
Abstract
Triple negative breast cancer (TNBC) is one of the most difficult of all types of breast cancer to treat. TNBC is characterized by the absence of estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2. The development of effective drugs can help to alleviate the suffering of patients. The novel nickel(II)-based coordination polymer (CP), [Ni2(HL)(O)(H2O)3·H2O] (1) (where H4L=[1,1’:2’,1’’-triphenyl]-3,3’’,4’,5’-tetracarboxylic acid), was synthesized via solvothermal reaction in this study. The overall structure of CP1 was fully identified by SXRD, Fourier transform infrared spectroscopy and elemental analysis. Using advanced chemical synthesis, we developed Hyaluronic Acid/Carboxymethyl Chitosan-CP1@Doxorubicin (HA/CMCS-CP1@DOX), a nanocarrier system encapsulating doxorubicin (DOX), which was thoroughly characterized using Scanning Electron Microscopy (SEM), Fourier Transform Infrared Spectroscopy (FTIR), and Thermogravimetric Analysis (TGA). These analyses confirmed the integration of doxorubicin and provided data on the nanocarriers’ stability and structure. In vitro experiments showed that this system significantly downregulated Tissue Inhibitor of Metalloproteinases-1 (TIMP-1) in triple-negative breast cancer cells and inhibited their proliferation. Molecular docking simulations revealed the biological effects of CP1 are derived from its carboxyl groups. Using reinforcement learning, multiple new derivatives were generated from this compound, displaying excellent biological activities. These findings highlight the potential clinical applications and the innovative capacity of this nanocarrier system in drug development.
Similar content being viewed by others

Introduction
Breast cancer (BC) is the cancer with the highest incidence rate in women and poses a serious threat to women’s health. The occurrence of BC is influenced by a variety of factors, including genetic and non-genetic factors1. Currently, the treatment for BC includes surgery, chemotherapy, radiotherapy, targeted therapy and immunotherapy. According to the expression levels of ER, PR, HER2, and Ki67 proteins, infiltrative BCs were categorized as hormone receptor-positive (HR+) (Luminal A, Luminal B), human epidermal growth factor receptor 2-positive (HER2), and Triple negative breast cancer (TNBC). TNBC is a subtype of BC that is highly aggressive, difficult to treat, and has a poor prognosis, and there is an urgent need for effective therapies. Tissue inhibitor of metalloproteinases-1 (TIMP-1) is a member of the TIMP family of proteins. TIMP-1 is overexpressed in a wide range of human cancers. TIMP-1 is considered an important biomarker of poor prognosis in patients with TNBC, and hence TIMP-1 is an attractive therapeutic target for TNBC2.
Now, an attractively research class of functional materials family, namely coordination polymers (CPs), have shined brilliantly on various areas such as chemical sensor, catalyst, optical, gas adsorption and desorption, drug delivery etc.because of their variable microstructures3,4,5,6,7,8,9. CPs are made up of organic ligands (with various functional groups) and metal units (single metal ions or multi-nuclear metal clusters). The structural diversities and numerous functionalities of CPs can be controlled via the adjustment of the coordination fashions and micrios geometric configuration of organic ligands and metal units10,11,12,13,14. Many chemical scientists are trying to explore how to formulate and synthesize CPs with required configurations and target characteristics on demand15,16,17,18. As we all know, during the assembly procedure of the CPs, the coordination configurations of the organic ligands and metal units are very easily affected by the internal and external synthesis conditions like the molar ratio of the organic ligands and metal units, the temperature and pH of the reaction and the solvent systems and so on19,20,21,22. Therefore, the directed approach to the design and synthesis of CPs with attractive characteristics continues to be a great difficulty. However, many experiments have demonstrated that the configuration of organic ligands is decisive for the prediction of the structure and properties of CPs23,24,25,26. Thus, how to choose the ideal ligand has become the top priority for obtaining the functional CPs.
Herein, followed the discussion above, a symmetrical rigid v-shaped carboxylic acid ligand [H4L=[1,1’:2’,1’’-terphenyl]-3,3’’,4’,5’-tetracarboxylic acid], containing various coordination sites, was selected as the organic linkers to construct the CPs with the Ni(II) ions via the solvothermal synthesis methods. Luckily, a 3D dense packing CPs, named [Ni2(HL)(O)(H2O)3·H2O] (1), has been obtained, which has been characterized by the SXRD. Diffraction data analysis indicated that CP1 crystallized in the monoclinic space group P21/c. The topological analysis is discussed in detail27.
Doxorubicin, commonly known as DOX, is an anthracycline antibiotic widely used in the treatment of breast cancer among other malignancies. This drug has a well-established history of clinical use and works by intercalating into DNA and blocking topoisomerase enzymes, thereby inhibiting the replication and transcription of cancer cells, effectively inducing their death. However, the use of doxorubicin is limited by its side effects, particularly the severe local tissue reactions or necrosis that can occur if the drug leaks outside the veins. Therefore, choosing an appropriate drug delivery system to target doxorubicin to the affected cells is a highly challenging task that is crucial for enhancing efficacy and reducing adverse effects.
Based on the research background, this study developed a new drug delivery system incorporating DOX into a nickel (II) coordination polymer (CP1@DOX). CP1 was chosen as the carrier due to its unique advantages of providing controlled release and high drug loading capacity. To overcome its limitations in biocompatibility and stability, hyaluronic acid (HA) and carboxymethyl chitosan (CMCS) hydrogel were used to encapsulate CP1, enhancing the system’s biocompatibility and stability in the body, and ensuring the effectiveness and stability of drug delivery (HA/CMCS-CP1@DOX). Subsequently, a series of biological tests were conducted to validate the safety and efficacy of the prepared material. The results showed that the DOX-loaded nanoparticle system effectively inhibited the proliferation of MDA-468 cells and significantly downregulated the expression of the key TNBC marker gene TIMP-1. Further exploration of the mechanism of action through molecular docking simulations and the use of reinforcement learning technologies to develop new drug molecules demonstrated the system’s potential for precise and efficient treatment. These findings provide a theoretical and experimental basis for constructing a safe and effective new drug delivery system using CP1 and HA/CMCS hydrogel.
Experimental
Chemicals and measurements
Except where indicated differently, the materials employed in these experiments were obtained from commercially available suppliers and were not refined further. IR spectra were documented in the 400 to 4000 cm−1 region using a KBr particle-based Nicolet Impact 410 FTIR spectrometer. Elemental analyses (C, H and N) were carried out using a Perkin Elmer 2400 analyzer.
Preparation and characterization for [Ni2(HL)(O)(H2O)3·H2O] (1)
A solution contain NiCl2·6H2O (0.0118 g, 0.05 mmol) and H4L (0.0406 g, 0.1 mmol) were added in the mixed solution system DMA/H2O (5/5 mL), stirred for 30 min. The mixture was then transferred to a Teflon-lined stainless steel vessel (15 mL); the vessel was sealed and allowed to heat at 120 °C for 48 h. The solution was returned to ambient conditions by filtration to obtain light-green, clear, rod-shaped crystals CP1, which were cleaned by DMA and allowed to dry in air to a 44% yield (on the basis of H4L). Elemental analysis for CP1: Anal. Calcd: C, 43.41, H, 3.15; Found: C, 43.47, H, 3.21.
X-ray measurements were performed with an Oxford Xcalibur E diffractometer. CrysAlisPro was employed to organize the analysis of the appropriate intensity measurements and to transform the data to HKL files. The initial structural models were prepared on the basis of the direct approach creating them with the SHELXS program28 and modified with the shelXL-2014 program with least squares approach. The whole non-hydrogen atoms were blended with anisotropic parameter. All the H-atoms were immobilized on the C-atoms attached to them with assistance of the AFIX commands. The refinement of CP1 and the crystallographic data are given below in Table 1.
Preparation of hydrogels containing ni(II) MOF of DOX
Initially, a doxorubicin (DOX)-loaded polymer was synthesized by immersing a nickel (II) CP1 in a 20 mg/mL solution of DOX for a duration of 24 h. Following this, solutions of HA at a concentration of 1 wt% and CMCS at concentrations of 3, 5, and 7 wt% were prepared. At ambient temperature, the EDC/NHS activation solution was gradually combined with the DOX-loaded polymer solution, and this mixture was then continuously stirred into the HA solution for 30 min to ensure homogeneity. Subsequently, the CMCS solution was blended with the HA solution in a 1:1 volume ratio within a mold to form the hydrogel. After the reaction had fully proceeded, the resultant hydrogels, encapsulating the Ni(II) metal-organic framework (MOF) with DOX, were thoroughly washed with deionized water to remove any unreacted substances. The micromorphology of the hydrogel samples was characterized using Scanning Electron Microscopy (SEM). Prior to SEM analysis, the samples were freeze-dried and coated with a thin layer of gold to enhance the electron conductivity and image clarity.
CCK8 assay
The TNBC cell line MDA-468 was cultured into 96-well cell culture plates and incubated for 24 h. The HA/CMCS-CP1@DOX at concentration of 20 and 50mM were diluted in DMEM (Gibco, USA) and added into MDA-468 cells. Subsequently, 10 µl of CCK-8 solution (Meilunbio, China) was added to cells at 24, 48, 72 and 96 h post treatment. After 2 h incubation, the absorbance at 450 nm was determined by microplate reader (Bio-Rad, USA).
Quantitative real-time PCR
The MDA-468 cells were culture into 24-well cell culture plates and incubated for 24 h. Then, cells were treated with HA/CMCS-CP1@DOX at concentration of 50mM for 48 h. The total RNA in cells was isolated using Trizol reagent (Invitrogen, USA) and cDNA was obtained by HiScript III 1st Strand cDNA Synthesis Kit (Vazyme, China). The quantitative Real-time PCR was performed using ChamQ Universal SYBR qPCR Master Mix (Vazyme, China). The relative mRNA expression of TIMP-1 was normalized to GAPDH.
Molecular docking
Molecular docking simulations were meticulously carried out using AutoDock 4, targeting the GABAA-R receptor, a critical component in our investigation of breast cancer mechanisms29. The crystal structure of the GABAA-R, essential for these simulations, was obtained from the Protein Data Bank, with the PDB ID being 6CDU, the water and small organic molecules were purged and the structure was set rigid. Precise localization within the receptor was defined with the docking pocket centered at coordinates X=-36.172, Y = 45.017, Z = 25.59 Å. The grid settings for the docking area were strategically set to 60 × 60 × 90 in each dimension to ensure comprehensive spatial coverage. Each compound tested in the simulation was permitted up to 20 binding poses, providing a diverse range of interaction profiles. The scoring of these interactions was conducted using the Lamarckian genetic algorithm, a robust method well-suited for complex molecular docking studies. This detailed setup allows for a nuanced exploration of potential drug-receptor interactions, enhancing our understanding of the molecular underpinnings of breast cancer.
Reinforcement learning for generating new molecules
In this investigation, the MolDQN (Molecule Deep Q-Networks) library (the detailed description about the MolDQN can be found in the original study30), integrated with TensorFlow 1.14, was deployed to facilitate a reinforcement learning-based approach, distinctively designed for the optimization of molecular structures. Diverging from traditional machine learning techniques that require extensive pre-labeled datasets, MolDQN leverages direct environmental interactions, employing a sophisticated reward system to forego the necessity of voluminous training data. A decaying ε-greedy strategy was adopted to delicately balance the exploration of novel actions against the exploitation of known advantageous actions, thereby optimizing the learning process. The system’s efficacy in predicting novel molecules was gauged using metrics such as binding affinity, synthetic accessibility (SA), and the quantitative estimate of drug-likeness (QED), which are pivotal for assessing the potential efficacy and manufacturability of the molecules.
Binding affinity assessments were efficiently conducted using QuickVina 231. SA scores were derived from the Gym_molecule library, while QED evaluations were performed using the RDKit library (https://www.rdkit.org/), providing a comprehensive dataset to inform the predictive capability of the model. The experimental framework encompassed 7000 episodes, each permitting up to 15 adjustments to the molecular structure based on the initial template. Besides the total number of episodes and the maximum allowed number of adjustments, all other parameters were used as the same to the original model of MolDQN. The optimized episode will be used as the input for the optimization of next episode. For each optimization process of episode, the weighted summation of QED and SA scores are used as reward for the reinforcement learning for the first 14 adjustments, while for the final adjustment, the binding affinity is used as reward. This iterative process enables the MolDQN system to refine molecular designs systematically, optimizing specified properties and interactions as dictated by the reward configuration. This methodological approach not only ensures an exhaustive exploration and exploitation of the chemical space but also significantly propels the development of pharmacologically viable new drug candidates, illustrating a robust application of machine learning in pharmaceutical research.
Results and discussion
Structural characterization
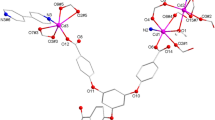
Analysis of SXRD data indicated that CP1 crystallized in the monoclinic space group P21/c, exhibiting a three-dimensional dense packed framework structure. The asymmetric unit of CP1 consists of three Ni(II) ions, one H4L organic ligand, and five coordinated water molecules. As shown in Fig. 1a, where all Ni(II) ions show distorted octahedral geometries: the Ni1 ion is connected by four O atoms from four different carboxylic acid moieties from four separate organic ligands and two O2- ions. The Ni2 ion is ligated by both oxygen atoms from two different carboxylic acid moieties of two distinct organic ligands, two ligand water molecules, and two O2- ions; the Ni3 ion is linked by four oxygen atoms from four distinct carboxylic acid moieties of four separate organic ligands and two ligand water molecules. The selected bond lengths have Ni-O spacings in the range of 2.063(6) to 2.164(7) Å and O-Ni-O angles between 84.3(12)° and 180.0°.
In CP1, the carboxylate moieties of organic ligands took the bidentate bridging and monodentate bridging coordination modes in CP1 (Fig. 1b) to connect the center metal ions. Each organic ligand binds six independent center Ni(II) ions. And the O2− connect two Ni1 ions and one Ni2 ion, which produce the 1D …Ni2…2Ni1… chain (Fig. 1c). The 1D chain were further connected by the organic ligand to produce the dense pacing 3D configuration (Fig. 2a). Topological profiling indicates that the Ni ions (Ni1, Ni2, and Ni3) can be considered as 6-, 4-, and 4-connected nodes, etc.; the O2- ions could be abbreviated to 3-connected nodes; and the organic ligands could be considered as 6-connected nodes. Therefore, the whole structure of 1 can be simplified to a 5-node (3,4,4,6,6-c) topological network with the point symbols {42·63·8}{43}2{44·62}2{44·66·85}2{44·67·84} (Fig. 2b).

Coordination conditions of Ni(II) ions in CP1. The symmetry codes: #1: -1-x, y, z; #2: 1-x, 1-y, 1-z; #3: -1 + x, y, -1 + z; #4: x, 0.5-y, -0.5 + z; #5: -1 + x, y, z (a); The coordination modes of L3− in CP1 (b); The 1D metal chains in CP1 (c).

Three-dimensional dense stacking configuration of CP1 observed along the b-axis (a); topological mesh of CP1 (b).
Micromorphology of hydrogels
After synthesizing HA/CMCS-CP1@DOX, the molecular structure was extensively characterized using FTIR. The FTIR spectrum showed several distinct absorption peaks, where a broad peak near 3500 cmcm−1 typically indicates O-H or N-H bond stretching vibrations, suggesting the presence of moisture or hydrogen bonds. The absorption peak around 1650 cm−1 corresponds to the stretching vibrations of the C = O bond, a characteristic feature of the carboxyl groups in doxorubicin or chitosan. Peaks around 1450 cm−1 and 1000–1100 cm−1 represent C-H bending and C-O stretching vibrations, revealing a complex organic framework (Fig. 3a). The appearance of these characteristic peaks confirmed the successful synthesis of HA/CMCS-CP1@DOX. Further characterization using SEM revealed its porous and irregular structure, with an average pore size of about 200 nm, enhancing the drug loading capacity and release efficiency while facilitating the storage and controlled release of DOX, ensuring effective delivery to targeted areas (Fig. 3b). Additionally, the thermal stability was studied using TGA, which showed that the material began to lose weight rapidly at about 250 °C due to the decomposition of organic components, and after 400 °C, the rate of weight loss plateaued, indicating that most of the organic material had decomposed, leaving potentially more stable inorganic components such as the metal framework (Fig. 3c). These characterization results collectively display the composite structure of the HA/CMCS-CP1@DOX nanocarrier system and its potential for drug release performance, providing crucial information for further application development.

Characterization of HA/CMCS-CP1@DOX nanomaterials: (a) FTIR, (b) SEM, (c) TGA.
To precisely evaluate the biocompatibility and potential cytotoxicity of nanoparticle carriers on human normal liver cells, this study selected two types of culture media, CP1 and HA/CMCS-CP1, and assessed cellular activity and toxicity using the CCK-8 assay (Fig. 4). The results demonstrated that HA/CMCS-CP1 had minimal impact on liver cells, showing excellent biocompatibility, while CP1 exhibited significant cytotoxicity. Specifically, after 12 h of treatment, the cell viability in the CP1 group decreased to 57.17% and showed a clear time-dependent stabilization as the duration increased. These findings highlight the limitations of using CP1 directly as a drug carrier, whereas the HA/CMCS coating significantly enhanced the biocompatibility of the nanoparticles. Additionally, HA/CMCS-CP1 demonstrated the ability to effectively control drug release, reducing non-specific damage to normal liver cells, thereby showcasing its potential and clinical value as a drug delivery system. This study not only deepens our understanding of the biocompatibility of nanoparticle carriers but also provides experimental and theoretical support for future targeted cancer drug delivery strategies.

Cell viability of liver cells treated with CP1 and HA/CMCS-CP1 for 12 h.
HA/CMCS-CP1@DOX inhibited TNBC cell proliferation
In order to investigate whether HA/CMCS-CP1@DOX framework could have an inhibitory effect on TNBC, we selected the TNBC cell line MDA-468 for drug experiments. Treatment of MDA-468 cells with HA/CMCS-CP1@DOX resulted in a significant down-regulation of cell viability of TNBC cells, and the inhibitory effect of 50 mM was stronger than that of 20 mM (Fig. 5A). Further we detected the mRNA level of TIMP-1 by quantitative Real-time PCR after treating MDA-468 cells with 50 mM of HA/CMCS-CP1@DOX for 48 h. As shown in Fig. 5B, HA/CMCS-CP1@DOX was able to significantly down-regulate the expression of TIMP-1, a major marker gene of TNBC. Our results suggest that the novel drug was able to inhibit the proliferation of TNBC cells by down-regulating the level of TIMP-1.

(A) The cell viability of MDA-468 after treatment with HA/CMCS-CP1@DOX was determined by CCK-8 assay. (B) The relative mRNA expression of TIMP-1 was determined by quantitative Real-time PCR. *P < 0.05.
Molecular docking
To elucidate the underlying mechanism of biological activity, we conducted a molecular docking study on the experimentally synthesized CP1 using the AutoDock 4 force field. The GABAA-R is the potential target receptor in the docking simulation for investigating the biological effects. As shown in Fig. 6a, the effective ligand from CP1 has four carboxyl groups, which indicates the potential capability of forming hydrogen bond interactions. And this effective ligand will be used as the input for the following reinforcement learning. In Fig. 6b the binding pose between CP1 and the receptor GABAA-R (6CDU) is presented. Indeed, one can see that a hydrogen bond has been formed between the carboxyl group and the hydroxy group from residue SER-299 (3.3 Å). Such molecular docking result suggests the underlying mechanism of the biological effect of CP1.

(a) The effective ligand on CP1 and (b) the binding pose between CP1 and the receptor 6CDU. The binding affinity of the presented binding pose was − 8.43 kcal/mol.
From the molecular docking simulation and the above experiment, the compound has been demonstrated to have excellent biological effect, the effective ligand of CP1, as shown in Fig. 7, can be used as the template for optimizing and generating new molecules that will have promising biological effect. In the current study, the effective ligand has been used as the input for the reinforcement learning method, and up to 7000 predicted episodes have been generated. In Fig. 7a, the binding affinity of each episode has been shown, one can see that the binding affinity decreases with rise in amount of predicted episodes and convergences around − 10.0 kcal/mol after 4000 episodes. The convergence of the binding affinity indicates the success of the reinforcement learning algorithm, further evidences can be seen in Fig. 7b and c, where the SA and QED scores are shown. Both SA and QED scores increase monotonically with increasing number of predicted episodes, and convergence around 0.65 and 0.60 for SA and QED scores, respectively.

(a) The binding affinity, (b) the SA score, and (c) the QED score of 7000 predicted episodes, the brown line is the average, and the green dashed line indicates the corresponding value of the effective ligand on compound 1.
Only some of the predicted episodes from the reinforcement learning method can be viewed as potential molecules that may have biological effect when binding with the Ni ion, just as CP1. In Fig. 8 the distributions of binding affinity, SA and QED scores are shown. In Fig. 8a, a peak between − 10.0 to -11.0 kcal/mol can be seen, such a peak indicates some of the predicted episode could have low binding affinities. Similarly, in Fig. 8b and c, it can be seen that the peaks of both SA and QED scores are between 0.6 and 0.7, and such findings suggest that some prediction sets may have low binding affinities and high SA and QED scores simultaneously, which is in line with the original intention of the reinforcement learning method. And the integration results of these peaks indicate that about 30% of the prediction sets can fulfill one of the following criteria: i: binding affinity lower than − 10.0 kcal/mol; ii: SA score higher than 0.6; iii: QED score above 0.6.

The distributions of (a): the binding affinity, (b) the SA score, and (c) the QED score of 7000 predicted episodes.
In Fig. 9, three optimized episodes have been chosen which satisfy all three criterions, meaning the chosen episode should have a binding affinity lower than − 10.0 kcal/mol, and the SA and QED scores are higher than 0.6. In Fig. 9a, the episode has a hydroxy group and an imide group, its binding affinity, SA and QED scores are − 10.3 kcal/mol, 0.62 and 0.67. The episode in Fig. 9b has a hydroxy group and a carboxyl group, its binding affinity, SA and QED scores are − 10.0 kcal/mol, 0.66 and 0.63. Lastly, the episode in Fig. 9c has two hydroxy groups, and its binding affinity, SA and QED scores are − 10.0 kcal/mol, 0.65 and 0.64. Moreover, the common feature of them is that all of them have a 7-membered ring structure. Our study focuses on theoretical predictions about the chemical properties and stability of compounds, highlighting the challenges of moving from models to practical synthesis due to potential instabilities in strained rings. The preliminary findings are designed to set the stage for future experimental research. To bridge the gap between theory and practice, we plan to collaborate with experts in synthetic chemistry, ensuring that our predictions are both validated and applicable to real-world scenarios.

Three optimized episodes that have been chosen among 7000 episodes, their SA scores are about 0.62 (a), 0.66 (b) and 0.65 (c), their QED scores are about 0.67, 0.63 and 0.64, and their binding affinities are about − 10.3, -10.0 and − 10.0 kcal/mol, respectively.
To assess the biological effects of the three optimized combinations, we conducted molecular docking simulations using their Ni ion-binding complexes with the receptor 6CDU. Figure 10 shows the binding sites with relatively low binding affinity. It can be seen that all three compounds could form two binding interactions with the receptor. Explicitly, as shown in Fig. 10a, the amide oxygen interacts with the hydroxy group on residue SER-270, the H-bond length is 3.2 Å. The hydroxy group interacts with the C = O group on residue ASP-287, the H-bond length is 2.7 Å. Similar to the first optimized compound, the second compound in Fig. 10b also exhibits two H-bond interactions, the amide oxygen interacts with the hydroxy group on residue SER-270, the H-bond length is 3.3 Å. The hydroxy group interacts with the C = O group on residue ASP-287, the H-bond length is 2.5 Å. However, the carboxyl group is not engaging into any binding interactions. In Fig. 10c, it can be seen that the third compound also has two H-bond interactions, and both of them are formed by the hydroxy groups. The interacting residues are SER-270 (2.9 Å) and LEU-232 (2.5 Å). The above results suggest that all three optimized episodes can be used as the ligand that would bind with the Ni ion and exhibit excellent biological effect. While our molecular docking simulations have demonstrated the formation of hydrogen bonds within the binding pocket—an important indicator of receptor activation or inhibition—the current limitations of our resources and technology have restricted a thorough exploration of the functional impacts of these interactions. Future research will employ advanced computational techniques, such as molecular dynamics simulations, to comprehensively assess how ligand binding affects receptor structure and functionality over time. These in-depth analyses will not only expand our understanding of ligand-receptor interactions but also explore their potential therapeutic implications. We plan to detail these considerations in subsequent studies, thereby laying a solid foundation for future research to build upon.

The binding poses between the optimized episodes when binding with the Ni and the receptor 6CDU. Their binding affinities are − 10.17 (a), -9.00 (b) and − 9.37 (c) kcal/mol, respectively.
Conclusion
In conclusion, a new CPs based on rigid v-shaped O-donor ligand [H4L=[1,1’:2’,1’’-terphenyl]-3,3’’,4’,5’-tetracarboxylic acid] and Ni(II) ions was obtained by solvothermal reaction. The whole structure of CP1 has been examined via XRD, FT-IR and elemental analysis. Structure analysis reveal that CP1 crystallizes in the monoclinic system with P21/c space group, which show a 3D dense packing structure. Topological analysis shows that the whole structure of CP1 can be simplified as 5-nodal (3,4,4,6,6-c) topological net with the point symbol of {42·63·8}{43}2{44·62}2{44·66·85}2{44·67·84}. Through chemical methods, we successfully synthesized HA/CMCS-CP1@DOX, a novel nanocarrier system loaded with DOX. This system was thoroughly characterized using SEM, FTIR, and TGA. In vitro cellular experiments demonstrated that this DOX-loaded nanocarrier system effectively inhibited the proliferation of MDA-468 breast cancer cells, potentially due to its mechanism of modulating the expression of the TIMP-1 gene in breast cancer cells. Although these experiments are currently limited to in vitro conditions, this study provides preliminary experimental support for the application of the DOX-loaded hydrogel and CP-based drug delivery system in clinical cancer treatment, highlighting the need for further development and evaluation of these advanced drug delivery systems for clinical use. We explicitly investigated the biological effect of CP1 by molecular docking simulations, in addition, using the effective ligand from CP1 as the template, more episodes are generated through reinforcement learning method and show excellent potentials in the application of breast cancer treatment, their activities are conferred binding affinity, SA and QED scores. The simulation study also sheds light on the new methodology for drug design.
Data availability
Supporting data derived from the results of this research are obtainable upon contact with the corresponding author.
References
Borri, F. & Granaglia, A. Pathology of triple negative breast cancer. Semin Cancer Biol. 72, 136–145 (2021).
Li, Z., Qiu, Y., Lu, W., Jiang, Y. & Wang, J. Immunotherapeutic interventions of Triple negative breast Cancer. J. Transl Med. 16, 147 (2018).
Wang, B., Cote, A. P., Furukawa, H., O’Keeffe, M. & Yaghi, O. M. Colossal cages in zeolitic imidazolate frameworks as selective carbon dioxide reservoirs. Nature 453, 207–211 (2008).
Wang, X. K. et al. Monometallic Catalytic Models Hosted in Stable Metal-Organic Frameworks for Tunable CO2 Photoreduction. ACS Catal. 9, 1726–1732 (2019).
Yang, Y. et al. Fabricating a super stable luminescent chemosensor with multi-stimuli-response to metal ions and small organic molecules through turn-on and turn-off effects. J. Mater. Chem. C. 5, 4511–4519 (2017).
Usov, P. M. et al. Cooperative electrochemical water oxidation by Zr nodes and Ni–porphyrin linkers of a PCN-224 MOF thin film. J. Mater. Chem. A 4, 16818–16823 (2016).
Myers, A. L. & Prausnitz, J. M. Thermodynamics of mixed-gas adsorption. AIChE J. 11, 121–127 (1965).
Pham, T. et al. Theoretical Investigations of CO2 and CH4 Sorption in an Interpenetrated Diamondoid Metal-Organic Material. Langmuir 30, 6454–6462 (2014).
Ru, J. et al. Enantiopure heterobimetallic single-chain magnets from the chiral RuIII building block. Dalton Trans. 43, 933–936 (2014).
Davis, S. E., Zope, B. N. & Davis, R. J. On the mechanism of selective oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over supported Pt and Au catalysts. Green Chem. 14, 143–147 (2012).
Pukdeejorhor, L. et al. Polymorphism of Mixed Metal Cr/Fe Terephthalate Metal-Organic Frameworks Utilizing a Microwave Synthetic Method. Cryst. Growth Des. 19, 5581–5591 (2019).
Xu, M. M., Chen, Q., Xie, L. H. & Li, J. R. Exchange reactions in metal-organic frameworks: New advances. Coordin. Chem. Rev. 421, 213421 (2020).
Padmanaban, S., Kim, M. & Yoon, S. Acid-mediated surface etching of a nano-sized metal-organic framework for improved reactivity in the fixation of CO2 into polymers. J. Ind. Eng. Chem. 71, 336–344 (2019).
Song, X. et al. Pure-Supramolecular-Linker Approach to Highly Connected Metal-Organic Frameworks for CO2 Capture. J. Am. Chem. Soc. 141, 14539–14543 (2019).
Costes, J. P., Vendier, L. & Wernsdorfer, W. Metalloligands for designing single-molecule and single-chain magnets. Dalton Trans. 39, 4886–4892 (2010).
Zhu, Q. L. & Xu, Q. Metal–organic framework composites. Chem. Soc. Rev. 43, 5468–5512 (2014).
Wei, L. Q. & Ye, B. H. Efficient Conversion of CO2 via Grafting Urea Group into a [Cu2(COO)4]-Based Metal-Organic Framework with Hierarchical Porosity. Inorg. Chem. 58, 4385–4393 (2019).
Li, M., Li, D., O’Keeffe, M. & Yaghi, O. M. Topological Analysis of Metal-Organic Frameworks with Polytopic Linkers and/or Multiple Building Units and the Minimal Transitivity Principle. Chem. Rev. 114, 1343–1370 (2014).
Cheng, M. et al. Direct Syntheses of Nanocages and Frameworks Based on Anderson-Type Polyoxometalates via One-Pot Reactions. Inorg. Chem. 58, 11988–11992 (2019).
Zhang, Z. J., Yao, Z. Z., Xiang, S. C. & Chen, B. L. Perspective of microporous metal–organic frameworks for CO2 capture and separation. Energy Environ. Sci. 7, 2868–2899 (2014).
Zhou, H. C., Long, J. R. & Yaghi, O. M. Introduction to Metal-Organic Frameworks. Chem. Rev. 112, 673–674 (2012).
Tang, K. et al. High CO2/N2 Selectivity and H2 Adsorption of a Novel Porous Yttrium Metal-Organic Framework Based on N, N′, N″-Tris(isophthalyl)-1,3,5-benzenetricarboxamide. Cryst. Growth Des. 13, 1382–1385 (2013).
Wei, Y. P., Liu, Y., Guo, F., Dao, X. Y. & Sun, W. Y. Different functional group modified zirconium frameworks for the photocatalytic reduction of carbon dioxide. Dalton Trans. 48, 8221–8226 (2019).
Zhu, J. et al. A New Class of Metal-Cyclam-Based Zirconium Metal-Organic Frameworks for CO2 Adsorption and Chemical Fixation. J. Am. Chem. Soc. 140, 993–1003 (2018).
Wang, B. et al. Tuning CO2 Selective Adsorption over N2 and CH4 in UiO-67 Analogues through Ligand Functionalization. Inorg. Chem. 53, 9254–9259 (2014).
Wu, Y. L. et al. A new porous Co(ii)-metal–organic framework for high sorption selectivity and affinity to CO2 and efficient catalytic oxidation of benzyl alcohols to benzaldehydes. CrystEngComm 23, 3717–3723 (2021).
Blatov, V. A., Shevchenko, A. P. & Proserpio, D. M. Applied Topological Analysis of Crystal Structures with the Program Package ToposPro. Cryst. Growth Des. 14, 3576 (2014).
Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. (eds) (a) OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 42, 339–341 ; (b) Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C: Struct. Chem. 71, 3–8 (2015). (2009).
Chen, Q. et al. Structural basis of neurosteroid anesthetic action on GABAA receptors. Nat. Commun. 9, 3972 (2018).
Zhou, Z., Kearnes, S., Li, L., Zare, R. N. & Riley, P. Optim. Molecules via Deep Reinforcement Learn. Sci. Rep. 9, 10752 (2019).
Alhossary, A., Handoko, S. D., Mu, Y. & Kwoh, C. Fast, accurate, and reliable molecular docking with QuickVina 2. Bioinformatics. 31, 2214–2216 (2015).
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Xiaojun Bai and Yanjun Xu synthesized and characterized the compounds; Yilun Liu performed other experiments.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bai, X., Xu, Y. & Liu, Y. Therapeutic effects of DOX-loaded hydrogel MOF nanocarriers on triple negative breast cancer and derivative design via reinforcement learning. Sci Rep 14, 23946 (2024). https://doi.org/10.1038/s41598-024-75466-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-75466-2
