies have shown that the interaction between ncRNA and mRNA can regulate target gene expression and regulate disease progression in several diseases, including DR6,7,8. And with the maturation of biotechnology, differentially expressed gene identification supported by gene sequencing technology is increasingly used to explore the pathogenesis of diseases, including DR9.
Intensive studies on diabetes in the last decade have revealed that immune inflammation is one of the major causes of type 2 diabetes and is driven by multiple types of immune cells10,11,12,13. For example, the accumulation of macrophages in the adipose tissue of diabetic patients alters the polarization of macrophages, leading to an increase in M1-type pro-inflammatory macrophages14; antigen-specific cytotoxic T lymphocytes are able to destroy pericytes, leading to vascular collapse in diabetic retinopathy15; intravitreal injections of anti-IL-17 A monoclonal antibodies are effective in improving Müller cell dysfunction, vascular leukocyte arrest and vascular leakage in the retina16. All of these indicate that immune cells play an important role in the development and progression of DR. Therefore, from the perspective of the immune system, evaluating the infiltration of immune cells in the DR microenvironment and determining the differences in the composition of infiltrating immune cells and immune-related genes (IRGs) are of great value for elucidating the molecular mechanism.
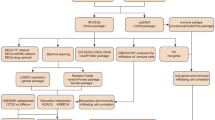
Therefore, in this study, we collected three datasets from the GEO database, which contains mRNA, miRNA, and lncRNA transcript expression matrix data from 55 DR samples and 23 healthy control samples. We aimed to analyse the predominantly infiltrating immune cells in DR by combining gene sequencing data with bioinformatics analysis and to obtain the central immune-related genes and their ceRNA networks with potential regulatory roles in DR. The results will provide new diagnostic markers and potential therapeutic targets for diabetic retinopathy patients. The overall flow chart of this study is shown in Fig. 1.

The overall workflow of this study. The figure illustrates the research flow of this paper. Among them, the blue text box represents the main research steps and results, and the orange font represents the databases and datasets used in the research process. There is no difference between the real and imaginary arrows in the figure. DE-mRNAs differentially expressed mRNAs,GO Gene Ontology, KEGG Kyoto Encyclopedia of Genes and Genomes, TSEGs tissue-specific expressed genes, IRGs immune-Related genes, IRHGs immune-related hub genes.
Materials and methods
Access to ncRNA and mRNA expression profiles of DR patients
From the National Institutes of Health (NIH)-National Center for Biotechnology Information (NCBI)-Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/), we retrieved and downloaded three DR datasets: GSE160306, GSE160308, and GSE102485. The data in datasets GSE160306 and GSE160308 are from the same samples but represent different transcript data, where dataset GSE160306 contains mRNA and lncRNA transcript data, and dataset GSE160308 contains miRNA data from retinal tissue. The datasets (GSE160306 and GSE160308) contained a total of 79 samples, from which we selected retinal tissue samples from 36 DRs and 20 healthy controls for the bioinformatics analyses in this study. Dataset GSE102485 collected neovascular proliferating membranes from 19 patients with type I and type II diabetes and 3 controls for transcriptome analysis.
Therefore, we used dataset GSE160306-mRNA as a test dataset to screen DE-mRNAs in the DR and for subsequent ceRNA network construction; dataset GSE102485, GSE160308, and GSE160306-lncRNA were used as a validation dataset to verify the results of the ceRNA network. The details of these GEO datasets are shown in Table 1. It is worth noting that our approach is reasonable. For example, as a common practice, we17,18,19 and others20 often studied gene expression in pathophysiological conditions.
Immune cell infiltrate analyses
ssGSEA enrichment analysis is a method for assessing immune cell infiltration in samples in a dataset based on the expression levels of immune cell-specific marker genes. ImmuCellAI (ImmuCellAI, http://bioinfo.life.hust.edu.cn/ImmuCellAI) is an online tool that uses ssGSEA enrichment analysis to study immune cell infiltration in different disease expression matrices, including DR21. Upload the gene expression matrix data to ImmuCellAI to obtain the immune cell infiltration matrix. The correlation heatmap shows the correlation of genes in 24 types of immune cell infiltration, and the violin plot shows the difference in immune cell infiltration.
Immune and stromal cells analyses for the datasets
Recently published xCell (xCell, http://xCell.ucsf.edu/) is a new gene signature-based analysis method for inferring the characteristic values of genes in 64 immune and stromal cell species. It has been extensively analyzed in silico and compared to cellular immunophenotypes22. By applying the gene expression matrix data to xCell, the proportion of immune and stromal cell types for each retinal sample gene can be obtained by the GSEA analysis method. The cut-off values for the cell analyses were P < 0.05. Cell types were categorized into lymphoid, myeloid, stromal, stem cells and others. Violin plots are used to visualize the expression of different cell types in the xCell analysis results.
Functional enrichment analyses
The biological functions of identified DE-mRNAs of interest were assessed using the Metascape bioinformatics resources (Metascape, a gene annotation & analysis resource, http://metascape.org, version 3.5)23,24. Briefly, screened DE-mRNAs were imported into Metascape, and GO and KEGG enrichment analyses were then conducted. For GO analyses, enriched biological processes (BPs), molecular functions (MFs), and cellular components (CCs) were assessed, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses. A minimum overlap value of 3, P-value cutoff of 0.05 and minimum enrichment of 1.5 were set as thresholds. Bubble charts are used for the visualisation of the results of the enrichment analysis.
Gene Set Enrichment Analysis (GSEA) is used to assess the distribution trend of genes of a predefined geneset in a table of genes sorted by phenotypic relevance to determine their contribution to the phenotype25. We obtained GSEA software (version 3.0) from the GSEA (http://software.broadinstitute.org/gsea/index.jsp) website, divided the samples into two groups according to disease type, and obtained the data from the Molecular Signatures Database (http://www.gsea-msigdb.org/gsea/downloads.jsp) and downloaded the c2.cp.kegg.v7.4.symbols.gmt subset for assessing related pathways and molecular mechanisms based on gene expression profiles and phenotypic grouping, setting the minimum gene set to 5 and the maximum to 5000, with one thousand resamplings and a P-value < 0.05 was considered statistically significant.
Protein-protein interaction analysis
Protein-protein interaction (PPI) networks were generated from the STRING database (STRING, https://string-db.org/). A score of 0.4 (medium confidence) was set as a threshold. Next, to access hub genes, the visualized regulatory network was constructed using the Cytoscape software (Cytoscape, https://cytoscape.org/)26. CytoHubba was used to identify significant genes in this network as hub genes27. The Maximal Clique Centrality (MCC) algorithm is used to obtain the clustered network and analyze the importance of the network by analyzing the correlation of the degree value of each node with the neighboring nodes, which can then result in the hub gene in the network with the highest value. Based on this algorithm we came to calculate the top 10 hub genes of the PPI network28,29.
Access to immune-related genes
To understand whether immune-related genes (IRGs) are involved in regulation in DR patients, we obtained IRGs from immune-related databases for subsequent studies. The ImmPort database (https://www.immport.org/) and InnateDB database (https://www.innatedb.com) are key databases in immunological studies, covering immune related genes in mammals. The list of immune genes was downloaded from the ImmPort and InnateDB databases separately. The downloaded data were then merged and duplicates removed, resulting in a final list of immune-related genes from the 2 databases. These genes were used for subsequent identification of DR-related immune genes.
Prediction of target miRNAs
The online database-miRcode (http://www.mircode.org/)30 was used to predict the interacting miRNAs with hub genes. Searched for hub genes in miRcode separately and then obtained a list of target miRNAs for each hub gene. The predicted miRNAs were then further validated using the dataset GSE160308 miRNA. According to the screening criteria of adj. P-value < 0.05 and |LogFC|>1, the miRNAs that met the requirements were used as candidate miRNAs.
Construction of ceRNA networks
DIANA-LncBase31 (https://diana.e-ce.uth.gr/lncbasev3, version 3.0) is used to predict lncRNAs that interacted with the selected miRNAs32. The predicted lncRNA was validated in the GSE160306-lncRNA database to obtain target lncRNAs with statistical significance in DR (adj.P-value < 0.05 and |LogFC |>1 as screening criteria). Then the ceRNA network based on the interaction between mRNAs, miRNAs and lncRNAs was constructed using Cytoscape.
Statistical analysis of gene expression matrix
The series matrix files for each of the three datasets were annotated with the official gene symbols and generated read count gene expression matrix files for each dataset. The datasets GSE160306 and GSE160308 were of high-throughput sequencing count data, so the R package DESeq2 (version 1.32.0) was used for the difference analysis of the 2 datasets to obtain the DE-mRNAs, DE-miRNAs, and DE-lncRNAs between the DR group and the control group33. Specifically, we obtained the expression profiling dataset by removing the genes with expression value of 0 greater than 50%, constructing the input matrix using the DESeqDataSetFromMatrix function, further normalising the data using the DESeq function, and finally performing the difference analysis using the results function to obtain the significance of the differences of each gene.
For the dataset GSE102485, the GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r/) platform was used in this study to identify differentially expressed mRNAs. GEO2R is an online application based on the R language34. After user-defined grouping, GEO2R directly reads the raw sequence matrix files and platform annotation files uploaded by the submitters and plots the box plots of the distribution of expression values of the selected samples via ‘boxplot’ and ‘limma’ R language scripts and gene table (including P-value, fold change, gene annotation, etc.). Adj.P-value < 0.05 and |LogFC |>1 were set as thresholds for screening differentially expressed genes.
Results
Identification of DE-RNAs in the human retina
The dataset GSE160306-mRNA was included in our study. As shown in the heatmap (Fig. 2A) and volcano plot (Fig. 2B), a total of 27 DE-mRNAs (12 up-regulated and 15 down-regulated) were identified in the GSE160306-mRNA dataset based on the screening criteria of adj.P-value < 0.05 and |LogFC |>1. The heatmap shows the relative differential expression of the 27 DE-mRNAs between DR retinal tissue and normal retinal tissue (Fig. 2A).

Differential expression of mRNAs and lncRNAs in the human retina dataset GSE160306. (A) Volcano plot of mRNA expression in dataset GSE160306-mRNA. The red triangles represent upregulated genes, the grey triangles represent nonsignificant genes, and the blue triangles represent downregulated genes. (B) Heatmap of DE-mRNAs expression in dataset GSE160306-mRNA. Vertical coordinates represent the 27 DE-mRNAs in the dataset GSE160306-mRNAs with the largest changes in expression. Each square in the graph represents the relative expression after Z-score transformation in the corresponding sample: red squares indicate genes with high expression, and the darker the red, the higher the gene expression level; blue squares indicate genes with low expression, and the darker the blue, the lower the gene expression level. The greater the color difference between the DR group and the control group, the greater the fluctuation in gene expression level. (C) Volcano plot of lncRNA expression in dataset GSE160306-lncRNA. The pink triangles represent upregulated genes, the grey triangles represent nonsignificant genes, and the green triangles represent downregulated genes. (D) Heatmap of DE-lncRNAs expression in dataset GSE160306-lncRNA. Vertical coordinates represent the 10 DE-lncRNAs in the dataset GSE160306-lncRNAs with the largest changes in expression. Pink squares indicate genes with high expression; green squares indicate genes with low expression. The greater the color difference between the DR group and the control group, the greater the fluctuation in gene expression level. DE-mRNAs differentially expressed mRNAs, DE-lncRNAs differentially expressed lncRNAs.
For the dataset GSE160306-lncRNA, we also identified DE-lncRNAs using the screening criteria of “adj.P-value < 0.05 and |LogFC|>1”. The results showed that 10 lncRNAs were differentially expressed in the dataset GSE160306-lncRNA, of which 3 were up-regulated and 7 were down-regulated. (Fig. 2C). The heatmap showed that these 10 DE-lncRNAs were differentially expressed between DR retinal tissues and normal retinal tissues (Fig. 2D).
Macrophages are immunomodulatory cells with major infiltration in DR
Data from dataset GSE160306-mRNA was uploaded to ImmucellAI for analysis of the degree of immune cell infiltration in DR retinal samples. The correlation heatmap of 24 immune cells showed a significant positive correlation between natural killer T (NKT) cells and exhausted T cells (Fig. 3A-B); and a significant negative correlation between NKT cells and mucosal-associated invariant T cells (MAIT) and macrophages (Fig. 3A and C-D). Violin plots of immune cell infiltration results showed significant macrophage infiltration in DR retinal tissues compared with normal retinas, while infiltration of natural killer T cells, exhausted T cells, and MAIT cells were reduced in DR retinal tissues (Fig. 3E).

Evaluation and visualization of immune cell infiltration. (A) Correlation heatmap of 24 types of immune cells. The size of the colored squares represents the strength of the correlation; blue represents a negative correlation, and red represents a positive correlation. The darker the color, the stronger the correlation. (B) Scatter plot of the correlation between natural killer T (NKT) cells and exhausted T cells. The blue bars represent the expression of NKT cells in each sample and the yellow bars represent the expression of exhausted T cells in each sample. (C) Scatter plot of the correlation between NKT cells and MAIT cells. The blue bars represent the expression of NKT cells in each sample and the yellow bars represent the expression of MAIT cells in each sample. (D) Scatter plot of the correlation between NKT cells and macrophages. The blue bars represent the expression of NKT cells in each sample and the yellow bars represent the expression of macrophages in each sample. (E) Violin diagram of the proportion of 24 types of immune cells. The red marks represent the difference in infiltration between the two groups.
Then, to further identify the cell types involved in the DR process, we used xCell to convert gene expression data into cell type enrichment scores. The xCell cell type results for the GSE160306 dataset are shown in Fig. 4A-E. Among the 64 types of immune cells, a total of 41 types of immune cells were present in DR. Compared to the control group, there were 5 types of immune cells with statistically significant enrichment in DR, including memory B-cells, eosinophils, lymphatic endothelial cells, plasmacytoid dendritic cells and M2 macrophages. Notably, we found that macrophages were present in the results of immune infiltration analysis by both ImmucellAI and xCell methods, further illustrating the important role of macrophages in the progression of DR (Fig. 4F).

xCell scores of 64 cell types between DR samples and control samples in the GSE160306 dataset. (A–E) Violin charts of lymphoid cells, myeloid cells, stem cells, stromal cells and other cells, respectively. (F) Venn diagram of immune cell intersection of ImmucellAI and xCell results.
CSMNs class-switched memory B-cells, CLP common lymphoid progenitor, HSCs hematopoietic stem cells, MEP megakaryocyte-erythroid progenitor, Ecs endothelial cells, SMCs smooth muscle cells, MSCs mesenchymal stem cells.
GO and GSEA functional enrichment analysis
The consequences of GO analysis indicated that these DE-mRNAs were associated with diverse biological processes, molecular functions and cellular components. The top 3 biological processes those DE-mRNAs were involved in were: negative regulation of adaptive immune response based on somatic recombination of immune receptors built from immunoglobulin superfamily domains, negative regulation of adaptive immune response, and inflammatory response; the top 3 significant cellular components related to those DE-mRNAs were: extracellular matrix, external encapsulating structure, and collagen-containing extracellular matrix; the top 2 significant molecular functions in which the DE-mRNAs participated were: immune receptor activity, and protein homodimerization activity (Fig. 5A).

GO/KEGG and GSEA enrichment analysis of DE-mRNA in the GSE160306 dataset. (A) Bubble plot of GO enrichment of DE-mRNAs in dataset GSE160306-mRNA. The most significant BP involved in negative regulation of adaptive immune response based on somatic recombination of immune receptors built from immunoglobulin superfamily domains, negative regulation of adaptive immune response, and inflammatory response; CC involved in extracellular matrix, external encapsulating structure, and collagen-containing extracellular matrix; and MF involved in immune receptor activity, and protein homodimerization activity. (B–H) GSEA enrichment analysis of DE-mRNAs from dataset GSE160306-mRNA. DE-mRNAs differentially expressed mRNAs, GO gene ontology, BP biological process, CC cellular components, MF molecular function, GSEA gene set enrichment analysis.
To evaluate the possible signaling pathways and molecular mechanisms in the gene expression matrix of dataset GSE160306, we performed GSEA analysis. The results showed that 7 gene sets were enriched from the c2.kegg subcollection, which are as follows: limonene and pinene degradation, dorso ventral axis formation, glycosphingolipid biosynthesis lacto and neolacto series, other glycan degradation, glycosaminoglycan biosynthesis chondroitin sulfate, ascorbate and aldarate metabolism and long term potentiation (Fig. 5B–H).
PPI network analysis and hub gene sreening
The interaction network between proteins coded by DE-mRNAs, which was comprised of 27 nodes and 6 edges, was constructed by STRING (Fig. 6A). Next, to obtain genes with significant positions in DR, we used the CytoHubba plugin to identify hub genes. Through the MCC algorithm, we identified the top 10 hub genes in the PPI network and visualized them by Cytoscape (Fig. 6B)27. Figure 6C shows the ranking and score of these hub genes under the MCC algorithm, and Table 2 summarises the expression of hub genes in the dataset GSE160306-mRNA.These genes have high values in the PPI network, which means that they may play an essential role in the pathogenesis of DR.

PPI network and hub genes of DE-mRNAs. (A) Interaction network between proteins encoded by DE-mRNA. The network consists of 27 nodes and 6 edges. Each node represents a protein and each edge represents a protein-protein association. (B) CytoHubba identified the top 10 hub gene networks. Red colour represents the gene scoring 2 and green colour represents the gene scoring 1. (C) CytoHubba ranking of top 10 hub genes.
Screening 5 Immune-related hub genes from the Immune Geneset
As the most common autoimmune disease, a better understanding of immune-related mechanisms in DR is an important part of current research. The discovery of differentially expressed immune genes in DR retina may contribute to the specific diagnosis and treatment of DR. Therefore, we collected and organized 5232 IRGs (Supplementary materials-Table S1) from the ImmPort and InnateDB databases, and intersected 10 hub genes of CytoHubba with 5232 IRGs (Fig. 7A). Eventually, we obtained five immune-related hub genes (four up-regulated and one down-regulated): SLPI (adj.P-value = 0.014, LogFC = 2.33) (Fig. 7B), ACAN (adj.P-value < 0.001, LogFC = 1.89) (Fig. 7C), FCGR2B (adj.P-value = 0.008, LogFC = 1.19) (Fig. 7D), C4A (adj.P-value = 0.041, LogFC = 1.08) (Fig. 7E) and CR2 (adj.P-value = 0.018, LogFC = -1.65) (Fig. 7F).

5 immune-related hub genes and their expression in the dataset GSE160306. (A) Violin diagram of immune-related genes and hub genes. (D–F) Violin plots of the expression of the 5 hub genes in dataset GSE160306. Pink represents the DR group and blue represents the control group.
3 Candidate miRNAs were identified
The miRCode online database was used to predict upstream miRNAs for SLPI, ACAN, FCGR2B, C4A and CR2, and we obtained 253 effector miRNAs for five hub genes (Supplementary materials-Table S2). The dataset GSE160308 miRNA contains 39 DR samples from humans and 20 healthy control samples. Then, differential expression analysis was performed on the GSE160308-miRNA dataset using the DESeq2 algorithm, and 17 DE-miRNAs were obtained (based on the screening criteria of adj.P value < 0.05 and |LogFC |>1). The volcano plot shows the miRNA distribution and expression levels in the dataset GSE160308, where red triangles represent up-expressed miRNAs and green triangles represent down-expressed miRNAs (Fig. 8A). The heatmap demonstrates the relative expression of the 17 DE-miRNAs between the DR group and the control group (Fig. 8B). Then, 253 predicted miRNAs were taken to intersect with the 17 DE miRNAs, and the validated candidate miRNAs were obtained: miR-10a-5p (adj.P-value < 0.001, LogFC = 2.43) (Fig. 9A), miR-507 (adj.P-value = 0.045, LogFC=-1.22) (Fig. 9B) and miR-23c (adj.P-value < 0.001, LogFC=-1.75) (Fig. 9C). These three shared miRNAs are considered regulatory miRNAs of hub genes.

Differential expression of miRNAs in dataset GSE160308. (A) Volcano plot of miRNA expression in dataset GSE160308. The red triangles represent upregulated genes, the black triangles represent nonsignificant genes, and the green triangles represent downregulated genes. (B) Heatmap of 17 DE-miRNAs expression in dataset GSE160308. Vertical coordinates represent the 17 DE-miRNAs in the dataset GSE160308 with the largest changes in expression. Red squares indicate genes with high expression; green squares indicate genes with low expression. The greater the color difference between the DR group and the control group, the greater the fluctuation in gene expression level.

Related gene expression in ceRNA networks. (A) Violin plot of miR-10a-5p expression in dataset GSE160308. (B) Violin plot of miR-507 expression in dataset GSE160308. (C) Violin plot of miR-23c expression in dataset GSE160308. (D) Venn diagram of the intersection of LncBaes-lncRNA with the dataset GSE160306-lncRNA. (E) Histogram of DDN-AS1 expression in dataset GSE160306. (F) Histogram of LINC01515 expressed in dataset GSE160306. (G) CeRNA network. The ceRNA network consists of 2 lncRNAs, 3 miRNAs and 5 immune-related hub genes. (H) FCGR2B expression in dataset GSE102485. (I) C4A expression in dataset GSE102485.
Identified 2 regulatory lncRNAs and constructed a ceRNA network
Similar to the prediction of miRNAs, we used the LncBase online database to predict lncRNAs that interacted with 3 candidate miRNAs. From the LncBase database, we predicted a total of 723 lncRNAs (Supplementary materials-Table S3). This implies that these lncRNAs have potential roles in regulating miR-10a-5p, miR-507, and miR-23c. However, it is still unclear whether these lncRNAs are present in the DR and whether these lncRNAs are expressed at statistically different levels in the DR. Therefore, we used the dataset GSE160306-lncRNA to validate the predicted lncRNAs in the DR. Of the 723 predicted lncRNAs, only 2 lncRNAs were found in the 10 DE-lncRNAs of the dataset GSE160306-lncRNAs, namely: DDN-AS1 (adj.P-value = 0.008, LogFC=-1.214) (Fig. 9E) and LINC01515 (adj.P-value = 0.013, LogFC = 1.383) (Fig. 9F). This suggests that DDN-AS1 and LINC01515 may regulate the expression of hub genes by interacting with candidate miRNAs in DR.
To this extent, through bioinformatics analysis, we have explored and identified 2 lncRNAs, 3 miRNAs, and 5 mRNAs. This suggests that both coding and non-coding RNAs may play a regulatory role in the progression of DR. We then used Cytoscape to concatenate these genes into a network and obtained a network of ceRNAs targeting 5 hub genes (Fig. 9G).
Validated and obtained the immune-related hub gene-FCGR2B and its ceRNA network
Then, to further validate the stability of the 5 candidate IRHGs in DR, we selected dataset GSE102485, which also derived from the human retina, to validated it.From Fig. 6-G, we found that SLPI lacked upstream miRNA and lncRNA molecules, while CR2 and ACAN did not have a complete ceRNA regulatory pathway, so we further verified the reliability of C4A and FCGR2B. The results showed that among C4A and FCGR2B, only FCGR2B was validated with adj.P-value < 0.001, LogFC = 1.827 (Fig. 9H-I). To this end, we obtained complete and validated ceRNA networks: the DDN-AS1/miR-10a-5p/FCGR2B and LINC01515/miR-10a-5p/FCGR2B networks. (Validation of SLPI, CR2 and ACAN in dataset GSE102485 is described in the Supplementary materials - Figure S1A-C.)
Discussion
As the most common microvascular complication of diabetes, patients with DR have a 25-fold higher risk of blindness than those without diabetes35. Due to the lack of detailed understanding of DR, patients with DR often lose the best opportunity for treatment, resulting in a poor prognosis. Therefore, the search for key regulatory processes during the progression of DR is of profound importance to improving the prognosis of DR patients. The regulation of gene expression is an important process of biological function. The abnormal expression of genes is often closely associated with a range of pathological conditions, including DR.With the rapid development of science and technology, bioinformatics technology can provide us with transcript information of plasma, atrial fluid, vitreous, and even neonatal retinal tissues, which provides a powerful strategy to study the specific pathogenesis of DR.
In this study, we performed bioinformatics analysis of DR datasets from the GEO database. The results showed that a total of 27 DE-mRNAs were identified in the dataset GSE160306, and GO/KEGG and GSEA enrichment analyses indicated that these DE-mRNAs were associated with cellular defence responses and immune responses. This study then used the ImmuCellAI and xCell platforms to analyse immune cell infiltration in DR retinal tissues, and the results of the 2 platforms showed that macrophages have a significant weight in DR disease. Subsequently, based on the STRING online database, we established a PPI network of DE-mRNAs from which we screened the top 10 hub genes. Whereas the list of immune-related genes (IRGs) (5235 IRGs) downloaded from ImmPort and InnateDB databases revealed that only five of the 10 hub genes, SLPI, ACAN, FCGR2B, C4A and CR2, were associated with immune regulation. This suggests that these 5 immune-related hub genes (IRHGs) may be critical nodes in DR and have the potential to determine the progression of DR. Finally, by predicting the upstream miRNAs and lncRNAs of IRHGs, 2 complete immune-associated ceRNA networks were obtained: DDN-AS1/miR-10a-5p/FCGR2B and LINC01515/miR-10a-5p/FCGR2B, and changes in the expression level of IRHG-FCGR2B were also validated in the dataset GSE102485 was validated. This further suggests that the immune-associated ceRNA network targeting FCGR2B may have a potential regulatory role in DR.
The immune response characterized by activation of classical immune cells is considered one of the most important mechanisms in DR, and currently there is still a lack of research on immune cell infiltration in DR. Therefore, it is important to clarify the types and functions of immune cells involved in DR for the treatment of DR. In this study, we used the ImmuCellAI and xCell platforms to analyse immune cell infiltration in DR and demonstrated that macrophages were present and statistically significant in the results of both analyses. Abu et al. found that the levels of CD163+M2 macrophages were positively correlated with the levels of VEGF in the epiretinal fibrovascular membrane tissue of patients with proliferative diabetic retinopathy (PDR), suggesting that M2 macrophages may be involved in the angiogenesis of PDR36; Van Hove et al. analyzed the transcriptome of single cells in mouse retina and revealed the existence of macrophages in DR37. Thus, our studies and those of the above scholars have demonstrated that macrophages take an important role in the immunomodulatory processes of DR, especially in pro-angiogenesis and fibrosis.
As well as macrophages, other types of immune cells have also been studied in diabetes. For example, a previous study showed a significant increase in vascular permeability of retinal tissue after artificial depletion of NKT cells in DR model mice, suggesting that the degree of NKT cell infiltration was positively correlated with the severity of DR38. MAIT is an important mucosal immune effector cell. Studies have shown that MAIT cells can reduce the positive proportion of diabetes-related autoantibodies39; additional to this, the diabetic state also induces Müller neuroglia cells to secrete vascular endothelial growth factor (VEGF) and TNF-α, which further aggravates the progression of DR40. These demonstrate that immune cells are important regulators of DR disease transformation and progression and cannot be ignored in DR progression.
GO functional enrichment analysis of DE-mRNAs showed that these DE-mRNAs are mainly involved in the adaptive immune, inflammatory response process in DR. Existing studies suggest that an imbalance (mainly a decrease) in adaptive immune Treg cells may be the main cause of bursting inflammation in DR41,42,43; in terms of inflammation, macrophage-restricted protein tyrosine phosphatase 1B (PTP1B) is a key regulator of inflammation in insulin-resistant metabolic syndrome, and dysregulation of PTP1B may underlie retinal microvascular disease44; Chondroitin sulphate (CS) is a naturally occurring sticky polysaccharide of animal cartilage origin.Qi et al. revealed that CS reduced serum levels of inflammatory cytokines (TNF-α, IL-1, IL-6 and MCP-1) and attenuated inflammation in diabetic rats45. ascorbic acid is used as an antioxidant, and it has been found that low levels of ascorbic acid may exacerbate DR lesions by affecting the function of transporter proteins46. The above studies provide support for the results of GSEA enrichment analysis in this paper and suggest us that DE-mRNAs affect DR progression mainly through inflammation-related pathways. Therefore, the study of DR inflammation and immune response is crucial for the treatment of DR.
In this study, we identified the immune-related hub gene FCGR2B in DR. The presence of IRHGs implies that these genes have significant regulatory roles in the immune mechanisms of DR. FCGR2B (Fc gamma receptor Iib, FCGR2B) is an inhibitory receptor gene encoding a protein that is a low-affinity receptor for the Fc region of the immunoglobulin γ complex47. The encoded protein is involved in the phagocytosis of immune complexes and in the regulation of antibody production by B-cells. Previous studies have shown that FCGR2B plays a regulatory role in various inflammatory diseases. For example, the deletion of FCGR2B reduces the T cell tolerance of type II collagen in BQ mice, allowing autoreactive T cells to be activated and develop arthritis48; FCGR2B may also inhibit allergic lung inflammation by attenuating allergen-specific IgE responses49. Unfortunately, the study of FCGR2B in diabetes retinopathy is relatively scarce. Only Wang et al. proposed that the expression of transcriptome FCGR2B significantly increases with the severity of DR, which is related to the inflammation and immune response of DR, and suggested that FCGR2B is a key regulatory factor of DR50. However, the above research still suggests that FCGR2B, as an important inflammatory regulator, may play a critical role in DR, which requires further exploration in the future.
miR-10a-5p also has abundant regulatory functions in the organism. Existing studies have demonstrated its regulatory role in a variety of diseases such as ovarian cancer51, osteoarthritis52, myocardial infarction53, and diabetes. Strycharz showed that miR-10a-5p expression is elevated in female visceral adipose tissue (VAT) and promotes oxidative stress, chronic inflammation and insulin signalling in T2DM54; Karam-Palos et al. found that miR-10a-5p was differentially expressed in tear fluid and blood of T2DM patients with DR, and further regulated the progression of DR by modulating the levels of apoptosis function genes BCL2L2L and TP5355.
DDN-AS1 and LINC01515 are relatively new lncRNAs, and current research on them has focused on cancer. For example, Liu’s study found that DDN-AS1 promotes cancer cell proliferation, migration and invasion through miR-15a/16-TCF3 feedback loop56; LINC01515 not only promotes nasopharyngeal carcinoma progression by acting as a sponge for miR-32557, but also regulates oxidative stress in airway epithelial cells58. Although there is a temporary lack of studies on DDN-AS1 and LINC01515 in DR, previous studies can still show that they have the ability to bind to miRNAs, which also provides indirect evidence for this study.
Conclusions
The results and discussion of this study suggest that immune-related hub genes and signaling pathways, including ceRNA networks, have important roles in the regulation of DR. Ultimately, we proposed new ceRNA networks that may be regulators of DR progression by bioinformatics analysis of gene expression matrices: DDN-AS1/miR-10a-5p/FCGR2B and LINC01515/miR-10a-5p/FCGR2B. However, some scholars have previously explored the variation of transcript levels in diabetic retinopathy. For example, De Silva K et al. explored miRNA biomarkers associated with type 2 diabetes by integrating data from five miRNA sequences59; Becker et al. explored miRNA/mRNA mechanisms in DR by genetic sequencing of retinal tissue from DR patients60. All these studies illustrate that transcriptomic analysis is an important tool to reveal the mechanism of DR progression. The present study on DR mechanism not only explored the regulatory mechanism of DR from the immune perspective by bioinformatics but also constructed a complete ceRNA network at the transcriptional level. The results of this study will further improve the understanding of the mechanism of DR progression and provide potential targets for clinical treatment of DR.
Although this study used the GEO dataset from human retinal tissue to explore immune-related hub genes in DR and identify their ceRNA networks, there are still some limitations to be addressed. First, the sample size used for analytical validation in this study was relatively small. In the future, it is necessary to continue to increase the sample size and conduct prospective cohort studies to further confirm our point of view. Second, the ssGSEA assay was used in this study to calculate the proportion of immune cells in DR tissues; therefore, the use of tissue-based flow cytometry to validate the assay results enhances the confidence of our findings. Finally, due to the lack of data including disease phenotype and disease activity status, the association between immune cells and related genes and disease severity requires further estimation.
Data availability
The datasets generated during and/or analysed during the current study are available in the National Institutes of Health (NIH)-National Center for Biotechnology Information (NCBI)-Gene Expression Omnibus (GEO) databases (https:/www.ncbi.nlm.nih.gov/gds/) with accession numbers GSE160306, GSE160308 and GSE102485, xCell (xCell, http://xCell.ucsf.edu/), ImmuCellAI (ImmuCellAI, http://bioinfo.life.hust.edu.cn/ImmuCellAI), Metascape (Metascape, a gene annotation & analysis resource, http://metascape.org, version 3.5), STRING database (STRING, https://string-db.org/), Cytoscape software (Cytoscape, https://cytoscape.org/), GSEA (http://software.broadinstitute.org/gsea/index.jsp), Molecular Signatures Database (http://www.gsea-msigdb.org/gsea/downloads.jsp), ImmPort database (https://www.immport.org/), InnateDB database (https://www.innatedb.com), miRcode (http://www.mircode.org/) and DIANA-LncBase (https://diana.e-ce.uth.gr/lncbasev3, version 3.0) databases. All data generated and analyzed in this study can be found in these databases.
Abbreviations
- DR:
-
Diabetic retinopathy
- IRGs:
-
Immune-related genes
- GEO:
-
Gene Expression Omnibus
- DE-mRNAs:
-
Differentially expressed mRNAs
- PPI:
-
Protein-protein interaction
- IRGs:
-
Immune-related genes
- IRHGs:
-
Immune-related hub genes
- ceRNA:
-
Competing endogenous RNA
- NKT:
-
Natural killer T
- MAIT:
-
Mucosa-associated invariant T
- MCC:
-
Maximal Clique Centrality
- DM:
-
Diabetes mellitus
- DE-lncRNAs:
-
Differentially expressed lncRNAs
- NIH:
-
National Institutes of Health
- BPs:
-
Biological processes
- MFs:
-
Molecular functions
- CCs:
-
Cellular components
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- MCC:
-
Maximal clique centrality
- VEGF:
-
Vascular endothelial growth factor
- PAGs:
-
Proangiogenic granulocytes
- PDR:
-
Proliferative diabetic retinopathy
- TNF:
-
Tumor necrosis factor
References
Simó-Servat, O., Hernández, C. & Simó, R. Usefulness of the vitreous fluid analysis in the translational research of diabetic retinopathy. Mediat. Inflamm. 2012, 872978. https://doi.org/10.1155/2012/872978 (2012).
Loukovaara, S. et al. Quantitative proteomics analysis of vitreous humor from Diabetic Retinopathy patients. J. Proteome Res. 14, 5131–5143. https://doi.org/10.1021/acs.jproteome.5b00900 (2015).
Yin, L., Zhang, D., Ren, Q., Su, X. & Sun, Z. Prevalence and risk factors of diabetic retinopathy in diabetic patients: a community based cross-sectional study. Medicine. 99, e19236. https://doi.org/10.1097/md.0000000000019236 (2020).
Reichard, P. et al. Intensified conventional insulin treatment retards the microvascular complications of insulin-dependent diabetes mellitus (IDDM): the Stockholm diabetes intervention study (SDIS) after 5 years. J. Intern. Med. 230, 101–108. https://doi.org/10.1111/j.1365-2796.1991.tb00415.x (1991).
Solomon, S. D. et al. Diabetic Retinopathy: A position Statement by the American Diabetes Association. Diabetes care 40, 412–418. https://doi.org/10.2337/dc16-2641 (2017).
Salmena, L., Poliseno, L., Tay, Y., Kats, L. & Pandolfi, P. P. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 146, 353–358. https://doi.org/10.1016/j.cell.2011.07.014 (2011).
Su, K. et al. The role of a ceRNA regulatory network based on lncRNA MALAT1 site in cancer progression. Biomed. Pharmacotherapy = Biomedecine Pharmacotherapie. 137, 111389. https://doi.org/10.1016/j.biopha.2021.111389 (2021).
Chu, Q. et al. Long noncoding RNA SNHG4: a novel target in human diseases. Cancer Cell Int. 21. https://doi.org/10.1186/s12935-021-02292-1 (2021).
Carr, H. L., Turner, J. D., Major, T., Scheel-Toellner, D. & Filer, A. New Developments in Transcriptomic Analysis of Synovial tissue. Front. Med. 7. https://doi.org/10.3389/fmed.2020.00021 (2020).
Cinkajzlová, A., Mráz, M. & Haluzík, M. Adipose tissue immune cells in obesity, type 2 diabetes mellitus and cardiovascular diseases. J. Endocrinol. 252, R1–r22. https://doi.org/10.1530/joe-21-0159 (2021).
Deng, T. et al. Class II major histocompatibility complex plays an essential role in obesity-induced adipose inflammation. Cell Metabol. 17, 411–422. https://doi.org/10.1016/j.cmet.2013.02.009 (2013).
Anderson, E. K., Gutierrez, D. A. & Hasty, A. H. Adipose tissue recruitment of leukocytes. Curr. Opin. Lipidol. 21, 172–177. https://doi.org/10.1097/MOL.0b013e3283393867 (2010).
Hevener, A. L. et al. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J. Clin. Investig. 117, 1658–1669. https://doi.org/10.1172/jci31561 (2007).
Prieur, X. et al. Differential lipid partitioning between adipocytes and tissue macrophages modulates macrophage lipotoxicity and M2/M1 polarization in obese mice. Diabetes 60, 797–809. https://doi.org/10.2337/db10-0705 (2011).
Adams, D. D. Autoimmune destruction of pericytes as the cause of diabetic retinopathy. Clin. Ophthalmol. (Auckland N Z) 2, 295–298. https://doi.org/10.2147/opth.s2629 (2008).
Qiu, A. W., Liu, Q. H., Wang, J. L. & Blocking IL-17A alleviates Diabetic Retinopathy in rodents. Cell. Physiol. Biochemistry: Int. J. Experimental Cell. Physiol. Biochem. Pharmacol. 41, 960–972. https://doi.org/10.1159/000460514 (2017).
Wang, L. et al. A comprehensive data mining study shows that most nuclear receptors act as newly proposed homeostasis-associated molecular pattern receptors. J. Hematol. Oncol. 10. https://doi.org/10.1186/s13045-017-0526-8 (2017).
Wang, L. et al. Novel extracellular and nuclear caspase-1 and inflammasomes propagate inflammation and regulate gene expression: a comprehensive database mining study. J. Hematol. Oncol. 9. https://doi.org/10.1186/s13045-016-0351-5 (2016).
Li, J. et al. Identification and validation of Immune-Related Biomarker Gene and Construction of ceRNA Networks in Septic Cardiomyopathy. Front. Cell. Infect. Microbiol. 12, 912492. https://doi.org/10.3389/fcimb.2022.912492 (2022).
Li, Y. et al. A Functional Genomics Approach to Understand Variation in Cytokine production in humans. Cell 167, 1099–1110e1014. https://doi.org/10.1016/j.cell.2016.10.017 (2016).
Miao, Y. R. et al. ImmuCellAI: a Unique Method for Comprehensive T-Cell subsets abundance prediction and its application in Cancer Immunotherapy. Adv. Sci. (Weinh) 7, 1902880. https://doi.org/10.1002/advs.201902880 (2020).
Aran, D., Hu, Z. & Butte, A. J. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18, 220. https://doi.org/10.1186/s13059-017-1349-1 (2017).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523. https://doi.org/10.1038/s41467-019-09234-6 (2019).
Global National incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the global burden of Disease Study 2017. Lancet (London England) 392, 1789–1858. https://doi.org/10.1016/s0140-6736(18)32279-7 (2018).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A 102, 15545–15550. https://doi.org/10.1073/pnas.0506580102 (2005).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. https://doi.org/10.1101/gr.1239303 (2003).
Chin, C. H. et al. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8(Suppl 4). https://doi.org/10.1186/1752-0509-8-s4-s11 (2014).
Luan, H. et al. Identification of Key Prognostic Biomarker and its correlation with Immune infiltrates in pancreatic ductal adenocarcinoma. Dis. Markers 2020, 8825997. https://doi.org/10.1155/2020/8825997 (2020).
Yang, X. et al. Study on the Multitarget mechanism and key active ingredients of Herba Siegesbeckiae and Volatile Oil against Rheumatoid Arthritis based on Network Pharmacology. Evidence-based Complement. Altern. Medicine: eCAM 2019, 8957245. https://doi.org/10.1155/2019/8957245 (2019).
Jeggari, A., Marks, D. S. & Larsson, E. miRcode: a map of putative microRNA target sites in the long non-coding transcriptome. Bioinf. (Oxford England) 28, 2062–2063. https://doi.org/10.1093/bioinformatics/bts344 (2012).
Karagkouni, D. et al. DIANA-LncBase v3: indexing experimentally supported miRNA targets on non-coding transcripts. Nucleic Acids Res. 48, D101–d110. https://doi.org/10.1093/nar/gkz1036 (2020).
Li, J. H., Liu, S., Zhou, H., Qu, L. H. & Yang, J. H. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 42, D92–97. https://doi.org/10.1093/nar/gkt1248 (2014).
Liu, S. et al. Three Differential expression analysis methods for RNA sequencing: limma, EdgeR, DESeq2. J. Visualized Experiments: JoVE https://doi.org/10.3791/62528 (2021).
Barrett, T. et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 41, D991-995. https://doi.org/10.1093/nar/gks1193 (2013).
Wykoff, C. C. et al. Risk of blindness among patients with diabetes and newly diagnosed Diabetic Retinopathy. Diabetes care 44, 748–756. https://doi.org/10.2337/dc20-0413 (2021).
Abu El-Asrar, A. M. et al. Interleukin-11 overexpression and M2 macrophage density are Associated with angiogenic activity in proliferative Diabetic Retinopathy. Ocul. Immunol. Inflamm. 28, 575–588. https://doi.org/10.1080/09273948.2019.1616772 (2020).
Van Hove, I. et al. Single-cell transcriptome analysis of the Akimba mouse retina reveals cell-type-specific insights into the pathobiology of diabetic retinopathy. Diabetologia 63, 2235–2248. https://doi.org/10.1007/s00125-020-05218-0 (2020).
Suvas, P., Liu, L., Rao, P., Steinle, J. J. & Suvas, S. Systemic alterations in leukocyte subsets and the protective role of NKT cells in the mouse model of diabetic retinopathy. Exp. Eye Res. 200, 108203. https://doi.org/10.1016/j.exer.2020.108203 (2020).
Gazali, A. M. et al. Mucosal-associated invariant T cell alterations during the development of human type 1 diabetes. Diabetologia 63, 2396–2409. https://doi.org/10.1007/s00125-020-05257-7 (2020).
Wang, J., Xu, X., Elliott, M. H., Zhu, M. & Le, Y. Z. Müller cell-derived VEGF is essential for diabetes-induced retinal inflammation and vascular leakage. Diabetes 59, 2297–2305. https://doi.org/10.2337/db09-1420 (2010).
Qiao, Y. C. et al. Changes of Regulatory T Cells and of Proinflammatory and Immunosuppressive cytokines in patients with type 2 diabetes Mellitus: a systematic review and Meta-analysis. J. Diabetes Res. 2016, 3694957. https://doi.org/10.1155/2016/3694957 (2016).
Yang, T. T. et al. Regulatory T cells in the pathogenesis of type 2 diabetes mellitus retinopathy by miR-155. Eur. Rev. Med. Pharmacol. Sci. 19, 2010–2015 (2015).
Yuan, N., Zhang, H. F., Wei, Q., Wang, P. & Guo, W. Y. Expression of CD4 + CD25 + Foxp3 + Regulatory T cells, Interleukin 10 and transforming growth factor β in newly diagnosed type 2 Diabetic patients. Exp. Clin. Endocrinol. Diabetes 126, 96–101. https://doi.org/10.1055/s-0043-113454 (2018).
Forrester, J. V., Kuffova, L. & Delibegovic, M. The role of inflammation in Diabetic Retinopathy. Front. Immunol. 11, 583687. https://doi.org/10.3389/fimmu.2020.583687 (2020).
Qi, S. S. et al. Chondroitin sulfate alleviates Diabetic osteoporosis and repairs bone microstructure via Anti-Oxidation, Anti-inflammation, and regulating bone metabolism. Front. Endocrinol. 12, 759843. https://doi.org/10.3389/fendo.2021.759843 (2021).
Salceda, R. & Contreras-Cubas, C. Ascorbate uptake in normal and diabetic rat retina and retinal pigment epithelium. Comp. Biochem. Physiol. Toxicol. Pharmacology: CBP146, 175–179. https://doi.org/10.1016/j.cbpc.2007.02.015 (2007).
Sun, Z. et al. FCGR2B as a prognostic and immune microenvironmental marker for gliomas based on transcriptomic analysis. Medicine 102, e35084. https://doi.org/10.1097/md.0000000000035084 (2023).
Li, Q. et al. Two major genes associated with autoimmune arthritis, Ncf1 and Fcgr2b, additively protect mice by strengthening T cell tolerance. Cell. Mol. Life Sci. 79, 482. https://doi.org/10.1007/s00018-022-04501-0 (2022).
Dharajiya, N. et al. FcgammaRIIb inhibits allergic lung inflammation in a murine model of allergic asthma. PloS One 5, e9337. https://doi.org/10.1371/journal.pone.0009337 (2010).
Wang, J. H., Wong, R. C. B. & Liu, G. S. Retinal transcriptome and Cellular Landscape in Relation to the progression of Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 63. https://doi.org/10.1167/iovs.63.9.26 (2022).
Gao, F., Wu, Q. & Lu, D. MicroRNA-10a-5p-mediated downregulation of GATA6 inhibits tumor progression in ovarian cancer. Hum. Cell 37, 271–284. https://doi.org/10.1007/s13577-023-00987-3 (2024).
Jiang, H. et al. LncRNA SNHG5 promotes chondrocyte proliferation and inhibits apoptosis in osteoarthritis by regulating miR-10a-5p/H3F3B axis. Connect. Tissue Res. 62, 605–614. https://doi.org/10.1080/03008207.2020.1825701 (2021).
Cao, X. et al. Silencing long non-coding RNA MIAT ameliorates myocardial dysfunction induced by myocardial infarction via MIAT/miR-10a-5p/EGR2 axis. Aging. 13, 11188–11206. https://doi.org/10.18632/aging.202785 (2021).
Strycharz, J. et al. Visceral adipose tissue of Prediabetic and Diabetic females Shares a set of similarly upregulated microRNAs functionally annotated to inflammation, oxidative stress and insulin signaling. Antioxid. (Basel Switzerland). 10 https://doi.org/10.3390/antiox10010101 (2021).
Karam-Palos, S. et al. Oxidative stress mediates epigenetic modifications and the expression of miRNAs and genes related to apoptosis in Diabetic Retinopathy patients. J. Clin. Med. 13. https://doi.org/10.3390/jcm13010074 (2023).
Liu, Z. et al. DDN-AS1-miR-15a/16-TCF3 feedback loop regulates tumor progression in cervical cancer. J. Cell. Biochem. 120, 10228–10238. https://doi.org/10.1002/jcb.28307 (2019).
Liu, D. et al. LINC01515 promotes nasopharyngeal carcinoma progression by serving as a sponge for miR-325 to up-regulate CDCA5. J. Mol. Histol. 52, 577–587. https://doi.org/10.1007/s10735-021-09969-x (2021).
Wang, X. et al. linc01515 regulates PM(2.5)-induced oxidative stress via targeting NRF2 in airway epithelial cells. Environ. Pollution (Barking Essex: 1987) 331, 121798. https://doi.org/10.1016/j.envpol.2023.121798 (2023).
De Silva, K. et al. A data-driven biocomputing pipeline with meta-analysis on high throughput transcriptomics to identify genome-wide miRNA markers associated with type 2 diabetes. Heliyon 8, e08886. https://doi.org/10.1016/j.heliyon.2022.e08886 (2022).
Becker, K. et al. In-depth transcriptomic analysis of human retina reveals molecular mechanisms underlying diabetic retinopathy. Sci. Rep. 11, 10494. https://doi.org/10.1038/s41598-021-88698-3 (2021).
Acknowledgements
A preprint of this study has been published online*. However, all authors of this article promise that this article does not involve multiple submissions, and the submission and publication of this article do not involve any conflict of interest. *Jingru Li, Chaozhong Li, Xinyu Wu et al. Identification of Key ncRNAs and ceRNA Network in DR via Bioinformation Analysis, 22 February 2022, PREPRINT (Version 1) available at Research Square [https://doi.org/10.21203/rs.3.rs-1291708/v1].
Funding
This study was supported by the National Natural Science Foundation of China (No. 82360076; 81860073; 81760074; 82160439; 81860265); Young and middle-aged academic leaders and reserve candidates of Kunming Medical University (J13397031); Yunnan Provincial Department of Science and Technology (No.202001AT070039); Yunnan Health Training Project of High-Level Talents (No.H-2018032); 100 Young and Middle-aged Academic and Technical Backbones of Kunming Medical University (No.60118260106); Young Talents of Yunnan Thousand Talents Plan (No.RLQN20200002).
Author information
Authors and Affiliations
Contributions
All authors made significant contributions to the manuscript and their contributions were recognized by all participating authors. JL and CL wrote the manuscript and drew the figures; XW and SY collected the original data; GS, PD, SL, and LZ collated the original data; PY and YP were responsible for the initial review of the manuscript; JF and LW provided the overall design of the manuscript and the final review of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.

Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, J., Li, C., Wu, X. et al. Bioinformatics analysis of immune infiltration in human diabetic retinopathy and identification of immune-related hub genes and their ceRNA networks. Sci Rep 14, 24003 (2024). https://doi.org/10.1038/s41598-024-75055-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-75055-3

{kind=link}